

Summary
Immunoglobulin (Ig)A is the most abundant immunoglobulin in humans, and in the airway mucosa secretory IgA (sIgA) plays a pivotal role in first‐line defense against invading pathogens and antigens. IgA has been reported to also have pathogenic effects, including possible worsening of the prognosis of idiopathic pulmonary fibrosis (IPF). However, the precise effects of IgA on lung fibroblasts remain unclear, and we aimed to elucidate how IgA activates human lung fibroblasts. We found that sIgA, but not monomeric IgA (mIgA), induced interleukin (IL)‐6, IL‐8, monocyte chemoattractant protein (MCP)‐1 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) production by normal human lung fibroblasts (NHLFs) at both the protein and mRNA levels. sIgA also promoted proliferation of NHLFs and collagen gel contraction comparable to with transforming growth factor (TGF)‐β, which is involved in fibrogenesis in IPF. Also, Western blot analysis and real‐time quantitative polymerase chain reaction (PCR) revealed that sIgA enhanced production of α‐smooth muscle actin (α‐SMA) and collagen type I (Col I) by NHLFs. Flow cytometry showed that NHLFs bound sIgA, and among the known IgA receptors, NHLFs significantly expressed CD71 (transferrin receptor). Transfection of siRNA targeting CD71 partially but significantly suppressed cytokine production by NHLFs co‐cultured with sIgA. Our findings suggest that sIgA may promote human lung inflammation and fibrosis by enhancing production of inflammatory or fibrogenic cytokines as well as extracellular matrix, inducing fibroblast differentiation into myofibroblasts and promoting human lung fibroblast proliferation. sIgA’s enhancement of cytokine production may be due partially to its binding to CD71 or the secretory component.
Keywords: fibroblasts, fibrosis, human, IgA, lung
Introduction
Immunoglobulin (Ig) A, produced at approximately 40–60 mg/kg of body weight/day, is the most abundant immunoglobulin in humans 1. IgA‐producing plasma cells reside mainly in mucosal tissues, such as in the respiratory, gastrointestinal and genitourinary tracts, sites that are directly exposed to the external environment. There, IgA comes into contact with pathogenic microorganisms as well as invading antigens and toxins and acts as the first line of defense 2. In the blood, approximately 90% of IgA exists as monomeric IgA (mIgA) 3. In contrast, approximately half of IgA in airway secretions is in the form of secretory IgA (sIgA). sIgA is a J chain‐containing dimeric IgA (dIgA), with a secretory component (SC) that is conferred during the process between binding to polymeric immunoglobulin receptor on the basolateral surfaces of mucosal epithelial cells and release into the lumen 4. sIgA entraps pathogens and antigens and serves to promote their clearance and removal through receptor blockade and immune exclusion. In addition, IgA has been shown to improve the viscoelastic properties of airway secretions, which would help mucocilliary transport 5. Thus, IgA is commonly thought of as a key immunoglobulin for human immunity.
In contrast to IgA’s protective role in human immunity against infectious, allergic and autoimmune disorders, IgA has also been shown to have somewhat unfavorable effects in human biology. For example, sIgA is a known eosinophil and basophil activator in terms of enhancing degranulation, which may have a protective effect against helminth infections but an accelerating effect on hypersensitivity disorders such as asthma 6, 7, 8. Similarly, aggregation of IgA induces activation of mesangial cells, which may be involved in initiation of the pathology of IgA nephropathy 9. Therefore, IgA might be unfavorable to human immunity in certain disease conditions.
A recent report showed that the level of serum IgA can be used as a prognostic marker for idiopathic pulmonary fibrosis (IPF): a high level of serum IgA was associated with poor survival of IPF patients 10. Because transforming growth factor (TGF)‐β, the most potent cytokine involved in fibrogenesis in IPF, is critical for mucosal IgA responses 11, it is conceivable that IgA might play an important role in the pathogenesis of IPF. Interactions between fibroblasts and alveolar epithelial cells appear to be the central mechanism in that pathogenesis 12, 13. In particular, progressive accumulation of activated fibroblasts and differentiated myofibroblasts in response to TGF‐β and deposition of extracellular matrix in the lung are observed in IPF 14. The effects of IgA on lung fibroblasts and myofibroblasts have not been well elucidated, and the pathogenic role of IgA in fibrosing diseases such as IPF remains largely unclear. Here, we sought to elucidate the effects of IgA on activation of human lung fibroblasts.
Materials and methods
Reagents
Purified human sIgA was obtained from MP Biomedicals (Fountain Parkway, Solon, OH, USA) 8, and mIgA from human serum was obtained from Oriental Yeast Co. (Tokyo, Japan). Before the experiments, we checked the molecular sizes of both mIgA and sIgA by electrophoresis followed by Western blotting using anti‐IgA antibody. By doing so, we confirmed that the mIgA used in the study positioned at around 150 kDa and sIgA at approximately 380 kDa, values that are consistent with a previous report 15 (data not shown). The following recombinant proteins were purchased: human TGF‐β1 (Pepro Tech, Rocky Hill, NJ, USA), human interleukin (IL)‐1β (Tonbo Biosciences, San Diego, CA, USA) and human tumor necrosis factor (TNF)‐α (R&D Systems, Minneapolis, MN, USA).
Cell culture
Normal human lung fibroblasts (NHLFs) obtained from Lonza (Walkersville, MD, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F‐12 medium (Wako Pure Chemical Industries, Osaka, Japan) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA), 100 U/ml penicillin and 100 µg/ml streptomycin at 37ºC, under a 5% CO2 atmosphere, until used for the experiments. NHLFs between passages 2 to 10 were used for the experiments.
Proliferation assay
NHLFs in DMEM/Ham’s F‐12 medium were seeded at 1 × 104/well in 96‐well plates for 24 h at 37ºC and then incubated with a stimulus and bromodeoxyuridine (BrdU) for 16 h. Cell proliferation was then assessed using a cell proliferation enzyme‐linked immunosorbent assay (ELISA) kit, BrdU (colorimetric) version 16 (Roche Applied Science, Indianapolis, IN, USA). Briefly, after the culture medium was removed, the cells were fixed and DNA was denatured. The cells were then labeled with anti‐BrdU‐POD antibody and developed by adding the substrate solution. DNA replication, assessed based on the incorporation of BrdU, was quantified by measuring the absorbance using a scanning multi‐well spectrophotometer (Bio‐Rad Laboratories, Hercules, CA, USA) at 450 nm with a reference wavelength of 690 nm.
Collagen gel contraction assay
Collagen gel contraction assay was used to evaluate contraction mediated by NHLFs embedded in collagen gel matrices by measuring the gel surface area 16, 17. First, NHLFs were mixed with Cellmatrix Type I‐A (Nitta Gelatin, Osaka, Japan) in DMEM (Nitta Gelatin) to a final cell density of 3 × 105 cells/ml. The mixture was placed in 24‐well plates in a volume of 500 µl containing 1.5 × 105 cells/well and allowed to gel at 37ºC for 30 min. After gelation, 500 µl of DMEM was dripped onto the gel in each well, and after 24 h the medium was replaced by fresh medium containing a stimulus. At the same time, the gel was carefully detached from the well wall using a spatula. The gel, still in the well, was then further cultured for up to 72 h at 37ºC in a 5% CO2 atmosphere. Images of the gels at 24, 48 and 72 h were then scanned through the bottom of the plates using a scanner (Pixus MP990; Canon, Tokyo, Japan) and analyzed by measuring the gel surface area using ImageJ software (National Institutes of Health, Bethesda, MA, USA).
