

Abstract
The development of small molecule kinase drugs is a rapidly evolving field and represents one of the most important research areas within oncology. This innovation letter provides an overview and analysis of approved kinase drugs according to their WHO registration (INN) dates, primary biological targets, and selectivity and structural similarities, which are also depicted in an associated poster. It also discusses new trends in kinase drug discovery programs such as new kinase targets, novel mechanisms of action, and diverse indications.
Keywords: Kinase drugs, small molecules, WHO, INN, FDA, imatinib
Kinases catalyze the transfer of the γ-phosphate of ATP onto a diverse range of hydroxyl substrates (e.g., proteins, lipids, sugars, nucleotides).1,2 Although kinases account for only ∼5% of the protein-coding genes, their activity mediates most cellular signal transductions and regulates a variety of cellular activities.1,3 Furthermore, despite their diverse primary sequences, functions, and substrates, they share a great deal of similarity in structure, especially in their catalytically active ATP binding domains.1 Based upon the nature of the phosphorylated hydroxyl groups these enzymes can be generally classified as serine/threonine kinases, tyrosine kinases, dual-specificity kinases, tyrosine-kinase-like, lipid kinases, sugar kinases, and nucleotide kinases.1,2
Kinases are one of the most intensively pursued targets, particularly in cancer drug discovery programs.4 Kinases have been successfully targeted by natural products, small molecules, antibodies and antibody conjugates.5 Although compounds that inhibit kinases have been used as drugs since the 1930s, the first characterized kinase drugs originated in the 1990s. The first of these was the small molecule ROCK inhibitor fasudil (Figure 1), which was approved in Japan in 1995 for the treatment of cerebral vasospasm. This was followed by the approval of sirolimus (Figure 1), a natural product targeting mTOR. Sirolimus was approved in 1999 for use during renal transplants in combination with cyclosporine to prevent organ rejection. Also in 1998, trastuzumab became the first approved monoclonal antibody kinase drug, targeting ErbB2 for the treatment of ErbB2-positive breast cancer.
Figure 1.

First three approved small molecule kinase drugs.
Biochemical assays have now been developed for more than four-fifths of the human kinome, small molecule kinase inhibitors have been identified for approximately one-fifth,3 and there are now 201 WHO registered (INN) small molecule kinase entities.6 Although each drug–protein interaction is unique, small molecule kinase inhibitors are commonly categorized by their mode of binding to the target protein, with several clear categories defined (Types I–VI).7
Drug binding can occur in the ATP binding pocket of the active enzyme conformation (Type I) or the inactive form (Type I1/2 & II). Type III and Type IV are allosteric inhibitors that bind next to the ATP binding pocket or at a nonsubstrate (ATP, peptide) site, respectively. Type V are classified as bivalent inhibitors, spanning two distinct sites of the kinase domain, and Type VI are covalent inhibitors usually containing an α,β-unsaturated carbonyl functionality, which irreversibly form a covalent bond with cysteines in or around the active site of the enzyme. The orientation of key kinase regions (e.g., DFG-Asp, activation segment, and spine) can be important for drug binding (for Types I, II, and III) or the orientation of such regions can be less defined or variable (for Types IV–VI).7 Further adding to the complexity, drugs can interchange between binding modes when interacting with different kinases, a phenomenon common among the Type I, I1/2, and II drugs.7 In addition, the presence of substrate and ATP in cells could result in binding profiles differing to those observed in vitro.8
Imatinib (Glivec)
Until the early 1980s, oncology-focused drug discovery programs identified mainly antimetabolites, alkylators, and microtubule destabilizers, with the aim of reducing the standard characteristics of cancer (e.g., replication and proliferation). Due to their lack of selectivity such drugs are usually highly toxic, which limits their therapeutic window, although the development of antihormonals (e.g., tamoxifen, 1973) is a specific early example of a more targeted approach for certain cancers such as breast cancer.9 The discovery of cancer-causing genes uniquely associated with cancerous cells (oncogenes) represented a radical departure from this paradigm. One such example was the BCR-Abl protein kinase (1986)—a product of an interchromosomal exchange (reciprocal translocation) between chromosomes 9 and 22—which displayed elevated tyrosine-kinase activity.10 Its production is a defining molecular pathogenic event in chronic myelogenous leukemia (CML) and provided the first potential drug target whose activity significantly differed between normal and leukemic cells.10 Approximately 15 years after the discovery of the BCR-Abl protein kinase, the first targeted small molecule kinase inhibitor (imatinib, 2001, BCR-Abl, Figure 1) was approved by the FDA for CML.10 The clinical development of imatinib (Glivec) was extremely rapid. The first patient was treated in 1998, and less than three years later in 2001, following three large, multinational trials, Glivec was approved by the FDA for CML.10 In 2002, Glivec was also approved for treatment of GIST with KIT mutations.10 Glivec revolutionized the treatment of CML and GIST4 and has been subsequently approved for the treatment of a variety of other indications including fibrosarcoma, hypereosinophilic syndrome, ALL, myelodysplastic/myeloproliferative diseases, and systemic mastocytosis. Mechanistically imatinib is a Type II inhibitor, binding to an inactive conformation of Abl.11 In 2016, imatinib entered the generic drug market in the USA.12
