

Summary
Behçet’s syndrome (BS) is a complex disease with different organ involvement. The vascular one is the most intriguing, considering the existence of a specific group of patients suffering from recurrent vascular events involving the venous and, more rarely, the arterial vessels. Several clinical clues suggest the inflammatory nature of thrombosis in BS, especially of the venous involvement, thus BS is considered a model of inflammation‐induced thrombosis. Unique among other inflammatory conditions, venous involvement (together with the arterial one) is currently treated with immunosuppressants, rather than with anti‐coagulants. Although many in‐vitro studies have suggested the different roles of the multiple players involved in clot formation, in‐vivo models are crucial to study this process in a physiological context. At present, no clear mechanisms describing the pathophysiology of thrombo‐inflammation in BS exist. Recently, we focused our attention on BS patients as a human in‐vivo model of inflammation‐induced thrombosis to investigate a new mechanism of clot formation. Indeed, fibrinogen displays a critical role not only in inflammatory processes, but also in clot formation, both in the fibrin network and in platelet aggregation. Reactive oxygen species (ROS)‐derived modifications represent the main post‐translational fibrinogen alterations responsible for structural and functional changes. Recent data have revealed that neutrophils (pivotal in the pathogenetic mechanisms leading to BS damage) promote fibrinogen oxidation and thrombus formation in BS. Altogether, these new findings may help understand the pathogenetic bases of inflammation‐induced thrombosis and, more importantly, may suggest potential targets for innovative therapeutic approaches.
Keywords: Behçet’s syndrome, fibrinogen, neutrophils, oxidative stress, thrombosis
Introduction
Behçet’s syndrome (BS) is a complex disease, accounting for several different organ involvements 1. The vascular one is perhaps the most intriguing, as BS is considered a model of inflammation‐induced thrombosis 2. BS is not a unique disease, as many different clinical phenotypes have been described 3. Among others, the ‘vascular cluster’ identifies a specific group of patients suffering from recurrent thrombotic events involving the venous and, more rarely, the arterial vessels 4.
Several pathophysiological mechanisms suggest the inflammatory nature of vascular manifestations in BS. Indeed, BS is a ‘neutrophilic vasculitis/perivasculitis’. Generally, vascular manifestations occur associated with signs of inflammatory activation (i.e. fever and constitutional symptoms). More importantly, both venous and arterial involvements are currently treated with immunosuppressants (both conventional and biotechnological), rather than with anti‐platelet or anti‐coagulant drugs 5.
Interestingly, only few specific traditional thrombophilic factors have been described in BS patients so far, while some immune‐mediated pathogenetic mechanisms have been suggested 2. Indeed, procoagulant mechanisms may, at least in part, link inflammation and thrombus formation in BS. In particular, specific components of the coagulation cascade (i.e. tissue factor, fibrinogen, thrombin and protein C) are able to hyperactivate the immune system in BS 6. Moreover, Factor V Leiden mutation is reported to be more prevalent in some BS populations, as well as the prothrombin gene mutation 7. However, clear and definite data on the contribution of such factors and mutations in thrombo‐inflammation in BS are still not available to date.
In this context the results of our recent investigation, performed on a large cohort of BS patients, highlighted a new mechanism in thrombus formation in this condition 8.
In this review, the main clues (both clinic and pathogenetic) dealing with the inflammatory nature of thrombo‐inflammation in BS will be briefly described. The main general mechanisms linking inflammation and thrombosis will be also outlined and, in particular, we will focus our attention particularly on fibrinogen modifications and consequent altered clot formation secondary to increased blood oxidative stress in BS.
Histological, clinical and therapeutic clues suggesting thrombo‐inflammation in Behçet’s syndrome
BS is classified among the vasculitides of variable vessel size 9. However, in contrast to other vasculitides, BS is usually characterized by the absence of granulomatous inflammatory lesions in the vessel wall. Despite the evidence of vasculitic lesions involving smaller arteries, arterioles and venules, neutrophils and lymphocytes can also have a perivascular localization in large vessel involvement in BS 7. Regarding other typical involvements 10, 11, 12, this histological feature suggests that BS is also a perivasculitis, rather than only a vasculitic process.
Deep vein thrombosis (DVT) and superficial venous thrombosis (SVT) are the most typical vascular involvements, affecting up to 40% of patients with BS, sometimes simultaneously 1, 2. Although more rare, the occurrence of venous thrombosis in atypical sites, the presence of arterial involvement (mainly aneurysms or pseudo‐aneurysms) and the co‐existence of venous and arterial involvement are more specific vascular manifestations in BS 2.
Several clinical features indirectly suggest the inflammatory nature of vascular involvement in BS. First, the vascular manifestations (both venous and arterial) are associated with signs of inflammatory activity (i.e. fever, constitutional symptoms and an increased acute phase response) 13, despite usually without the occurrence of other typical disease manifestations (e.g. ocular, neurological, etc.). Secondly, 18F‐fluorodeoxyglucose positron emission tomography (FDG‐PET) reveals a significant uptake in the arterial vessel wall, mainly of aneurysmatic and pseudoaneurysmatic lesions 14. Arterial involvement is less frequent compared with venous involvement in BS, but the formation of aneurysms and pseudoaneuryms, especially of the pulmonary arteries, is a quite specific feature of the disease 2. According to the more recent European League Against Rheumatism (EULAR) recommendations, this kind of vascular involvement should always be treated with immunosuppressants, in particular with corticosteroids and cyclophosphamide as first‐line treatment, or with anti‐tumor necrosis factor (TNF)‐α in refractory cases 15.
However, these findings can also be found in other systemic vasculitides, and are not sufficient to claim BS as the model of thrombo‐inflammation. Indeed, in ANCA‐associated vasculitis (AAV) and in large‐vessel vasculitis (LVV), the atherothrombotic events occur especially during disease activity 2. Moreover, FDG‐PET is an important diagnostic tool in LVV, due to the inflammatory process affecting the aorta and the consequent uptake of the arterial wall 16. Finally, the vasculitic process of the aorta and its branches in LVV (either giant cell arteritis and Takayasu arteritis) is currently treated with high‐dose glucocorticoids and/or biologicals [anti‐interleukin (IL)‐6Ra or anti‐TNF‐α, respectively] 17.
Immunosuppressive treatment for venous thrombotic events in Behçet’s syndrome: a clinical proof‐of‐concept of thrombo‐inflammation
A unique clinical feature suggesting BS as the best model of thrombo‐inflammation is certainly the treatment of the venous thrombotic events. Indeed, according to the pathogenetic concept that thrombotic events in BS are sustained by an inflammatory process rather than a thrombophilic state, resolution of venous thrombosis is mainly achieved with immunosuppressants, rather than with anti‐coagulants. Except for the treatment of cerebral vein thrombosis 18, the use of anti‐coagulants is generally not considered effective in BS for preventing recurrent venous thrombotic events 15.