Western blotting
NHLFs in D‐MEM/Ham’s F‐12 medium were seeded at 4 × 105/well in 6‐well plates, incubated for 24 h at 37ºC, and then incubated with a stimulus for 48 h. The cells were then lysed with a lysis buffer consisting of radioimmunoprecipitation assay (RIPA) buffer (Wako Pure Chemical Industries), phenylmethylsulfonyl fluoride (PMSF) as a protease inhibitor (Nacalai Tesque, Kyoto, Japan) and the production of α‐smooth muscle actin (α‐SMA) and collagen type 1 (Col I) by NHLFs was assessed via Western blotting. Briefly, equal amounts of protein (20 µg) from the lysates, as determined by bicinchoninic acid (BCA) assay (TaKaRa BCA Protein Assay Kit; Takara Bio Inc., Shiga, Japan), were separated on NuPAGE Bis‐Tris protein gels (Thermo Fisher Scientific) and then transferred onto polyvinylidene difluoride membranes (Thermo Fisher Scientific). After blocking with WesternBreeze Blocker (Thermo Fisher Scientific) for 30 min, anti‐human α‐SMA antibody (1 : 1250, mouse IgG2a, clone 1A4; Sigma‐Aldrich, St Louis, MO, USA), goat anti‐human collagen type I antibody (1 : 2000; Southern Biotech, Birmingham, AL, USA) and anti‐human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) antibody (1 : 10000, mouse IgG1, clone 686613; R&D Systems) were used for detection of the proteins. The blots were developed using horseradish peroxidase (HRP)‐conjugated anti‐mouse or anti‐goat secondary antibodies (1 : 10000) and detected using a chemiluminescent system (Amersham Imager 600; GE Healthcare, Piscataway, NJ, USA). The results were expressed as the relative ratio of the band density of the target protein to that of GAPDH using ImageQuant TL software, Version 8.1 (GE Healthcare).
Flow cytometry
To examine whether sIgA binds to NHLFs, cells were incubated with sIgA for 1 h at 4ºC. The cells were then stained with allophycocyanin (APC)‐conjugated mouse anti‐human IgA antibody (clone IS11‐8E10; Miltenyi Biotec, Bergisch‐Gladbach, Germany) or isotype control for 30 min at 4ºC. To assess expression of receptors on NHLFs, 1 × 105 NHLFs were incubated with a stimulus for 48 h. The cells were then incubated with 5 µg/ml of fluorescein isothiocyanate (FITC)‐conjugated mouse anti‐human CD71 monoclonal antibody (mAb) (IgG2aκ, clone CY1G4; BioLegend, San Diego, CA, USA) or isotype control. For detection of other receptors, cells were first labeled with anti‐human CD89 mAb (mouse IgG1κ, clone A59; BioLegend), anti‐human CD351 mAb (mouse IgG1κ, clone TX61; BioLegend), anti‐human asialoglycoprotein receptor I (ASGPR1) mAb (mouse IgG1κ, clone 8D7; BD Pharmingen, San Jose, CA, USA) or isotype control followed by labeling with phycoerythrin (PE)‐conjugated goat anti‐mouse IgG (Thermo Fisher Scientific). The stained cells were analyzed using FACSVerse (BD Biosciences, Franklin Lakes, NJ, USA), and dot‐plots and histograms were generated using FlowJo (Tree Star Inc., Ashland, OR, USA).
Transfection of NHLFs with siRNA targeted against CD71
NHLFs in D‐MEM/Ham’s F‐12 medium were seeded at 0.8 × 105/well in 24‐well plates and incubated for 2 h at 37ºC. SiRNA against CD71 (5′rGrGUUrArCUrGrGrGrCrArAUUUrCUrATT3′ and 5′UrArGrArArAUUrGrCrCrCrArGUrArArCrCTT3′; Sigma‐Aldrich) was mixed with HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) to a final concentration of 10 nM, which was then added to the cells. An equal concentration of a non‐targeting, scrambled siRNA was used as a control. The cells were incubated for 24 h at 37ºC, washed with culture medium and used for the experiments.
Measurement of cytokines
NHLFs in D‐MEM/Ham’s F‐12 medium were seeded at 1 × 105/well in 24‐well plates and cultured for 24 h at 37ºC. The cells were then incubated with a stimulus for 48 h, and the cell‐culture supernatants were collected and stored at –20ºC until assay. For anti‐IgA SC antibody (anti‐SC antibody) (mouse IgG1, clone SC‐05; Abcam, Cambridge, UK) treatment, sIgA and anti‐SC antibody were first reacted for 1 h at 37ºC. NHLFs were then incubated with the resultant complexes for 24 h at 37ºC, followed by collection of the cell‐culture supernatants. The levels of IL‐6, IL‐8, monocyte chemoattractant protein (MCP)‐1 and granulocyte macrophage colony‐stimulating factor (GM‐CSF) in the cell culture supernatants were determined by cytometric bead array (CBA; BD Biosciences), according to the manufacturer’s instructions.
Real‐time quantitative polymerase chain reaction (RT–qPCR) analysis
NHLFs in D‐MEM/Ham’s F‐12 medium were seeded at 1 × 105/well in 24‐well plates for 24 h at 37ºC. The cells were then incubated with a stimulus for the indicated times. The total RNA was extracted from the cultured cells using an RNeasy Mini kit (Qiagen), according to the manufacturer’s instructions. The extracted mRNA was reverse‐transcribed to cDNA using a cDNA Synthesis Kit (Bio‐Rad Laboratories). RT–PCR was performed using primers and Taqman probes designed by Thermo Fisher Scientific. Data were calculated by the ΔΔCt method, using β‐actin as a reference. Relative quantitation (RQ) values were calculated using the following equation: RQ = 2–ΔΔCt.
Immunohistochemistry
NHLFs in D‐MEM/Ham’s F‐12 medium were seeded at 2×104 cells/well in chamber slide II (Iwaki, Tokyo, Japan) and incubated for 24 h at 37ºC. The cells were then fixed with cold acetone for 2 min and blocked with PBS containing 5% BSA and horse normal serum (Vectastain ABC kit; Vector Laboratories, Burlingame, CA, USA). Mouse anti‐human transferrin receptor (1 : 500, mouse IgG1, clone H68.4; Thermo Fisher Scientific) and purified mouse IgG1κ isotype control (1 : 500, mouse IgG1κ; BioLegend) were used as primary antibodies. Bound primary antibodies were detected with biotin‐conjugated anti‐mouse IgG antibody (Vectastain ABC kit) for 30 min at 37ºC. All chamber slides were developed with peroxidase substrate kit DAB. Immunostained slides were visualized, and images were captured using a fluorescence microscope BZ‐X710 (Keyence, Osaka, Japan).
Statistical analysis
Statistical differences between two groups were evaluated using the Mann–Whitney U‐test. To evaluate statistical differences among three or more groups, the Kruskal–Wallis test was performed. We used GraphPad Prism, version 7 (GraphPad Software, San Diego, CA, USA) for statistical analysis. Statistical significance was defined as a P‐value of less than 0·05. Data are expressed as the mean ± standard error of mean (s.e.m.).
Results
sIgA, but not mIgA, induced production of IL‐6, IL‐8, MCP‐1 and GM‐CSF by NHLFs
First, we investigated the effect of IgA on cytokine synthesis by NHLFs using flow cytometry and RT–PCR, as lung fibroblasts produce abundant inflammatory mediators 18, 19. As shown in Fig. 1a, production of each of IL‐6, IL‐8, MCP‐1 and GM‐CSF by NHLFs was induced dose‐dependently by sIgA at 100–300 μg/ml. Conversely, mIgA did not show any effect on production of those cytokines by NHLFs (Fig. 1b). When time–course analysis was performed, production of the cytokines gradually increased from 24 h and reached a maximum at 72 h (data not shown). Expression levels of mRNA for these cytokines by NHLFs exposed to sIgA started to increase at 2 h and reached statistical significance at 4 h with sIgA at 100–300 μg/ml (Fig. 1c), but mIgA showed no effect (Fig. 1d). We conclude that sIgA, but not mIgA, induced production of IL‐6, IL‐8, MCP‐1 and GM‐CSF by NHLFs.
