

Abstract
BPSM1 (Bone phenotype spontaneous mutant 1) mice develop severe polyarthritis and heart valve disease as a result of a spontaneous mutation in the Tnf gene. In these mice, the insertion of a retrotransposon in the 3' untranslated region of Tnf causes a large increase in the expression of the cytokine. We have found that these mice also develop inducible bronchus‐associated lymphoid tissue (iBALT), as well as nodular lymphoid hyperplasia (NLH) in the bone marrow. Loss of TNFR1 prevents the development of both types of follicles, but deficiency of TNFR1 in the hematopoietic compartment only prevents the iBALT and not the NLH phenotype. We show that the development of arthritis and heart valve disease does not depend on the presence of the tertiary lymphoid tissues. Interestingly, while loss of IL‐17 or IL‐23 limits iBALT and NLH development to some extent, it has no effect on polyarthritis or heart valve disease in BPSM1 mice.
Keywords: Arthritis; BPSM1; heart valve disease; iBALT; IL‐17; IL‐23; NLH; nodular lymphoid hyperplasia; tertiary lymphoid organs, bronchus‐associated; TNF
Constitutive TNF overexpression predisposes mice to rheumatoid arthritis and heart valve disease, and causes development of tertiary lymphoid organs in the lungs and bone marrow. We show that the development of arthritis and heart valve disease does not depend on the presence of the tertiary lymphoid tissues, and that loss of IL‐17 or IL‐23 limits tertiary lymphoid organ development while having development no effect on polyarthritis or heart valve disease.

Introduction
Tertiary lymphoid organs (TLO) such as bronchus‐associated lymphoid tissue (BALT) are follicular aggregates containing primarily B and T cells.1 Unlike secondary lymphoid organs such as Peyer's patches and nasal‐associated lymphoid tissue, the development of BALT is not preprogrammed and commences after birth. While BALT is common in rabbits and rats, it is virtually absent from healthy humans and mice.1 BALT is often found in the peri‐vascular space next to bronchi, but it can also develop in small airspaces that are apparently not adjacent to an artery or an airway. BALT can be induced in a number of pathological situations such as chronic inflammation, infection or autoimmunity, and for this reason is often referred to as inducible BALT (iBALT). While some of these clusters are only poorly organized aggregates containing mostly B cells, others have a complex structure containing B cells, T cells, follicular dendritic cells and specialized stroma, reminiscent of the lymphoid follicles found in secondary lymphoid organs. While iBALT appears to be the most common TLO, lymphocytic nodules have also been described in the joints of rheumatoid arthritis patients and in the salivary glands of Sjögren's syndrome patients.2 The presence of lymphocyte nodules in the bone marrow has been associated with systemic autoimmune diseases, chronic myeloproliferative disorders, and viral infection, and it can be benign or neoplastic. The causes underlying TLO development have not been completely elucidated but a number of chemokines (CXCL13, CCL19, CCL21) and their receptors (CXCR5, CCR7) have been implicated in the development of iBALT.1
In BPSM1 (Bone phenotype spontaneous mutant 1) mice, the insertion of a retrotransposon into the 3' unstranslated region (3' UTR) of the Tnf gene results in the generation of a mRNA lacking several motifs that are responsible for the posttranscriptional control of its stability.3 This mutation is dominant and causes Tnf mRNA to accumulate. As a result, TNF protein is overexpressed and found at very high levels in the serum of these mice, which develop severe polyarthritis and heart valve disease.3 Interestingly, iBALT is present in the lungs of all BPSM1 mice two weeks after birth and persists throughout their life. Similarly, all BPSM1 mice have benign lymphoid nodules in the bone marrow, first observed between four (BPSM1 m/m ) and six (BPSM1 +/m ) weeks after birth. No TLO was observed in the joints or salivary glands of BPSM1 mice. We have generated a number of compound mutant mice on the BPSM1 background, and report here the consequences of these additional mutations on the development of TLO. Remarkably, the development of polyarthritis and heart valve disease appears to be entirely independent of the presence of TLO.
Results
BPSM1 mice develop iBALT and nodular lymphoid hyperplasia
BPSM1 mice have a mutation in the Tnf gene causing constitutive overproduction of TNF, particularly from monocyte/macrophage populations.3 Heterozygous BPSM1 m/+ animals show the first overt symptoms of arthritis around 10 weeks of age. They put on weight more slowly than their wildtype (WT) littermates, and their arthritis gradually becomes more severe until they become paralyzed around 200 days. Homozygote BPSM1 m/m animals display clear signs of severe arthritis by 2 weeks of age and become paralyzed between 4 and 6 weeks after birth. Histological analysis also revealed the presence of valvular inflammation in the heart of these mice, a condition whose severity depends on the genetic background and proved fatal in BPSM1‐BALB/c mice.3 We noticed that the lungs of all affected BPSM1 mice contained lymphocyte aggregates not only in close proximity to major airways, but also in places quite distant from them (Figure 1a). These lymphocyte foci were not observed in the first 5 days after birth but were clearly present in BPSM1 m/+ mice at postnatal day 10, and were a distinctive feature of all mutant animals thereafter (not shown). The development of iBALT in BPSM1 mice distinctly preceded the development of arthritis or heart disease symptoms. Homozygote BPSM1 m/m animals had significantly higher levels of circulating TNF than BPSM1 m/+ littermates, and their arthritis and heart disease was much more severe.3 In the lungs, lymphocyte foci were larger in size in BPSM1 m/m animals than in BPSM1 m/+ animals, but not more numerous (Figure 1a, see also Figure 3).
Figure 1.
