

Abstract

Allosteric HIV-1 integrase inhibitors (ALLINIs) are a new class of potential antiretroviral therapies with a unique mechanism of action and drug resistance profile. To further extend this class of inhibitors via a scaffold hopping approach, we have synthesized a series of analogues possessing an isoquinoline ring system. Lead compound 6l binds in the v-shaped pocket at the IN dimer interface and is highly selective for promoting higher-order multimerization of inactive IN over inhibiting IN-LEDGF/p75 binding. Importantly, 6l potently inhibited HIV-1NL4–3 (A128T IN), which confers marked resistance to archetypal quinoline-based ALLINIs. Thermal degradation studies indicated that at elevated temperatures the acetic acid side chain of specific isoquinoline derivatives undergo decarboxylation reactions. This reactivity has implications for the synthesis of various ALLINI analogues.
Keywords: HIV-1 integrase, allosteric inhibitor, protein multimerization
Integrase (IN) is a key HIV-1 enzyme that catalyzes the integration of the viral cDNA into the host genome. In addition, IN has a second noncatalytic function, which involves IN binding to the viral RNA genome in virions to ensure correct maturation of infectious virus particles.1 Structurally, IN functions as an oligomeric complex, with each protomer being composed of three unique domains: N-terminal domain (NTD), catalytic core domain (CCD), and C-terminal domain (CTD).2 Because of the essential roles of IN in the HIV-1 life cycle, IN has been widely explored and exploited as a viable drug target for the treatment of HIV-1 infections. Initially, drug development efforts focused on disruption of the active site of IN, leading to a class of compounds referred to as IN strand transfer inhibitors (INSTIs). Three members of this class, raltegravir, elvitegravir, and dolutegravir, have ultimately become highly successful FDA approved drugs.3−5 However, similar to other HIV therapeutics, these compounds are susceptible to resistance mutations.6−8 Therefore, there is a need for the continued development of novel therapies with alternative mechanisms of action to treat HIV-1 infections.
More recently, the v-shaped pocket at the IN CCD dimer interface, which provides the principal binding site for cellular cofactor Lens Epithelium Derived Growth Factor (LEDGF/p75), has been exploited for the design of allosteric HIV-1 IN inhibitors (ALLINIs, Figure 1), which have also been alternatively referred to as LEDGINs,9,10 NCINIs,11 and INLAIs.12 The primary antiviral activity of these compounds is seen during virion maturation where ALLINIs induce higher-order IN multimerization and consequently impair IN-RNA interactions, which ultimately yields eccentric noninfectious virions.1,13−15 ALLINIs also exhibit secondary, albeit significantly reduced, activity during the early phase of HIV-1 infection, where these compounds adversely affect IN-LEDGF/p75 interactions.10,16
Figure 1.

Chemical structures of the representative ALLINI compounds BIB-II and KF116.
ALLINIs contain conserved structural elements attached to a substituted heteroaromatic core that allows them to mimic key interactions between LEDGF/p75 and IN dimer. The most critical ALLINI substituent is an ether-containing acetic acid side chain that engages in both hydrogen bonding and electrostatic interactions with the E170, H171, and T174 residues located in the LEDGF binding pocket of HIV-1 IN.17,18 An additional interaction is created by an adjacent aromatic ring that projects into a relatively narrow channel in the dimer interface capped by W132. Core ring systems such as quinoline19 and pyridine14 from which these substituents project do not provide significant binding interactions with the protein, but instead serve as rigid scaffolding units to efficiently project the substituents into the binding pocket. In addition, the decoration of these cores with methyl groups likely plays an important role in the relative orientation of the adjacent aromatic and acetic acid functional groups through conformational blocking. Despite the fact that the core ring systems may not have direct binding interactions with IN, the cores themselves have been implicated in the development of resistance mutations due to their relative size and positioning in the binding pocket. For example, a single A128T IN substitution emerges in cell culture and confers substantial resistance to archetypal quinoline-based compounds including BIB-II (Figure 1).6,9 Thus, these central rings impart key geometric features in addition to obvious electronic and steric properties, thereby influencing the overall potency of the ALLINIs.