Figure 1.
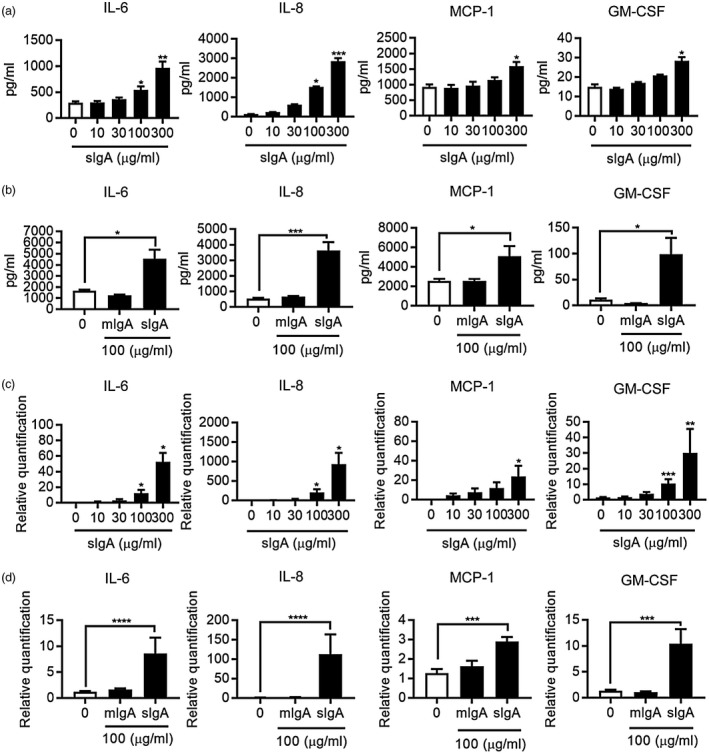
Secretory immunoglobulin A (sIgA) up‐regulates cytokine production by normal human lung fibroblasts (NHLFs). (a,b) NHLFs were cultured in the presence and absence of sIgA or monomeric IgA (mIgA) at the indicated concentrations for 48 h. The levels of interleukin (IL)‐6, IL‐8, monocyte chemoattractant protein (MCP)‐1 and granulocyte macrophage colony‐stimulating factor (GM‐CSF) in the NHLF culture supernatants were determined by cytometric bead array (CBA). Bars represent the mean ± standard error of the mean (s.e.m.) (n = 3 independent experiments). *P < 0·05, ***P < 0·01, ****P < 0·001 versus cells without stimulus. (c,d) NHLFs were cultured in the presence and absence of sIgA or mIgA at the indicated concentrations for 4 h. The relative expression levels of mRNA for IL‐6, IL‐8, MCP‐1 and GM‐CSF were determined by real‐time polymerase chain reaction (PCR) and expressed as the mean ± s.e.m. (n = 3 independent experiments). *P < 0·05; **P < 0·01; ***P < 0·001; ****P < 0.0001 versus cells without stimulus.
Treatment with anti‐SC antibody suppressed cytokine production by NHLFs stimulated with sIgA
sIgA is a J chain‐containing dIgA with an SC; it is different from mIgA. CBA was used to examine whether the SC was critical to activation of NHLFs by sIgA and induction of cytokine production. Anti‐SC inhibited IL‐8 and MCP‐1 production statistically significantly by NHLFs (Fig. 2). IL‐6 production reached statistical significance in terms of its percentage of the control MFI data (data not shown).
Figure 2.
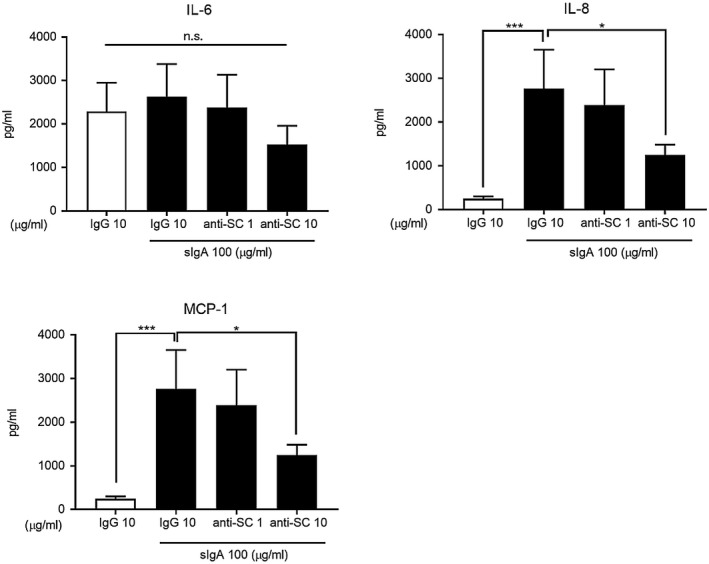
Effect of anti‐IgA secretory component antibody (anti‐SC) treatment on cytokine production by normal human lung fibroblasts (NHLFs) with secretory immunoglobulin A (sIgA) stimulation. NHLFs were cultured for 24 h with sIgA at 0 and 100 µg/ml, followed by incubation with anti‐SC at 0, 1 and 10 µg/ml for 1 h at room temperature. The levels of interleukin (IL)‐6, IL‐8 and monocyte chemoattractant protein (MCP)‐1 in the NHLF culture supernatants were determined by cytometric bead array (CBA). Bars represent the mean ± standard error of the mean (s.e.m.) (n = 3 independent experiments). *P < 0·05; ***P < 0·001 versus cells without stimulus; n.s. = not significant.
sIgA, but not mIgA, enhanced proliferation of NHLFs
Next, we examined effect of IgA on NHLF proliferation using an ELISA kit to measure BrdU incorporation during DNA synthesis. sIgA at 30 and 100 μg/ml significantly enhanced NHLF proliferation, and the effect was as strong as that of TGF‐β1, a well‐known enhancer of fibroblast proliferation 20 (Fig. 3a). However, mIgA at 100 μg/ml did not affect NHLF proliferation (Fig. 3a).
Figure 3.