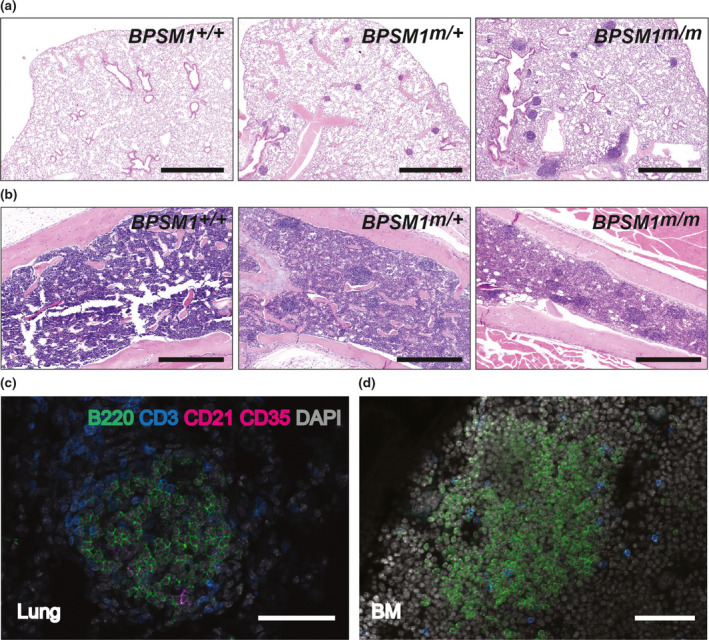
Spontaneous iBALT and NLH in BPSM1 mice. (a) Lung tissue stained with Hematoxylin and Eosin (H&E) showing iBALT in BPSM1 m/+ and BPSM1 m/m mice. Scale bars, 1 mm. (b) H&E‐stained femoral bone marrow showing spontaneous nodular lymphoid hyperplasia in BPSM1 m/+ and BPSM1 m/m mice. Scale bars, 0.5 mm. (c) Confocal image showing the presence B cells (B220), T cells (CD3) and follicular dendritic cells (CD21/CD35) in iBALT of BPSM1 m/+ mice. Scale bar, 50 μm. (d) Confocal image showing the presence of B cells and T cells in bone marrow lymphoid nodules of BPSM1 m/+ mice. Scale bar, 50 μm.
Figure 3.
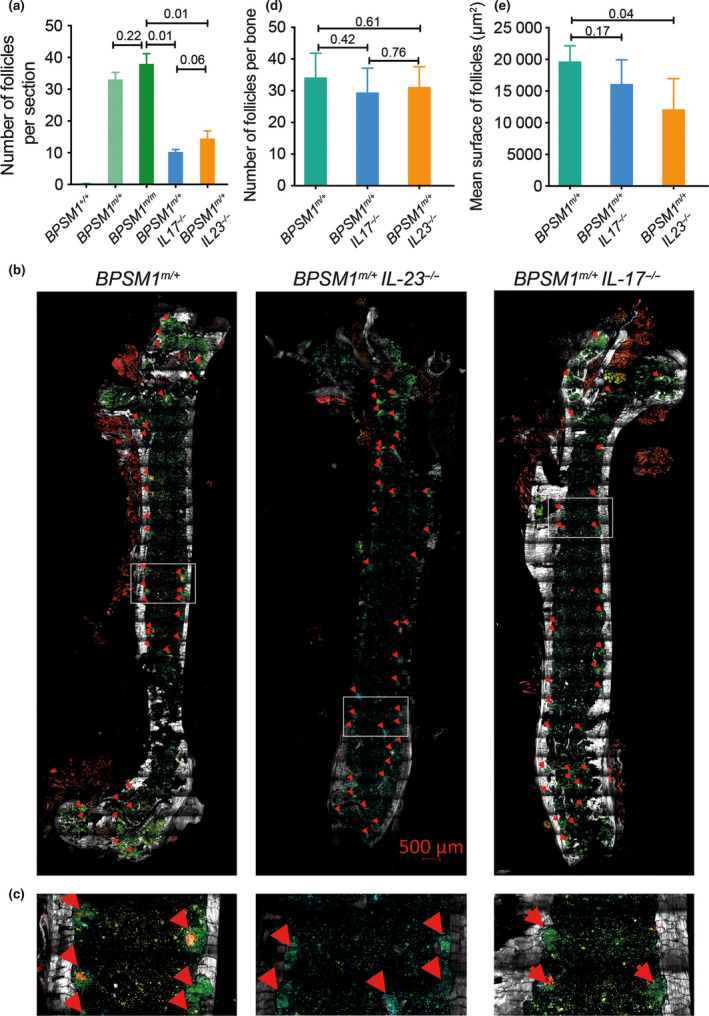
Loss of IL‐17 or IL‐23 influences iBALT and NLH formation in BPSM1 mice. (a) Quantification of iBALT follicles on H&E‐stained lung sections from WT , BPSM1 m/+ , BPSM1 m/m , BPSM1 m/+ IL‐23 −/− and BPSM1 m/+ IL‐17 −/− animals (5 sections per animal, n ≥ 5 animals for each genotype, mean ± s.d.). (b) Dual confocal/2‐photon microscopy showing the presence B cells (B220, green), T cells (CD3, blue), collagen (grey) and follicular dendritic cells (CD21, CD35, red) in nondecalcified femoral bone marrow of BPSM1 m/+ , BPSM1 m/+ IL‐17 −/− and BPSM1 m/+ IL‐23 −/− animals. Red arrows indicate lymphocyte follicles. (c) Magnified picture of the highlighted area in the white rectangle in b. (d) Total number of follicles/section in BPSM1 m/+ , BPSM1 m/+ IL‐17 −/− and BPSM1 m/+ IL‐23 −/− animals as determined from 2 sections of 2 mice per genotype. (e) Mean surface of follicles was calculated by adding the surface of each iBALT detected and dividing by the number of follicles per section. P‐values were calculated using a two‐tailed Student's t‐test performed using Prism (GraphPad) to determine statistical significance.