There are three main retrospective studies suggesting that in DVT the use of immunosuppressants is able to significantly reduce thrombotic recurrences, whereas anti‐coagulants do not reduce the risk of DVT relapse 19, 20, 21. In particular, for acute DVT, azathioprine, cyclophosphamide or cyclosporin, together with corticosteroids, are strongly recommended 15.
More recently, anti‐TNF‐α antibodies have been emerging as valuable treatment for different organ involvements in BS patients 22, 23, 24, 25, 26, 27, 28, 29. Of note, we found in a recent retrospective study that the anti‐TNF‐α adalimumab (ADA), alone or associated with other traditional immunosuppressive treatments, was significantly more effective than disease‐modifying anti‐rheumatic drugs (DMARDs) alone in resolving venous thrombosis, either DVT or SVT 28. Notably, no additional benefits from anti‐coagulation therapy were shown in our study in patients treated with the ADA‐based regimen or with DMARDs alone, again suggesting the inflammatory nature of venous thrombosis in BS 28.
Inflammation and thrombosis: an emerging relationship
Inflammation and coagulation are two tightly linked interdependent processes, and each one is able to activate and propagate the other 30, 31. However, the mechanisms underlying this phenomenon are still to be elucidated.
Inflammation imbalances pro‐ and anti‐coagulant equilibrium promoting coagulation through several processes. First, inflammation involves the activation of several cell types (including platelets, leukocytes and endothelial cells) and the production of inflammatory molecules as cytokines, chemokines, adhesion molecules, tissue factor expression and microparticles. Proteases derived from activated leukocytes inhibit anti‐thrombin and thrombomodulin promoting a procoagulant state in the endothelium. Secondly, inflammation increases procoagulant factors and inhibits anti‐coagulant pathways and fibrinolytic activity causing a thrombotic state. Finally, it is also responsible for endothelial damage by an increased expression of tissue factor, superoxide‐dependent nitric oxide inactivation and inhibition of the protein C pathway, resulting in the loss of physiological anti‐coagulant/anti‐aggregant function of endothelium. Interestingly, it has been shown that inflammation might directly promote clot formation, even in the absence of endothelial damage 32, 33. In this context, inflammation‐dependent platelet activation plays a fundamental role by enhancing tissue factor expression, thrombin production and activation of coagulation factors leading to a hypercoagulable state 34. This concept is supported by accumulating evidence highlighting the role of inflammation in venous thromboembolism (VTE), a condition where endothelial damage is not mandatory and can occur without the traditional risk factors for atherothrombosis 32, 33, 35.
The existence of an increased thrombotic risk in patients with systemic inflammatory diseases (BS, systemic lupus erythematosus, AAV, Takayasu arteritis, inflammatory bowel diseases, rheumatoid arthritis, Sjögren’s syndrome and systemic sclerosis) underlines the link between inflammation and thrombosis 2, 36, 37, 38, 39. Collectively, the results of basic science and clinical epidemiological studies confirm the strict relation between inflammation and thrombosis 40, 41, 42.
The results of clinical trials with anti‐inflammatory agents reinforce this concept, demonstrating the anti‐thrombotic effect of anti‐inflammatory treatment during the active phases of several types of diseases. It is now accepted that the anti‐thrombotic properties of statins, beyond their effect on lipids, are likely to be linked to their anti‐inflammatory properties that are independent of changes in cholesterol profile 43. Indeed, patients who achieve lower C‐reactive protein (CRP) levels on statin therapy have better clinical outcomes regardless of low‐density lipoprotein (LDL) cholesterol levels 44.
To address the hypothesis that lowering inflammation will lower vascular event rates, the CANTOS (Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study) trial was performed. CANTOS was a large‐scale placebo‐controlled trial which used canakinumab as targeted anti‐inflammatory agent for the secondary prevention of myocardial infarction. This trial, which enrolled more 10 000 thousand patients with a previous history of myocardial infarction, demonstrated that canakinumab, a human monoclonal antibody that selectively neutralizes IL‐1β, significantly reduces the rates of recurrent myocardial infarction, stroke and cardiovascular death in the absence of lipid lowering 45. In particular, in CANTOS, canakinumab treatment significantly decreased high‐sensitivity C‐reactive protein (hsCRP) levels and major adverse cardiovascular events in comparison with placebo, despite having no effect on LDL cholesterol 46. Clinical benefits were smaller among individuals who achieved less robust hsCRP reductions, suggesting a key role for inflammation in thrombotic process 46.
This, together with other ongoing trials 47 could provide fundamental clinical confirmation of the direct role played by inflammation in the pathogenesis of vascular events.
Fibrinogen oxidation as a new link between venous and arterial thrombosis
Although inflammation‐induced arterial thrombosis has been known for many years, the relationship between inflammation and venous thrombosis has only recently been clarified 32, 33. Venous and arterial thrombi have been historically considered as being very different in terms of composition and structure. While fibrin and erythrocytes are the major components of the ‘red clot’ in venous thrombosis, ‘white clot’ arterial thrombus has been traditionally proposed to be composed mainly of fibrin and platelets. In recent years, much evidence suggests that arterial and venous thrombi have similar composition, with a complex fibrin network with entrapped erythrocytes, platelets and leukocytes. Only the relative content of these elements seems to represent a distinguishing feature between venous and arterial clots 48, 49, 50, 51. Several studies have demonstrated high erythrocyte content in post‐myocardial infarction intracoronary thrombi 48, 49, 52 and that erythrocyte content may influence thrombus stability 53, 54. At the same time, platelets, traditionally considered important components in arterial clot, are now also accepted as a main component in venous thrombi 55. These data are supported by evidence that treatments usually associated with the prevention of arterial thrombosis may also have a role in venous thrombosis 56, 57, 58, 59. As a consequence, the classical view of separate mechanisms for arterial and venous thrombosis has recently been deeply challenged.
Patients with arterial thrombosis have been shown to also be at increased risk for venous thrombosis, and overlapping risk factors (age, obesity, hypertension, diabetes, metabolic syndrome, hypertriglyceridemia) have been found to be associated with both arterial and venous thrombotic events 32, 33. Furthermore, it has been shown that inflammation and platelet activation are also involved in the pathogenesis of venous thrombosis 33, 60. In line with these observations, many diseases are characterized by both venous and arterial thrombosis, such as cancer 61, 62, 63, 64 and infections 65, as well as anti‐phospholipid antibody syndrome 66, 67, AAV 68, 69, 70, LVV 71, 72, 73 and BS 74, 75.
Therefore, it can be speculated that the two vascular complications are simultaneously triggered by common biological stimuli responsible for activating coagulation and inflammatory pathways in both the arterial and venous systems.
Fibrinogen and clot formation
Clot formation involves thrombin‐mediated cleavage of soluble fibrinogen to insoluble polymerized fibrin (Fig. 1a). Fibrinogen displays a critical role in the formation of the clot, both in the fibrin network and in platelet aggregation. Clot biochemical features, including its rate of formation, structure and mechanical fibrinolytic stability, are strictly dependent on fibrinogen.