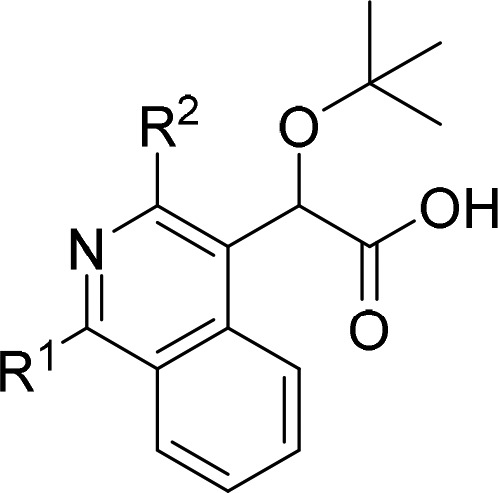
In an effort to expand the scope of current ALLINIs, an alternative central ring scaffold was sought that would (1) maintain the critical C4-substituent and simultaneously facilitate substitution at the C1 and C3 positions (see numbering in Scheme 1) to capture previously unexploited contacts at the IN dimer interface, (2) exhibit full potency against HIV-1 containing the A128T IN substitution, which confers substantial resistance to quinoline-based ALLINIs, and (3) provide a platform for the rapid assembly of these compounds. Utilizing these criteria as a guide, an isoquinoline core was identified that could be accessed through methodology reported for the synthesis of 1,3,4-trisubstituted systems.20 This isoquinoline scaffold could not only accommodate the desired substitution of the key aryl and acetic acid substituents, but the electron deficient heterocycle to which the ALLINI substituents would be attached is expected to provide a similar geometry and electronic environment to the highly potent quinoline and pyridine analogues.
Scheme 1. Synthesis of Isoquinoline Analogues 6a–l.

The application of this approach was realized starting from the commercially available 1,3-dichloroisoquinoline (1). As part of a sequence to introduce the fully elaborated acetic acid side chain onto the isoquinoline, halogenation of 1 at the C4 position was affected via treatment with LDA and iodine.20 The α-ketoester precursor to the acetic acid side chain was then introduced at this position via reaction of the corresponding cuprate, generated using isopropylmagnesium chloride and copper(I) bromide dimethyl sulfide complex, with ethyl chlorooxoacetate following a previously reported procedure for the functionalization of quinoline and pyridine cores.21 In contrast, direct metalation and acylation of 1 provided only low yields (∼18%) of the desired product. Once installed, however, the ketoester moiety could selectively be reduced with sodium borohydride at 0 °C to provide alcohol 2. Utilizing standard conditions for the introduction of the t-butyl group onto the secondary alcohol, 2 could then be converted to the desired ether 3 with the fully elaborated side chain.21
At this stage, 3 could be functionalized either successively or concurrently through application of appropriate coupling conditions to generate a variety of analogues. Thus, using a standard Suzuki coupling reaction, compounds 4a–i were formed regiospecifically. More forceful coupling conditions using the more reactive bis(trit-butylphosphine)palladium(0) or a higher loading of Pd(PPh3)4 (40 mol % vs 20 mol %) at higher temperature could then be employed to install aryl substituents at the more sterically hindered and less reactive C3 position of the isoquinoline (R2) to form compounds 5a–j. Alternatively, directly subjecting compound 3 to these “more forcing” conditions in the presence of excess boronate resulted in successive coupling at both the C1 and C3 positions in a single pot, albeit with the introduction of identical substituents at both positions. With the ethyl esters 5a–l in hand, saponification of the ethyl group and subsequent acidification provided the desired carboxylic acids 6a–l, representing either a six- or seven-step sequence to access the desired analogues from 1. In nearly all cases, the carboxylic acids conveniently precipitated from solution upon acidification and could be obtained cleanly through subsequent filtration. The only exceptions were compound 6l, a benzyl-pyrazole containing analogue, which failed to precipitate and required an additional extraction step during the course of the workup, and compound 6a, which required a subsequent chromatographic purification step.