Distinctive lymphocyte nodules were also present in the bone marrow of all BPSM1 mice. These nodules appeared around 6 weeks after birth in heterozygote BPSM1 m/+ animals. In homozygote BPSM1 m/m animals, the nodules appeared around 4 weeks after birth and were more numerous than in BPSM1 m/+ animals (Figure 1b, see also Figure 3). Immunohistochemical analysis showed that these lung and bone marrow follicles contained predominantly B cells, a feature quite common in iBALT,1 but that T cells were also present (Figure 1c, d). In addition, follicular dendritic cells were associated with iBALT but absent from the bone marrow nodules (Figure 1c, d). Nodular lymphoid hyperplasia (NLH) in the bone marrow has been reported in human patients and is associated with various chronic inflammatory conditions and blood neoplasms.4, 5, 6 In contrast, there are very few reports of these benign lymphoid follicles in mouse bone marrow. Interestingly, while iBALT was present in CCR7‐deficient mice as reported,7 no bone marrow nodules were present in these animals (Supplementary figure 1a).
Development of TLO in the salivary glands and arthritic joints have been reported to be associated with chronic inflammation.2 However, we never observed TLO in either of these locations in BPSM1 m/+ or BPSM1 m/m mice, even at a very advanced stage of disease (not shown).
We conclude that BPSM1 mice develop lymphocytic aggregates in the lungs and bone marrow, and that the size of these aggregates appears to be related to the level of serum TNF present in these mice and their age (Figures 1, 3).
iBALT and NLH require the presence of B cells, but not T cells
Certain models of polyarthritis clearly require the presence of B and/or T cells as drivers of the pathology. In order to determine the role of B and T cells in the pathology of BPSM1 mice, we crossed them with Mb1‐cre knockin8 and CD3ε knockout9 mice, respectively. The absence of B cells (due to the homozygous knockin of the Mb1‐cre allele) or T cells (due to the loss of CD3ε) did not change the kinetics or severity of arthritis and heart disease in BPSM1 mice.3 In contrast, loss of B cells prevented the development of TLO in both the lungs and the bone marrow of BPSM1 m/+ Mb1‐cre KI/KI mice (Figure 2a), whereas lymphocyte follicles were still present in both these locations in T cell‐deficient animals (in BPSM1 m/+ CD3ε −/− mice) (Figure 2b). This is consistent with the fact that B cells are the prominent cell type in iBALT and NLH and indicates that T cells are not required for the organization of these structures. Nevertheless, T cells likely participate in generating a robust immune response in iBALT.1, 2 Arthritis and heart disease developed independently from the presence of B cells in BPSM1 m/+ Mb1‐cre KI/KI animals, indicating that iBALT and NLH play no part in these diseases in the BPSM1 model.
Figure 2.
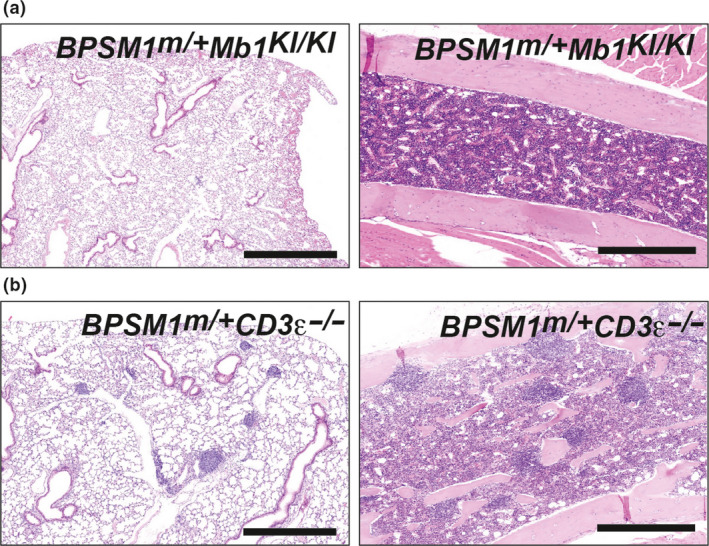
Loss of B cells prevents iBALT and NLH, loss of T cells does not. (a) H&E‐stained lung tissue (left hand panels) and femoral bone marrow (right hand panels) from a) BPSM1 m/+ Mb1 Ki/Ki and (b) BPSM1 m/+ CD3ε −/− mice. Scale bars, 1 mm for lung panels, 0.5 mm for bone marrow panels.
We have previously reported that loss of Myd88 or GM‐CSF did not influence the development of arthritis in BPSM1 mice.3 Similarly, no difference in iBALT and NLH was noticed in BPSM1 m/+ Myd88 −/− and BPSM1 m/+ GMCSF −/− animals (Supplementary figure 1b, c). In contrast, loss of TNFR1 was sufficient to completely prevent development of arthritis and heart disease in BPSM1 mice. Indeed, even homozygote BPSM1 m/m animals had no sign of these diseases in the absence of TNFR1 at 1 year of age, despite having very high levels of TNF in their blood.3 Likewise, iBALT and NLH were completely absent in these animals (Supplementary figure 1d), indicating that TNFR1, but not TNFR2, is required for the development of TLO in BPSM1 mice.