Figure 1.
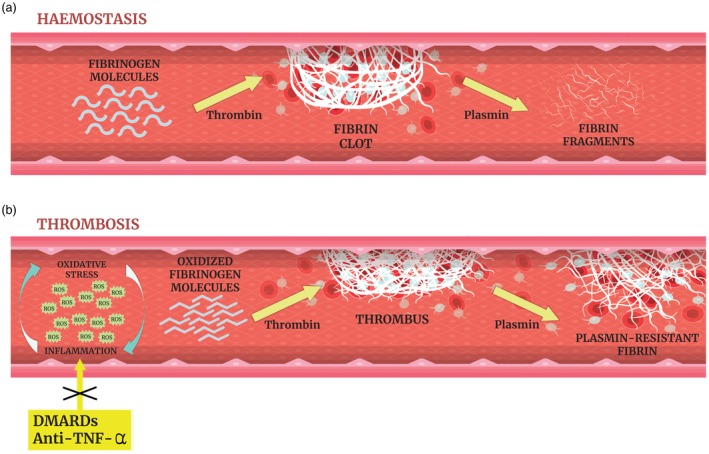
(a) Maintenance of blood fluidity is essential to preserve physiological function of tissues supplying the body with oxygen and other nutrients and removing waste products. Hemostasis consists of a series of enzymatic steps activated in response of vessel injury by forming a fibrin plug that serves to limit bleeding/hemorrhage. It is affected by many factors, including cellular and plasma components. It starts with platelets adhesion to damaged endothelium, and concludes with clot retraction and finally fibrinolysis. Numerous circulating proteins constitutively survey the vasculature to prevent unnecessary clot formation or its premature degradation. Under normal physiological conditions a delicate equilibrium is maintained between the pathological states of hypercoagulability and hypocoagulability in the circulating blood. (b) Oxidative stress and inflammation as interconnected processes that co‐exist in the inflamed milieu. Reactive oxygen species (ROS) are released by vascular and inflammatory cells at the site of inflammation leading to oxidative damage; conversely, ROS production enhances proinflammatory responses. Our experimental data indicate that ROS promote fibrinogen oxidation (carbonylation) leading to fibrinogen secondary structure modifications which affect its biological activity. Fibrinogen oxidation induces the build‐up of an altered thrombogenic clot mainly characterized by a tight fibrin network composed of filaments with slightly decreased fiber size that are resistant to plasmin‐induced lysis. This oxidized fibrin network persists in the vascular bed and contributes to vascular occlusion and thrombus development. In BS, the use of traditional disease‐modifying anti‐rheumatic drugs (DMARDs) (mainly azathioprine and cyclophosphamide) and/or anti‐tumor necrosis factor (TNF)‐α (namely infliximab and adalimumab) is effective for the treatment of both venous and arterial manifestations. The efficacy of immunosuppressants might be due partly to their ability to interfere with the mechanisms described above.
Fibrinogen is a trimeric340‐kDa glycoprotein, primarily synthesized in hepatocytes. Upon thrombin‐mediated cleavage of short N‐terminal peptides from the Aα (FpA) and Bβ (FpB) chains of fibrinogen, fibrin polymerization induces double‐stranded protofibril formation followed by thickening of protofibril chains and, finally, the formation of a fibrin clot 76 which can be monitored spectrophotometrically easily and reliably 77. The initial formation of protofibrils – monitored as a ‘lag’ phase – is characterized by no increase in turbidity. Subsequent lateral aggregation of fibrin protofibrils induces a turbidity increase whose magnitude is related to the structure of the formed clot. Formation of thicker fibers corresponds to a greater increase in final turbidity 77.
Several variables such as ionic strength, pH, calcium, fibrinogen and thrombin concentrations can deeply influence clot formation, structure and stability 78, 79. Different clot structures are generated in the presence of different thrombin concentrations. Highly permeable fibrin clots with thick, loosely woven fibrin strands or less permeable clots composed of dense networks of relatively thin fibrin strands have been reported, depending on thrombin concentrations 80. However, the absence of a general consensus on this issue can, at least in part, be ascribed to the different experimental conditions used in the experiments.
Several studies have shown that clot structure also strongly influences the viscoelastic clot properties. In particular a complex relationship, depending on fiber properties (such as thickness, length, density, degree of branching and extent of cross‐linking) between fibrin structure and clot stiffness, has been shown 81. However, discrepancies in the results of the various studies exist. Fibrin structure also influences clot susceptibility to fibrinolysis. It has been reported that thin fibrin fibers show a slower rate of tissue‐type plasminogen activator (tPA)‐mediated plasmin generation, reducing the rate of fibrinolysis 82. Moreover, while thin fibers are lysed more quickly than thick fibers, clots composed of thin fibers are more resistant to fibrinolysis than clots composed of thick fibrin fibers 83. Interestingly, clot stiffness results increased both in coronary artery disease patients and in subjects presenting with a high risk of thrombotic events (smokers and diabetic patients). Importantly, stiffer clots, characterized by increased fiber density and lower fibrin diameter, have been shown to exhibit delayed lysis 84, 85. As a whole, these findings indicate the complex achievement of reliable experimental conditions for clot structural and functional studies.
Among hemostatic proteins, fibrinogen is the main target of different types of post‐translational modifications such as phosphorylation, glycosylation and nitration. In addition, reactive oxygen species (ROS)‐derived modifications represent the main post‐translational fibrinogen alterations responsible for structural and functional changes 86.
It is known that biological systems are continuously exposed to endogenous and/or exogenous ROS, which at low doses display crucial roles in cell signalling processes, while at high doses induce oxidative stress and consequently serious metabolic dysfunctions and damage to biological macromolecules 87.
In‐vitro evidence has suggested that blood coagulation is activated by oxidative stress, although the mechanisms that link these events have not been clarified in humans 88. A recent study performed on a group of young healthy volunteers exposed to acute hypoxia, maximal physical exercise (in condition of ‘oxidative stress’), with or without anti‐oxidant supplementation, found that anti‐oxidant prophylaxis increased thrombin generation which was normalized only in the presence of oxidizing conditions.
It is well known that ROS can damage proteins through carbonyl group formation, hydrogen ion abstraction, protein–protein cross‐linkages formation and protein fragmentation 89 causing marked alterations in their structure and function. Indeed, oxidatively damaged proteins accumulate during ageing and as result of a variety of diseases 90, 91.
Fibrinogen is a probable target for oxidants relative to other plasma proteins 92, and specific sites in each of its chains are subjected to oxidative modifications. Accordingly, some authors observed the formation of thin‐fibered fibrin clots and the inhibition of fibrin protofibrils lateral aggregation 93 upon treatment with hypochlorus acid (a molecule predominantly generated in the plasma by neutrophil lysosomal myeloperoxidase) 94. The formed altered clots appear mechanically weak and are paradoxically less susceptible to fibrinolysis in vitro due to decreased clot porosity 95.