Small Molecule Kinase Drugs (2001–2017)
Following the FDA approval of imatinib in 2001, the main primary targets of all small molecule kinase entities registered by the WHO before 2011 (2001–2010) were tyrosine kinases; more specifically BCR-Abl, along with the VEGFR and ErbB family. Of such, 19 small molecule kinase entities have been marketed for various cancers: ErbB – erlotinib (INN 2002, OSI Pharmaceuticals), gefitinib (INN 2002, AstraZeneca), lapatinib (INN 2003, Novartis), vandetanib (INN 2004, Genzyme), neratinib (INN 2007, Puma Biotechnology), afatinib, (INN 2010, Boehringer Ingelheim); BCR-Abl – nilotinib (INN 2005, Novartis), bosutinib (INN 2005, Wyeth), dasatinib (INN 2005, Bristol-Myers Squibb), ponatinib (INN 2010, Ariad Pharmaceuticals), radotinib (INN 2010, Daewoong Il-Yang); VEGFR – sunitinib (INN 2005, Pfizer), sorafenib (INN 2003, Bayer), pazopanib (INN 2005, GlaxoSmithKline), axitinib (INN 2005, Pfizer), regorafenib (INN 2009, Bayer), lenvatinib (INN 2010, Eisai Lnc), tivozanib (INN 2009, Kyowa Hakko Kirin) (Figure 5). During this period, small molecule entities targeting several other kinases such as JAK2, MET, SYK, and CDK were also registered. However, only one has been marketed to date: fostamatinib (INN 2009, Rigel, but not FDA approved until 2018) targets SYK for the treatment of immune thrombocytopenic purpura (ITP). As of 2018, these drugs are still on the market, with six of the 19 (erlotinib, imatinib, nilotinib, dasatinib, sunitinib, and pazopanib) making the “Top 200 drugs by worldwide sales” in 201613 and three (imatinib, nilotinib, and dasatinib) listed on the “WHO model list of essential medicines”. Of the 40 drug substances that have not received approval and were assessed during this time frame, three drug substances have been discontinued, 33 are in clinical phase II or higher, and two are in preregistration. For nine of the clinical phase compounds, INN registration took place between 2001 and 2005.
Figure 5.

This figure depicts the INN drug names, INN registration dates, chemical structures and classification by relevant biological targets of FDA approved small molecule kinase inhibitors. Structural similarities within the respective groups are highlighted. *Reduced selectivity; **Highly promiscuous kinase inhibitors.
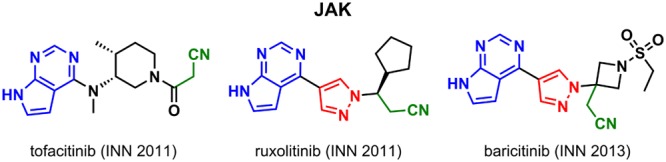
Post-2010, approval of additional WHO registered small molecule entities targeting EGFR and VEGFR continued: EGFR – icotinib (INN 2011, Beta Pharma), olmutinib (INN 2015, Hanmi), brigatinib (INN 2016, Ariad Pharmaceuticals), osimertinib (INN 2016, AstraZeneca), VEGFR – cabozantinib (INN 2011, Exelixis), nintedanib (INN 2011, Boehringer Ingelheim), rivoceranib (INN 2017, Advenchen Laboratories & Jiangsu Hengrui). All are approved solely for the treatment of cancer except for nintedanib, which was also approved for use as an idiopathic pulmonary fibrosis (IPF) treatment. Approvals for new targets including serine-threonine kinases and lipid kinases also began to appear: BRAF – dabrafenib (INN 2012, GlaxoSmithKline), vemurafenib (INN 2011, Plexxikon, Daiichi Sankyo), encorafenib (INN 2013, Novartis); JAK – ruxolitinib (INN 2011, Incyte), tofacitinib (INN 2011, Pfizer), baricitinib (INN 2013, Incyte); MAP2K/MEK – trametinib (INN 2011, Japan Tobacco), cobimetinib (INN 2013, Exelixis), binimetinib (INN 2013, Array BioPharma); ALK– crizotinib (INN 2011, Pfizer), alectinib (INN 2012, Chugai Pharmaceutical), ceritinib (INN 2013, Novartis); BTK – ibrutinib (INN 2013, Pharmacyclics), acalabrutinib (INN 2016, Acerta Pharma, AstraZeneca); PI3Kδ – idelalisib (INN 2012, Novartis & Icos), copanlisib (INN 2012, Bayer); CDK – palbociclib (INN 2013, Pfizer), ribociclib (INN 2014, Novartis & Astex Pharmaceuticals), abemaciclib (INN 2014, Lilly) (Figure 5). Furthermore, several of these drugs were marketed for new indications beyond cancer including IPF, arthritis, psoriatic, ulcerative colitis, graft-versus-host disease, and immune thrombocytopenia. For these later drugs, as of 2018, all are still on the market, with four of the 26 (ibrutinib, nintedanib, palbociclib, and tofacitinib) making the “Top 200 drugs by worldwide sales” in 2016, with three of these top drugs developed against new targets.13
Between 2001 and 2017, in total 201 small molecule kinase entities were registered with the WHO (Figure 2). Forty-six drugs made it to the market of which 26 compounds were commercialized within the last five years (2013–2018). Thirty-seven are solely used in the treatment of cancer and 35 of the 46 target the tyrosine kinase family;7 however, several “first in class” inhibitors have recently been commercialized, e.g., trametinib (MEK, 2013), palbociclib (CDK, 2015), and idelalisib (PI3Kδ, 2014).