We next attempted to introduce a chlorobenzimidazole ring system at C1 to generate compound 9 based on previous observations indicating a critical role of the benzimidazole substituent for high potency of pyridine-based KF116 (Figure 1).14 While introduction of aromatic and heteroaromatic ring systems at the C1 position of the isoquinoline had been achieved through direct coupling reactions (Scheme 1), the introduction of a chlorobenzimidazole ring system at this position could not be accomplished efficiently in a single coupling step. Therefore, starting from the common intermediate 3, the nitrile group necessary for elaboration to the benzimidazole substituent was first installed via palladium-mediated coupling (Scheme 2).10 A subsequent Suzuki coupling reaction introduced the chromane system at the C3 position, providing compound 7. In addition to the desired product, initial conditions employed for this Suzuki reaction, which included the addition of water in the presence of either sodium carbonate or sodium bicarbonate as the base, resulted in the formation of the ether byproduct S3 (Supporting Information) produced via hydrolysis and decarboxylation of 7 along with the desired product in a 1:1.2 ratio. This decarboxylation could be eliminated under anhydrous reaction conditions while still using sodium bicarbonate as the base, producing compound 7 in a 47% yield. In order to elaborate the nitrile into the benzimidazole, a three-step sequence was carried out to affect hydrolysis of the nitrile moiety while simultaneously “maintaining” the ester functionality on the acetic acid side chain through a series of hydrolysis/esterification events to provide carboxylic acid 8. Coupling of this acid with 4-chloro-o-phenylenediamine in the presence of HATU resulted in a mixture of regioisomeric amides that could be cyclized to form the benzimidazole upon heating in acetic acid. Acid 9 was then ultimately obtained through straightforward hydrolysis of the remaining ester.
Scheme 2. Synthesis of Compound 9.

The synthesized isoquinoline compounds 6a–l and 9 were initially evaluated for their ability to inhibit IN catalytic activities in the absence of LEDGF/p75 (Table 1). The “parent” diphenyl derivatives 6a and 6c, both of which possess a phenyl ring at the C3 position, exhibited relatively weak inhibitory activity (IC50 > 10 μM). Replacement of these phenyl substituents with chromane rings in compounds 6b and 6d resulted in approximately 2- and 10-fold increases in potency over 6a and 6c, respectively. This data parallels those seen in previous SAR studies of quinoline-based ALLINIs, which also indicated improved potencies resulting from introduction of the larger chromane substituent at the C3 position.12,22
Table 1. Chemical Structures and Inhibitory Activities (IC50, μM) of Analogues Tested in LEDGF/p75 Independent Integration Assaysa.

The standard errors from two independent experiments are indicated.
The remaining analogues, 6e–l and 9, all possess the chromane substituent at the C3 position but vary with regard to the substitution at the C1 position. This structural consistency has facilitated direct comparison of the R1 substituents. With this in mind, the R1 phenyl substituents containing electron-deficient groups (6e, 6f, and 6g) demonstrated considerably less potency than the rings with electron-donating substituents (6h and 6j). Moreover, regioisomeric compounds 6d and 6h suggest that the variation of the substituents on the phenyl ring from the meta- to para-position did not substantially affect the inhibitory activities of these compounds. The heterocycle-containing systems (6k, 6l, and 9) all showed reasonable potency as well. Among these heterocycles, the larger substituents showed greater potency. The most potent of these, the benzyl pyrazole analogue 6l, displayed a potency of 0.48 μM. Compound 9, which possesses the benzimidazole substituent, was approximately 2-fold less potent (IC50 of 0.81 μM) than 6l.