TNFR1 is required for iBALT and NLH development
To further dissect the requirement of TNFR1 in the establishment of TLO, arthritis and heart disease, we performed bone marrow reconstitution experiments. All WT C57BL/6 Ly5.1 recipient mice were lethally irradiated (2× 5.5 Gy) at 6 weeks of age, then injected with 2 × 106 bone marrow cells of the indicated genotypes and examined 5 months posttransplant unless otherwise indicated. BPSM1 m/+ recipients were irradiated at 4 weeks of age before they developed any sign of arthritis and injected with 2 × 106 WT bone marrow cells.
WT mice that received BPSM1 m/+ bone marrow failed to develop iBALT but had bone marrow nodules that were larger than those of BPSM1 m/+ mice of the corresponding age. Additionally, these mice developed arthritis and heart valve disease (Table 1 and Supplementary figure 2a, b). These results suggest that the niche that allows iBALT development in BPSM1 m/+ mice is established early postnatally in response to high TNF levels, but that high TNF levels are not sufficient to establish the niche in older mice. In contrast, BPSM1 m/+ bone marrow alone was sufficient for the development of NLH. WT recipients of BPSM1 m/m bone marrow also did not develop iBALT, although rare lymphocyte aggregates could sometimes be observed loosely attached to major airways (Supplementary figure 2c). These mice developed numerous large lymphocyte follicles in their bone marrow (Supplementary figure 2d). It is important to note that, because of severe arthritis, these animals could not be examined any later than two months posttransplant. It is therefore possible that the few aggregates observed in the lungs of BPSM1 m/m into WT chimeric mice would have developed into more well‐organized iBALT if the reconstitution had lasted 5 months.
Table 1.
Summary of the results of bone marrow reconstitution experiments
| Donor/Recipient | iBALT | BM NLH | Arthritis | HVD |
|---|---|---|---|---|
| BPSM1 m/+ to WT (5 month) | NO | YES, increased | YES | YES |
| WT to BPSM1 m/+ (5 month) | YES | NO or very reduced | NO | NO |
| BPSM1 m/+ to TNFR1 −/− (5 month) | NO | NO | NO | NO |
| BPSM1 m/m to WT (2 month) | NO | YES | YES | YES |
| BPSM1 m/m TNFR1 −/− to WT (2 month) | NO | YES | YES | YES |
The development of iBALT, bone marrow NLH, arthritis and heart valve disease (HVD) was examined following transplantation of 2 × 106 donor bone marrow cells to lethally‐irradiated recipients. At least five recipients were examined in each transplantation experiment. Note that recipients of BPSM1 m/m and BPSM1 m/m TNFR1 −/− bone marrow were analysed 2 months after transplant because of severe arthritis.
BPSM1 m/+ mice that received WT bone marrow still had iBALT 5 months after the transplant (Table 1 and Supplementary figure 2e, f), despite having normal TNF levels in their serum.3 In contrast, bone marrow nodules were rare or absent in these mice. These animals did not develop arthritis or heart valve disease. This indicates that the iBALT niche established early in BPSM1 m/+ animals is still active after irradiation and is also capable of attracting WT cells in the presence of normal levels of circulating TNF. The near‐complete absence of bone marrow lymphocytic nodules in these mice indicated the efficiency of the reconstitution, and the requirement of high levels of hematopoietically‐derived TNF for the development of NLH. Importantly, both these points were corroborated by the absence of arthritis and heart disease in BPSM1 m/+ recipients of WT bone marrow.
TNFR1 −/− recipients of BPSM1 +/− bone marrow had neither iBALT nor NLH (Table 1 and Supplementary figure 2g, h), and did not develop arthritis or heart disease. While no conclusion can be made about iBALT development from this experiment since WT recipients of BPSM1 m/+ bone marrow also had no iBALT, this result suggests a requirement for TNFR1 in non‐hematopoietic cells for the development of NLH, since the transplant of BPSM1 m/+ marrow to irradiated WT recipients led to large lymphocyte nodules in the bone marrow. It thus appears that the establishment of the niche that allows NLH development requires the participation of nonhematopoietic cells expressing TNFR1.
To determine whether TNFR1 was needed in the hematopoietic compartment for the development of iBALT and NLH, we transplanted the bone marrow of BPSM1 m/m TNFR1 −/− donors into irradiated WT recipients and examined the mice 2 months later (again, time‐limited because of the development of severe arthritis). Only a few poorly organized lymphocyte aggregates could be observed in these animals (Table 1 and Supplementary figure 2). In contrast, and despite the fact that BPSM1 m/m TNFR1 −/− donors themselves never develop NLH (Supplementary figure 1d), WT recipients of BPSM1 m/m TNFR1 −/− bone marrow had NLH (Supplementary figure 2j), demonstrating that overexpression of TNF triggers NLH by acting on a nonhematopoietic component, and that the expression of TNFR1 in the hematopoietic compartment is not necessary for NLH, arthritis or heart valve disease.