Figure 2.

INN registrations for small molecule kinase inhibitors (2001–2017).
Most function through Type I and Type II binding mechanisms.7 A few Type I 1/2 inhibitors have also managed to make it to the market (e.g., lapatinib, vemurafenib, sunitinib, dasatinib, erlotinib, lenvatinib).7 In addition, non-ATP competitive inhibitors of MEK that target a site adjacent to the ATP binding pocket (e.g., trametinib)7 and inhibitors that act through irreversible mechanisms (acalabrutinib, afatinib, ibrutinib, neratinib, osimertinib and olmutinib) targeting BTK and EGFR are also among the marketed compounds.7,14−16 To date, no drugs displaying Type IV or Type V behaviors have been marketed.
The structure to function relationship of numerous FDA approved kinase inhibitors has recently been thoroughly reviewed.3 With regards to their origin, most approved small molecule kinase drugs were inspired by previously approved structural motifs, leading to some common structural features. As covered in the review,3 these recurring motifs often bind to well-defined regions of a typical protein kinase active site, for example, to the adenine binding pocket (the hinge region), adjacent hydrophobic regions such as the selectivity pocket or allosteric pocket, or the solvent exposed channel. In several cases, this strategy has resulted in closely related compounds displaying differing kinase mutant targeting profiles (see Present and Future Kinase Drug Development).
Figure 5 provides an overview of approved drugs classified by a primary biological target and chemical structures, allowing for assessment of structural similarities. The ErbB family targeting drugs contain a heterocyclic pharmacophore fragment consisting of a N-phenyl 4-anilino quinazoline (blue) or a N-(pyrimidinyl)benzene-1,4-diamine moiety (green), with the exception of neratinib, which bears a 4-(phenylamino)quinoline-3-carbonitrile core instead. Erlotinib and icotinib possess a strikingly similar chemical structure differing solely by a macrocyclization. Neratinib is the first WHO registered member of this group possessing a dimethylamino-but-2-enamide Michael acceptor, which is responsible for its irreversible binding mode (purple). This α,β-unsaturated amide group (purple) is also present in afatinib, and a similar irreversible group without the base is present in olmutinib and osimertinib. A further recurrent motif is the ether substitution pattern (red) in erlotinib, gefitinib, vandetanib, neratinib, afatinib, and icotinib. The VEGFR inhibitors sorafenib, regorafenib, tivozanib, and lenvatinib have a (4-(pyridin-4-yloxy)phenyl)urea motif (blue) in common, whereas cabozantinib possesses an amide functionality rather than an urea. Sorafenib and regorafenib are almost congruent and differ solely by one single arylfluoride substituent. Pazopanib and axitinib share a heterocyclic indazole (green). Tivozanib, lenvatinib, and cabozantinib share the 7-methoxyquinoline element (purple), whereas sunitinib and nintedanib have a 3-methyleneindolin-2-one moiety (red) in common. Rivoceranib does not show any distinct structural similarities to the other compounds in the group. Imatinib was the first in class compound targeting BCR-Abl. The successor compounds nilotinib and radotinib are structurally related to imatinib bearing a common N-(o-tolyl)pyrimidin-2-amine motif (blue). Nilotinib and radotinib solely differ by one heterocyclic substituent; nilotinib bears a 3-pyridine substituent, whereas radotinib possesses a 2-pyrazine instead. Nilotinib, ponatinib, and radotinib all contain a N-(3-(trifluoromethyl)phenyl) amide substructure (green); however, the overall ponatinib scaffold is significantly different to both that of nilotinib and radotinib. Bosutinib and dasatinib show no obvious structural similarities to the imatinib template.
Among the more recent kinases to be targeted, the CDK4/6 drug palbociclib served as a template for ribociclib. Both compounds differ slightly in the ring size of the heterocyclic core: palbociclib contains a six-membered pyrido-pyrimidin-one, while in ribociclib this is replaced by a five-membered pyrrolo-pyrimidine. Furthermore, palbociclib and ribociclib have the N-cyclopentyl substituent (green) in common. Structural elements common to all three CDK4/6 compounds (ribociclib, palbociclib, and abemaciclib) are the N-(pyridin-2-yl)pyrimidin-2-amine (blue) and the piperazine moiety (red). The BTK drugs, ibrutinib and acalabrutinib, both possess a Michael acceptor functionality (purple), which is required for the irreversibile binding of these compounds; ibrutinib has an acrylamide and acalabrutinib has a but-2-ynamide. In ibrutinib, this α,β-unsaturated motif is linked to the central core via a piperidine, whereas in acalabrutinib it is linked via a pyrrolidine. The JAK compounds tofacitinib, ruxolitinib, and baricitinib bear a common 7H-pyrrolo[2,3-d]pyrimidine structural motif (blue) and an aliphatic nitrile functionality (green). In ruxolitinib and baricitinib, these two fragments are connected via a shared pyrazole moiety (red) resulting in a common pyrrolo[2,3-pyrimidin-4-yl)-pyrazol) propanenitrile substructure. Structural similarity of the BRAF compounds vemurafenib, dabrafenib, and encorafenib is limited to the shared N-(2-fluorophenyl) sulfonamide fragment (blue) and a 2-amino pyrimidine moiety (red) in dabrafenib and encorafenib. In contrast, the only similarity between the MAP2K/MEK drugs trametinib, binimetinib, and cobimetinib is a single 2,4-dihaloaniline substructure (blue). No similarity is observed between the approved compounds targeting PI3Kδ or ALK.