Our subsequent efforts have focused on elucidating the mode of action of 6l, the most potent compound in the isoquinoline series (Table 2). For comparison, archetypal quinoline-based BIB-II was examined in parallel experiments. As expected, BIB-II19 exhibited a dual mode of action by inducing higher-order IN multimerization and inhibiting IN-LEDGF/p75 binding with comparable potency in vitro (Table 2). In contrast from BIB-II, 6l displayed marked preference for promoting higher-order IN multimerization (EC50 of <1.79 μM) compared with its ability to inhibit IN-LEDGF/p75 binding (IC50 of ∼37 μM). ALLINI-induced higher-order IN multimerization can in turn impair IN catalytic activities and IN–RNA interactions.1,19 Accordingly, BIB-II exhibited comparable EC50/IC50 values in all assays indicated in Table 2, whereas 6l displayed selectivity for those assays related to higher-order IN multimerization. We noted that increasing concentrations of 6l resulted in quenching of the donor (Europium-cryptate)-IN signal, which in turn adversely affected the measured HTRF signal increases due to inhibitor induced higher-order multimerization of IN. Therefore, 6l could be promoting higher-order IN multimerization with even higher potency than the EC50 value of 1.79 μM determined by the HTRF assay. Consistent with this notion, 6l displayed improved IC50 values of ∼0.21 and ∼0.48 μM for inhibiting IN-RNA binding and LEDGF/p75 independent IN catalytic activities, respectively. Compound 6l was significantly less potent (IC50 of ∼23 μM) when IN activities were monitored in the presence of LEDGF/p75. The differential potencies seen in LEDGF/p75 independent vs dependent assays provide another line of evidence for marked specificity of 6l for inducing higher-order IN multimerization over interfering with IN-LEDGF/p75 binding.
Table 2. Inhibitory Activities of 6l and BIB-II in Vitroa.
| inhibitor | IN multimerization, EC50 (μM) | IN-LEDGF/p75 binding, IC50 (μM) | LEDGF/p75 independent IN activity, IC50 (μM) | LEDGF/p75 dependent IN activity, IC50 (μM) | IN-RNA binding, IC50 (μM) |
|---|---|---|---|---|---|
| 6l | <1.79 ± 0.19 | 36.7 ± 6.2 | 0.48 ± 0.06 | 23.4 ± 0.78 | 0.21 ± 0.03 |
| BIB-II | 0.146 ± 0.014 | 0.15 ± 0.01 | 0.12 ± 0.04 | 0.15 ± 0.01 | 0.136 ± 0.02 |
Standard errors from two independent experiments are shown.
Cell culture assays compared antiviral activities of 6l with respect to WT vs BIB-II resistant viruses. Compound 6l inhibited WT HIV-1NL4–3 with EC50 of ∼1.1 μM and exhibited slightly higher potency (EC50 of ∼0.73 μM) with respect to HIV-1NL4-3 (A128T IN), which confers substantial resistance to BIB-II (Table 3).
Table 3. Antiviral Activities of 6l (EC50 values in μM)a.
| inhibitor | HIV-1NL4–3 | HIV-1NL4–3 (A128T IN) |
|---|---|---|
| 6l | 1.10 ± 0.07 | 0.73 ± 0.07 |
| BIB-II | 0.63 ± 0.3019 | 12.2 ± 6.419 |
Standard errors from two independent experiments are shown.
To elucidate the structural basis for interactions of these isoquinoline compounds with IN, we have solved the crystal structures of 6l and its less potent predecessor 6b bound to the CCD dimer (Figure 2). The comparative analysis of these two compounds was carried out to better understand the SAR for this series. Indeed, our structural studies allowed us to elucidate both similarities and differences between these two isoquinoline compounds. The carboxylic acid moiety of both compounds forms a bidentate interaction with the backbone amides of H171 and E170 of subunit 2. Additional hydrogen bonding interactions occur between H171 and T174 of subunit 2 and the ether oxygen of both compounds. The chromane rings and t-butoxy moieties of both compounds are situated in the hydrophobic pocket that is generated at the interface between subunits 1 and 2 and are formed by several hydrophobic residues and capped by W132.
Figure 2.
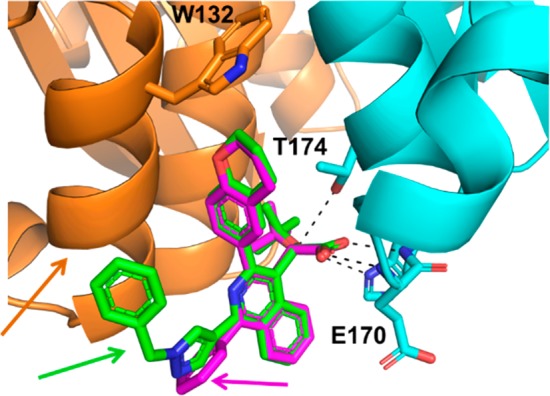
Crystal structures of 6b (magenta) and 6l (green) bound to the CCD dimer (subunits 1 and 2 are colored orange and cyan, respectively).