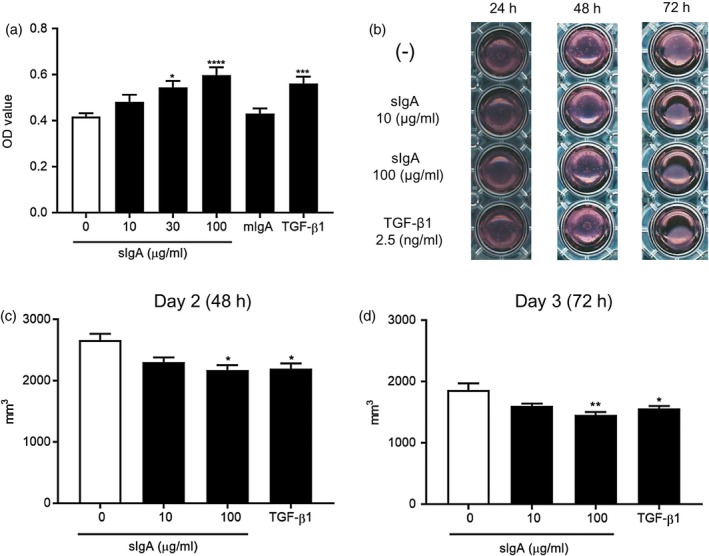
Secretory immunoglobulin A (sIgA) induces proliferation of normal human lung fibroblasts (NHLFs) and collagen gel contraction by NHLFs. (a) NHLFs were cultured with sIgA or monomeric IgA (mIgA) at 100 µg/ml or transforming growth factor (TGF)‐β1 at 10 ng/ml, with bromodeoxyuridine (BrdU) for 16 h, and BrdU incorporation was analyzed using a cell proliferation enzyme‐linked immunosorbent assay (ELISA) kit. Bars represent the mean ± standard error of the mean (s.e.m.) (n = 3 independent experiments). *P < 0·05; ***P < 0·001; ****P < 0·0001 versus cells without stimulus. (b) Representative images of collagen gel contraction by NHLFs in the presence and absence of sIgA or TGF‐β1 at 24, 48 and 72 h. Data are representative of four independent experiments. (c) The gel surface areas in the presence and absence of the indicated concentrations of sIgA or TGF‐β1 at 2·5 ng/ml, at 48 h (left panel) and 72 h (right panel), were measured and analyzed using Image J software. Bars represent the mean ± standard error of mean (s.e.m.) (n = 4 independent experiments). *P < 0·05; **P < 0·01 versus cells without stimulus.
sIgA significantly induced collagen gel contraction by NHLFs
To further evaluate the direct effect of sIgA on NHLF function, we next performed a gel contraction assay to determine NHLFs’ ability to cause contraction of collagen matrices. We found that sIgA, but not mIgA, at 100 μg/ml significantly induced collagen gel contraction by NHLFs, and the effect was as strong as, or even stronger than, that of TGF‐β1 at 2·5 ng/ml (Fig. 3b and c).
NHLFs produced α‐SMA and Col I in response to sIgA, but not mIgA
Activated lung fibroblasts are known to differentiate into myofibroblasts, for which enhanced expression of α‐SMA is frequently used as a qualitative marker. Myofibroblasts play important roles, especially in tissue remodeling, wound healing and other fibrotic disorders, including IPF. Accordingly, we examined production of α‐SMA by NHLFs in response to sIgA. As shown in Fig. 4a and c, incubation with sIgA enhanced synthesis of α‐SMA by NHLFs at both the mRNA and protein levels. When activated myofibroblasts migrate to a damaged tissue site they synthesize extracellular matrix proteins, including collagen. As shown in Fig. 4b and d, sIgA 100 μg/ml also induced Col I production by NHLFs at both mRNA and protein levels; however, mIgA did not influence NHLFs’ production of either α‐SMA or Col I (data not shown).
Figure 4.
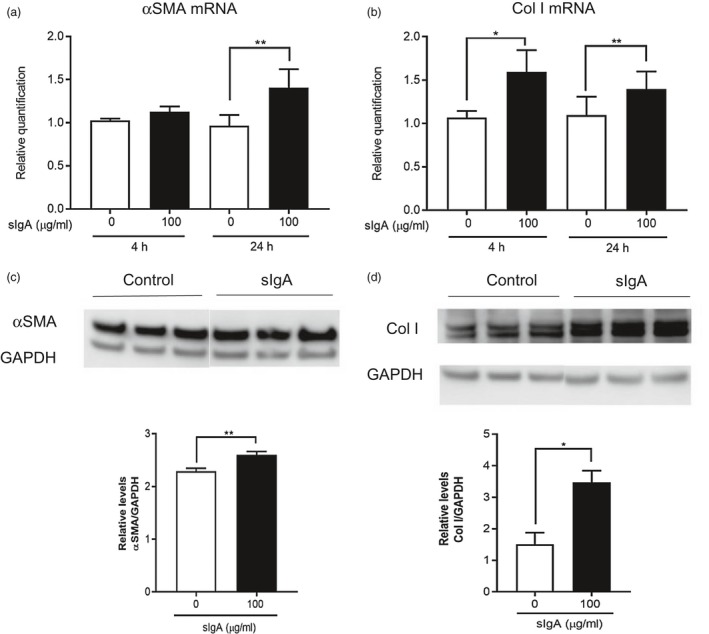
Secretory immunoglobulin A (sIgA) enhances production of α‐smooth muscle actin (SMA) and collagen type 1 (Col I) by normal human lung fibroblasts (NHLFs). (a,b) NHLFs were cultured in the presence and absence of sIgA at the indicated concentrations for 4 h (α‐SMA) and 24 h (Col I). The relative expression levels of mRNA for α‐SMA and Col I were determined by real‐time polymerase chain reaction (PCR) and expressed as the mean ± standard error of the mean (s.e.m.) (n = 5 independent experiments). *P < 0·05; **P < 0·01 versus cells without stimulus. (c,d) NHLFs were cultured in the presence and absence of sIgA at 100 µg/ml for 48 h, and the cell lysates were collected to quantitate α‐SMA (c) and Col I (d) by Western blot analysis. Three independent samples from separate experiments were applied onto each gel for electrophoresis, followed by Western blot analysis. The expression of each was normalized to that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and expressed as the mean ± s.e.m. (n = 3, 11 independent experiments). *P < 0·05; **P < 0·01.
NHLFs bind sIgA on their cell surface and express a known IgA‐receptor, CD71, on their surface
To examine whether sIgA binds directly to NHLFs, we used fluorochrome‐conjugated anti‐IgA antibody and flow cytometry. sIgA at 100 μg/ml showed significant binding to NHLFs (Fig. 5a and b), while binding of mIgA was weaker (data not shown). Based on this result, we next sought to identify the receptor(s) to which sIgA bound. Others reported that more than one receptor can bind IgA 21. Among the known IgA receptors, NHLFs expressed CD71 on their cell surface (Fig. 5c and d) and also at the mRNA level, as assessed by RT–PCR (data not shown). We also confirmed expression of CD71 by cultured NHLFs using immunochemistry (Fig. 5e). Interestingly, CD71 expression on NHLFs was strongly induced by incubation with two inflammatory cytokines, TNF‐α and IL‐1β, which are known to be involved in inflammatory disorders as well as fibroproliferative disorders 22 (Fig. 5f and g). Three other known receptors; namely, CD89 (Fc receptor for IgA, FcαR), ASGPR and CD351 (Fc receptor for IgM and IgA, Fcα/μR), were not detected on NHLFs at either the mRNA (data not shown) or protein level (Fig. 5h). These results suggest that sIgA binds to CD71 expressed on the cell surface of NHLFs.
Figure 5.