IL‐23 and Il‐17 play a role in lymphoid neogenesis
The IL‐23/IL‐17 axis is involved in the pathogenesis of several inflammatory diseases such as psoriasis, rheumatoid arthritis (RA), ankylosing spondylitis and inflammatory bowel disease.10, 11 Moreover, IL‐17, and to a lesser extent IL‐23, have been shown to contribute to iBALT formation.12 To examine the roles of the Il‐23/IL‐17 axis in the different pathologies present in BPSM1 mice, we crossed them to IL‐23p19‐13 and IL‐17A‐deficient14 mice, respectively. Loss of either IL‐23 or IL‐17 reduced iBALT in BPSM‐1 m/+ mice, although a small number of iBALT foci were still present in both BPSM1 m/+ IL‐23 −/− and BPSM1 m/+ IL‐17 −/− animals (Figure 3; Supplementary figure 3a). To quantify this effect, we counted lymphocyte foci on 5 H&E‐stained lung sections of multiple (n ≥ 5 for each genotype) WT, BPSM1 m/+ , BPSM1 m/m , BPSM1 m/+ IL‐23 −/− and BPSM1 m/+ IL‐17 −/− animals (Figure 3a). While the numbers of iBALT follicles were similar in BPSM1 m/m and BPSM1 m/+ mice, these numbers were halved in BPSM1 m/+ IL‐23 −/− and BPSM1 m/+ /IL‐17 −/− animals. To study the effects of IL‐23 and IL‐17 loss on the bone marrow lymphoid nodules in the BPSM1 m/+ mice, we used dual confocal/multiphoton imaging (Figure 3b, c). The number of BM lymphoid nodules was not reduced in the absence of either IL‐23 or IL‐17, but their size was significantly decreased in the absence of IL‐23 (Figure 3b–e).
Mutations in the IL‐23 receptor gene have been associated with the development of ankylosing spondylitis.15 BPSM1 mice are a model of ankylosing spondylitis since they show inflammation of their spine as well as their sacroiliac joints. In contrast to the marked effects in the lungs and bone marrow, loss of neither IL‐23 nor IL‐17 had any effect on the kinetics or the severity of polyarthritis and heart disease in BPSM1 mice (Supplementary figure 3c, d). This is in agreement with our previous observation that loss of T cells did not modify the course of the disease in BPSM1 mice.3
Altogether, these results suggest that the IL‐23/IL‐17 axis plays an important role in the development of iBALT and NLH in BPSM1 mice, but these cytokines have little to no influence on the development of polyarthritis and heart disease in these animals.
High systemic TNF levels and accompanying iBALT in BPSM1 mice are associated with improved control of Mycobacterium tuberculosis pulmonary infection
The elevated systemic levels of TNF in BPSM1 mice drives their overt phenotype including the development of iBALT. We speculated that the elevated levels of TNF in BPSM1 mice may also impact on the outcomes of infectious diseases. TNF is essential for the control of Mycobacterium tuberculosis (Mtb) in humans and mice.16, 17 Deficiency in TNF signaling causes rapid tuberculosis disease progression. However, it has been postulated that excessive TNF may equally promote Mtb disease pathogenesis by causing collateral tissue damage.18 To examine the impact of high systemic TNF levels, we infected BPSM1 m/+ and control animals with Mtb. We found that the high TNF levels, and accompanying lung phenotype, enhanced Mtb control and reduced lung bacterial loads in BPSM1 m/+ mice compared to littermate controls (Figure 4a). Lung histology showed a mild increase in pulmonary inflammation (Figure 4b, c), which correlated with an enrichment of F4/80+ macrophages within inflammatory lesions (Figure 4d). These data indicate that systemic elevations in TNF do not promote Mtb disease, as has been suggested by others,18, 19 but rather they enhance pulmonary clearance of the organism, perhaps in part by stimulating a beneficial macrophage response.
Figure 4.
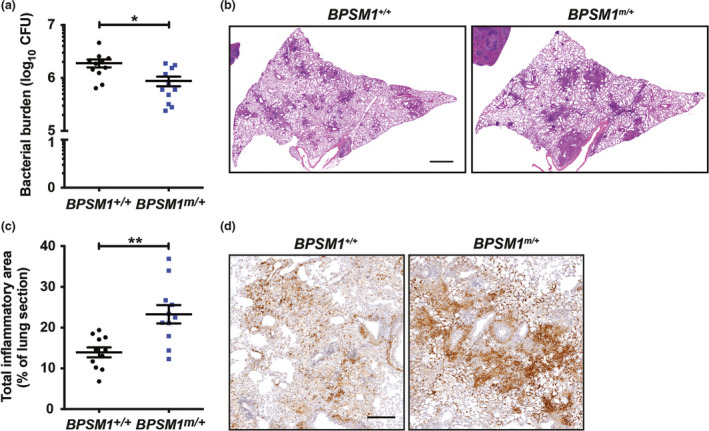
BPSM1 mice show improved ability to control M. tuberculosis infection. (a) Bacterial burdens in the lungs of BPSM1 +/+ and BPSM1 m/+ mice enumerated 5 weeks postinfection with aerosolized Mtb. (b) Lung histology of mice 5 weeks postinfection. Sections of the left lobe were stained with H&E. Scale bars, 1 mm. (c) Quantitation of the extent of pulmonary inflammation in H&E‐stained lung sections. (d) Immunohistochemical staining of F4/80 in lung inflammatory lesions. Scale bars, 0.2 mm. Graphs show mean and s.e.m. and each point represents one mouse (n = 11 in each group). Data were analyzed by a Student's t‐test (*P < 0.05; **P < 0.01).