There are ∼1500 ATP requiring enzymes, in addition to the ∼500 serine/threonine and tyrosine kinases. As the most common site of interaction for kinase drugs is the kinase ATP-binding pocket, selectivity issues or poly pharmacology are frequently observed. In general, kinase drug poly pharmacology is not this clear-cut. Drugs can bind tightly to kinases with modest sequence similarities or on occasions may distinguish between kinases closely related by sequence, especially when specifically designed to do so.17 Therefore, experimental profiling (kinome-wide and proteome-wide screening) is crucial for assessing selectivity for kinase and nonkinase targets. Several studies have utilized kinase profiling (cell based, binding or catalytic) to assess drug selectivity across the kinome, with applications directed toward understanding kinome selectivity, mutant responses and providing opportunities in drug repurposing.8,17,18 “Selectivity” within these studies is defined using a variety of approaches and different approaches will produce subtle differences in selectivity profiles. “Group selective” kinase drugs interact broadly with one kinase group but are selective outside of the target group(s).19 They can consist of compounds with diverse chemical structures and modes of action (including both type I and II inhibitors).19
For many marketed small molecule kinase drugs, their tightest interactions are with the primary targets of which the drugs were developed against (Figure 5). Some can be referred to as unispecific, whereas others target more than 100 kinases simultaneously, making it difficult to attribute their biological effects to any mode of action.18,20 Examples with exceptional selectivity include gefitinib, lapatinib, tofacitinib, and afatinib.18,19,21 The differences in affinity between the intended target and off targets vary. For example, a ∼10-fold difference can be observed between the intended target of erlotinib (EGFR) and other off targets, yet for an alternate EGFR inhibitor vandetenib this difference is 2-fold; neither of which seem to demonstrate group selectivity.17 Furthermore, related drugs erlotinib and gefitinib with similar selectivity profiles can have differing numbers of primary targets.17 Several of the marketed drugs display obvious group selectivity. Tyrosine kinase group selectivity is clearly observed for vandetanib, dasatinib, nilotinib, and imatinib and is present but less obvious for tamatinib, crizotinib, ruxolitinib, pazopanib, and erlotinib.17,19,21 Furthermore, bosutinib, axitinib, and neratinib display selectivity across both the tyrosine kinase and serine/threonine kinases groups.17,19,21 On the contrary, although sorafenib and sunitinib act as pan-kinase inhibitors,19,21 they do hit their primary target in the clinic but with some off-target related toxicity. Kinases can be classified according to their tendency to bind to chemical compounds, as exemplified by a recent study highlighting a number of kinases, which are highly susceptible to chemical inhibition, e.g., FLT3, TRKC, and HGK/MAP4K4, while others were less susceptible, e.g., COT1, NEK6/7, and p38-γ.20
Present and Future Kinase Drug Development
Although the development of kinase drugs is now an established field, and still growing, problems with drug resistance, toxicity, and selectivity are evident.5 In this section we will review some emerging trends in kinase drug discovery that may address some of these challenges.
Allosteric inhibitors are mechanistically diverse, as they may occupy binding pockets or inhibit different protein–protein interactions that only apply to particular kinases or small families of kinases. Consequently, they often give high kinome selectivity.22 For natural products this concept is not new as exemplified for rapamycin (sirolimus) and the subsequently designed rapamycin analogues (rapalogues)23 such as everolimus and temsirolimus that target a specific allosteric mechanism to inhibit mTOR. However, for small molecules, FDA approvals of allosteric inhibitors are recent, as seen for the MEK inhibitors cobimetinib,24 binimetinib, and trametinib,25 with other clinical candidates following (e.g., selumetinib); Figures 3 and 5. There is also an interest in the use of allosteric inhibitors as a way of providing a differentiated mechanism to ATP pocket binders. One interesting example is the progression of BCR-Abl inhibitor asciminib (Figure 3) into clinical studies.26 Asciminib is being clinically combined with Type I/II BCR-Abl inhibitors such as nilotinib, imatinib, dasatinib, and bosutinib as an example of a “double hit” strategy where two kinase inhibitors with orthogonal mechanisms target the same kinase, potentially providing a more durable response, with reduced potential for relapse due to point resistance mutations. The screening methods for identifying allosteric kinase inhibitors have become increasingly sophisticated, and therefore, there has been a gradual shift from serendipitously discovered inhibitors (e.g., from cell-based screens) to biochemical cascades that are set up to screen for such inhibitors, for example, by varying the ATP concentration to screen out ATP-competitive inhibitors. However, overall allosteric inhibitors present a design challenge to the medicinal chemist. The productivity of kinase drug discovery thus far has benefited from ATP-pocket binders having multiple activities across the kinome. Thus, historical project compounds for target X in corporate collections often provide a good starting point for new target Y, and recurring structural motifs have proved to be useful across multiple kinase targets.27 As well as requiring a different screening cascade, larger and more structurally diverse compound collections and/or alternative screening strategies (e.g., structurally guided fragment-based programmes28) may be required to be equally productive in identifying potent allosteric hit molecules.
