

Abstract
Background:
Severe Cushing’s syndrome (SCS) is associated with acute cardiovascular, metabolic and infectious complications. It is considered an emergency, requiring an immediate diagnosis, together with a broad spectrum of supportive and hypocortisolaemic treatments. Surgical intervention, aimed at removing the source of cortisol or adrenocorticotropic hormone (ACTH), is the optimal treatment in most cases of Cushing’s syndrome. However, in hypercortisolaemic states, surgical intervention has high rates of perioperative mortality and morbidity. Oral adrenal steroidogenesis inhibitors, even if more effective in combination, are not always efficient enough or well tolerated. Despite their common use, a more potent, parental, immediate, and thus life-saving, therapy is necessary.
Methods:
The authors present three different clinical scenarios of etomidate treatment in patients hospitalized in the third reference endocrinological centre in Poland between 2016 and 2017.
Results:
Patients with Cushing’s disease, ectopic Cushing’s syndrome and adrenocortical carcinoma presented with severe hypercortisolaemia and exacerbated cortisol-dependent comorbidities. In these three cases, etomidate acted as an accurate, well tolerated and effective cortisol-lowering drug for several days or even months. Patients were monitored in a general ward setting, and no side effects of the therapy were observed.
Conclusions:
In doses far lower than those used for anaesthesia, etomidate works as a useful cortisol-lowering therapy in patients intolerant to or unable to take oral medications. Additionally, if urgent, the most potent and effective medical intervention is necessary, and clinicians should be aware of such a therapeutic option.
Keywords: adrenal steroidogenesis inhibitors, adrenocortical carcinoma, Cushing’s disease, ectopic Cushing’s syndrome, etomidate, hypercortisolaemia, pituitary corticotropinoma
Introduction
Cushing’s syndrome (CS) is characterized by a chronic excess of cortisol, and in its overt form, it includes demonstrable clinical features. Excessive cortisol may result from endogenous or exogenous sources, with the latter being the first to exclude.
The most common cause of endogenous CS is Cushing’s disease with the incidence of 1.2–2.4 cases per million people per year.1 However, in 10–15% of cases, adrenocorticotropic hormone (ACTH)-dependent CS is caused by nonpituitary tumours (ectopic CS; ECS).2 ACTH-independent CS, caused by adrenal cortisol-secreting neoplasms or hyperplasia, accounts for 15–20% of all cases. It is important to note that hypercortisolaemia (alone or together with excessive androgens) is also the most frequent hormonal hypersecretion associated with adrenocortical carcinoma (ACC).3
Severe CS (SCS) is characterized by massively elevated random serum cortisol levels [>36–41 μg/dl (1000–1100 nmol/l) at any time] or a 24-h urinary free cortisol that is more than four-fold higher than the upper limit of the normal range or severe hypokalaemia (<3.0 mmol/l); SCS is generally associated with the recent onset of one or more of the following: sepsis, opportunistic infection, intractable hypokalaemia, uncontrolled hypertension, heart failure, gastrointestinal haemorrhage, glucocorticoid-induced acute psychosis, progressive debilitating myopathy, thromboembolism or uncontrolled hyperglycaemia and ketoacidosis.4 SCS is considered an emergency situation, requiring immediate supportive and hypocortisolaemic therapy. Such treatment has to take precedence over investigations into the cause and localization of CS, which can be indeterminable and time-consuming.
Initial resection of the primary lesion underlying CS of all aetiologies is the treatment of choice.5 In patients with overt CS, cortisol-lowering therapy, as well as adjunctive treatment for cortisol-related comorbidities, is needed both pre- and postoperatively. Chronic severe hypercortisolaemia is associated with immunosuppression, thus predisposing the individual to life-threatening infections or sepsis of bacterial, fungal and opportunistic pathogens. These complications can be especially unfavourable and be a barrier to initiating or continuing the often ‘last-chance’ option for patients with malignant tumours, chemotherapy.6 Moreover, hypercortisolism alters coagulation-factor profiles, thus resulting in an increased risk of cardiovascular events, such as myocardial infarction, stroke or venous thromboembolism.7
Etomidate is the only medical treatment available for seriously ill patients who are not candidates for immediate surgery and cannot take oral medications. Etomidate exerts its action via reversible inhibition of 11-beta-hydroxylase, CYP17A1 and cholesterol side-chain cleavage enzyme.8,9 It can be used to quickly alleviate severe hypercortisolism and thus be the bridge to other medical or surgical therapies if the patient’s condition improves.