Discussion
iBALT is a normal feature of certain mammals (e.g. rabbits and rats) while it is absent from healthy mice and humans. In humans and mice, the formation of iBALT appears intimately linked to chronic inflammation. The generation of disorganized lymphoid clusters in the lungs is easy to obtain in C57BL/6 mice upon intranasal administration of lipopolysaccharide (LPS).12 However, the formation of structured iBALT, containing many B cell follicles separated by well‐defined T cell areas, required that LPS be administered soon after birth. LPS administration to 2 or 3‐week‐old mice only produced disorganized B cell clusters lacking defined T cell areas.12 Thus, the competence of the mouse lungs to respond to high TNF levels and establish an iBALT niche occurs early after birth and is limited in time. BPSM1 mice overexpress TNF, but their levels of other inflammatory cytokines are normal.3 While it is tempting to speculate that TNF is involved in the response of the lung tissue to LPS, it is likely that other cytokines are also elevated in this model of BALT induction. Involvement of other cytokines may explain why it is relatively easy to induce iBALT with LPS when transplantation of BPSM1 m/+ or BPSM1 m/m bone marrow could not. Of note, most of the conditions that induce the formation of BALT (such as response to Influenza virus, asthma, RA, Sjögren's syndrome, chronic obstructive pulmonary disease) involve a strong inflammatory response and multiple cytokines. Interestingly, BPSM1 m/+ recipients transplanted with WT bone marrow could develop iBALT and maintain it while having normal serum levels of TNF posttransplantation.3 This indicates that the iBALT niche already established at the time of irradiation survives the treatment and is capable of recruiting wildtype B, T and dendritic cells to maintain the tissue over long periods. This suggests that the niche is made of and maintained by either nonhematopoietic cells (maybe the functional equivalent of mesenchymal stromal organizer cells required for the development of secondary lymphoid organs,20 or radio‐resistant tissue‐resident hematopoietic cells. Interestingly, emerging evidence suggests that dendritic cells are likely to be important organizers of iBALT formation,21 and some dendritic cell populations have been shown to be radio‐resistant (e.g. Langerhans cells22).
The presence or absence of iBALT in different species may be the result of differences in the expression of the Tnf gene around or soon after birth. However, the time restriction most likely is not absolute, since properly organized iBALT is found in adults in association with chronic diseases such as RA or viral infections. In most cases, the destruction of lung tissue is extensive, and it is possible that the healing process brings the tissue back to a state similar to that found in the few days following birth.
At first glance, the situation in the bone marrow appears different, since the transplant of BPSM1 m/+ bone marrow into irradiated adult WT recipients readily triggers the development of lymphoid nodules in the bone marrow, more extensive in fact than in the original BPSM1 m/+ donors. However, this may also occur because of the severe injury caused to the recipient's bone marrow by irradiation, which most likely also triggers healing processes similar to those evoked earlier. It is interesting to note that expression of TNFR1 in a nonhematopoietic cell type (or a radio‐resistant hematopoietic cell type) in the host, but not in the donor bone marrow cells, is necessary for the development of the lymphoid nodules in the bone marrow. Once again, mesenchymal stromal organizer cells come to mind.20 TNFR2 does not compensate for the loss of TNFR1 in this instance and this may simply reflect the absence of TNFR2 on this cell type, rather than a difference in signaling between the two receptors.
Whether bone marrow lymphoid nodules perform any particular function remains elusive. They have been described in patients suffering from a variety of infectious or inflammatory diseases, as well as in patients with chronic lymphocytic leukemia.4, 5, 6 In all cases, the presence of BM follicles seems to be associated with high circulating TNF levels, even in the case of chronic lymphocytic leukemia. Since benign lymphoid follicles are not so rare in human patients,23, 24 it is somewhat puzzling that their presence in mice is barely ever mentioned in the literature. An interesting difference with iBALT is that BM nodules evidently require persistent high levels of TNF to be maintained, whereas iBALT does not. In that regard, iBALT could be considered as an optional secondary lymphoid organ that sometimes develops after birth.
Rangel‐Moreno et al.12 have described a role for IL‐17 and IL‐23 in the formation of iBALT in response to intranasal LPS administration. Since mutations of IL‐23 receptor have been associated with the development of ankylosing spondylitis in humans, we wanted to examine whether loss of IL‐23 or IL‐17 would also decrease the severity of arthritis and the inflammation of the heart valves in BPSM1 mice. However, loss of either of these cytokines had no effect on the extent of bone erosion or valve thickening. This once more highlights the independence between iBALT development and arthritis.
Whether iBALT is beneficial or detrimental to lung immunity remains controversial. Indeed, the extent of iBALT correlates with disease severity in patients with chronic obstructive pulmonary disease25 or RA.26 Conversely, iBALT is normally present in perfectly healthy rats and rabbits and has been shown to help clear influenza virus infection in mice devoid of secondary lymphoid organs.27 Of note, none of our mice harboring iBALT has ever shown symptoms associated with chronic obstructive pulmonary disease, indicating that iBALT alone is unlikely to be a driver of this disease. It is tempting to speculate that iBALT contributes to defense against pulmonary pathogens, based on the reduced Mtb burden observed in BPSM1 m/+ mice. However, this requires further investigation.
Since the presence or absence of iBALT does not modify the course and severity of arthritis and heart disease development in BPSM1 mice, we suggest that it plays no part in these diseases, and that the extent of iBALT in chronic obstructive pulmonary disease and RA patients merely represents the severity of the inflammation in these patients, and possibly the amount of TNF produced in their lungs. Likewise, the presence of lymphoid nodules in the bone marrow of BPSM1 mice appears more like a developmental accident because of persistently high levels of TNF and the presence of a TNFR1‐expressing cell type capable of organizing these structures rather than a consequence of the arthritis or the heart valve disease.
Methods
Mice
All animal experiments were conducted with the approval of the Animal Ethics Committee of the Walter and Eliza Hall Institute. BPSM1 mice were the result of a spontaneous mutation.3 The generation of mice devoid of CD3ε, Myd88, GM‐CSF, TNFR1, IL‐17, IL‐23, CCR7, as well as that of Mb1‐cre knockin mice has been described.8, 9, 13, 14, 28, 29, 30, 31 All mice were on the C57BL/6 genetic background. At least ten mice of each genotype were examined.