Figure 3.

Advanced allosteric clinical candidates.
While the range of binding modes (Types I–IV) may be increasingly diverse, the molecules shown in Figure 5 are all inhibitors. Another interesting modality that is rapidly gaining traction is degradation or indirect downregulation as an alternative paradigm to inhibition. While this concept is yet to be tested in a clinical setting, the significant investment in this area in pharmaceutical discovery means that it will likely have an impact on the clinic in the future and may even yield marketed drugs. Degradation may be achieved via various modalities, but the recent interest in proteolysis-targeting chimeras (PROTACS) is particularly noteworthy29 and has resulted in preclinical reports of degraders of a number of kinases, including BTK,30,31 BCR-Abl,32 and CDK9.33 This approach offers great potential in the rational design of kinase degraders, as in theory most kinase ligands could be converted to a PROTAC if a suitable vector and linker out to solvent can be designed to attach the E3 ligase binder. However, the size of the resulting molecule may mean that pharmacokinetic properties have to be carefully optimized if oral administration is desired. Alternatively, there are reports of small molecule inhibitors for certain kinases acting as downregulators. One such example is PF-956980, which was reported to be a pan-JAK inhibitor but a downregulator of only JAK2 and JAK3 (not JAK1 and TYK2), which affects via mRNA downregulation.34 Rational design for downregulation with a small molecule may be challenging, but broader screening of compounds to investigate possible effects on cellular protein levels by serendipity offers an opportunity to discover additional examples. One advantage of this approach is that such rule of five compliant molecules are more likely to have suitable pharmacokinetic properties for oral administration.
A topic that is particularly significant for oncology has been an increased focus on designing kinase inhibitors that target specific oncogenic mutations to enhance the efficacy for patients whose tumors harbor such mutations. An example of this approach is EGFR, where it was found that inhibitors such as gefitinib and erlotinib are more efficacious with patients that harbor activating mutations such as L858R and Exon 19 deletion, although this understanding came from postanalysis of the data from clinical trials.35 Resistance to EGFR inhibitors can also occur, most commonly due to the T790M mutation, which has an increased affinity with ATP.36 Furthermore, compounds such as afatinib have been designed with a covalent warhead that binds irreversibly to C797, thus conferring increased cellular activity for the activating mutations and T790M, but also for wild type EGFR. The high activity against wild-type EGFR limits clinical utility against T790M due to toxicity. More recently, medicinal chemistry programs have combined targeting of the double mutants (activating mutation plus resistance mutation), as well as designing in selectivity versus wild type EGFR to widen therapeutic margin, which has resulted in the approval of osimertinib.37 Targeting specific mutations have resulted in a number of other approvals, notably for ALK, where targeting of the EML4-ALK4 fusion with drugs such as crizotinib38 has evolved into an increased focus on gatekeeper resistance mutation L119M and a host of other mutations in the ATP pocket, and activity against some or all of these mutations39 has been reported for approved drugs such as ceritinib, alectinib, and brigatinib.40−43 Successor compounds of imatinib, such as nilotinib and ponatinib, are examples of other drugs that display distinct kinase mutant-targeting profiles.44
Another potential topic for the future of kinase modulation is the targeting of pseudokinases, which represent a significant portion of the kinome (∼10%)45 and yet has relatively few reported inhibitors. The pseudokinases are structurally similar to catalytically active kinases and consequently may retain some affinity for ATP. They do not catalyze the transfer of a phosphate moiety from ATP, but they may have other functions in the cell, e.g., through protein–protein interactions. In a similar vein, catalytically active kinases may also have noncatalytic functions that could be modulated by medicinal chemists, which represents a further opportunity. A number of clinical and preclinical compounds have been reported,46 which give promise for the future.
In terms of medical applications, as discussed above, most approved drugs and clinical candidates within the kinase field continue to be for oncology indications. One noteworthy recent development is a significantly increased focus on immuno-oncology (IO), both in the clinic and preclinically. IO can be defined as the stimulation of an immune response to a tumor, either directly by affecting immune cell function or indirectly via modulation of the tumor microenvironment.47 While the IO field initially focused on checkpoint inhibitors of targets such as PD-1 and PD-L1, many of the emerging IO targets are kinases, which have given renewed interest in such targets and inhibitors as CSF1R48 (e.g., pexidartinib, Phase 3), ALK549 (e.g., galunisertib, Phase 2/3), and PI3Kδ/γ50 (e.g., idelalisib) to name a few (Figures 4 and 5). From a medicinal chemistry perspective, the challenges of IO are not specific to the field, indeed many examples of compounds being positioned for IO were discovered earlier and have been subsequently repositioned. However, the ability to combine with other drugs may be an even greater consideration for IO kinase inhibitors, which may favor compounds with no drug–drug interaction potential and high selectivity, and the ability to deliver flexible dosing schedules (continuous or intermittent dosing, or “priming”) may be important for some targets and thus may lead to specific requirements for pharmacokinetic properties.