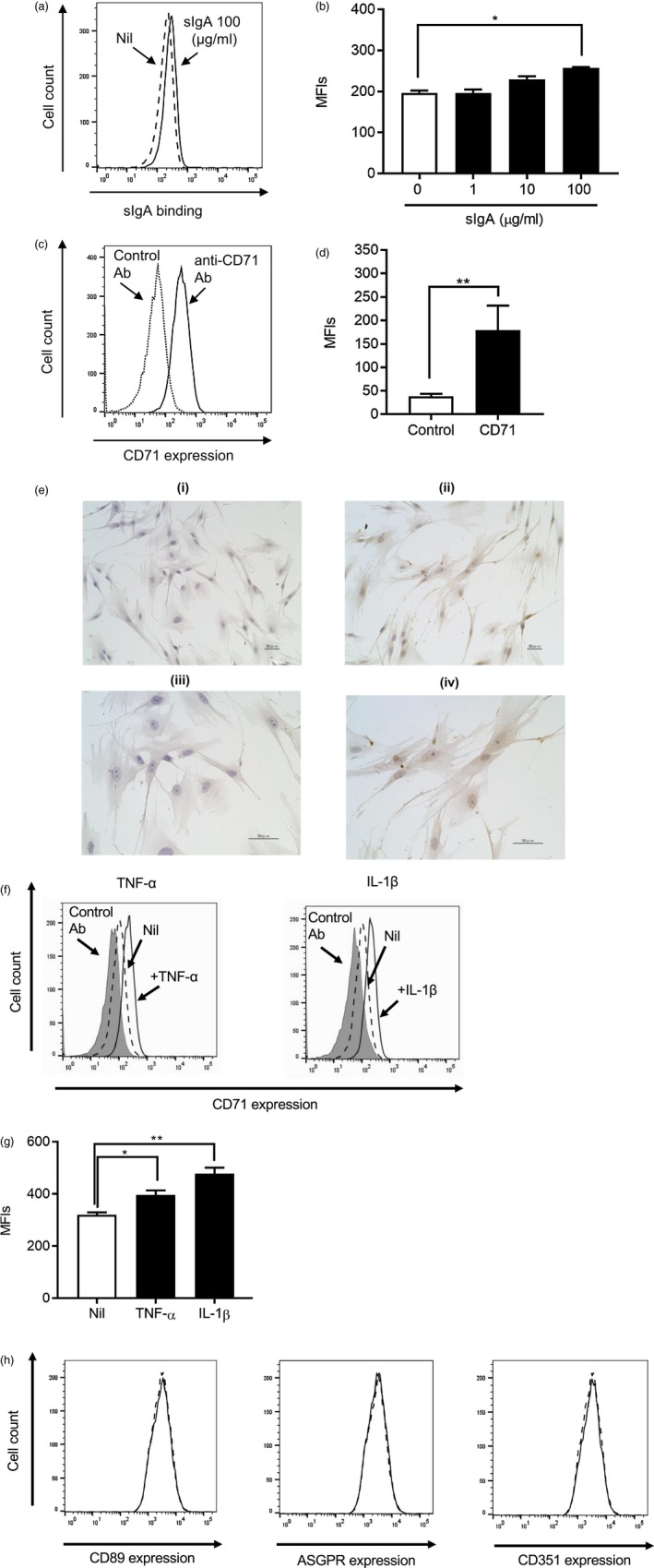
Normal human lung fibroblasts (NHLFs) bind secretory immunoglobulin A (sIgA) and express a known IgA receptor, CD71, on their cell surface. (a,b) NHLFs were incubated with (solid line) and without (dashed line) IgA at the indicated concentrations for 1 h and stained with allophycocyanin (APC)‐conjugated anti‐human IgA antibody or isotype control for analysis by flow cytometry. (a) Representative histograms from three independent experiments are shown, and (b) mean fluorescence intensities (MFIs) from three experiments are expressed as the mean ± standard error of the mean (s.e.m.) (n = 3 independent experiments). *P < 0·05. (c,d) NHLFs were stained with fluorescein isothiocyanate (FITC)‐conjugated anti‐human CD71 monoclonal antibody (mAb) or isotype‐matched control antibody and analyzed by flow cytometry. (c) Representative histograms from five independent experiments are shown, and (d) MFIs from five experiments are expressed as the mean ± s.e.m. (n = 5 independent experiments). **p < 0·01. (e) NHLFs were stained with purified mouse IgG1κ isotype control (left panels) and mouse anti‐human CD71 antibody (right panels). (f,g) NHLFs were incubated with and without tumour necrosis factor (TNF)‐α or interleukin (IL)‐1β at 10 ng/ml for 48 h, and then the expression level of CD71 was analyzed by flow cytometry. (f) Representative histograms from three independent experiments are shown. The dashed lines show the cells without stimulus, and the solid lines show the cells with indicated stimuli. The gray areas show the cells with control stain. (g) MFIs from three experiments in (f) are expressed as the mean ± s.e.m. (n = 3 independent experiments). *P < 0·05; **P < 0·01. (h) NHLFs were stained with phycoerythrin (PE)‐conjugated anti‐human ASGPR1 mAb, anti‐human CD89 mAb, anti‐human CD351 mAb or isotype‐matched control antibody and analyzed by flow cytometry. Representative histograms from three independent experiments are shown. The dashed lines show the cells stained with control antibody, and the solid lines show the cells stained with specific antobody against each receptor.
sIgA affects NHLF functions, at least partly, through CD71
To elucidate the importance of CD71 expressed by NHLFs to the effects of sIgA, we examined whether siRNA knock‐down of CD71 on NHLFs inhibited binding of sIgA. As shown in Fig. 6a, siRNA knock‐down of CD71 was efficient at the protein level (as well as the mRNA level; data not shown), and the level of sIgA bound to NHLFs was reduced by CD71 siRNA transfection (Fig. 6b). We also examined the effect of CD71 siRNA on production of IL‐8 and MCP‐1 by NHLFs stimulated with sIgA. We found that CD71 siRNA partially but significantly suppressed NHLFs’ production of both of those chemokines (Fig. 6c). These results suggest that, at least to some extent, sIgA may activate NHLFs through binding to CD71.
Figure 6.
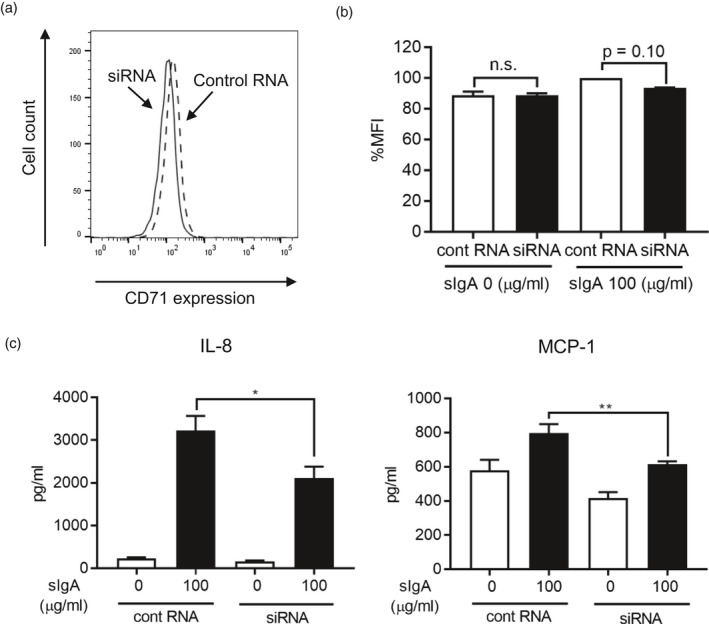
Transfection of siRNA targeting CD71 inhibited IgA binding to normal human lung fibroblasts (NHLFs), and partly reduced up‐regulation of cytokine‐synthesis induced by sIgA. (a) NHLFs were transfected with CD71 siRNA (solid line) or control RNA (dashed line) at 10 nM for 24 h, and the expression level of CD71 on the cell surface is shown. Data are representative of three independent experiments. (b) NHLFs transfected with CD71 siRNA (black bar) or control RNA (white bar) at 10 nM for 24 h were incubated with sIgA at 100 for 1 h and then stained with anti‐human IgA antibody for analysis by flow cytometry. Mean fluorescent intensities (MFIs) are expressed as the mean ± standard error of the mean (s.e.m.) (n = 4 independent experiments); n.s. = not significant. (c) NHLFs transfected with CD71 siRNA or control RNA at 10 nM for 24 h were cultured in the presence and absence of sIgA for 24 and 48 h. The levels of interleukin (IL)‐8 and monocyte chemoattractant protein (MCP)‐1 in NHLF culture supernatants were determined by cytometric bead array (CBA). Bars represent the mean ± s.e.m. (n = 3 independent experiments). *P < 0·05; **P < 0·01.