Bone marrow reconstitution
To perform bone marrow reconstitution, recipient mice (all on a C57BL/6 genetic background) were lethally irradiated (2× 5.5 Gy) at 4–6 weeks of age, then injected with 2 × 106 bone marrow cells of the indicated genotypes, and examined 5 months posttransplant, except for the recipients of BPSM1 m/m TNFR1 −/− bone marrow, which were analysed 2 months after transplant.
Histology and immunochemistry
5‐mm‐thick sections of formalin‐fixed paraffin‐embedded tissue were stained with Haematoxylin and Eosin (H&E). Images shown are representative of 10 or more animals analysed per genotype. For immunohistochemistry, tissues were fixed in 4% paraformaldehyde and infiltrated with sucrose (increasing concentrations; 10%, 20% and 30%) before being embedded in optimal cutting temperature compound freezing media (Sakura Fineteck). Frozen sections of 12‐μm thickness were fixed in acetone for 10 min and blocked with phosphate‐buffered saline (PBS) containing 3% BSA and Fc block. Sections were stained with antibodies for the detection of B220 (RA3‐6B2), CD3 (145‐2C11) and CD21/35 (8C12) from BD Biosciences (Franklin Lakes, NJ, USA). Images were acquired on a LSM780 confocal microscope (Zeiss, Oberkochen, Germany).
Immunofluorescence of undecalcified long bone sections
Immunofluorescence of undecalcified bone was performed as described previously.32 20‐μm bone sections were cut using a Cryojane tape transfer system (Leica microsystems, Wetzlar, Germany). Antibodies used were as above. After washing in PBS, slides were incubated with secondary antibodies, counter‐stained with DAPI, and mounted with Prolong Diamond anti‐fade (Invitrogen, Waltham, MA, USA). Imaging of sections was performed on an upright Zeiss LSM 880 with the following lasers: Argon (458‐, 488‐ and 514‐nm), a diode‐pumped solid‐state 561‐nm laser and a Helium‐Neon 633‐nm, a tunable infrared multiphoton laser (Mai Tai DeepSee 690–1020 nm, Spectraphysics, Santa Clara, CA, USA). Dual confocal/multiphoton imaging was performed by combining nondescanned and internal detectors. Bone was detected using second harmonic generation with collagen excited at 840–860 nm. Signal was detected using 420–480 nm NDD detectors. Multi Z‐plane tilescans were captured and stitched using Zen software (Zeiss) and data analyzed using Fiji/ImageJ. Quantification was performed by researchers blinded from genotype of mice.
M. tuberculosis infection
Mice were infected with 50‐100 colony forming units of Mtb strain H37Rv (Nicholas P West, University of Queensland, Australia) by aerosol, using an Inhalation Exposure System (Glas‐Col, Terre Haute, IN, USA). Mice were euthanised 5 weeks postinfection. Lungs were inflated by intratracheal infusion of PBS and were aseptically removed. Left lobes were fixed overnight at 4°C in 4% paraformaldehyde, while right lobes and spleens were homogenised with steel beads in PBS+0.05% Tween‐80. Homogenates were serially diluted and spread on Middlebook 7H11 agar (BD Biosciences) supplemented with 0.5% (v/v) glycerol and 10% (v/v) OADC supplements [50 g L−1 bovine serum albumin, 20 g L−1 dextrose, 0.04 g L−1 catalase and 0.5 g L−1 oleic acid (Sigma‐Aldrich, St Louis, MO, USA)]. Colonies were counted following incubation for 21 days at 37°C, and were expressed as colony forming units/organ. Fixed lungs were embedded in paraffin, sectioned (5‐μm) and stained with either haematoxylin and eosin (H&E), or immunohistochemically with rat anti‐mouse F4/80 (WEHI) using the automated Omnis EnVision G2 template (Dako, Glostrup, Denmark). Slides were scanned with a Pannoramic SCAN II scanner (3D Histech, Budapest, Hungary) and analyzed quantitatively with custom‐written macros using FIJI software.
Supporting information
Acknowledgments
We thank T Kitson, C Gatt and J Stanley for animal care and expertise; B Malissen (Marseille Luminy, France) for CD3ε KO mice; M Reth (Max‐Planck Institute of Immunobiology) for Mb1‐cre KI mice; S Akira (Osaka University Immunology Frontier Research Center) for Myd‐88 KO mice; T Mak (The Campbell Family Institute for Breast Cancer Research) for TNFR1 KO mice; I Wicks (WEHI) for GM‐CSF KO mice, Y Iwakura (Institute of Medical Science, University of Tokyo) for IL‐17A KO mice, Genentech for IL‐23p19KO mice. This work was supported by the Australian NHMRC (Program Grants 461221 and 1054925, Research Fellowships 1042629 (PB) and 1122277(GTB), Career Development Fellowships 1123000 (CS) and 1086977 (EDH), Project Grants 1051506 and 1127885 (PB)), the Leukemia and Lymphoma Society (Specialised Center of Research grant 7015), the Sylvia & Charles Viertel Charitable Foundation, the Arthritis Australia Zimmer Fellowship, and infrastructure support from the NHMRC (IRISS) and the Victorian State Government (OIS).
Conflict of Interest
The authors declare no conflict of interest.