Patients and methods
We describe three cases of recent etomidate treatment. All patients were hospitalized bet-ween 2016 and 2017 at the Department of Endocrinology, Bielański Hospital in Warsaw, which is the third reference endocrinological centre in Poland.
Ethics statement: This case series did not need approval from an ethical committee, as it is simply a retrospective review of case notes with no additional intervention or other investigation.
Informed consent for publication
Informed written consent for the publication of patient information in the present manuscript was obtained from Patient no. 2. Because Patient no. 1 and Patient no. 3 died before the preparation of the manuscript, informed written consent was obtained from legally authorized representatives.
Case series description and results
Case 1
A 23-year-old female with rapidly progressing CS due to metastatic ACC was admitted to the Department of Endocrinology. She had undergone right-sided adrenalectomy 2 years before with no additional mitotane treatment. Imaging revealed a mass of 65 × 40 mm in the side of the previous resection and massive liver metastases (Figure 1). In addition to the second palliative surgery for the recurrent tumour, hypocortisolaemic treatment was introduced as soon as she had been diagnosed with the relapse of ACC. Ketoconazole at a dose of 800 mg daily and mitotane at a dose of 6 g per day were provided, with poor clinical and biochemical effects. Thus, in the next step, metyrapone at a dose of 750 mg daily was added. At the subsequent fourth admission, her state had substantially declined with uncontrolled diabetes and hypertension, severe hypokalaemia and hypocalcaemia. She also presented the symptoms of an upper respiratory tract infection. Her cortisol levels were as high as 90–128.2 µg/dl. Due to the complete lack of any effect, the use of oral steroidogenesis inhibitors (ketoconazole and metyrapone) was discontinued. The patient was put on a continuous intravenous infusion of propylene glycol etomidate preparation (Hypnomidate), followed by a decrease in cortisol level to approx. 40 µg/dl. After one week of treatment, the patient was well enough to be referred to the Oncology Department for the first course of chemotherapy. Therefore, etomidate was stopped. At 8 days later, she was admitted again with post-chemotherapy, life-threatening leukopaenia, agranulocytosis, anaemia and diarrhoea with a recurrence of electrolyte disturbances and a rapid increase in cortisol levels. Thus, the infusion of etomidate was administered again. Unfortunately, computed tomography imaging showed rapid progression of the disease. Therefore, she was disqualified from the next course of chemotherapy and died within 2 months.
Figure 1.
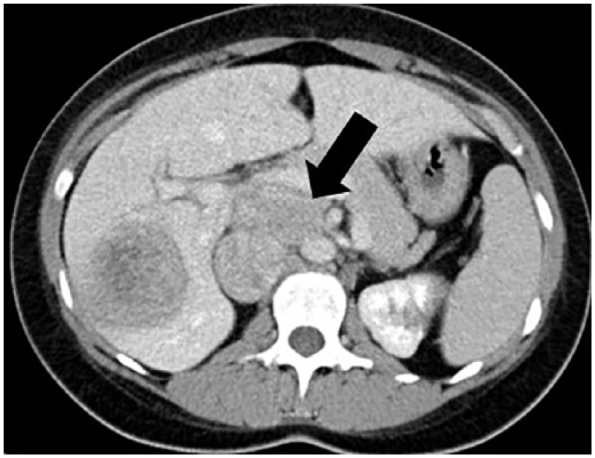
Abdomen computed tomography of a young female patient with florid Cushing’s syndrome (2 years after initial surgery with no subsequent mitotane treatment), showing relapsed adrenocortical carcinoma (65 × 40 mm, black arrow) despite previous resection, with massive liver metastasis also present.
Case 2
A 45-year-old male with a history of recently diagnosed insulin-independent diabetes mellitus, refractory hypokalaemia, myopathy, spinal pain and mild cushingoid features was admitted to the Department of Endocrinology. A hormonal evaluation was performed, revealing ACTH-dependent CS. The corticotropin-releasing factor stimulation test was suggestive for pituitary adenoma. However, the MRI of the pituitary gland was not evident; moreover, an abdominal computed tomography scan showed a suspicious lesion in his pancreas. The next investigations were planned, but, due to severe hypokalaemia, we had to start treatment with ketoconazole at an initial dose of 400 mg per day. We observed an elevation in the transaminases, which made it risky to increase the ketoconazole dose. On the 6th day of ineffectual oral therapy, the patient’s clinical condition deteriorated with fever and severe exhaustion. The patient was diagnosed with Staphylococcus aureus sepsis, and antibiotics were introduced. Despite ketoconazole ingestion, the patient’s cortisol level reached 160 µg/dl. Therefore, etomidate-lipuro was given intravenously with a consequent rapid lowering of hormone concentrations. With short breaks of a few to several hours to perform localization studies (endoscopic ultrasound, somatostatin receptor scintigraphy, positron emission tomography), the etomidate infusion was continued for 50 days, as the diagnostic process turned out to be longer than expected. During that time, we observed a gradual improvement in the patient’s condition. To our puzzlement, eventually, bilateral inferior petrosal sinus sampling helped to confirm the diagnosis of pituitary corticotropinoma (Figure 2) and the patient underwent effective trans-sphenoidal surgical resection of the tumour.
Figure 2.
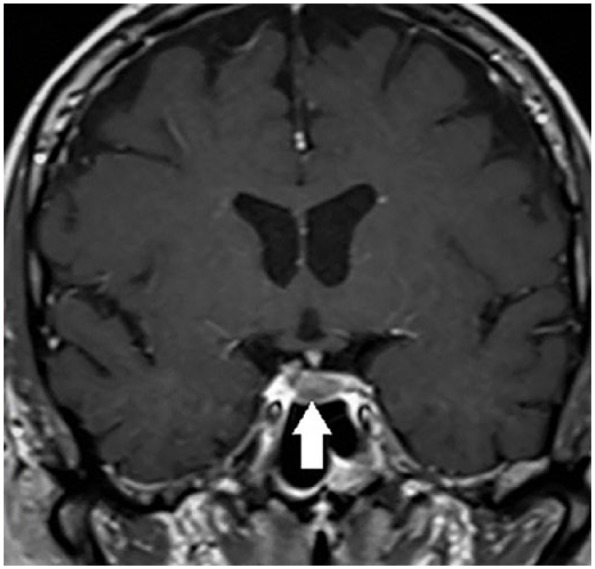
Magnetic resonance imaging of the pituitary region, showing microcorticotropinoma (white arrow) in a young male patient with severe Cushing’s syndrome.
Case 3
A 66-year-old female with ectopic CS due to a metastatic lung carcinoid with liver lesions underwent a left adrenalectomy three months earlier as the first step of complete adrenal gland removal and was referred to the Department of Endocrinology with severe hypercortisolaemia, decompensation of diabetes, severe hypokalaemia and mood disorders (Figure 3). Upon admission, she was negative and aggressive, revealing the decision to discontinue the use of all her oral drugs a few weeks earlier (ketoconazole and antipsychotics among others). As she was mentally and physically unstable, she was put on an Etomidate-Lipuro infusion with an immediate improvement in her status. However, her cortisol levels were unsteady, although we were unsure about increasing the dose of etomidate. Thus, we decided to add low-dose ketoconazole to the etomidate, although it did not stabilize her cortisol levels. This lack of stabilization was probably because the course of her hospitalization was repeatedly complicated, with cephalic vein thrombosis, candidiasis, sepsis and post-antibiotic enterocolitis pseudomembranacea. Fortunately, after 58 days of etomidate treatment, she was able to undergo complete adrenalectomy without further complications.
Figure 3.
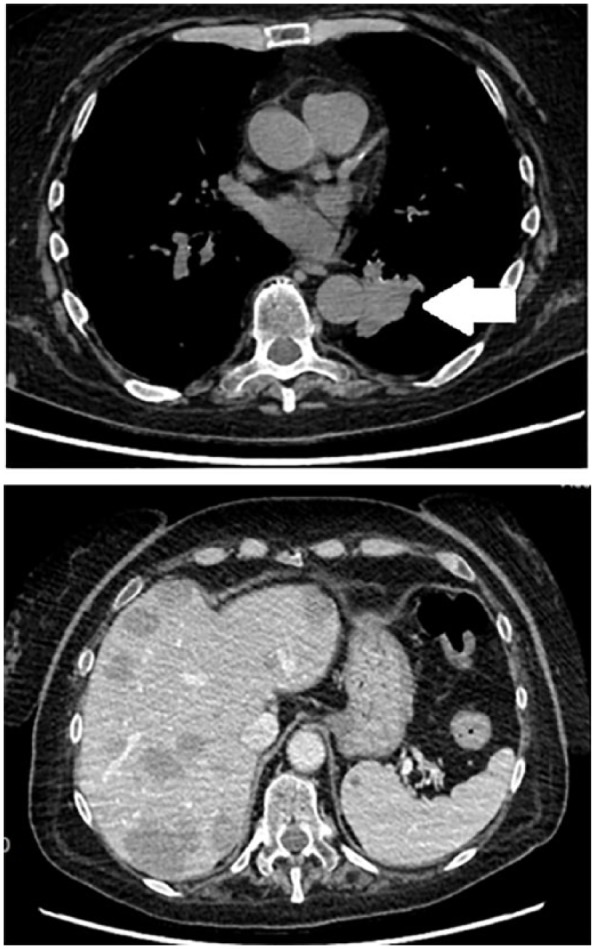
Chest and abdomen computed tomography scanning of metastatic, inoperable ACTH-secreting lung carcinoid (white arrow) in a 66-year-old female with severe ectopic Cushing’s syndrome.
ACTH, adrenocorticotropic hormone.
Discussion
As an anaesthetic, etomidate has gained increasing popularity due to its ideal pharmacokinetics (fast onset of action and short duration of hypnotic effect), favourable cardiovascular profile, and respiratory and central nervous system effects. These observations promoted its use in a wide variety of indications in adult and paediatric populations.10 However, with evidence of reversible and dose-dependent adrenocortical suppression, another field of etomidate usage has emerged.
For CS, surgical removal of the source of cortisol or ACTH excess is the first-line treatment. Occasionally, in cases of ACTH-dependent CS, bilateral adrenalectomy has to be considered.11,12 Medical therapeutic options for CS include agents that block adrenal steroidogenesis at various enzymatic steps, with ketoconazole and metyrapone being the most commonly used. However, these agents in monotherapy have been proven effective in only half of patients13; metyrapone starts its action within hours, but ketoconazole takes days or even weeks to have an effect.14 Conventional cortisol-lowering treatment is usually ineffective in the setting of very severe hypercortisolaemia. Combination therapy may be useful in some cases and can also lower individual drug doses and, consequently, the possibility of adverse effects.15–17 The use of mitotane with its specific properties is mainly limited to cases of ACC. Recent data suggest that cortisol-secreting ACCs have the poorest prognosis, whether in an advanced or localized stage.18 Although mitotane is the medical treatment of choice, its slowly emerging antisecretory effect means that other agents are necessary to achieve more rapid and effective control of hypercortisolism.19 In the study by Corcuff and colleagues,15 metyrapone at a dose of 750–4000 mg per day and ketoconazole at a dose of 800–1200 mg per day were prescribed to patients with ECS and ACC with severe hypercortisolaemia. Mitotane at a dose of 1500–6000 mg per day was introduced simultaneously or later, ‘as some patients could hardly ingest the total amount of pellets prescribed’. The combinative treatment resulted in decreased urinary free cortisol within 1 week of treatment and the control of hypercortisolism at 1 month in 73% and 86% of patients with ECS and ACC, respectively. In 2 patients out of 22 (9%), plasma transaminase elevations resulted in ketoconazole withdrawal. In the work of Claps and colleagues,17 three patients with metastatic or inoperable ACC and full-blown CS were treated with a combination of metyrapone, mitotane and chemotherapy (etoposide, doxorubicin and cisplatin, EDP-M scheme). In all three patients, the therapy resulted in marked clinical improvement and urinary free cortisol decrease when carried out for 8, 12 and 16 weeks (with a few days of metyrapone interruption due to nausea and asthenia in one patient). Despite the encouraging data presented above, combinative therapy of mitotane, ketoconazole and metyrapone was completely ineffective in the presented young female patient with ACC (Case 1). With rapid deterioration of her general condition and the onset of the infection, we were uncertain that, even in increased doses, it would not act as fast as needed. Thus, etomidate presents the only option left for patients in whom combinative treatment was ineffective, too slow or withdrawn. Especially in patients with ACC, some authors observed worse effects of combination therapy in normalizing kalaemia, which may be the result of the production of different mineralocorticoid precursors from the tumour.20 It has been suggested that etomidate may also have anti-proliferative and anti-tumourigenic effects on adrenal cortical cells, as well as metastatic adrenal tumours.21 However, no studies have compared etomidate with other, combined or not, oral therapies according to its efficacy or safety profiles. In addition, the data regarding the ineffectiveness of etomidate is sparse. In the already mentioned study by Corcuff and colleagues,15 15% of patients displayed adrenal insufficiency as a result of the combinative therapy, so it may be assumed that every treatment potent enough to result in substantial cortisol reduction may end in this complication. For this reason, care should be taken in each situation in order to implement hydrocortisone replacement therapy on time. Moreover, the ‘block and replace’ protocol can be the desired therapy mode. It is aimed at reducing cortisol to minimally detectable levels with concomitant glucocorticoid replacement therapy. There are some considerable risks, for example imperfect substitution (with supraphysiological doses of hydrocortisone being implemented or, contrarily, doses too small for the ongoing stress), as well as the fact that higher doses or multiple drugs may be necessary. However, this strategy may be preferable if significant cyclicity in cortisol levels is present.5 In our series, this strategy was not used. ‘Blocking’ etomidate infusion speed rates are higher than ‘nonblocking’, although with a high level of inter-study variability and inconsistency.22 This finding may indicate the necessity to maximally individualize the choice of strategy and dose to the clinical scenario and the patient’s reaction to the drug(s). Thus, one may be aware of the danger of the induction of sedation with ‘blocking’ doses.
Recent guidelines5 suggest that a loading dose of 3–5 mg should be followed by a continuous infusion of 0.03–0.10 mg/kg/h (~2.5–3.0 mg/h). It is supposed to effectively reduce serum cortisol within 12 h with the dose titration according to serum cortisol (to a maximum dose of 5 mg/h in selected cases). However, in many cases, etomidate was proven to be effective in smaller doses. In the case report by Castilla and colleagues,23 an infant with CS resistant to ketoconazole was effectively treated with etomidate at a dose of 0.02–0.03 mg/kg/h. Reza-Albarran and colleagues24 proposed starting with higher doses of etomidate (0.04 mg/kg/h) and then gradually decreasing the dosage to maintain an infusion rate of 0.01 mg/kg/h. In our patients [Figure 4(a–c)], we used a loading bolus of 2.5 milligrams and an infusion of 0.01–0.02 mg/kg/h (1–2 mg/h), which increased by 0.5 mg/h to slowly and gradually let the patient adjust to lower levels of cortisol and to let the receptors regain normal sensitivity. We up-titrated the dose to a maximum of 5 mg/h in one case. This is not the first time such regimen has been proposed.25 It should be noted that no side effects of etomidate were observed during long-term treatment in any of our patients. Studies have reported no sedative symptoms in doses smaller than 0.1–0.3 mg/kg/h (<7–8 mg/h).26,27 Although the current recommendations emphasize the need for close monitoring of patients undergoing etomidate treatment, it may be mostly true in the situations in which greater doses of etomidate are needed, the general condition of patient requires close supervision or due to co-administration of a sedative or tranquilizing medications. Our experience thus supports the point of view of Soh and colleagues25 to conduct, or at least continue, etomidate treatment in the general ward setting. The two patients (Cases 1 and 2) had etomidate introduced in the intensive care unit (ICU) with 24-h monitoring for possible side effects. The continuation of the infusion was carried out in the general ward without any side effects. A patient (Case 3) was given etomidate in the Department of Endocrinology, with the assistance of an anaesthesiologist. This situation was imposed by the circumstances, because the ICU lacked available critical care beds.
Figure 4.

Serum cortisol levels and etomidate doses in the three presented cases. (a) In Case 1, etomidate was introduced in a bolus of 2.5 mg, and the infusion velocity was gradually increased to reach 1 mg/h in the first 24 h and a maximum of 2.8 mg/h on day 8. The cortisol levels fell adequately. (b) In Case 2, the dosing regimen was similar to that in Case 1, with the infusion speed rates presented in the table. The target cortisol levels were reached in a week and, as the patient’s state improved, the infusion velocity was increased to achieve more physiological levels. (c) In Case 3, the presented fluctuating levels of cortisol were probably due to several complications during hospitalization and day-by-day alterations in the patients’ condition. Higher doses of etomidate (up to 5 mg/h) were necessary to control cortisol overproduction.
Some data have reported that prolonged infusion of etomidate can be successfully carried out for several weeks, if a patient’s condition imposes such necessity. In the case report by Krakoff and colleagues,28 the patient was kept on an infusion for 5.5 months, because of an inability to take oral drugs (due to parenteral nutrition). The main uncertainty of the authors applied to the solvent, propylene glycol, which is known to cause nephrotoxicity as well as thrombophlebitis and pain of injection. With the lipid formula, these side effects are reduced, and it can even be given via a peripheral route.25 The longest reported use of lipid formula is 56 days.29 Krakoff and colleagues warned of a possible prolonged effect of etomidate on cortisol suppression after an infusion lasting for a few months and the possibility of developing adrenal insufficiency after discontinuation of the drug. They explain this effect by etomidate re-equilibrating to the plasma compartment of the adipose tissue storage. In our patients, etomidate-lipuro was given for 50 and 58 days, and we did not observe any negative effects of the prolonged etomidate treatment.
Conclusion
Our experience presented in this case series supports the concept of etomidate as a useful and well tolerated cortisol-lowering therapy. Etomidate constitutes one of the most potent and almost always effective drugs for severe hypercortisolaemia. Despite its potency, when administered skilfully and cautiously, the infusion of etomidate can be carried out, even in the general ward setting, for the required periods of time. In several situations, for example in severe or critically ill patients who are intolerant of or unable to take oral medications, clinicians of different specialties should be aware of such a therapeutic option.
Acknowledgments
A. Łebek-Szatańska prepared, analyzed and interpreted data and wrote the manuscript. L. Papierska, W. Zgliczyński and E. Baum analyzed and interpreted data and revised the manuscript. K. Nowak and A. Żyłka prepared data for the manuscript and revised the manuscript. All authors approved the final version of the manuscript and take full responsibility for its content.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
ORCID iD: Agnieszka Łebek-Szatańska  https://orcid.org/0000-0002-3804-0273
https://orcid.org/0000-0002-3804-0273
Contributor Information
Agnieszka Łebek-Szatańska, Department of Endocrinology, Centre of Postgraduate Medical Education, Bielański Hospital in Warsaw, Cegłowska 80 Street, 01-809 Warsaw, Poland.
Karolina M. Nowak, Department of Endocrinology, Centre of Postgraduate Medical Education, Bielański Hospital in Warsaw, Poland
Wojciech Zgliczyński, Department of Endocrinology, Centre of Postgraduate Medical Education, Bielański Hospital in Warsaw, Poland.
Elżbieta Baum, Intensive Care Unit, Bielański Hospital in Warsaw, Poland.
Agnieszka Żyłka, Department of Endocrinological Oncology and Nuclear Medicine, The Maria Skłodowska Curie Cancer Centre and Institute of Oncology, Warsaw, Poland.
Lucyna Papierska, Department of Endocrinology, Centre of Postgraduate Medical Education, Bielański Hospital in Warsaw, Poland.
References
- 1. Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing’s disease. Endocr Rev 2015; 36: 385–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Isidori AM, Kaltsas GA, Pozza C, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab 2006; 91: 371–377. [DOI] [PubMed] [Google Scholar]
- 3. Puglisi S, Perotti P, Pia A, et al. Adrenocortical carcinoma with hypercortisolism. Endocrinol Metab Clin North Am 2018; 47: 395–407. [DOI] [PubMed] [Google Scholar]
- 4. Alexandraki KI, Grossman AB. Therapeutic strategies for the treatment of severe Cushing’s syndrome. Drugs 2016; 76: 447–458. [DOI] [PubMed] [Google Scholar]
- 5. Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing’s syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2015; 100: 2807–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malandrino P, Ghuzlan AA, Castaing M, et al. Prognostic markers of survival after combined mitotane- and platinum-based chemotherapy in metastatic adrenocortical carcinoma. Endocr Relat Cancer 2010; 17: 797–807. [DOI] [PubMed] [Google Scholar]
- 7. Pas R van der, Leebeek FWG, Hofland LJ, et al. Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol 2013; 78: 481–488. [DOI] [PubMed] [Google Scholar]
- 8. Fleseriu M, Castinetti F. Updates on the role of adrenal steroidogenesis inhibitors in Cushing’s syndrome: a focus on novel therapies. Pituitary 2016; 19: 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hahner S, Stürmer A, Fassnacht M, et al. Etomidate unmasks intraadrenal regulation of steroidogenesis and proliferation in adrenal cortical cell lines. Horm Metab Res 2010; 42: 528–534. [DOI] [PubMed] [Google Scholar]
- 10. Erdoes G, Basciani RM, Eberle B. Etomidate – a review of robust evidence for its use in various clinical scenarios. Acta Anaesthesiol Scand 2014; 58: 380–389. [DOI] [PubMed] [Google Scholar]
- 11. Castinetti F, Morange I, Conte-Devolx B, et al. Cushing’s disease. Orphanet J Rare Dis 2012; 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guerin C, Taieb D, Treglia G, et al. Bilateral adrenalectomy in the 21st century: when to use it for hypercortisolism? Endocr Relat Cancer 2016; 23: 131–142. [DOI] [PubMed] [Google Scholar]
- 13. Nieman LK. Recent updates on the diagnosis and management of Cushing’s syndrome. Endocrinol Metab 2018; 33: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab 2014; 99: 1623–1630. [DOI] [PubMed] [Google Scholar]
- 15. Corcuff JB, Young J, Masquefa-Giraud P, et al. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole. Euro J Endocrinol 2015; 172: 473–481. [DOI] [PubMed] [Google Scholar]
- 16. Kamenický P, Droumaguet C, Salenave S, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2011; 96: 2796–2804. [DOI] [PubMed] [Google Scholar]
- 17. Claps M, Cerri S, Grisanti S, et al. Adding metyrapone to chemotherapy plus mitotane for Cushing’s syndrome due to advanced adrenocortical carcinoma. Endocrine 2018; 61: 169–172. [DOI] [PubMed] [Google Scholar]
- 18. Nowak KM, Samsel R, Cichocki A, et al. Prognostic factors in adrenocortical carcinoma: data from a large Polish series. Pol Arch Intern Med 2018; 128: 371–378. [DOI] [PubMed] [Google Scholar]
- 19. Puglisi S, Perotti P, Pia A, et al. Adrenocortical carcinoma with hypercortisolism. Endocrinol Metab Clin North Am 2018; 47: 395–407. [DOI] [PubMed] [Google Scholar]
- 20. Arlt W, Biehl M, Taylor AE, et al. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab 2011; 96: 3775–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fassnacht M, Hahner S, Beuschlein F, et al. New mechanisms of adrenostatic compounds in a human adrenocortical cancer cell line. Euro J Clin Invest 2000; 30(Suppl. 3): 76–82. [DOI] [PubMed] [Google Scholar]
- 22. Preda VA, Sen J, Karavitaki N, et al. Therapy in endocrine disease: etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Euro J Endocrinol 2012; 168: X1–X1. [DOI] [PubMed] [Google Scholar]
- 23. Yun Castilla C, Rodríguez Amuedo F, Morales Martínez A, et al. Usefulness of ethomidate in patients with Cushing syndrome with severe arterial hypertension and hypopotassemia. Med Intensiva 2017; 41: 321–322. [DOI] [PubMed] [Google Scholar]
- 24. Reza-Albarrán AA, Andino Ríos GG, Gómez Herrera LG. Etomidate in the control of severe Cushing’s syndrome by neuroendocrine carcinoma. Clin Case Rep 2018; 6: 851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soh LM, Gunganah K, Akker SA, et al. Etomidate in the emergency management of hypercortisolemia. Euro J Endocrinol 2012; 167: 727–728. [DOI] [PubMed] [Google Scholar]
- 26. Allolio B, Schulte HM, Kaulen D, et al. Nonhypnotic low-dose etomidate for rapid correction of hypercortisolaemia in Cushing’s syndrome. Klin Wochenschr 1988; 66: 361–364. [DOI] [PubMed] [Google Scholar]
- 27. Schulte HM, Benker G, Reinwein D, et al. Infusion of low dose etomidate: correction of hypercortisolemia in patients with Cushing’s syndrome and dose-response relationship in normal subjects. J Clin Endocrinol Metab 1990; 70: 1426–1430. [DOI] [PubMed] [Google Scholar]
- 28. Krakoff J, Koch CA, Calis KA, et al. Use of a parenteral propylene glycol-containing etomidate preparation for the long-term management of ectopic Cushing’s syndrome. J Clin Endocrinol Metab 2001; 86: 4104–4108. [DOI] [PubMed] [Google Scholar]
- 29. Drake WM, Perry LA, Hinds CJ, et al. Emergency and prolonged use of intravenous etomidate to control hypercortisolemia in a patient with Cushing’s syndrome and peritonitis. J Clin Endocrinol Metab 1998; 83: 3542–3544. [DOI] [PubMed] [Google Scholar]