Discussion
In this study we have demonstrated, by both RT–PCR and CBA, that sIgA, but not mIgA, induced IL‐6, IL‐8, MCP‐1 and GM‐CSF production by NHLFs. sIgA also enhanced proliferation of NHLFs as assessed by cellular incorporation of BrdU and collagen gel contraction, both at levels comparable to with TGF‐β1. We should note that because sIgA is much larger in size than TGF‐β1, its potency on a molar basis is inferior to that of TGF‐β1. Nevertheless, as the biological concentration of sIgA is much higher than that of TGF‐β1, the potency we observed in our experiments is biologically possible. In addition, Western blotting showed that Col I, a component of the extracellular matrix, and α‐SMA, a commonly used marker for myofibroblast differentiation, were also up‐regulated by sIgA. Finally, we demonstrated that sIgA binds to NHLFs, presumably to CD71, known as a transferrin receptor, and that knock‐down of CD71 at least partly inhibited cytokine production by NHLFs.
The protective role of sIgA has long been of interest, whereas other activities of sIgA have not been focused upon, at least in the pathogenesis of lung diseases. For example, patients with selective IgA deficiency, the most common primary immunodeficiency, often develop recurrent sinopulmonary infections and allergic as well as autoimmune diseases 23, suggestive of a definite role for IgA in protection against pathogens and antigens. Conversely, some reports indicated pathophysiological roles for IgA, especially in other organs such as the kidney, liver and intestinal tract 24, 25, 26, 27, suggesting that it may have effects on various human diseases. Those reports led us to hypothesize that IgA may have effects on other human diseases. In order to test that hypothesis we conducted the present study, which generated evidence of a new pathological activity for sIgA. That is, whereas biological levels of sIgA of approximately 10–200 μg/ml in the human epithelial lining fluid 28 induced activation of lung fibroblasts that may be beneficial in tissue repair and regeneration, at the same time such activation may be detrimental in fibroproliferative disorders such as IPF.
Several cytokines are known to act in combination to induce epithelial damage and persistent inflammation, resulting in tissue fibrosis in fibrosing disorders, including IPF, in the lung. For example, IL‐6 plays an important role in induction of leukocyte infiltration during acute inflammation that may lead to fibrosis 29, 30, 31, 32. Similarly, IL‐8 exhibits a wide range of proinflammatory activities, especially in neutrophil‐mediated acute inflammation. IL‐8 and MCP‐1 have been well documented to act as chemotactic factors for neutrophils, monocytes, mast cells, fibrocytes and T helper type 17 (Th17) cells in the lung 33, 34, 35, 36, 37. Accumulation of neutrophils in the alveolitis of IPF is a characteristic feature of that disease 38, and high levels of neutrophil‐origin elastase have been observed in the bronchoalveolar lavage fluid (BALF) of IPF patients 39. Taken together, sIgA may enhance both inflammation and fibrosis in the lung by inducing IL‐6, IL‐8 and MCP‐1 production by NHLFs.
However, the roles of GM‐CSF in tissue repair and fibrosis remain controversial. GM‐CSF deficiency reportedly resulted in enhanced fibrogenesis in a bleomycin‐induced pulmonary fibrosis model in mice, suggesting a protective role for GM‐CSF 40. Conversely, GM‐CSF stimulated keratinocyte proliferation 41, and over‐expression of GM‐CSF in the lung resulted in lung fibrosis and inflammation by recruiting macrophages and eosinophils into the lung, indicating a pathogenic role for GM‐CSF in lung fibrosis 42. The roles of GM‐CSF may differ in humans compared to mice, and also may depend on the inflammatory status of each individual. However, as GM‐CSF is known to recruit granulocytes and macrophages to sites of inflammation 43, sIgA‐induced GM‐CSF production by NHLFs may accelerate the pathogenesis of IPF.
Fibroblasts in IPF, i.e. activated fibroblasts and myofibroblasts, are identified by high expression levels of α‐SMA and have distinct properties: high ability to proliferate and to produce extracellular matrix, proteases and fibrogenic cytokines and contractile activity 44, 45. Therefore, differentiation of fibroblasts to myofibroblasts is thought to be the key step in the development of tissue fibrosis. Our present study demonstrated that sIgA enhanced expression of α‐SMA by NHLFs, indicating that sIgA might induce myofibroblast differentiation. Our data further support the importance of sIgA for NHLF activation and differentiation by showing that it induced proliferation, collagen gel contraction and Col I production by NHLFs. In particular, the effects of sIgA on proliferation and collagen gel contraction are interesting because they were comparable to those of TGF‐β1, one of the most potent fibrogenic cytokines.
Activation of fibroblasts is essential for tissue repair and regeneration as well as for maintenance of biological homeostasis, and here we may have identified another protective role of sIgA. Conversely, tissue fibrosis is a critical pathogenic step in various lung diseases, including IPF, pulmonary hypertension 46, asthma 47 and chronic obstructive pulmonary disease (COPD) 48. Among those diseases, IPF is a chronic, progressive lung disease with a poor prognosis, characterized by fibroblast proliferation and extracellular matrix deposition. Accumulating evidence shows that IPF results from an excessive wound‐healing process that leads to aberrant fibrosis in response to alveolar epithelial injury 13. The main steps in the pathogenesis of IPF may be migration, proliferation and activation of lung fibroblasts and their differentiation into myofibroblasts, induced mainly by TGF‐β 49. In the present study, we showed that sIgA is another potent inducer of fibroblast activation. Because the airway surface as well as the surfaces of the intestines and urogenital tract are always anatomically exposed to the external environment, the function of sIgA observed in the present study may have developed to protect against tissue damage caused by invading pathogens and antigens.
Under normal conditions, lung fibroblasts are separated from airway mucus by an epithelial cell layer having firm tight‐junctions 50. However, in the initiation of lung fibrosis in IPF, deprivation of alveolar epithelial cells with basement membrane denudation is reported to occur, and the damaged area often overlies fibroblastic foci 51, 52. We confirmed that fibroblastic foci in the IPF lung had sites where a stretch of epithelial cells had collapsed – as assessed by absence of cells stained by anti‐cytokeratin antibody – and was replaced by cells positively stained by anti‐vimentin antibody (Supporting information, Fig. S1). Therefore, under certain conditions such as IPF, sIgA in airway mucus may make contact with lung fibroblasts. In addition, although the origin of accumulated fibroblasts in IPF has not been fully determined, epithelial–mesenchymal transition (EMT) was suggested to be one possible mechanism 53. In EMT, airway epithelial cells located adjacent to airway lumen acquire mesenchymal features 54. Epithelial cells that had undergone EMT and acquired characteristics similar to those of mesenchymal cells may respond to sIgA, in the same manner as the NHLFs we used in our experiments. Especially under conditions with increased levels of IgA, such as when a respiratory infection co‐exists, lung fibroblasts may come into direct contact with sIgA in the airway mucus, resulting in exacerbated airway inflammation and fibrosis 4, 55.
The effects of sIgA on NHLFs were sIgA‐specific, as mIgA showed no effects. The molecular importance of the SC and the different roles of mIgA and sIgA have been reported 56, 57. The SC may be the crucial molecule in the process of sIgA’s activation of NHLFs in the present setting. Indeed, we found that anti‐SC antibody treatment significantly inhibited cytokine production by NHLFs (Fig. 2). However, it must be noted that a serum IgA level in which the majority exists in the form of mIgA was reported to be a prognostic marker for IPF 10. Although we did not detect any pathogenic role or mIgA on activation of NHLFs, the level of mIgA may be linked to the level of sIgA because both mIgA and dIgA, a component of sIgA, are produced by plasma cells. Moreover, sIgA is especially present in the blood in certain conditions such as inflammatory diseases 58, 59.