Differences between 6b and 6l are observed at R1. The benzene substituent of 6b is directed out of the dimer interface into space and has minimal interactions with CCD dimer. In contrast, the R1 substituent of 6l forms hydrophobic interactions with the surface of α-helix (124–133) of subunit 1, which could help to explain the substantially enhanced inhibitory activity of 6l over 6b.
To better understand the structural basis for differential effects of the A128T substitution on 6l and BIB-II, we subsequently superimposed the crystal structures of these compounds (Figure S1, Supporting Information). The rigid quinoline core of BIB-II extends toward A128 and is therefore affected by the bulkier and more polar A128T substitution.19 In contrast, the isoquinoline core is substantially distanced and projects away from A128. Interestingly, the benzyl moiety on the R1 substituent of 6l is positioned relatively close to A128. However, the highly flexible nature of this R1 substituent presumably allows 6l to effectively negate the effects of the A128T substitution.
In addition to these SAR studies, the stability of the acetic acid side chains of this class of compounds was also examined. This investigation was based upon both the unexpected decarboxylation of compound 7 mentioned above and the fact that new peaks, albeit minor, were observed in the 1H NMR spectra of several of the final carboxylic acid-containing compounds upon storage, indicating some type of decomposition. Most notably, these 1H NMR spectra showed what appeared to be an upfield shift of the methine proton on the acetic acid side chain. This data suggested that these compounds may be prone to decarboxylation of the acetic acid side chain. To test this hypothesis, a variation of forced degradation23 was employed to investigate the stability of selected compounds. In this case, samples were heated in DMSO-d6 in an NMR tube to expedite this degradative process and allow rapid assessment of the conversion and compound identity. Compounds 6d and 6e, possessing electron-deficient and electron-rich groups at the C1 position, respectively, as well as compound 6i were initially heated at 60 °C for 17 h. The 1H NMR spectra of each of these compounds at this time point indicated very little to no degradation (Supporting Information). After this initial period, compound 6i was further heated by increasing the temperature to 100 °C and monitored at various time points over a period of 17 days. Interestingly, a sharp singlet at 10.12 ppm in the 1H NMR spectrum increased in intensity over time relative to the starting material. A carbon signal at 193.6 ppm in the 13C NMR spectra was also observed, indicating the presence of an aldehyde peak. In the HSQC NMR spectrum, a strong correlation of the carbon signal at 193.6 ppm with the proton at 10.12 ppm also supported this assignment. Upon further examination of the 1H NMR spectrum, a shift in the t-butyl ether protons from 0.90 to 1.11 ppm could also be observed, correlating with loss of the ether group, likely as t-butanol, during the course of the reaction. The use of high-resolution mass spectrometry in conjunction with the other spectroscopic data further supported the assignment of this compound as aldehyde 10 seen in Scheme 3.
Scheme 3. Degradation of 6i under Thermal Conditions.

The 1H NMR spectra for compounds 6d and 6e, which were stored in DMSO-d6 for 17 days at room temperature after the initial heating at 60 °C, revealed a similar aldehyde peak observed around ∼10.1 ppm. During subsequent heating of both of these compounds to 100 °C for 3 days, the presumed aldehyde peak grew rapidly in intensity, suggesting that this degradation occurs slowly at room temperature but much faster at elevated temperatures. Surprisingly, reinvestigation of the T0 (time-zero, prior to heating) 1H NMR spectra for compound 6e revealed the presence of a small amount of the sharp singlet at ∼10.1 ppm, among other minor impurities, suggesting that the analogues with electron-withdrawing groups may be more prone to degradation upon compound storage, even at lower temperatures. To our knowledge, this is the first time that this phenomenon has been observed for a t-butyl ether-containing acetic acid side chain in an ALLINI compound.