Figure 4.

Emerging immune-oncology inhibitors.
Overall, it is an exciting time for the kinase field, with novel targets, mechanisms of drug action, and drug binding sites. The field has followed a fresh direction, moving beyond standard ATP mimic inhibitors. This strategy promises to yield fundamental insights into novel kinase biology and targeting, facilitating game changing breakthroughs in kinase drug development.
Acknowledgments
We would like to thank Gottfried Sedelmeier for his support in composing this innovation article, as well as Carolyn Blackett, Mark Wigglesworth, Richard Ward and Garry Pairaudeau for reading the manuscript and providing valuable comments. Approval dates/relevant companies and targets for marketed drugs were obtained from Thomson Reuters Integrity and Adis Insight, respectively (30th July 2018).
Glossary
ABBREVIATIONS
- ALK
anaplastic lymphoma kinase
- ATP
adenosine triphosphate
- BCR-Abl
BCR-ABL fusion
- BRAF
serine/threonine-protein kinase
- BTK
Bruton’s tyrosine kinase
- CDKs
cyclin-dependent kinase
- CML
chronic myelogenous leukemia
- COT1
serine/threonine-protein kinase cot-1
- EGFR
epidermal growth factor receptor
- ErbB
erythroblastic leukemia viral oncogene homologue (ERBB) receptors
- FDA
Food and Drug Administration
- FLT3
Fms-related tyrosine kinase
- GIST
gastrointestinal stromal tumors
- IGF1R
insulin-like growth factor-1 receptor
- INN
International Nonproprietary Names
- IPF
idiopathic pulmonary fibrosis
- ITK
IL-2-inducible T-cell kinase
- ITP
immune thrombocytopenic purpura
- IO
immuno-oncology
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinases
- KI
kinase inhibitor
- MAP2K
mitogen-activated protein kinase kinase
- MAP4K4
mitogen-activated protein kinase kinase kinase kinase-4
- MAPK
mitogen-activated protein kinase
- MET
tyrosine-protein kinase Met
- NEK6/7
NIMA-related kinases 6/7
- PI3Kδ
phosphatidylinositol-4,5-bisphosphate 3-kinase delta
- PROTACS
proteolysis-targeting chimeras
- RET
receptor tyrosine-protein kinase
- SYK
spleen tyrosine kinase
- TRKC
tropomyosin receptor kinase C
- VEGFR
vascular endothelial growth factor receptor
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00445.
Poster depicting approved kinase inhibitors with INN registration dates (PDF)
Author Contributions
∥ Authors contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fabbro D.; Cowan-Jacob S. W.; Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172 (11), 2675–700. 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rompay A. R.; Johansson M.; Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther. 2000, 87 (2–3), 189–98. 10.1016/S0163-7258(00)00048-6. [DOI] [PubMed] [Google Scholar]
- Wu P.; Nielsen T. E.; Clausen M. H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36 (7), 422–39. 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Alessi D. R. Kinase drug discovery–what’s next in the field?. ACS Chem. Biol. 2013, 8 (1), 96–104. 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhullar K. S.; Lagaron N. O.; McGowan E. M.; Parmar I.; Jha A.; Hubbard B. P.; Rupasinghe H. P. V. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 2018, 17 (1), 48. 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelmeier G.; Lightfoot H.; Sedelmeier J. WHO Listed Small Molecule Kinase Inhibitors 2001–2017. Chimia 2018, 72 (72), 518. 10.2533/chimia.2018.518. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 2018, 17 (5), 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Jordan V. C. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Br. J. Pharmacol. 2006, 147 (Suppl 1), S269–76. 10.1038/sj.bjp.0706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discovery 2002, 1 (7), 493–502. 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery?. ACS Chem. Biol. 2014, 9 (6), 1230–41. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. T.; Kesselheim A. S. Journey of Generic Imatinib: A Case Study in Oncology Drug Pricing. J. Oncol Pract 2017, 13 (6), 352–355. 10.1200/JOP.2016.019737. [DOI] [PubMed] [Google Scholar]
- Sedelmeier G.; Sedelmeier J. Top 200 Drugs by Worldwide Sales 2016. Chimia 2017, 71 (10), 730. 10.2533/chimia.2017.730. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Progress with covalent small-molecule kinase inhibitors. Drug Discovery Today 2018, 23 (3), 727–735. 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- Liao B. C.; Lin C. C.; Lee J. H.; Yang J. C. Update on recent preclinical and clinical studies of T790M mutant-specific irreversible epidermal growth factor receptor tyrosine kinase inhibitors. J. Biomed. Sci. 2016, 23 (1), 86. 10.1186/s12929-016-0305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Zhang M.; Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J. Hematol. Oncol. 2016, 9, 21. 10.1186/s13045-016-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23 (3), 329–36. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Klaeger S.; Heinzlmeir S.; Wilhelm M.; Polzer H.; Vick B.; Koenig P. A.; Reinecke M.; Ruprecht B.; Petzoldt S.; Meng C.; Zecha J.; Reiter K.; Qiao H.; Helm D.; Koch H.; Schoof M.; Canevari G.; Casale E.; Depaolini S. R.; Feuchtinger A.; Wu Z.; Schmidt T.; Rueckert L.; Becker W.; Huenges J.; Garz A. K.; Gohlke B. O.; Zolg D. P.; Kayser G.; Vooder T.; Preissner R.; Hahne H.; Tonisson N.; Kramer K.; Gotze K.; Bassermann F.; Schlegl J.; Ehrlich H. C.; Aiche S.; Walch A.; Greif P. A.; Schneider S.; Felder E. R.; Ruland J.; Medard G.; Jeremias I.; Spiekermann K.; Kuster B. The target landscape of clinical kinase drugs. Science 2017, 358 (6367), eaan4368. 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29 (11), 1046–51. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29 (11), 1039–45. 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26 (1), 127–32. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Wu P.; Clausen M. H.; Nielsen T. E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. 10.1016/j.pharmthera.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Benjamin D.; Colombi M.; Moroni C.; Hall M. N. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discovery 2011, 10 (11), 868–80. 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- Rice K. D.; Aay N.; Anand N. K.; Blazey C. M.; Bowles O. J.; Bussenius J.; Costanzo S.; Curtis J. K.; Defina S. C.; Dubenko L.; Engst S.; Joshi A. A.; Kennedy A. R.; Kim A. I.; Koltun E. S.; Lougheed J. C.; Manalo J. C.; Martini J. F.; Nuss J. M.; Peto C. J.; Tsang T. H.; Yu P.; Johnston S. Novel Carboxamide-Based Allosteric MEK Inhibitors: Discovery and Optimization Efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012, 3 (5), 416–21. 10.1021/ml300049d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H.; Kikuchi S.; Hayakawa K.; Iida T.; Nagahashi N.; Maeda K.; Sakamoto J.; Matsumoto N.; Miura T.; Matsumura K.; Seki N.; Inaba T.; Kawasaki H.; Yamaguchi T.; Kakefuda R.; Nanayama T.; Kurachi H.; Hori Y.; Yoshida T.; Kakegawa J.; Watanabe Y.; Gilmartin A. G.; Richter M. C.; Moss K. G.; Laquerre S. G. Discovery of a Highly Potent and Selective MEK Inhibitor: GSK1120212 (JTP-74057 DMSO Solvate). ACS Med. Chem. Lett. 2011, 2 (4), 320–4. 10.1021/ml200004g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie A. A.; Schoepfer J.; Jahnke W.; Cowan-Jacob S. W.; Loo A.; Furet P.; Marzinzik A. L.; Pelle X.; Donovan J.; Zhu W.; Buonamici S.; Hassan A. Q.; Lombardo F.; Iyer V.; Palmer M.; Berellini G.; Dodd S.; Thohan S.; Bitter H.; Branford S.; Ross D. M.; Hughes T. P.; Petruzzelli L.; Vanasse K. G.; Warmuth M.; Hofmann F.; Keen N. J.; Sellers W. R. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543 (7647), 733–737. 10.1038/nature21702. [DOI] [PubMed] [Google Scholar]
- Kettle J. G.; Wilson D. M. Standing on the shoulders of giants: a retrospective analysis of kinase drug discovery at AstraZeneca. Drug Discovery Today 2016, 21 (10), 1596–1608. 10.1016/j.drudis.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Mukherjee P.; Bentzien J.; Bosanac T.; Mao W.; Burke M.; Muegge I. Kinase Crystal Miner: A Powerful Approach to Repurposing 3D Hinge Binding Fragments and Its Application to Finding Novel Bruton Tyrosine Kinase Inhibitors. J. Chem. Inf. Model. 2017, 57 (9), 2152–2160. 10.1021/acs.jcim.7b00213. [DOI] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem., Int. Ed. 2016, 55 (6), 1966–73. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Zorba A.; Nguyen C.; Xu Y.; Starr J.; Borzilleri K.; Smith J.; Zhu H.; Farley K. A.; Ding W.; Schiemer J.; Feng X.; Chang J. S.; Uccello D. P.; Young J. A.; Garcia-Irrizary C. N.; Czabaniuk L.; Schuff B.; Oliver R.; Montgomery J.; Hayward M. M.; Coe J.; Chen J.; Niosi M.; Luthra S.; Shah J. C.; El-Kattan A.; Qiu X.; West G. M.; Noe M. C.; Shanmugasundaram V.; Gilbert A. M.; Brown M. F.; Calabrese M. F. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (31), E7285–E7292. 10.1073/pnas.1803662115Som. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhimschi A. D.; Armstrong H. A.; Toure M.; Jaime-Figueroa S.; Chen T. L.; Lehman A. M.; Woyach J. A.; Johnson A. J.; Byrd J. C.; Crews C. M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57 (26), 3564–3575. 10.1021/acs.biochem.8b00391. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–10. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson C. M.; Jiang B.; Erb M. A.; Liang Y.; Doctor Z. M.; Zhang Z.; Zhang T.; Kwiatkowski N.; Boukhali M.; Green J. L.; Haas W.; Nomanbhoy T.; Fischer E. S.; Young R. A.; Bradner J. E.; Winter G. E.; Gray N. S. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2017, 14 (2), 163–170. 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field S. D.; Arkin J.; Li J.; Jones L. H. Selective Downregulation of JAK2 and JAK3 by an ATP-Competitive pan-JAK Inhibitor. ACS Chem. Biol. 2017, 12 (5), 1183–1187. 10.1021/acschembio.7b00116. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350 (21), 2129–39. 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Yun C. H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K. K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (6), 2070–5. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay M. R.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D. A.; Currie G. S.; Grist M.; Hassall L.; Hill G. B.; James D.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamont S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. L. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57 (20), 8249–67. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54 (18), 6342–63. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Katayama R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Wiley Cancer Science 2018, 109, 572–580. 10.1111/cas.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsilje T. H.; Pei W.; Chen B.; Lu W.; Uno T.; Jin Y.; Jiang T.; Kim S.; Li N.; Warmuth M.; Sarkisova Y.; Sun F.; Steffy A.; Pferdekamper A. C.; Li A. G.; Joseph S. B.; Kim Y.; Liu B.; Tuntland T.; Cui X.; Gray N. S.; Steensma R.; Wan Y.; Jiang J.; Chopiuk G.; Li J.; Gordon W. P.; Richmond W.; Johnson K.; Chang J.; Groessl T.; He Y. Q.; Phimister A.; Aycinena A.; Lee C. C.; Bursulaya B.; Karanewsky D. S.; Seidel H. M.; Harris J. L.; Michellys P. Y. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J. Med. Chem. 2013, 56 (14), 5675–90. 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- Das M. Brigatinib effective in ALK-positive non-small-cell lung cancer. Lancet Oncol. 2017, 18 (6), e310 10.1016/S1470-2045(17)30342-X. [DOI] [PubMed] [Google Scholar]
- Kinoshita K.; Asoh K.; Furuichi N.; Ito T.; Kawada H.; Hara S.; Ohwada J.; Miyagi T.; Kobayashi T.; Takanashi K.; Tsukaguchi T.; Sakamoto H.; Tsukuda T.; Oikawa N. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg. Med. Chem. 2012, 20 (3), 1271–80. 10.1016/j.bmc.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Friboulet L.; Li N.; Katayama R.; Lee C. C.; Gainor J. F.; Crystal A. S.; Michellys P. Y.; Awad M. M.; Yanagitani N.; Kim S.; Pferdekamper A. C.; Li J.; Kasibhatla S.; Sun F.; Sun X.; Hua S.; McNamara P.; Mahmood S.; Lockerman E. L.; Fujita N.; Nishio M.; Harris J. L.; Shaw A. T.; Engelman J. A. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discovery 2014, 4 (6), 662–673. 10.1158/2159-8290.CD-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossari F.; Minutolo F.; Orciuolo E. Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11 (1), 84. 10.1186/s13045-018-0624-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L. J.; Linley A.; Hammond D. E.; Hood F. E.; Coulson J. M.; MacEwan D. J.; Ross S. J.; Slupsky J. R.; Smith P. D.; Eyers P. A.; Prior I. A. New Perspectives, Opportunities, and Challenges in Exploring the Human Protein Kinome. Cancer Res. 2018, 78 (1), 15–29. 10.1158/0008-5472.CAN-17-2291. [DOI] [PubMed] [Google Scholar]
- Byrne D. P.; Foulkes D. M.; Eyers P. A. Pseudokinases: update on their functions and evaluation as new drug targets. Future Med. Chem. 2017, 9 (2), 245–265. 10.4155/fmc-2016-0207. [DOI] [PubMed] [Google Scholar]
- Dhanak D.; Edwards J. P.; Nguyen A.; Tummino P. J. Small-Molecule Targets in Immuno-Oncology. Cell Chem. Biol. 2017, 24 (9), 1148–1160. 10.1016/j.chembiol.2017.08.019. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Knolhoff B. L.; Meyer M. A.; Nywening T. M.; West B. L.; Luo J.; Wang-Gillam A.; Goedegebuure S. P.; Linehan D. C.; DeNardo D. G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74 (18), 5057–69. 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S.; Turley S. J.; Nickles D.; Castiglioni A.; Yuen K.; Wang Y.; Kadel E. E. III; Koeppen H.; Astarita J. L.; Cubas R.; Jhunjhunwala S.; Banchereau R.; Yang Y.; Guan Y.; Chalouni C.; Ziai J.; Senbabaoglu Y.; Santoro S.; Sheinson D.; Hung J.; Giltnane J. M.; Pierce A. A.; Mesh K.; Lianoglou S.; Riegler J.; Carano R. A. D.; Eriksson P.; Hoglund M.; Somarriba L.; Halligan D. L.; van der Heijden M. S.; Loriot Y.; Rosenberg J. E.; Fong L.; Mellman I.; Chen D. S.; Green M.; Derleth C.; Fine G. D.; Hegde P. S.; Bourgon R.; Powles T. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554 (7693), 544–548. 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali K.; Soond D. R.; Pineiro R.; Hagemann T.; Pearce W.; Lim E. L.; Bouabe H.; Scudamore C. L.; Hancox T.; Maecker H.; Friedman L.; Turner M.; Okkenhaug K.; Vanhaesebroeck B. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 2014, 510 (7505), 407–411. 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.