We also found that CD71, a transferrin receptor, was at least partially responsible for the effects of sIgA on NHLFs. However, it does not seem to be the only receptor involved, as its knock‐down did not completely eliminate sIgA’s binding to NHLFs or its effects on cytokine production by NHLFs. Also, because CD71 is a critical receptor for human life, as it is required for iron import into cells 60 and is essential for cell maintenance, targeting CD71 to abrogate the effects of sIgA on certain fibrogenic diseases does not seem realistic. Further investigation of possible involved receptors and the intracellular signaling pathway is warranted.
In summary, we have demonstrated that sIgA, but not mIgA, induced various responses of NHLFs: IL‐6, IL‐8, MCP‐1 and GM‐CSF production, proliferation and collagen gel contraction. sIgA was also able to enhance Col I and α‐SMA production by NHLFs, and we showed that it binds to NHLFs, at least partly via CD71. Overall, our experiments demonstrate the important role of sIgA in activation of fibroblasts which may be due partially to its binding to CD71. However, additional – as yet undetermined – receptors may also be involved in the pathway and need to be elucidated.
Disclosure
The authors have no conflicts of interest to declare.
Author contributions
S. A., M. S., K. K., H. M., H. N., T. N. and K. O. designed the experiments. S. A. and K. W. performed the experiments. S. A., M. S. and K. O. wrote the paper.
Supporting information
Fig. S1. An example of mesenchymal cells directly facing the airway lumen in the human lung with fibrosis.After deparaffinization of lung sections followed by antigen retrieval at pH 9 for 30 min,the sections were incubated with mouse anti‐cytokeratin monoclonal antibody (IgG1, clone AE1/AE3, Nichirei, Tokyo, Japan) (a, b) or anti‐vimentin monoclonal antibody (IgG2a, κ, clone Vim 3B4, Dako, CA, USA) (c, d) at 37ºC for 1 h.The primary antibody was visualized using the Histofine Simple Stain PO kit (Nichirei) according to the manufacturer’s instructions. Sections were developed with DAB, and images were captured using a fluorescent microscope BZ‐X710 (Keyence, Osaka, Japan).
Acknowledgements
The authors thank Drs Hiroyuki Tashimo, Osamu Narumoto and Akira Hebisawa for participating in fruitful discussions throughout the research. The authors also thank Ms Sayaka Igarashi, Mrs Isao Asari and Kenji Urata for their skilled technical assistance and Ms Mariko Yoshizawa for her efficient secretarial assistance. We further thank Lawrence W. Stiver (Quality Translation Co., Ltd, Tokyo, Japan; qualityt@gol.com) for his careful reading of our manuscript. This work was supported by JSPS KAKENHI Grant no. JP 17K09629 to M.S.
Contributor Information
M. Suzukawa, Email: suzukawam@tokyo-hosp.jp
K. Ohta, Email: fueta-tky@umin.ac.jp
References
- 1. Mestecky J, Russell MW, Jackson S, Brown TA. The human IgA system: A reassessment. Clin Immunol Immunopathol 1986; 40:105–14. [DOI] [PubMed] [Google Scholar]
- 2. Stockley RA, Mistry M, Bradwell AR, Burnett D. A study of plasma proteins in the sol phase of sputum from patients with chronic bronchitis. Thorax 1979; 34:777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Newkirk MM, Klein MH, Katz A, Fisher MM, Underdown BJ. Estimation of polymeric IgA in human serum: An assay based on binding of radiolabeled human secretory component with applications in the study of IgA nephropathy, IgA monoclonal gammopathy, and liver disease. J Immunol (Balt) 1983; 130:1176–81. [PubMed] [Google Scholar]
- 4. Stockley RA, Afford SC, Burnett D. Assessment of 7S and 11S immunoglobulin A in sputum. Am Rev Respir Dis 1980; 122:959–64. [DOI] [PubMed] [Google Scholar]
- 5. Puchelle E, Jacqot J, Zahm JM. In vitro restructuring effect of human airway immunoglobulins A and lysozyme on airway secretions. Eur J Respir Dis Suppl 1987; 153:117–22. [PubMed] [Google Scholar]
- 6. Abu‐Ghazaleh RI, Fujisawa T, Mestecky J, Kyle RA, Gleich GJ. IgA‐induced eosinophil degranulation. J Immunol (Balt) 1989; 142:2393–400. [PubMed] [Google Scholar]
- 7. Iikura M, Yamaguchi M, Fujisawa T et al Secretory IgA induces degranulation of IL‐3‐primed basophils. J Immunology (Balt) 1998; 161:1510–5. [PubMed] [Google Scholar]
- 8. Bartemes KR, Cooper KM, Drain KL, Kita H. Secretory IgA induces antigen‐independent eosinophil survival and cytokine production without inducing effector functions. J Allergy Clin Immunol 2005; 116:827–35. [DOI] [PubMed] [Google Scholar]
- 9. Diven SC, Caflisch CR, Hammond DK, Weigel PH, Oka JA, Goldblum RM. IgA induced activation of human mesangial cells: Independent of FcalphaR1 (CD 89). Kidney Int 1998; 54:837–47. [DOI] [PubMed] [Google Scholar]
- 10. ten Klooster L, van Moorsel CH, Kwakkel‐van Erp JM, van Velzen‐Blad H, Grutters JC. Immunoglobulin A in serum: An old acquaintance as a new prognostic biomarker in idiopathic pulmonary fibrosis. Clin Exp Immunol 2015; 181:357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borsutzky S, Cazac BB, Roes J, Guzmán CA. TGF‐β receptor signaling is critical for mucosal IgA responses. J Immunol 2004; 173:3305–9. [DOI] [PubMed] [Google Scholar]
- 12. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 2007; 117:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakai N, Tager AM. Fibrosis of two: Epithelial cell–fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta 2013; 1832:911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378:1949–61. [DOI] [PubMed] [Google Scholar]
- 15. Moldt B, Saye‐Francisco K, Schultz N, Burton DR, Hessell AJ. Simplifying the synthesis of SIgA: Combination of dIgA and rhSC using affinity chromatography. Methods 2014; 65:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakota Y, Ozawa Y, Yamashita H, Tanaka H, Inagaki N. Collagen gel contraction assay using human bronchial smooth muscle cells and its application for evaluation of inhibitory effect of formoterol. Biol Pharm Bull 2014; 37:1014–20. [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi T, Liu X, Kim HJ et al TGF‐beta1 and serum both stimulate contraction but differentially affect apoptosis in 3D collagen gels. Respir Res 2005; 6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith RS, Fedyk ER, Springer TA, Mukaida N, Iglewski BH, Phipps RP. IL‐8 production in human lung fibroblasts and epithelial cells activated by the pseudomonas autoinducer N‐3‐oxododecanoyl homoserine lactone is transcriptionally regulated by NF‐κB and activator protein‐2. J Immunol 2001; 167:366–74. [DOI] [PubMed] [Google Scholar]
- 19. Azghani AO, Neal K, Idell S et al Mechanism of fibroblast inflammatory responses to Pseudomonas aeruginosa elastase. Microbiology 2014; 160:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sheppard D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc 2006; 3:413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woof JM, Kerr MA. The function of immunoglobulin A in immunity. J Pathol 2006; 208:270–82. [DOI] [PubMed] [Google Scholar]
- 22. Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta 2013; 1832:979–88. [DOI] [PubMed] [Google Scholar]
- 23. Yel L. Selective IgA deficiency. J Clin Immunol 2010; 30:10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tumlin JA, Lohavichan V, Hennigar R. Crescentic, proliferative IgA nephropathy: Clinical and histological response to methylprednisolone and intravenous cyclophosphamide. Nephrol Dialysis Transpl 2003; 18:1321–9. [DOI] [PubMed] [Google Scholar]
- 25. Matysiak‐Budnik T, Moura IC, Arcos‐Fajardo M et al Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med 2008; 205:143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lebreton C, Menard S, Abed J et al Interactions among secretory immunoglobulin A, CD71, and transglutaminase‐2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology 2012; 143:698–707 e4. [DOI] [PubMed] [Google Scholar]
- 27. Oortwijn BD, van der Boog PJ, Roos A et al A pathogenic role for secretory IgA in IgA nephropathy. Kidney Int 2006; 69:1131–8. [DOI] [PubMed] [Google Scholar]
- 28. van de Graaf EA, Out TA, Kobesen A, Jansen HM. Lactoferrin and secretory IgA in the bronchoalveolar lavage fluid from patients with a stable asthma. Lung 1991; 169:275–83. [DOI] [PubMed] [Google Scholar]
- 29. Fielding CA, Jones GW, McLoughlin RM et al Interleukin‐6 signaling drives fibrosis in unresolved inflammation. Immunity 2014; 40:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fielding CA, McLoughlin RM, McLeod L et al IL‐6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol (Balt) 2008; 181:2189–95. [DOI] [PubMed] [Google Scholar]
- 31. Hurst SM, Wilkinson TS, McLoughlin RM et al Il‐6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001; 14:705–14. [DOI] [PubMed] [Google Scholar]
- 32. McLoughlin RM, Witowski J, Robson RL et al Interplay between IFN‐gamma and IL‐6 signaling governs neutrophil trafficking and apoptosis during acute inflammation. J Clin Invest 2003; 112:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ribeiro RA, Flores CA, Cunha FQ, Ferreira SH. IL‐8 causes in vivo neutrophil migration by a cell‐dependent mechanism. Immunology 1991; 73:472–7. [PMC free article] [PubMed] [Google Scholar]
- 34. Lee YG, Jeong JJ, Nyenhuis S et al Recruited alveolar macrophages, in response to airway epithelial‐derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol 2015; 52:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collington SJ, Hallgren J, Pease JE et al The role of the CCL2/CCR35 axis in mouse mast cell migration in vitro and in vivo . J Immunology (Balt) 2010; 184:6114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singh SR, Sutcliffe A, Kaur D et al CCL2 release by airway smooth muscle is increased in asthma and promotes fibrocyte migration. Allergy 2014; 69:1189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang A, Wang Z, Cao Y et al CCL2/CCR37‐dependent recruitment of Th17 cells but not Tc17 cells to the lung in a murine asthma model. Int Arch Allergy Immunol 2015; 166:52–62. [DOI] [PubMed] [Google Scholar]
- 38. Hunninghake GW, Gadek JE, Lawley TJ, Crystal RG. Mechanisms of neutrophil accumulation in the lungs of patients with idiopathic pulmonary fibrosis. J Clin Invest 1981; 68:259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Obayashi Y, Yamadori I, Fujita J, Yoshinouchi T, Ueda N, Takahara J. The role of neutrophils in the pathogenesis of idiopathic pulmonary fibrosis. Chest 1997; 112:1338–43. [DOI] [PubMed] [Google Scholar]
- 40. Moore BB, Coffey MJ, Christensen P et al GM‐CSF regulates bleomycin‐induced pulmonary fibrosis via a prostaglandin‐dependent mechanism. J Immunol (Balt) 2000; 165:4032–9. [DOI] [PubMed] [Google Scholar]
- 41. Kaplan G, Walsh G, Guido LS et al Novel responses of human skin to intradermal recombinant granulocyte/macrophage‐colony‐stimulating factor: Langerhans cell recruitment, keratinocyte growth, and enhanced wound healing. J Exp Med 1992; 175:1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xing Z, Ohkawara Y, Jordana M, Graham F, Gauldie J. Transfer of granulocyte‐macrophage colony‐stimulating factor gene to rat lung induces eosinophilia, monocytosis, and fibrotic reactions. J Clin Invest 1996; 97:1102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shiomi A, Usui T. Pivotal roles of GM‐CSF in autoimmunity and inflammation. Mediators Inflamm 2015; 2015:568543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007; 132:1311–21. [DOI] [PubMed] [Google Scholar]
- 45. Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol 1991; 138:1257–65. [PMC free article] [PubMed] [Google Scholar]
- 46. Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: Remodelling, rarefaction or angiogenesis? J Anat 2002; 201:335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1989; 1:520–4. [DOI] [PubMed] [Google Scholar]
- 48. Salazar LM, Herrera AM. Fibrotic response of tissue remodeling in COPD. Lung 2011; 189:101–9. [DOI] [PubMed] [Google Scholar]
- 49. Fernandez IE, Eickelberg O. The impact of TGF‐beta on lung fibrosis: From targeting to biomarkers. Proc Am Thorac Soc 2012; 9:111–6. [DOI] [PubMed] [Google Scholar]
- 50. Nishioka M, Venkatesan N, Dessalle K et al Fibroblast–epithelial cell interactions drive epithelial–mesenchymal transition differently in cells from normal and COPD patients. Respir Res 2015; 16:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: A new therapeutic pathway in fibrosing lung disease? Trends Mol Med 2016; 22:303–16. [DOI] [PubMed] [Google Scholar]
- 52. Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J 2008; 32:1631–8. [DOI] [PubMed] [Google Scholar]
- 53. Chapman HA. Epithelial–mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol 2011; 73:413–35. [DOI] [PubMed] [Google Scholar]
- 54. Kevin K, Matthias CK, Paul JW et al Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci 2006; 103:13180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stockley RA, Burnett D. Local IgA production in patients with chronic bronchitis: Effect of acute respiratory infection. Thorax 1980; 35:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonetti PJ, Corthesy B. Secretory component: A new role in secretory IgA‐mediated immune exclusion in vivo. Immunity 2002; 17:107–15. [DOI] [PubMed] [Google Scholar]
- 57. Reterink TJF, Schroeijers WEM, Van Es LA, Daha MR. Dimeric and polymeric IgA, but not monomeric IgA, enhance the production of IL‐6 by human renal mesangial cells. Mediat Inflamm 1996; 5:191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mestecky J, McGhee JR. Immunoglobulin A (IgA): Molecular and cellular interactions involved in IgA biosynthesis and immune response. Adv Immunol 1987; 40:153–245. [DOI] [PubMed] [Google Scholar]
- 59. Delacroix D, Vaerman JP. Reassessment of levels of secretory IgA in pathological sera using a quantitative radioimmunoassay. Clin Exp Immunol 1981; 43:633–40. [PMC free article] [PubMed] [Google Scholar]
- 60. Graham RM, Chua AC, Herbison CE, Olynyk JK, Trinder D. Liver iron transport. World J Gastroenterol 2007; 13:4725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. An example of mesenchymal cells directly facing the airway lumen in the human lung with fibrosis.After deparaffinization of lung sections followed by antigen retrieval at pH 9 for 30 min,the sections were incubated with mouse anti‐cytokeratin monoclonal antibody (IgG1, clone AE1/AE3, Nichirei, Tokyo, Japan) (a, b) or anti‐vimentin monoclonal antibody (IgG2a, κ, clone Vim 3B4, Dako, CA, USA) (c, d) at 37ºC for 1 h.The primary antibody was visualized using the Histofine Simple Stain PO kit (Nichirei) according to the manufacturer’s instructions. Sections were developed with DAB, and images were captured using a fluorescent microscope BZ‐X710 (Keyence, Osaka, Japan).