The mechanism by which this decomposition happens is as of yet unknown, but it is worth noting that other groups have reported the decarboxylation of α-hydroxy carboxylic acids through chemical and enzymatic methods.24−28 Although 100 °C is acknowledged to not be a biologically relevant temperature, this type of degradation warrants further investigation because it could have potential implications for chemical stability of other ALLINIs as the t-butoxy acetic acid side chain is an important pharmacophore for all potent ALLINIs.
In conclusion, a series of isoquinoline ALLINI analogues were synthesized. Their SAR studies have revealed that sterically bulkier R1 and R2 substituents resulted in enhanced inhibitory activities of these compounds. Compound 6l, the most active compound, exhibited markedly higher potency for inducing higher-order IN multimerization than inhibiting IN-LEDGF/p75 binding. Significantly, 6l was highly potent against the HIV-1 variant containing the A128T IN substitution, which confers marked resistance to a number of quinoline-based inhibitors. The relative stability of selected isoquinoline compounds was also investigated, revealing lability in the acetic acid side chain. These observations will inform ongoing efforts in the field to synthesize various ALLINI derivatives.
Acknowledgments
We would like to thank Dr. C. McElroy, Dr. C. Yuan, and the OSU CCIC for assistance with acquisition of NMR data.
Glossary
ABBREVIATIONS
- IN
integrase
- ALLINIs
allosteric HIV-1 integrase inhibitors
- LEDGF/p75
Lens Epithelium Derived Growth Factor
- LEDGINs
LEDGF integrase inhibitors
- NCINIs
noncatalytic site integrase inhibitors
- INLAIs
integrase-LEDGF allosteric inhibitors
- HTRF
homogeneous time-resolved fluorescence
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00633.
Experimental procedures and characterization data for all compounds, thermal stability studies of compounds 6d, 6e, and 6i, conditions for biological assays, and crystal structure information (PDF)
Accession Codes
PDB numbers for compounds 6b, 6EB1, and 6l, 6EB2.
Author Contributions
J.R.F., M.K., and R.C.L. conceived and designed experiments. T.A.W., P.C.K., S.V.R., J.J.L., M.J.K., N.T.C., and D.A.A. performed the experiments and analyzed the results. T.A.W., J.R.F., P.C.K., and M.K. wrote the manuscript with contributions from all other authors.
This work was supported by National Institutes of Health grants U54 GM103368 (to M.K. and J.R.F.), R21 AI138775 (to J.R.F. and R.C.L.), R01 AI062520 (to M.K.), KL2 TR001068 (to R.C.L.), and a fellowship from 5T32 AT007533 (to T.A.W.).
The authors declare no competing financial interest.
Supplementary Material
References
- Kessl J. J.; Kutluay S. B.; Townsend D.; Rebensburg S.; Slaughter A.; Larue R. C.; Shkriabai N.; Bakouche N.; Fuchs J. R.; Bieniasz P. D.; Kvaratskhelia M. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268. 10.1016/j.cell.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos D. O.; Li M.; Yang R.; Rebensburg S. V.; Ghirlando R.; Jeon Y.; Shkriabai N.; Kvaratskhelia M.; Craigie R.; Lyumkis D. Cryo-EM Structures and Atomic Model of the HIV-1 Strand Transfer Complex Intasome. Science 2017, 355, 89–92. 10.1126/science.aah5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl D. J.; Chen X. Strand Transfer Inhibitors of HIV-1 Integrase: Bringing IN a New Era of Antiretroviral Therapy. Antiviral Res. 2010, 85, 101–118. 10.1016/j.antiviral.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Gonzalez Paz O.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- Raffi F.; Wainberg M. A. Multiple Choices for HIV Therapy with Integrase Strand Transfer Inhibitors. Retrovirology 2012, 9, 110–113. 10.1186/1742-4690-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Wang H.; Huang X.; Qin Z.; Deng Z.; Luo J.; Wang B.; Li M. Therapy-Emergent Drug Resistance to Integrase Strand Transfer Inhibitors in HIV-1 Patients: A Subgroup Meta-Analysis of Clinical Trials. PLoS One 2016, 11, e0160087. 10.1371/journal.pone.0160087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijting I. E. A.; Lungu C.; Rijnders B. J. A.; van der Ende M. E.; Pham H. T.; Mesplede T.; Pas S. D.; Voermans J. J. C.; Schuurman R.; van de Vijver D. A. M. C.; Boers P. H. M.; Gruters R. A.; Boucher C. A. B.; van Kampen J. J. A. HIV-1 Resistance Dynamics in Patients With Virologic Failure to Dolutegravir Maintenance Monotherapy. J. Infect. Dis. 2018, 218, 688–697. 10.1093/infdis/jiy176. [DOI] [PubMed] [Google Scholar]
- Wijting I.; Rokx C.; Boucher C.; van Kampen J.; Pas S.; de Vries-Sluijs T.; Schurink C.; Bax H.; Derksen M.; Andrinopoulou E.-R.; van der Ende M.; van Gorp E.; Nouwen J.; Verbon A.; Bierman W.; Rijnders B. Dolutegravir as Maintenance Monotherapy for HIV (DOMONO): A Phase 2, Randomised Non-Inferiority Trial. Lancet HIV 2017, 4, e547–e554. 10.1016/S2352-3018(17)30152-2. [DOI] [PubMed] [Google Scholar]
- Desimmie B. A.; Demeulemeester J.; Christ F.; Debyser Z. Rational Design of LEDGINs as First Allosteric Integrase Inhibitors for the Treatment of HIV Infection. Drug Discovery Today: Technol. 2013, 10, e517–e522. 10.1016/j.ddtec.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Christ F.; Voet A.; Marchand A.; Nicolet S.; Desimmie B. A.; Marchand D.; Bardiot D.; Van der Veken N. J.; Van Remoortel B.; Strelkov S. V.; De Maeyer M.; Chaltin P.; Debyser Z. Rational Design of Small-Molecule Inhibitors of the LEDGF/P75-Integrase Interaction and HIV Replication. Nat. Chem. Biol. 2010, 6, 442–448. 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- Balakrishnan M.; Yant S. R.; Tsai L.; O’Sullivan C.; Bam R. A.; Tsai A.; Niedziela-Majka A.; Stray K. M.; Sakowicz R.; Cihlar T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS One 2013, 8, e74163. 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic E.; Bonnard D.; Chasset S.; Bruneau J.-M.; Chevreuil F.; Le Strat F.; Nguyen J.; Beauvoir R.; Amadori C.; Brias J.; Vomscheid S.; Eiler S.; Lévy N.; Delelis O.; Deprez E.; Saïb A.; Zamborlini A.; Emiliani S.; Ruff M.; Ledoussal B.; Moreau F.; Benarous R. Dual Inhibition of HIV-1 Replication by Integrase-LEDGF Allosteric Inhibitors Is Predominant at the Post-Integration Stage. Retrovirology 2013, 10, 144–163. 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado K. A.; Wang H.; Slaughter A.; Feng L.; Kessl J. J.; Koh Y.; Wang W.; Ballandras-Colas A.; Patel P. A.; Fuchs J. R.; Kvaratskhelia M.; Engelman A. Allosteric Integrase Inhibitor Potency Is Determined through the Inhibition of HIV-1 Particle Maturation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8690–8695. 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Slaughter A.; Jena N.; Feng L.; Kessl J. J.; Fadel H. J.; Malani N.; Male F.; Wu L.; Poeschla E.; Bushman F. D.; Fuchs J. R.; Kvaratskhelia M. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLoS Pathog. 2014, 10, e1004171. 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch N. G.; Hart A. P.; Hume J. D.; Sun J.; McNeely K. A.; Lama C.; Pigza J. A.; Donahue M. G.; Kessl J. J. Synthesis and Evaluation of Aryl Quinolines as HIV-1 Integrase Multimerization Inhibitors. ACS Med. Chem. Lett. 2018, 9, 1007–1012. 10.1021/acsmedchemlett.8b00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J.; Jena N.; Koh Y.; Taskent-Sezgin H.; Slaughter A.; Feng L.; de Silva S.; Wu L.; Le Grice S. F. J.; Engelman A.; Fuchs J. R.; Kvaratskhelia M. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. 10.1074/jbc.M112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado K. A.; Engelman A. Multimodal Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. Expert Rev. Mol. Med. 2013, 15, e14. 10.1017/erm.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter A.; Jurado K. A.; Deng N.; Feng L.; Kessl J. J.; Shkriabai N.; Larue R. C.; Fadel H. J.; Patel P. A.; Jena N.; Fuchs J. R.; Poeschla E.; Levy R. M.; Engelman A.; Kvaratskhelia M. The Mechanism of H171T Resistance Reveals the Importance of Nδ-Protonated His171 for the Binding of Allosteric Inhibitor BI-D to HIV-1 Integrase. Retrovirology 2014, 11, 100. 10.1186/s12977-014-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Sharma A.; Slaughter A.; Jena N.; Koh Y.; Shkriabai N.; Larue R. C.; Patel P. A.; Mitsuya H.; Kessl J. J.; Engelman A.; Fuchs J. R.; Kvaratskhelia M. The A128T Resistance Mutation Reveals Aberrant Protein Multimerization as the Primary Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2013, 288, 15813–15820. 10.1074/jbc.M112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. A. Facile Synthesis of 1,3,4-Trisubstituted Isoquinolines. Tetrahedron Lett. 2009, 50, 3081–3083. 10.1016/j.tetlet.2009.04.040. [DOI] [Google Scholar]
- Fandrick K. R.; Li W.; Zhang Y.; Tang W.; Gao J.; Rodriguez S.; Patel N. D.; Reeves D. C.; Wu J.-P.; Sanyal S.; Gonnella N.; Qu B.; Haddad N.; Lorenz J. C.; Sidhu K.; Wang J.; Ma S.; Grinberg N.; Lee H.; Tsantrizos Y.; Poupart M.-A.; Busacca C. A.; Yee N. K.; Lu B. Z.; Senanayake C. H. Concise and Practical Asymmetric Synthesis of a Challenging Atropisomeric HIV Integrase Inhibitor. Angew. Chem., Int. Ed. 2015, 54, 7144–7148. 10.1002/anie.201501575. [DOI] [PubMed] [Google Scholar]
- Patel P. A.; Kvaratskhelia N.; Mansour Y.; Antwi J.; Feng L.; Koneru P.; Kobe M. J.; Jena N.; Shi G.; Mohamed M. S.; Li C.; Kessl J. J.; Fuchs J. R. Indole-Based Allosteric Inhibitors of HIV-1 Integrase. Bioorg. Med. Chem. Lett. 2016, 26, 4748–4752. 10.1016/j.bmcl.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessy M.; Patel R. D.; Prajapati P. N.; Agrawal Y. K. Development of Forced Degradation and Stability Indicating Studies of Drugs-A Review. J. Pharm. Anal. 2014, 4, 159–165. 10.1016/j.jpha.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe T. R.; Adkins R. L.; Belcher A. I.; Choy T.; Fuller A. E.; Morgan V. L.; Sencherey B. B.; Russell L. J.; Yates S. W. J. Org. Chem. 1982, 47, 3006–3008. 10.1021/jo00136a041. [DOI] [Google Scholar]
- Cassani C.; Bergonzini G.; Wallentin C.-J. Photocatalytic Decarboxylative Reduction of Carboxylic Acids and Its Application in Asymmetric Synthesis. Org. Lett. 2014, 16, 4228–4231. 10.1021/ol5019294. [DOI] [PubMed] [Google Scholar]
- Pink J. M.; Stewart R. Mechanism of Oxidative Decarboxylation of α-Hydroxy Acids by Bromine Water. Part I. Oxidation in Neutral and Alkaline Medium. Can. J. Chem. 1971, 49, 649–653. 10.1139/v71-104. [DOI] [Google Scholar]
- Mead J. F.; Levis G. M. Enzymatic Decarboxylation of the alpha-Hydroxy Acids by Brain Microsomes. Biochem. Biophys. Res. Commun. 1963, 11, 319–324. 10.1016/0006-291X(63)90564-3. [DOI] [PubMed] [Google Scholar]
- Paine T. K.; Paria S.; Que L. Oxidative Decarboxylation of α-Hydroxy Acids by a Functional Model of the Nonheme Iron Oxygenase, CloR. Chem. Commun. 2010, 46, 1830–1832. 10.1039/b925389k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.