Among the oxidative post‐translational modifications of fibrinogen, dityrosine cross‐links formation and marked protein carbonyl content have been also detected. These alterations exert a deep impact on the kinetics of fibrin formation as well as on the structure and biomechanical properties of fibrin, ultimately producing dysfunctional hemostatic clots.
ROS‐induced fibrinogen modification in Behçet’s syndrome: an in‐vivo proof‐of‐concept of inflammatory thrombus
A recent study, aimed at elucidating the mechanisms of inflammation‐induced thrombosis, investigated fibrinogen oxidative‐derived structural and functional modifications in a large population of BS patients which represented an excellent model of inflammation‐induced thrombosis (Fig. 1b). Systemic oxidative stress (lipid peroxidation markers and protein carbonyls) has been suggested as a prognostic factor in vasculitis, particularly in BS 96. Considering the existing relationship among oxidative stress, inflammation and endothelial dysfunction 97, fibrinogen structure and its possible relationship with neutrophil‐dependent ROS production was also explored. Nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase‐derived ROS play fundamental roles both in oxidative stress and in inflammation, and in phagocytic activity of neutrophils and monocytes 98, 99. Consequently, this study not only revealed significantly increased oxidative stress markers in blood and in fibrinogen fractions purified from BS patients, but also a dramatic enhancement in NADPH oxidase activity in the neutrophil population of these patients. In line with this, in‐vitro experiments showed that purified fibrinogen resulted markedly carbonylated when incubated with neutrophils, but not with monocytes or lymphocytes, from BS patients. In BS patients, fibrinogen carbonyl content significantly correlated with neutrophil‐derived ROS, but not with lymphocyte‐ or monocyte‐derived ROS. These findings are in line with the concept that fibrinogen is more prone to oxidation than albumin, and upon oxidation clot formation rate tends to decrease 100.
In the same recent study involving a group of BS patients the assessment of thrombin catalysed fibrin polymerization revealed a slower rate and turbidity (compared with healthy controls), which resulted significantly and inversely correlated with fibrinogen carbonyl content, thus suggesting a direct influence of carbonylation on fibrin polymerization. Moreover, in patients, only neutrophil (but not lymphocyte or monocyte) ROS production inversely and significantly correlated with the polymerization kinetic parameters. Fibrinogen secondary structure analysis revealed a decrease in α‐helix content, with consequent effects on the biological activity of fibrinogen. This is in line with other authors, who reported that fibrinogen oxidation impairs the capacity of isolated fibrinogen to form a fibrin clot under the effect of thrombin 101.
To examine another important feature of fibrinogen function in relation to carbonylation, in the same study fibrin resistance to plasmin‐induced lysis was determined both in BS patients and controls.
Interestingly, fibrin from BS patients was characterized by a marked resistance to plasmin‐induced lysis with respect to healthy controls. Moreover, fibrin resistance to lysis significantly correlated with fibrinogen carbonyl content and with neutrophil ROS production (but not with lymphocyte‐ or monocyte‐derived ROS). These findings are consistent with previous studies performed in patients with acute coronary syndrome, where it was shown that clots composed of dense networks were more resistant to lysis. These parameters also correlated with inflammation and oxidative stress 102.
In BS patients, clot structure was analysed by electron and differential interference contrast microscopy. The results of these investigations revealed an altered clot architecture, mainly characterized by a tight fibrin network composed of filaments with slightly decreased average fiber size, which was resistant to plasmin‐induced lysis compared to control subjects (Fig. 2). Thin fibers and small pores have been suggested to be typical features of thrombogenic clots, but the mechanisms underlying the formation of these abnormal fibrin clots have not yet been established 103. Undoubtedly, fibrinogen oxidative modification could play an important role in this context.
Figure 2.
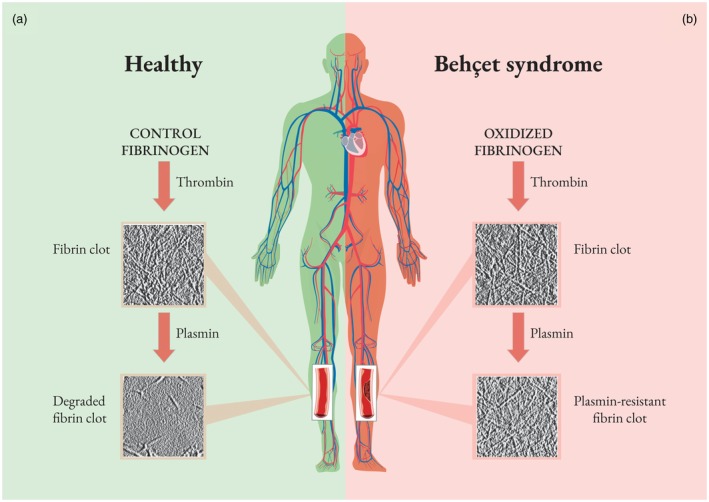
Unoxidized fibrinogen from healthy subjects forms a plasmin‐susceptible fibrin clot. Conversely, in Behçet’s syndrome (BS) patients, ROS promote fibrinogen oxidation and fibrinogen structure modifications, which are responsible for altered fibrinogen clotting ability and reduced fibrin susceptibility to plasmin‐induced lysis. This evidence offers a new model of inflammation‐induced thrombosis and supports current therapeutic concepts regarding the use of immunosuppressive (rather than anti‐coagulation) therapy in BS‐related thrombosis.
Conclusions
BS is a systemic vasculitis characterized by different disease phenotypes, the vascular one being the most intriguing for histological and clinical features. Only a few pathogenetic mechanisms suggest the relationship between thrombosis (especially of the venous district) and inflammation. Recently, we suggested a new in‐vivo mechanism of thrombo‐inflammation in a large BS patient population. These new data point out that neutrophil activation promotes fibrinogen oxidation and thrombus formation in BS. In particular, neutrophil activation leads to NADPH oxidase‐derived ROS production, which is associated with altered fibrinogen structure and impaired fibrinogen function. Interestingly, all these data were influenced neither by the activity status of the disease nor by the presence of vascular involvement in the BS cohort of patients, suggesting that BS is per se a model of inflammation‐induced thrombosis.
Author contributions
G. E., M. B. and C. F. conceived the structure of manuscript and drafted the paper. A. B., E. S., G. D. S., N. T. and D. P. critically revised the manuscript. All the authors approved the final version of the manuscript.
Disclosures
All authors have no conflicts of interest.
Acknowledgements
The authors wish to thank Stefano Salvati and Javier Hernández Plasencia for their help in preparing the figures of this manuscript. This research did not receive any specific grant from funding agencies in the public, commercial or not‐for‐profit sectors.
References
- 1. Emmi G, Silvestri E, Squatrito D et al Behçet’s syndrome pathophysiology and potential therapeutic targets. Intern Emerg Med 2014;9:257–65. Available at: http://link.springer.com/10.1007/s11739-013-1036-5. [DOI] [PubMed] [Google Scholar]
- 2. Emmi G, Silvestri E, Squatrito D et al Thrombosis in vasculitis: from pathogenesis to treatment. Thromb J 2015;13:15 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25883536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yazici H, Ugurlu S, Seyahi E. Behçet syndrome: is it one condition? Clin Rev Allergy Immunol 2012;43:275–80. [DOI] [PubMed] [Google Scholar]
- 4. Seyahi E. Behçet’s disease: how to diagnose and treat vascular involvement. Best Pract Res Clin Rheumatol 2016;30:279–95. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1521694216300420. [DOI] [PubMed] [Google Scholar]
- 5. Yazici H, Seyahi E, Hatemi G, Yazici Y. Behçet syndrome: a contemporary view. Nature Reviews. Rheumatology 2018. [DOI] [PubMed] [Google Scholar]
- 6. Aksu K, Donmez A, Keser G. Inflammation‐induced thrombosis: mechanisms, disease associations and management. Curr Pharm Des 2012;18:1478–93. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22364132. [DOI] [PubMed] [Google Scholar]
- 7. Chamorro A‐J, Marcos M, Hernández‐García I et al Association of allelic variants of factor V Leiden, prothrombin and methylenetetrahydrofolate reductase with thrombosis or ocular involvement in Behçet’s disease: a systematic review and meta‐analysis. Autoimmun Rev 2013;12:607–16. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1568997212002807. [DOI] [PubMed] [Google Scholar]
- 8. Becatti M, Emmi G, Silvestri E et al Neutrophil activation promotes fibrinogen oxidation and thrombus formation in Behçet disease. Circulation 2016;133:302–11. Available at: http://circ.ahajournals.org/lookup/doi/10.1161/CIRCULATIONAHA.115.017738. [DOI] [PubMed] [Google Scholar]
- 9. Jennette JC, Falk RJ, Bacon PA et al 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1–11. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23045170. [DOI] [PubMed] [Google Scholar]
- 10. Hirohata S, Kikuchi H. Histopathology of the ruptured pulmonary artery aneurysm in a patient with Behçet’s disease. Clin Exp Rheumatol 2013;27(2 Suppl 53):S91–5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19796542. [PubMed] [Google Scholar]
- 11. Ergun T, Gürbüz O, Harvell J, Jorizzo J, White W. The histopathology of pathergy: a chronologic study of skin hyperreactivity in Behçet’s disease. Int J Dermatol 1998;37:929–33. Available at: http://www.ncbi.nlm.nih.gov/pubmed/9888335. [DOI] [PubMed] [Google Scholar]
- 12. Boyd SR, Young S, Lightman S. Immunopathology of the noninfectious posterior and intermediate uveitides. Surv Ophthalmol 2001;46:209–33. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11738429. [DOI] [PubMed] [Google Scholar]
- 13. Seyahi E, Yurdakul S. Behçet’s syndrome and thrombosis. Mediterr J Hematol Infect Dis 2011;3:e2011026 Available at: http://www.mjhid.org/index.php/mjhid/article/view/2011.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bin Cho S, Yun M, Lee J‐H, Kim J, Shim W‐H, Bang D. Detection of cardiovascular system involvement in Behçet’s disease using fluorodeoxyglucose positron emission tomography. Semin Arthritis Rheum 2011;40:461–6 http://linkinghub.elsevier.com/retrieve/pii/S0049017210000818. [DOI] [PubMed] [Google Scholar]
- 15. Hatemi G, Christensen R, Bang D et al 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis 2018;77:808–18. Available at: http://ard.bmj.com/lookup/doi/10.1136/annrheumdis-2018-213225. [DOI] [PubMed] [Google Scholar]
- 16. Pipitone NAM, Versari A, Salvarani C. Usefulness of PET in recognizing and managing vasculitides. Curr Opin Rheumatol 2018;30:24–9. Available at: http://insights.ovid.com/crossref?an=00002281-201801000-00005. [DOI] [PubMed] [Google Scholar]
- 17. Pazzola G, Muratore F, Pipitone N, Salvarani C. Biotherapies in large vessel vasculitis. La Rev Med interne 2016;37:274–8. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0248866315006347. [DOI] [PubMed] [Google Scholar]
- 18. Saadoun D, Wechsler B, Resche‐Rigon M et al Cerebral venous thrombosis in Behçet’s disease. Arthritis Rheum 2009;61:518–26. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19333987. [DOI] [PubMed] [Google Scholar]
- 19. Ahn JK, Lee YS, Jeon CH, Koh E‐M, Cha H‐S. Treatment of venous thrombosis associated with Behcet’s disease: immunosuppressive therapy alone versus immunosuppressive therapy plus anticoagulation. Clin Rheumatol 2008;27:201–5. Available at: http://link.springer.com/10.1007/s10067-007-0685-z. [DOI] [PubMed] [Google Scholar]
- 20. Desbois AC, Wechsler B, Resche‐Rigon M et al Immunosuppressants reduce venous thrombosis relapse in Behçet’s disease. Arthritis Rheum 2012;64:2753–60. Available at: http://doi.wiley.com/10.1002/art.34450. [DOI] [PubMed] [Google Scholar]
- 21. Alibaz‐Oner F, Karadeniz A, Ylmaz S et al Behçet disease with vascular involvement. Medicine (Balt) 2015;94:e494 Available at: http://www.ncbi.nlm.nih.gov/pubmed/25674739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vitale A, Emmi G, Lopalco G et al Adalimumab effectiveness in Behçet’s disease: short and long‐term data from a multicenter retrospective observational study. Clin Rheumatol 2017;36:451–5. Available at: http://link.springer.com/10.1007/s10067-016-3417-4. [DOI] [PubMed] [Google Scholar]
- 23. Fabiani C, Vitale A, Emmi G et al Efficacy and safety of adalimumab in Behçet’s disease‐related uveitis: a multicenter retrospective observational study. Clin Rheumatol 2017;36:183–9. Available at: http://link.springer.com/10.1007/s10067-016-3480-x. [DOI] [PubMed] [Google Scholar]
- 24. Vitale A, Rigante D, Lopalco G et al New therapeutic solutions for Behçet’s syndrome. Expert Opin Invest Drugs 2016; 25:827–840. [DOI] [PubMed] [Google Scholar]
- 25. Fabiani C, Sota J, Rigante D et al Efficacy of adalimumab and infliximab in recalcitrant retinal vasculitis inadequately responsive to other immunomodulatory therapies. Clin Rheumatol 2018; 37: 2805–2809. [DOI] [PubMed] [Google Scholar]
- 26. Fabiani C, Sota J, Vitale A et al Cumulative retention rate of adalimumab in patients with Behçet’s disease‐related uveitis: a four‐year follow‐up study. Br J Ophthalmol 2018;102:637–41. Available at: http://bjo.bmj.com/lookup/doi/10.1136/bjophthalmol-2017-310733. [DOI] [PubMed] [Google Scholar]
- 27. Fabiani C, Vitale A, Rigante D et al Comparative efficacy between adalimumab and infliximab in the treatment of non‐infectious intermediate uveitis, posterior uveitis, and panuveitis: a retrospective observational study of 107 patients. Clin Rheumatol 2018. Available at: http://link.springer.com/10.1007/s10067-018-4228-6. [DOI] [PubMed] [Google Scholar]
- 28. Emmi G, Vitale A, Silvestri E et al Adalimumab‐based treatment versus DMARDs for venous thrombosis in Behçet syndrome. A retrospective study of 70 patients with vascular involvement. Arthritis Rheumatol (Hoboken, NJ) 2018; Available at: http://doi.wiley.com/10.1002/art.40531. [DOI] [PubMed] [Google Scholar]
- 29. Fabiani C, Vitale A, Rigante D et al Predictors of sustained clinical response in patients with Behçet’s disease‐related uveitis treated with infliximab and adalimumab. Clin Rheumatol 2018;37:1715–20. Available at: http://link.springer.com/10.1007/s10067-018-4092-4. [DOI] [PubMed] [Google Scholar]
- 30. GBD 2013 Mortality and Causes of Death Collaborators . Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;385:117–71. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0140673614616822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Becatti M, Mannucci A, Taddei N, Fiorillo C. Oxidative stress and inflammation: new molecular targets for cardiovascular diseases. Intern Emerg Med 2018;13:647–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29858969. [DOI] [PubMed] [Google Scholar]
- 32. Poredos P, Jezovnik MK. The role of inflammation in venous thromboembolism and the link between arterial and venous thrombosis. Int Angiol 2007;26:306–11. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18091697. [PubMed] [Google Scholar]
- 33. Di Minno MND, Tufano A, Ageno W, Prandoni P, Di Minno G. Identifying high‐risk individuals for cardiovascular disease: similarities between venous and arterial thrombosis in perspective. A 2011 update. Intern Emerg Med 2012;7:9–13. Available at: http://link.springer.com/10.1007/s11739-011-0582-y. [DOI] [PubMed] [Google Scholar]
- 34. Levi M, van der Poll T. Two‐way interactions between inflammation and coagulation. Trends Cardiovasc Med 2005;15:254–9. Available at: http://linkinghub.elsevier.com/retrieve/pii/S1050173805001234. [DOI] [PubMed] [Google Scholar]
- 35. Branchford BR, Carpenter SL. The role of inflammation in venous thromboembolism. Front Pediatr 2018;6:142 Available at: https://www.frontiersin.org/article/10.3389/fped.2018.00142/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee D‐K, Kim H‐J, Lee D‐H. Incidence of deep vein thrombosis and venous thromboembolism following TKA in rheumatoid arthritis versus osteoarthritis: a meta‐analysis. PLOS ONE 2016;11:e0166844 Available at: http://dx.plos.org/10.1371/journal.pone.0166844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andrade AR, Barros LL, Azevedo MFC et al Risk of thrombosis and mortality in inflammatory bowel disease. Clin Transl Gastroenterol 2018;9:142 Available at: http://www.ncbi.nlm.nih.gov/pubmed/29618721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cugno M, Tedeschi A, Borghi A et al Activation of blood coagulation in two prototypic autoimmune skin diseases: a possible link with thrombotic risk. PLOS ONE 2015;10:e0129456 Available at: http://dx.plos.org/10.1371/journal.pone.0129456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yusuf HR, Hooper WC, Grosse SD, Parker CS, Boulet SL. Ortel TL. Risk of venous thromboembolism occurrence among adults with selected autoimmune diseases: a study among a U.S. cohort of commercial insurance enrollees. Thromb Res 2015;135:50–7 https://linkinghub.elsevier.com/retrieve/pii/S0049384814005684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leung LLK, Morser J. Carboxypeptidase B2 and carboxypeptidase N in the crosstalk between coagulation, thrombosis, inflammation, and innate immunity. J Thromb Haemost 2018;16:1474–86. Available at: http://doi.wiley.com/10.1111/jth.14199. [DOI] [PubMed] [Google Scholar]
- 41. Yago T, Liu Z, Ahamed J, McEver RP. Cooperative PSGL‐1 and CXCR41 signaling in neutrophils promotes deep vein thrombosis in mice. Blood 2018;132:1426–37. Available at: http://www.bloodjournal.org/lookup/doi/10.1182/blood-2018-05-850859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu H, Li R, Pei L‐G et al Emerging role of high mobility group box‐1 in thrombosis‐related diseases. Cell Physiol Biochem 2018;47:1319–37. Available at: https://www.karger.com/Article/FullText/490818. [DOI] [PubMed] [Google Scholar]
- 43. Kunutsor SK, Seidu S, Khunti K. Statins and primary prevention of venous thromboembolism: a systematic review and meta‐analysis. Lancet Haematol 2017;4:e83–93. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28089655. [DOI] [PubMed] [Google Scholar]
- 44. Ridker PM, Cannon CP, Morrow D et al C‐reactive protein levels and outcomes after statin therapy. N Engl J Med 2005;352:20–8. Available at: http://www.ncbi.nlm.nih.gov/pubmed/15635109. [DOI] [PubMed] [Google Scholar]
- 45. Ridker PM, Everett BM, Thuren T et al Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–31. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28845751. [DOI] [PubMed] [Google Scholar]
- 46. Ridker PM, MacFadyen JG, Everett BM et al Relationship of C‐reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 2018;391:319–28. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0140673617328143. [DOI] [PubMed] [Google Scholar]
- 47. Everett BM, Pradhan AD, Solomon DH et al Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J 2013;166:199–207.e15. Available at: http://linkinghub.elsevier.com/retrieve/pii/S000287031300224X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quadros AS, Cambruzzi E, Sebben J et al Red versus white thrombi in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention: clinical and angiographic outcomes. Am Heart J 2012;164:553–60. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0002870312005224. [DOI] [PubMed] [Google Scholar]
- 49. Silvain J, Collet J‐P, Guedeney P et al Thrombus composition in sudden cardiac death from acute myocardial infarction. Resuscitation 2017;113:108–14. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28212919. [DOI] [PubMed] [Google Scholar]
- 50. Zalewski J, Bogaert J, Sadowski M et al Plasma fibrin clot phenotype independently affects intracoronary thrombus ultrastructure in patients with acute myocardial infarction. Thromb Haemost 2015;113:1258–69. Available at: http://www.thieme-connect.de/DOI/DOI?10.1160/TH14-09-0801. [DOI] [PubMed] [Google Scholar]
- 51. Kovács A, Sótonyi P, Nagy AI et al Ultrastructure and composition of thrombi in coronary and peripheral artery disease: correlations with clinical and laboratory findings. Thromb Res 2015;135:760–6. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0049384815000596. [DOI] [PubMed] [Google Scholar]
- 52. Sadowski M, Ząbczyk M, Undas A. Coronary thrombus composition: links with inflammation, platelet and endothelial markers. Atherosclerosis 2014;237:555–61. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0021915014014555. [DOI] [PubMed] [Google Scholar]
- 53. Fokkema ML, Vlaar PJ, Svilaas T et al Incidence and clinical consequences of distal embolization on the coronary angiogram after percutaneous coronary intervention for ST‐elevation myocardial infarction. Eur Heart J 2009;30:908–15. Available at: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehp033. [DOI] [PubMed] [Google Scholar]
- 54. Yunoki K, Naruko T, Inoue T et al Relationship of thrombus characteristics to the incidence of angiographically visible distal embolization in patients with ST‐segment elevation myocardial infarction treated with thrombus aspiration. JACC Cardiovasc Interv 2013;6:377–85. Available at: http://linkinghub.elsevier.com/retrieve/pii/S1936879813004342. [DOI] [PubMed] [Google Scholar]
- 55. Burches B, Karnicki K, Wysokinski W, McBane RD. Immunohistochemistry of thrombi following iliac venous stenting: a novel model of venous thrombosis. Thromb Haemost 2006;96:618–22. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17080219. [PubMed] [Google Scholar]
- 56. Sobieszczyk P, Fishbein MC, Goldhaber SZ. Acute pulmonary embolism: don’t ignore the platelet. Circulation 2002;106:1748–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12356622. [DOI] [PubMed] [Google Scholar]
- 57. Simes J, Becattini C, Agnelli G et al Aspirin for the prevention of recurrent venous thromboembolism: the INSPIRE collaboration. Circulation 2014;130:1062–71. Available at: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.114.008828. [DOI] [PubMed] [Google Scholar]
- 58. Weitz JI, Lensing AWA, Prins MH et al Rivaroxaban or aspirin for extended treatment of venous thromboembolism. N Engl J Med 2017;376:1211–22. Available at: http://www.ncbi.nlm.nih.gov/pubmed/28316279. [DOI] [PubMed] [Google Scholar]
- 59. Marik PE, Extended CR. Anticoagulant and aspirin treatment for the secondary prevention of thromboembolic disease: a systematic review and meta‐analysis. PLOS ONE 2015;10:e0143252 Available at: http://www.ncbi.nlm.nih.gov/pubmed/26587983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lacut K, van der Maaten J, Le Gal G, Cornily G, Mottier D, Oger E. Antiplatelet drugs and risk of venous thromboembolism: results from the EDITH case–control study. Haematologica 2008;93:1117–8. Available at: http://www.haematologica.org/cgi/doi/10.3324/haematol.12331. [DOI] [PubMed] [Google Scholar]
- 61. Hisada Y, Geddings JE, Ay C, Mackman N. Venous thrombosis and cancer: from mouse models to clinical trials. J Thromb Haemost 2015;13:1372–82. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25988873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ay C, Pabinger I, Cohen AT. Cancer‐associated venous thromboembolism: burden, mechanisms, and management. Thromb Haemost 2017;117:219–30. Available at: http://www.thieme-connect.de/DOI/DOI?10.1160/TH16-08-0615. [DOI] [PubMed] [Google Scholar]
- 63. Aronson D, Brenner B. Arterial thrombosis and cancer. Thromb Res 2018;164(Suppl 1):S23–8. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0049384818300033. [DOI] [PubMed] [Google Scholar]
- 64. Tuzovic M, Herrmann J, Iliescu C, Marmagkiolis K, Ziaeian B, Yang EH. Arterial thrombosis in patients with cancer. Curr Treat Options Cardiovasc Med 2018;20:40 Available at: http://link.springer.com/10.1007/s11936-018-0635-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Donze JD, Ridker PM, Finlayson SRG, Bates DW. Impact of sepsis on risk of postoperative arterial and venous thromboses: large prospective cohort study. BMJ 2014;349:g5334–g5334. Available at: http://www.bmj.com/cgi/doi/10.1136/bmj.g5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ruiz‐Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010;376:1498–509. Available at: http://linkinghub.elsevier.com/retrieve/pii/S014067361060709X. [DOI] [PubMed] [Google Scholar]
- 67. Emmi G, Urban ML, Scalera A et al Repeated low‐dose courses of rituximab in SLE‐associated antiphospholipid syndrome: data from a tertiary dedicated centre. Semin Arthritis Rheum 2017;46:e21–3. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0049017216301718. [DOI] [PubMed] [Google Scholar]
- 68. Ames PRJ, Margaglione M, Mackie S, Alves JD. Eosinophilia and thrombophilia in Churg Strauss syndrome: a clinical and pathogenetic overview. Clin Appl Thromb Hemost 2010;16:628–36. Available at: http://journals.sagepub.com/doi/10.1177/1076029609348647. [DOI] [PubMed] [Google Scholar]
- 69. Stassen PM, Derks RPH, Kallenberg CGM, Stegeman CA. Venous thromboembolism in ANCA‐associated vasculitis – incidence and risk factors. Rheumatology 2007;47:530–4. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18356178. [DOI] [PubMed] [Google Scholar]
- 70. Faurschou M, Mellemkjaer L, Sorensen IJ et al Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009;60:1187–92. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19333952. [DOI] [PubMed] [Google Scholar]
- 71. Aviña‐Zubieta JA, Bhole VM, Amiri N, Sayre EC, Choi HK. The risk of deep venous thrombosis and pulmonary embolism in giant cell arteritis: a general population‐based study. Ann Rheum Dis 2016;75:148–54. Available at: http://ard.bmj.com/lookup/doi/10.1136/annrheumdis-2014-205665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Samson M, Jacquin A, Audia S et al Stroke associated with giant cell arteritis: a population‐based study. J Neurol Neurosurg Psychiatry 2015;86:216–21. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24780954. [DOI] [PubMed] [Google Scholar]
- 73. de Souza AWS, Machado NP, Pereira VM et al Antiplatelet therapy for the prevention of arterial ischemic events in Takayasu arteritis. Circ J 2010;74:1236–41. Available at: http://www.ncbi.nlm.nih.gov/pubmed/20467149. [DOI] [PubMed] [Google Scholar]
- 74. La Regina M, Gasparyan AY, Orlandini F, Prisco D. Behçet’s disease as a model of venous thrombosis. Open Cardiovasc Med J 2010;4:71–7. Available at: http://www.bentham-open.org/pages/content.php?TOCMJ/2010/00000004/00000002/71TOCMJ.SGM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brenière C, Blanc C, Devilliers H et al Associated arterial and venous cerebral manifestations in Behçet’s disease. Rev Neurol (Paris) 2018;174:337–41. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0035378716302806. [DOI] [PubMed] [Google Scholar]
- 76. Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood 2013;121:1712–9. Available at: http://www.bloodjournal.org/cgi/doi/10.1182/blood-2012-09-306639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wolberg AS, Gabriel DA, Hoffman M. Analyzing fibrin clot structure using a microplate reader. Blood Coagul Fibrinolysis 2002;13:533–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12192305. [DOI] [PubMed] [Google Scholar]
- 78. Nair CH, Shah GA, Dhall DP. Effect of temperature, pH and ionic strength and composition on fibrin network structure and its development. Thromb Res 1986;42:809–16. Available at: http://www.ncbi.nlm.nih.gov/pubmed/3726801. [DOI] [PubMed] [Google Scholar]
- 79. Blombäck B, Carlsson K, Fatah K, Hessel B, Procyk R. Fibrin in human plasma: gel architectures governed by rate and nature of fibrinogen activation. Thromb Res 1994;75:521–38. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7992253. [DOI] [PubMed] [Google Scholar]
- 80. Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci 2008;38:15–23. Available at: http://www.ncbi.nlm.nih.gov/pubmed/18282807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu W, Jawerth LM, Sparks EA et al Fibrin fibers have extraordinary extensibility and elasticity. Science 2006;313:634–634. Available at: http://www.ncbi.nlm.nih.gov/pubmed/16888133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gabriel DA, Muga K, Boothroyd EM. The effect of fibrin structure on fibrinolysis. J Biol Chem 1992;267:24259–63. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1447176. [PubMed] [Google Scholar]
- 83. Collet JP, Park D, Lesty C et al Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol 2000;20:1354–61. Available at: http://www.ncbi.nlm.nih.gov/pubmed/10807754. [DOI] [PubMed] [Google Scholar]
- 84. Collet JP, Allali Y, Lesty C et al Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol 2006;26:2567–73. Available at: https://www.ahajournals.org/doi/10.1161/01.ATV.0000241589.52950.4c. [DOI] [PubMed] [Google Scholar]
- 85. Pieters M, Covic N, van der Westhuizen FH et al Glycaemic control improves fibrin network characteristics in type 2 diabetes – a purified fibrinogen model. Thromb Haemost 2008;99:691–700. Available at: http://www.thieme-connect.de/DOI/DOI?10.1160/TH07-11-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Karlaftis V, Perera S, Monagle P, Ignjatovic V. Importance of post‐translational modifications on the function of key haemostatic proteins. Blood Coagul Fibrinolysis 2016;27:1–4. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26484638. [DOI] [PubMed] [Google Scholar]
- 87. Ray PD, Huang B‐W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012;24:981–90. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fall L, Brugniaux JV, Davis D et al Redox‐regulation of haemostasis in hypoxic exercising humans: a randomised double‐blind placebo‐controlled antioxidant study. J Physiol 2018; 596:4879–4891. Available at: http://doi.wiley.com/10.1113/JP276414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stadtman ER, Levine RL. Free radical‐mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003;25:207–18. Available at: http://www.ncbi.nlm.nih.gov/pubmed/14661084. [DOI] [PubMed] [Google Scholar]
- 90. Bruschi M, Candiano G, Santucci L. Ghiggeri GM. Oxidized albumin. The long way of a protein of uncertain function. Biochim Biophys Acta 2013;1830:5473–9 http://linkinghub.elsevier.com/retrieve/pii/S0304416513001451. [DOI] [PubMed] [Google Scholar]
- 91. Becatti M, Fiorillo C, Gori AM et al Platelet and leukocyte ROS production and lipoperoxidation are associated with high platelet reactivity in non‐ST elevation myocardial infarction (NSTEMI) patients on dual antiplatelet treatment. Atherosclerosis 2013;231:392–400. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0021915013005893. [DOI] [PubMed] [Google Scholar]
- 92. Shacter E, Williams JA, Lim M, Levine RL. Differential susceptibility of plasma proteins to oxidative modification: examination by Western blot immunoassay. Free Radic Biol Med 1994;17:429–37. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7835749. [DOI] [PubMed] [Google Scholar]
- 93. Weigandt KM, White N, Chung D et al Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys J 2012;103:2399–407. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0006349512011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Rosen H, Klebanoff SJ, Wang Y, Brot N, Heinecke JW, Fu X. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc Natl Acad Sci USA 2009;106:18686–91. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.0909464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Burney PR, White N, Pfaendtner J. Structural effects of methionine oxidation on isolated subdomains of human fibrin D and αC regions. PLOS ONE 2014;9:e86981 Available at: http://dx.plos.org/10.1371/journal.pone.0086981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Akar A, Arca E, Serdar MA et al Correlation between erythrocyte antioxidant activity, lipid peroxidation, and disease activity in patients with Behçet’s disease. J Eur Acad Dermatol Venereol 2003;17:482–3. Available at: http://www.ncbi.nlm.nih.gov/pubmed/12834474. [DOI] [PubMed] [Google Scholar]
- 97. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014;20:1126–67. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23991888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bedard K, Krause K‐H. The NOX Family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313. Available at: http://www.ncbi.nlm.nih.gov/pubmed/17237347. [DOI] [PubMed] [Google Scholar]
- 99. Barygina VV, Becatti M, Soldi G et al Altered redox status in the blood of psoriatic patients: involvement of NADPH oxidase and role of anti‐TNF‐α therapy. Redox Rep 2013;18:100–6. Available at: http://www.tandfonline.com/doi/full/10.1179/1351000213Y.0000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Becatti M, Marcucci R, Bruschi G et al Oxidative modification of fibrinogen is associated with altered function and structure in the subacute phase of myocardial infarction. Arterioscler Thromb Vasc Biol 2014;34:1355–61. Available at: http://atvb.ahajournals.org/cgi/doi/10.1161/ATVBAHA.114.303785. [DOI] [PubMed] [Google Scholar]
- 101. Shacter E, Williams JA, Levine RL. Oxidative modification of fibrinogen inhibits thrombin‐catalyzed clot formation. Free Radic Biol Med 1995;18:815–21. Available at: http://www.ncbi.nlm.nih.gov/pubmed/7750804. [DOI] [PubMed] [Google Scholar]
- 102. Fatah K, Hamsten A, Blombäck B, Blombäck M. Fibrin gel network characteristics and coronary heart disease: relations to plasma fibrinogen concentration, acute phase protein, serum lipoproteins and coronary atherosclerosis. Thromb Haemost 1992;68:130–5. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1384157. [PubMed] [Google Scholar]
- 103. Fatah K, Silveira A, Tornvall P, Karpe F, Blombäck M, Hamsten A. Proneness to formation of tight and rigid fibrin gel structures in men with myocardial infarction at a young age. Thromb Haemost 1996;76:535–40. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8902992. [PubMed] [Google Scholar]