References
- 1. Randall TD. Bronchus‐associated lymphoid tissue (BALT) structure and function. Adv Immunol 2010; 107: 187–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Foo SY, Phipps S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol 2010; 3: 537–544. [DOI] [PubMed] [Google Scholar]
- 3. Lacey D, Hickey P, Arhatari BD, et al. Spontaneous retrotransposon insertion into TNF 3'UTR causes heart valve disease and chronic polyarthritis. Proc Natl Acad Sci USA 2015; 112: 9698–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thiele J, Zirbes TK, Kvasnicka HM, et al. Focal lymphoid aggregates (nodules) in bone marrow biopsies: differentiation between benign hyperplasia and malignant lymphoma–a practical guideline. J Clin Pathol 1999; 52: 294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Engels K, Oeschger S, Hansmann ML, et al. Bone marrow trephines containing lymphoid aggregates from patients with rheumatoid and other autoimmune disorders frequently show clonal B‐cell infiltrates. Hum Pathol 2007; 38: 1402–1411. [DOI] [PubMed] [Google Scholar]
- 6. Naemi K, Brynes RK, Reisian N, et al. Benign lymphoid aggregates in the bone marrow: distribution patterns of B and T lymphocytes. Hum Pathol 2013; 44: 512–520. [DOI] [PubMed] [Google Scholar]
- 7. Kocks JR, Davalos‐Misslitz AC, Hintzen G, et al. Regulatory T cells interfere with the development of bronchus‐associated lymphoid tissue. J Exp Med 2007; 204: 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hobeika E, Thiemann S, Storch B, et al. Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci USA 2006; 103: 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malissen M, Gillet A, Ardouin L, et al. Altered T cell development in mice with a targeted mutation of the CD3‐ e gene. EMBO J 1995; 14: 4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sherlock JP, Joyce‐Shaikh B, Turner SP, et al. IL‐23 induces spondyloarthropathy by acting on ROR‐gammat+ CD3 + CD4‐CD8‐ entheseal resident T cells. Nat Med 2012; 18: 1069–1076. [DOI] [PubMed] [Google Scholar]
- 11. Frieder J, Kivelevitch D, Haugh I, et al. Anti‐IL‐23 and Anti‐IL‐17 biologic agents for the treatment of immune‐mediated inflammatory conditions. Clin Pharmacol Ther 2018; 103: 88–101. [DOI] [PubMed] [Google Scholar]
- 12. Rangel‐Moreno J, Carragher DM, de la Luz Garcia‐Hernandez M, et al. The development of inducible bronchus‐associated lymphoid tissue depends on IL‐17. Nat Immunol 2011; 12: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghilardi N, Kljavin N, Chen Q, et al. Compromised humoral and delayed‐type hypersensitivity responses in IL‐23‐deficient mice. J Immunol 2004; 172: 2827–2833. [DOI] [PubMed] [Google Scholar]
- 14. Nakae S, Komiyama Y, Nambu A, et al. Antigen‐specific T cell sensitization is impaired in IL‐17‐deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002; 17: 375–387. [DOI] [PubMed] [Google Scholar]
- 15. Australo‐Anglo‐American Spondyloarthritis Consortium , Reveille JD, Sims AM, et al. Genome‐wide association study of ankylosing spondylitis identifies non‐MHC susceptibility loci. Nat Genet 2010; 42: 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha‐neutralizing agent. N Engl J Med 2001; 345: 1098–1104. [DOI] [PubMed] [Google Scholar]
- 17. Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor‐alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995; 2: 561–572. [DOI] [PubMed] [Google Scholar]
- 18. Dorhoi A, Kaufmann SH. Tumor necrosis factor alpha in mycobacterial infection. Semin Immunol 2014; 26: 203–209. [DOI] [PubMed] [Google Scholar]
- 19. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013; 153: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Genovese L, Brendolan A. Lymphoid tissue mesenchymal stromal cells in development and tissue remodeling. Stem Cells Int 2016; 2016: 8419104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. GeurtsvanKessel CH, Willart MA, Bergen IM, et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus‐infected mice. J Exp Med 2009; 206: 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merad M, Manz MG, Karsunky H, et al. Langerhans cells renew in the skin throughout life under steady‐state conditions. Nat Immunol 2002; 3: 1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rywlin AM, Ortega RS, Dominguez CJ. Lymphoid nodules of bone marrow: normal and abnormal. Blood 1974; 43: 389–400. [PubMed] [Google Scholar]
- 24. Johnston A, Brynes RK, Naemi K, et al. Differentiating benign from malignant bone marrow B‐cell lymphoid aggregates: a statistical analysis of distinguishing features. Arch Pathol Lab Med 2015; 139: 233–240. [DOI] [PubMed] [Google Scholar]
- 25. Hogg JC, Chu F, Utokaparch S, et al. The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- 26. Rangel‐Moreno J, Hartson L, Navarro C, et al. Inducible bronchus‐associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006; 116: 3183–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moyron‐Quiroz JE, Rangel‐Moreno J, Kusser K, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 2004; 10: 927–934. [DOI] [PubMed] [Google Scholar]
- 28. Adachi J, Ookawa K, Kohno T, et al. Phenotypic alterations of small cell lung carcinoma induced by different levels of wild‐type p53 expression. Cell Death Differ 1998; 5: 148–155. [DOI] [PubMed] [Google Scholar]
- 29. Stanley E, Lieschke GJ, Grail D, et al. Granulocyte/macrophage colony‐stimulating factor‐deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 1994; 91: 5592–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pfeffer K, Matsuyama T, Kündig TM, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993; 73: 457–467. [DOI] [PubMed] [Google Scholar]
- 31. Forster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999; 99: 23–33. [DOI] [PubMed] [Google Scholar]
- 32. Hawkins ED, Duarte D, Akinduro O, et al. T‐cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016; 538: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials