

Abstract
Environmental exposure to cadmium (Cd) links to neurodegenerative disorders. Autophagy plays an important role in controlling cell survival/death. However, how autophagy contributes to Cd’s neurotoxicity remains enigmatic. Here, we show that Cd induced significant increases in autophagosomes with a concomitant elevation of LC3-II and p62 in PC12 cells and primary neurons. Using autophagy inhibitor 3-MA, we demonstrated that Cd-increased autophagosomes contributed to neuronal apoptosis. Impairment of Cd on autophagic flux was evidenced by co-localization of mCherry and GFP tandem-tagged LC3 puncta in the cells. This is further supported by the findings that administration of chloroquine (CQ) potentiated the basic and Cd-elevated LC3-II and p62 levels, autophagosome accumulation and cell apoptosis, whereas rapamycin relieved the effects in the cells in response to Cd. Subsequently, we noticed that Cd evoked the phosphorylation of Akt and BECN1. Silencing BECN1 and especially expression of mutant BECN1 (Ser295A) attenuated Cd-increased autophagosomes and cell death. Of note, inhibition of Akt with Akt inhibitor X, or ectopic expression of dominant negative Akt (dn-Akt), in the presence or absence of 3-MA, significantly alleviated Cd-triggered phosphorylation of Akt and BECN1, autophagosomes, and apoptosis. Importantly, we found that Cd activation of Akt functioned in impairing autophagic flux. Collectively, these results indicate that Cd results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Our findings underscore that inhibition of Akt to improve autophagic flux is a promising strategy against Cd-induced neurotoxicity and neurodegeneration.
Keywords: Cadmium, LC3, p62, BECN1, Akt, Autophagic flux
Graphical abstract
1. Introduction
Autophagy, an evolutionarily conserved intracellular degradation system, involves a sequential set of events including double membrane formation, elongation, vesicle maturation and finally delivery of the targeted materials to the lysosome [1; 2]. Because of its degradation of long-lived proteins, organelles and other cellular contents, autophagy plays a pivotal role in maintaining cellular homeostasis and protects cells from varying insults, including misfolded and aggregated proteins and damaged organelles, which is particularly crucial in neuronal survival [2; 3; 4]. For example, genetic inactivation of essential autophagy genes, such as autophagy-related 5 (ATG5) or 7 (ATG7), in neurons causes accumulation of protein aggregates and spontaneous neurodegeneration [5; 6]. Defected autophagic process may be one of the factors contributing to neuronal cell death [7]. Mounting evidence has pointed to autophagic dysfunction as a potential pathogenesis of several major neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD), where failure to eliminate abnormal and toxic protein aggregates promotes cellular stress and death [2; 4; 5; 6; 7]. Accordingly, much more attention has been paid to the relationship between autophagy and neurodegenerative disorders.
Autophagy induction is a double-edged sword in the pathogenesis of many human diseases [4]. Autophagy can play a protective role in many instances, or lead to cell death under certain circumstances [4; 8]. Cadmium (Cd), a toxic heavy metal pollutant in the environment, can easily traverse the blood-brain barrier and consequentially accumulate in the brain, thereby leading to neuronal cell dysfunction/apoptosis in the central nervous system (CNS) [9; 10; 11]. Cd neurotoxicity is closely associated with the development of PD, AD, HD and amyotrophic lateral sclerosis (ALS) [11; 12; 13; 14; 15]. Recently, a study has reported that Cd induces autophagy in PC12 cells, which plays a cytoprotective role in the cells exposed to Cd [16]. However, other studies have shown that Cd-induced autophagy or Cd-mediated overactivation of mitophagy contributes to cytotoxicity or cell death in mesangial cells [17], epidermal cells [18], mouse kidney cells [19] and brain neurons [20]. The discrepant findings prompted us to further explore and clarify the relationship between autophagy and apoptosis in neuronal cells induced by Cd, as well as the underlying mechanisms.
There are many important ATG genes related to autophagosome initiation and formation. The microtubule-associated protein 1 light chain 3 (LC3), a mammalian homologue of the yeast protein ATG8, has been found to be a specific biochemical marker for autophagy [1; 21]. LC3 is conjugated to phosphatidylethanolamine (PE) through an enzymatic cascade involving ATG7 (as an E1-like enzyme), ATG3 (as an E2-like enzyme) and ATG5-12-16 complex, and and is located on autophagosomal membranes after posttranslational modifications [18; 21; 22]. LC3 exists in two molecular forms with LC3-I and LC3-II. LC3-I is the unconjugated form in the cytosol, whereas LC3-II is the conjugated form that binds to autophagosomes and directly correlates with the number of autophagosomes [21; 23]. Thus, the level of LC3-II or GFP-LC3-II is widely used as a marker for monitoring the status of autophagy. However, of note, since autophagy is a dynamic process, the accumulation of LC3-II or autophagosomes could be related to either the induction of autophagy or the blockage of lysosomal function and/or fusion of autophagosomes with lysosomes [21; 24]. Multiple reports have described the term “autophagic flux”, which is used to represent the dynamic process of autophagy. In detail, autophagic flux refers to the whole process of cargo moving through the autophagic system, including autophagosome formation, maturation, fusion with lysosomes, the delivery of cargo to lysomsomes, the cargo degradation by lysosomal hydrolases, and the release of degraded products into the cytosol [25; 26]. Additionally, autophagy adaptor p62 protein, also called sequestosome 1 (SQSTM1), binds to ubiquitinated substrates and LC3, and is degraded along with its cargo [25]. The decreased p62 protein level indicates the enhancement of autophagy flux, so when autophagy flux is inhibited, the p62 protein level increases [27]. Therefore, analysis of the p62 protein level in the cells is essential to assess the status of autophagic flux, i.e. to determine whether autophagy is eventually executed or blocked [21].
Akt, a serine/threonine protein kinase, is a major regulator of neuronal cell survival [28]. Beclin 1 (BECN1), an essential core protein in autophagy, is a target of Akt [29]. Studies have shown that Akt suppression of autophagy can be mediated by activation of mTOR, which inhibits the autophagy-initiating Unc-51-like kinase 1 (ULK1) kinase complex [29; 30]. Akt can also directly phosphorylate BECN1 leading to suppression of autophagy [29]. Our recent studies have documented that Cd induces activation of Akt/mTOR signaling pathway contributing to apoptosis in neuronal cells [31]. It has been described that Cd induces autophagy in neuronal cells [16], whether and how Cd activation of Akt links to this event is largely unknown. Here, for the first time, we demonstrate that Cd induced impaired autophagic flux leading to accumulation of LC3-II and autophagosomes and consequential apoptotic cell death in neuronal cells. Cd activation of Akt and BECN1 linked to the increase of autophagosomes and apoptosis. Furthermore, Cd activation of Akt functioned in impairing autophagic flux. Our findings highlight that inhibition of Akt to improve autophagic flux is a promising approach against Cd-induced neurotoxicity.
2. Materials and methods
2.1. Materials
Cadmium chloride, poly-D-lysine (PDL), 3-methyladenine (3-MA), chloroquine diphosphate (CQ), monodansylcadaverine (MDC), 4′,6-diamidino-2-phenylindole (DAPI), and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA), whereas Akt inhibitor X was provided by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rapamycin were from ALEXIS Biochemicals Corporation (San Diego, CA, USA). Dulbecco’s modified Eagle medium (DMEM), 0.05% Trypsin-EDTA, NEUROBASALTM Media, and B27 Supplement were purchased from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
2.2. Cell culture
Rat pheochromocytoma (PC12) cell line (RRID: CVCL_0481) was obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA), which was used for no more than 10 passages. For culture, PC12 cells, seeded in a 6-well or 96-well plate pre-coated with PDL (0.2 μg/ml), were maintained in antibiotic-free DMEM supplemented with 10% horse serum and 5% FBS and incubated at 37°C in a humidified incubator containing 5% CO2. Primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in female ICR mice (being pregnant) as described [32], and seeded in a 6-well plate or 96-well plate coated with 10 μg/ml PDL for experiments after 6 days of culture. All procedures used in this study were approved by the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
2.3. Recombinant adenoviral constructs and infection of cells
Recombinant adenovirus encoding HA-tagged dominant negative Akt (Ad-dn-Akt, T308A/S473A) was generously provided from Dr. Kenneth Walsh (Boston University, Boston, MA), and the control virus expressing the green fluorescent protein (GFP) (Ad-GFP) or β-galactosidase (Ad-LacZ) were described previously [33]. Adenovirus expressing GFP-LC3 fusion protein (Ad-GFP-LC3) and a tandem adenovirus expressing mCherry-GFP-LC3 fusion protein (Ad-mCherry-GFP-LC3) were purchased from Sciben Biotech Company (Nanjing, China). For experiments, PC12 cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Ad-GFP or Ad-LacZ served as a control. Expression of HA-tagged dn-Akt was determined by Western blotting with an antibody to HA.
2.4. Lentiviral cloning, production, and infection
To generate lentiviral shRNAs to BECN1, oligonucleotides containing the target sequences were synthesized, annealed and inserted into FSIPPW lentiviral vector via the EcoR1/BamH1 restriction enzyme site [34]. The sequences of oligonucleiotides used were: BECN1 sense: 5′-AATTCCCGGACAACAAGTTTGACCATGCTGCAAGAGAGCATGGTCAAA CTTGTTGTCCTTTTTG-3′, anti-sense: 5′-GATCCAAAAAGGACAACAAGTTTGACCATGCTCTCTTGCAGCATGGTCAAACTTGTTGTCCGGG-3′. Lentiviral shRNA to GFP (for control) was generated as described [35]. To make FLAG-tagged mutant BECN1 (S295A) construct, site-directed mutagenesis was performed, as described [36]. The primers used were: Fragment 1 sense: 5’-CCGGAATTCATGGATTACAAGGATGACGACGATAAGATGGAAGGGTCT AAGACGTCCAA-3’, anti-sense: 5’-TGCGGGCAGGCGACCCAGCCTGAAGTTATTGATTGTGC-3’; Fragment 2 sense: 5’-GGCTGGGTCGCCTGCCCGCAGTTCCCGTGGAATGGAATGAGATTAATG C-3’, anti-sense: 5’-CGCGGATCCTCATTTGTTATAAAATTGTGAGGACACCCA-3’. The PCR product of FLAG-BECN1 (S295A) was cloned into pSin4-EF2-IRES-Pur vector. To produce lentiviral particles, above constructs were co-transfected together with pMD2G and psPAX2 (Addgene, Cambridge, MA, USA) to 293TD cells using MegaTran 1.0 reagent (OriGene Technologies, Rockville, MD, USA). Each virus-containing medium was collected 48 h and 60 h post-transfection, respectively. For use, monolayer PC12 cells, when grown to about 70% confluence, were infected with above lentivirus-containing medium in the presence of 8 μg/ml polybrene for 12 h twice at an interval of 6 h. Uninfected cells were eliminated by exposure to 2 μg/ml puromycin for 48 h before use. After 5 days of culture, cells were used for experiments.
2.5. MDC-labeled autophagic vacuoles
Intracellular autophagic status was monitored based on the incorporation of the autofluorescent drug MDC, which has been described as a selective marker for autophagic vacuoles [37; 38]. In brief, PC12 cells infected with lentiviral shRNA to BECN1 or GFP, respectively, were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. The next day, cells were treated with/without Cd (10 and 20 μM) for 8 h, 12 h and 24 h, or pretreated with/without 3-MA (4 mM) or CQ (25 μM) for 1 h, or pretreated with/without Akt inhibitor X (20 μM) for 2 h and then 3-MA (4 mM) for 1 h, followed by Cd (10 and/or 20 μM) for 12 h, with 5 replicates of each treatment. Subsequently, the cells were labeled with 0.05 mM MDC in PBS for 10 min at 37°C, and then washed 3 times with PBS, followed by cell imaging under a fluorescence microscopy (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. For quantitative analysis of the fluorescence intensity using MDC-labeled vacuoles, the integral optical density (IOD) was measured by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.6. GFP-LC3 or mCherry-GFP-LC3 assay
PC12 cells and primary neurons, PC12 cells infected with lentiviral shRNA to FLAG-BECN1 (S295A) or GFP, or PC12 cells infected with Ad-dn-Akt or Ad-LacZ, respectively, were infected with Ad-GFP-LC3 or Ad-mCherry-GFP-LC3 and seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. Next day, cells were treated with/without Cd (10 and/or 20 μM) for 8 h, 12 h and 24 h, or with/without Cd (10 μM) for 12 h following pretreatment with/without 3-MA (4 mM) for 1 h, with 5 replicates of each treatment. Afterwards, the cells were fixed with 4% paraformaldehyde in PBS for 30 min at 4°C, and then washed 3 times with PBS. For the cells infected with Ad-mCherry-GFP-LC3, after treatments, the cells were stained by adding DAPI (4 μg/ml in deionized water) as described [39]. Finally, slides were mounted in glycerol/PBS (1:1, v/v) containing 2.5% 1,4-diazabiclo-(2,2,2)octane. All the cell images were obtained using a fluorescence microscope (Leica DMi8, Germany) equipped with a digital camera. For the cells infected with Ad-GFP-LC3, autophagosome formation was estimated by counting the number of the cells with GFP-LC3 (green). At least 50 cells were scored in each experiment. For the cells infected with Ad-mCherry-GFP-LC3, autophagosome and autolysome status was evaluated by counting cells with GFP+/mCherry+ (yellow) and GFP–/Cherry-LC3+ (red) puncta, respectively. In tandem mCherry-GFP-LC3 assay, when an autophagosome fuses with the lysosome to form an autolysosome under acidic environments, GFP fluorescence is quenched in the autolysosome, whereas mCherry fluorescence is more stable. Therefore, when an autophagosome has not yet fused with a lysosome or when the degradation’s function of lysosome with acidification is impaired, co-localization of both GFP and mCherry fluorescence shows GFP+/mCherry+-LC3 (yellow) puncta in the merged image. In contrast, mCherry alone (without GFP) fluorescence presents GFP–/mCherry+-LC3 (red) puncta, which corresponds to an autolysosome.
2.7. Co-localization assay for autophagosomes/lysosomes
PC12 cells and primary neurons were infected with Ad-GFP-LC3 and seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. Next day, cells were treated with/without Cd (10 μM) for 12 h following pretreatment with/without rapamycin (0.2 μg/ml) for 48 h, with 5 replicates of each treatment. Afterwards, the cells were washed 3 times with serum-free medium, followed by labeling with LysoTracker® Red DND-99 (Invitrogen, Grand Island, NY, USA) at 37°C for 1 h. Next, the cells were washed 3 times with serum-free medium and then visualized under a fluorescence microscope (Leica DMi8, Germany) equipped with a digital camera. Autophagosomes were labeled by GFP-LC3 green fluorescence, lysosomes were stained by LysoTracker red fluorescence, and co-localization of both autophagosome and lysosome fluorescence showed autolysosome puncta (yellow) in the merged images. At least 50 cells per group were included for the counting of LysoTracker/GFP-LC3 and GFP-LC3 puncta/cell, respectively.
2.8. Live cell assay by trypan blue exclusion
PC12 cells infected with lentiviral shRNA to BECN1 or GFP, or PC12 cells infected with Ad-dn-Akt or Ad-GFP, respectively, were seeded at a density of 5 × 105 cells/well in a PDL-coated 6-well plate. The next day, cells were treated with/without Cd (10 and 20 μM) for 24 h, or with/without Cd (10 μM) for 24 h following pre-incubation with/without 3-MA (4 mM) for 1 h with 5 replicates of each treatment. Subsequently, live cells were monitored by counting viable cells using trypan blue exclusion test.
2.9. DAPI and TUNEL staining
PC12 cells and primary neurons, PC12 cells infected with lentiviral shRNA to BECN1, FLAG-BECN1 (S295A) or GFP, or PC12 cells infected with Ad-dn-Akt or Ad-GFP, respectively, were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. The next day, cells were treated with/without Cd (10 and 20 μM) for 24 h, or pretreated with/without 3-MA (4 mM) or CQ (25 μM) for 1 h, or pretreated with/without rapamycin (0.2 μg/ml) for 48 h, or pretreated with/without Akt inhibitor X (20 μM) for 2 h and then 3-MA (4 mM) for 1 h, followed by Cd (10 and/or 20 μM) for 24 h, with 5 replicates of each treatment. Subsequently, the cells with fragmented and condensed nuclei were stained by adding DAPI (4 μg/ml in deionized water) as described [39]. For the cells pretreated with/without 4 mM of 3-MA for 1 h and then exposed to 10 μM of Cd for 24 h, after DAPI staining, the following staining was performed by adding TUNEL reaction mixture (TdT enzyme solution and labeling solution) according to the manufacturer’s protocols of In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). Finally, photographs were taken under a fluorescence microscope (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. IOD for fluorescence intensity was quantitatively analyzed by Image-Pro Plus 6.0 software as described above.
2.10. Western blot analysis
The indicated cells, after treatments, were washed with cold PBS, and then on ice, lysed in the radioimmunoprecipitation assay buffer [50 mM Tris, pH 7.2; 150 mM NaCl; 1% sodium deoxycholate; 0.1% sodium dodecyl sulfate (SDS); 1% Triton X-100; 10 mM NaF; 1 mM Na3VO4; protease inhibitor cocktail (1:1000)]. Lysates were sonicated for 10 s and centrifuged at 16000 × g for 2 min at 4°C. The supernatants were collected and then Western blotting was performed as described previously [39]. In brief, lysates containing equivalent amounts of protein were separated on 7–12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were incubated with PBS containing 0.05% Tween 20 and 5% nonfat dry milk to block nonspecific binding, and then with primary antibodies against phosphorylated Akt (p-Akt) (Thr308), p-Akt (Ser473), cleaved-caspase-3, poly (ADP-ribose) polymerase (PARP) (Cell Signaling Technology, Danvers, MA, USA), Akt (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-BECN1 (Ser295) (Abcam, Cambridge, UK); LC3, SQSTM1/p62, BECN1, β-tubulin (Sciben Biotech Company) overnight at 4°C, respectively, followed by incubating with appropriate secondary antibodies including horseradish peroxidase-coupled goat anti-rabbit IgG, goat anti-mouse IgG, or rabbit anti-goat IgG (Pierce, Rockford, IL, USA) overnight at 4°C. Immunoreactive bands were visualized by using enhanced chemiluminescence solution (Sciben Biotech Company).
2.11. Statistical analysis
All data were expressed as mean values ± standard error (mean ± SE). Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. The criterion for the statistical significance was P < 0.05
3. Results
3.1. Cd induces increase in autophagosomes with a concomitant elevation of LC3-II and p62 in neuronal cells
To determine autophagic manifestation in Cd-exposed neuronal cells, the autofluorescent drug MDC, a specific autophagolysosome marker [37; 38], was employed. We observed that the accumulation of MDC was significantly induced by exposure to Cd dose-dependently in PC12 and primary neurons, as evaluated by the fluorescence intensity (in green) and quantification based on the incorporation of MDC (Fig. 1A and B). To corroborate the finding, we extended the studies by analyzing autophagic vacuoles with GFP-LC3 localization. Imaged and quantified results revealed that when PC12 cells and primary neurons, infected with Ad-GFP-LC3, were exposed to Cd (10 and 20 μM) for 8–24 h, the ratio of the cells with large LC3 puncta structures significantly increased compared to that of the vehicle-treated cells (Fig. 1C and D).
Fig. 1.

Cd induces increase in autophagosomes with a concomitant elevation of LC3-II and p62 in neuronal cells. PC12 cells and primary neurons, or PC12 cells and primary neurons infected with Ad-GFP-LC3 were treated with Cd (2.5, 5, 10 and/or 20 μM) for 2, 4, 6, 8, 10, 12 h and/or 24 h. (A and B) The cells were labeled using a specific autophagolysosome marker MDC staining and then the fluorescence intensity (in green) for MDC-labeled vacuoles was imaged (A) and quantified (B) as described in Materials and Methods. Scale bar: 20 μm. (C and D) Overlay of large LC3 puncta (in green) was shown (C) and the ratio of cells with large LC3 punctate structures was counted and calculated (D). Scale bar: 2 μm. (E and G) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (F and H) The blots for LC3-II and p62 were semi-quantified. For (B), (D) (F) and (H), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group.
Furthermore, our Western blot analysis showed that Cd elicited robust LC3-II expression in a concentration- and time-dependent manner in PC12 cells and primary neurons (Fig. 1E–H). Interestingly, Cd substantially increased the level of p62 protein (a substrate that is degraded by autophagy) dose- and time-dependently in the cells as well (Fig. 1E–H). Hence, these results indicate that Cd induced increases in autophagosomes with a concomitant elevation of LC3-II and p62 in neuronal cells, suggesting that Cd might block autophagic flux, leading to expansion of abnormal autophagsomes with large LC3 puncta in neuronal cells.
3.2. Cd triggers accumulation of LC3-derived autophagosomes contributing to neuronal cell death
To unveil whether Cd-induced apoptosis is attributed to Cd-evoked increase in the number of autophagosomes, PC12 cells and primary neurons were pretreated with/without 3-MA (4 mM), an autophagy inhibitor via suppressing class III PI3K,34 for 1 h, followed by exposure to Cd (10 μM) for 8 h, 12 h or 24 h. As shown in Fig. 2A and B, Cd-induced accumulation of intracellular MDC (Fig. 2A), and the ratio of the cells with large LC3 puncta (Fig. 2B) were significantly reversed by 3-MA in PC12 and primary neurons. Of note, 3-MA alone slightly increased the protein level of LC3-II, and decreased the cleavages of caspase-3 and PARP in the cells (Fig. 2C and D). Pretreatment with 3-MA exhibited a stronger inhibitory effect on Cd-elevated LC3-II, cleaved-caspase-3 and cleaved-PARP, whereas Cd-induced p62 was not attenuated by 3-MA in the cells (Fig. 2C and D). Consistently, using DAPI and concurrently TUNEL staining (Fig. 2E), we also observed that 3-MA potently attenuated the percentage of the cells with nuclear fragmentation and condensation (arrows) and the number of TUNEL-positive cells with fragmented DNA (in green) in PC12 cells and primary neurons induced by Cd exposure (Fig. 2E–G). The data suggest that Cd triggers accumulation of LC3-derived autophagosomes contributing to neuronal cell death.
Fig. 2.

Cd triggers accumulation of autophagosomes contributing to neuronal cell death. PC12 cells and primary neurons, or PC12 cells and primary neurons infected with Ad-GFP-LC3, were pretreated with/without 3-MA (4 mM) for 1 h and then exposed to Cd (10 μM) for 8 h (for Western blotting), 12 h (for MDC staining and GFP-LC3 assay) and 24 h (for DAPI and TUNEL staining). (A) The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. (B) The ratio of cells with large LC3 punctate structures was counted and calculated. (C) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (D) The blots for LC3-II, p62, cleaved-caspase-3, and cleaved-PARP were semi-quantified. (E) Apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining (upper panel) and concurrently by in situ detection of fragmented DNA (in green) using TUNEL staining (lower panel). Scale bar: 20 μm. (F and G) The percentages of cells with fragmented nuclei and the number of TUNEL-positive cells were quantified. For (A), (B), (D), (F) and (G), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, difference with 10 μM Cd group.
3.3. Cd impairs autophagic flux leading to accumulation of autophagosomes-dependent neuronal apoptosis
It is known that increase in the number of autophagosomes may result from either induction of autophagy or inhibition of autophagosome clearance [40]. To determine which stage of the autophagic process in the neuronal cells is affected by Cd, a tandem mCherry-GFP-LC3 assay was employed. For this, PC12 cells or primary neurons, infected with Ad-mCherry-GFP-LC3, were exposed to Cd (10 μM) for indicated time. As shown in Fig. 3A (merged), the cells treated with Cd for 0 h contained only red puncta (autolysosomes), whereas the cells treated with Cd for 8 h had both yellow (autophagosomes) and red puncta (autolysosomes). However, when exposed to Cd for 12 h and 24 h, cells had both green and yellow puncta (autophagosomes) (Fig. 3A). Time-dependent decrease of GFP–/mCherry+-LC3 (red) puncta per cell in response to Cd was quantified (Fig. 3B). These results point out that Cd-inhibited autophagosome clearance causes increased autophagosomes.
Fig. 3.

Cd-impaired autophagic flux results in accumulation of autophagosomes leading to neuronal apoptosis. PC12 cells and primary neurons, or PC12 cells and primary neurons infected with Ad-mCherry-GFP-LC3, were treated with Cd (10 μM) for 0–24 h (for mCherry-GFP-LC3 assay and DAPI staining), or pretreated with/without CQ (5–50 μM or 25 μM) for 1 h and then exposed to Cd (10 and/or 20 μM) for 8 h (for Western blotting), 12 h (MDC staining) and 24 h (for DAPI staining). (A) Co-localization of both GFP and mCherry fluorescence for GFP+/mCherry+-LC3 (yellow) and GFP–/mCherry+-LC3 (red) puncta was shown in the cells. Scale bar: 2 μm. (B) GFP–/mCherry+-LC3 (red) puncta per cell was quantified. (C and E) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (D and F) The blots for LC3-II, p62, cleaved-caspase-3, cleaved-PARP were semi-quantified. (G) The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. (H) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. For (B), (D), (F), (G) and (H), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, difference with 10 μM Cd group, †P < 0.05, – CQ group versus + CQ group.
To elucidate the role of impaired autophagic flux in Cd-induced apoptosis in neuronal cells, PC12 cells and primary neurons were subjected to pretreatment with/without 5–50 μM of CQ, an agent capable of effectively inhibiting the fusion of autophagosomes and lysosomes [41], for 1 h and then treated with/without 10 and/or 20 μM of Cd for 8 h, 12 h or 24 h. As shown in Fig. 3C and D, CQ concentration-dependently elevated Cd-induced expression of LC3-II and p62 in the cells. At 25 μM, CQ was able to potentiate the basic and Cd-increased LC3-II and p62 almost to a maximal level in PC12 cells and primary neurons (Fig. 3C–F). Consistently, CQ further enhanced the accumulation of autophagosomes in the cells exposed to Cd, as evidenced by MDC staining (Fig. 3G) and punctuation/aggregation of large LC3 (data not shown). The presence of CQ also further increased the cleavages of caspase-3 and PARP, as well as neuronal apoptosis induced by Cd (Fig. 3E, F and H). The data indicate that Cd treatment results in accumulation of autophagosomes and consequential cell apoptosis by impairing autophagic flux in neuronal cells
Rapamycin, a known mTOR inhibitor and autophagy inducer, has recently been demonstrated to prevent from Cd neurotoxicity by targeting both mTORC1 and mTORC2 pathways [31]. It has been reported to induce autophagy with enhanced autophagic flux and promote the fusion of autophagosomes and lysosomes in various cells [42; 43; 44; 45]. To provide more evidence that support association of impaired autophagic flux with accumulated autophagosomes-dependent cell apoptosis, we next tested the manifestation of Cd-induced LC3-II and p62 levels in the presence of rapamycin, and checked the co-localization between autophagosome and lysosome in neuronal cells exposed to Cd. As expected, pretreatment with rapamycin alone for 48 h increased LC3 but reduced p62 protein level, compared with the vehicle treatment (Fig. 4A and B), indicating intact autophagic flux. Of importance, pretreatment with rapamycin profoundly attenuated Cd-induced expression of LC3-II and accumulation of p62 in PC12 cells and primary neurons, implying a recovery of the authophagic flux (Fig. 4A and B). We also observed that Cd-induced cleaved-caspase-3 and apoptosis in the cells was obviously blocked by rapamycin (Fig. 4A–C), in line with our previous findings [31; 39]. In another experiment, PC12 cells and primary neurons infected with Ad-GFP-LC3, after exposed to Cd (10 μM) for 12 h following pretreatment with/without rapamycin (0.2 μg/ml) for 48 h, were stained with LysoTracker. We found that the majority of the LC3-decored vesicles (green) due to Cd exposure were not labeled by LysoTracker in the merged images (Fig. 4D). Interestingly, rapamycin alone or co-treatment with rapamycin/Cd elicited the autolysosome vesicles (yellow) co-labeled by LysoTracker/GFP-LC3 (Fig. 4D). The number of the vesicles labeled by LysoTracker/GFP-LC3 and GFP-LC3 was quantified (Fig. 4E). Taken together, our findings that rapamycin-induced reduction of p62 level and acceleration of autophagosome-lysosome fusion prevent Cd-induced neuronal apoptosis strongly support that Cd impairs autophagic flux by inhibiting autophagosome-lysosome fusion, leading to accumulated autophagosomes-dependent neuronal apoptosis.
Fig. 4.

Rapamycin rescues Cd-impaired autophagic flux from accumulation of autophagosomes and neuronal apoptosis. PC12 cells and primary neurons, or PC12 cells and primary neurons infected with Ad-GFP-LC3 were pretreated with/without rapamycin (Rap, 0.2 μg/ml) for 48 h and then exposed to Cd (10 μM) for 8 h (for Western blotting), 12 h (for Lysotracker/GFP-LC3 assay) and 24 h (for DAPI staining). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for LC3-II, p62, cleaved-caspase-3 were semi-quantified. (C) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. (D) Autophagosomes were labeled by GFP-LC3 green fluorescence, lysosomes were stained by LysoTracker red fluorescence, and co-localization of both autophagosome and lysosome fluorescence showed autolysosome puncta (yellow) in the merged images. Scale bar: 2 μm. (E) The vesicles labeled by LysoTracker/GFP-LC3 and GFP-LC3 was quantified. At least 50 cells per group were included for the counting in each condition. For (B), (C) and (E), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, difference with 10 μM Cd group.
3.4. Cd activates Akt-mediated BECN1 phosphorylation promoting autophagosome-dependent neuronal apoptosis
BECN1, an essential protein for autophagy, is a target of Akt [29]. Akt can inhibit autophagy [46]. Our recent studies have shown that Cd induces activation of Akt/mTOR pathway contributing to apoptosis in neuronal cells [31]. Here, we also observed that Cd induced the phosphorylation of Akt (Ser473) in a concentration- and time-dependent manner in PC12 cells and primary neurons (Fig. 5A–D). Interestingly, Cd also evoked the phosphorylation of BECN1 (Ser295) in a similar pattern (Fig. 5A–D). Accordingly, we asked whether BECN1 acts as a key molecule responsible for Cd-induced autophagosome’s increase and neuronal apoptosis. For this, we knocked down BECN1 and also ectopically overexpressed FLAG-tagged BECN1 (S295A) mutant in PC12 cells. As shown in Fig. 6A, the protein levels of BECN1 and p-BECN1 (Ser295) were downregulated by ~ 90% in the cells infected with lentiviral shRNA to BECN1. Silencing BECN1 obviously attenuated the basal and Cd-induced phosphorylation of BECN1 (Ser295) (Fig. 6A and B). Similarly, overexpression of mutant FLAG-BECN1 (S295A) also suppressed Cd-induced p-BECN1 (Ser295) (Fig.6G and H). However, downregulation of BECN1 (Fig. 6A and B) or overexpression of mutant BECN1 (S295A) (Fig. 6G and H) did not obviously alter the expression of p62. Of importance, downregulation of BECN1 or overexpression of mutant BECN1 (S295A) markedly attenuated Cd-induced LC3-II expression (Fig. 6A, B, G and H), MDC fluorescence (Fig. 6C and D) and large LC3 puncta formation (Fig. 6I) in the cells. Consistent with this, downregulation of BECN1 or overexpression of mutant BECN1 (S295A) potently blocked Cd-induced caspase-3 cleavage (Fig. 6A, B, G and H), cell viability reduction (Fig. 6E) and apoptosis (Fig. 6F and J). These findings imply that BECN1, especially the phosphorylation level of BECN1 on Ser295, is required for autophagosome-dependent apoptosis in Cd-exposed neuronal cells.
Fig. 5.

Cd-activated Akt is associated with BECN1 phosphorylation in neuronal cells. PC12 cells and primary neurons were treated with Cd (0–20 μM) for 8 h or with Cd (10 μM) for 0–24 h. (A and C) Total cell lysates were subjected to Western blotting using indicated antibodies, showing that Cd evoked the phosphorylation of Akt and BECN1 dose- and time-dependently in the cells. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (B and D) The blots for p-BECN1 (Ser295) and p-Akt (Ser473) were semi-quantified. All data were expressed as means ± SE (n = 3). *P < 0.05, difference with control group.
Fig. 6.

Downregulation of BECN1 or mutant BECN1 (S295A) relieves Cd-induced accumulation of autophagosomes and neuronal apoptosis. PC12 cells, infected with lentiviral shRNA to BECN1, FLAG-BECN1 (S295A) or GFP (as control) and/or infected with Ad-GFP-LC3, respectively, were treated with Cd (10 and 20 μM) for 8 h (for Western blotting), 12 h (MDC staining and GFP-LC3 assay) and 24 h (for live cell analysis and DAPI staining). (A and G) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (B and H) The blots for p-BECN1 (Ser295) LC3-II, p62, cleaved-caspase-3 were semi-quantified. (C and D) The cells were labeled using MDC staining and then the fluorescence intensity (in green) for MDC-labeled vacuoles was imaged (C) and quantified (D) as described in Materials and Methods. Scale bar: 20 μm. (E) Live cells were detected by counting viable cells using trypan blue exclusion. (F and J) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. (I) The ratio of cells with large LC3 punctate structures was counted and calculated. For (C), (D) and (E), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, BECN1 shRNA group or BECN1 (S295A) shRNA group versus GFP shRNA group.
Next, we sought to determine whether Cd-activated Akt mediates BECN1 phosphorylation, leading to autophagosome-dependent neuronal apoptosis. To answer the question, PC12 cells and primary neurons were pre-incubated with/without Akt inhibitor X alone, or in combination with 3-MA. We found that pretreatment with Akt inhibitor X (20 μM) or 3-MA (4 mM) alone substantially suppressed the basal and Cd-induced p-Akt, p-BECN1 and cleaved-caspase-3 in the cells (Fig. 7A and B). Especially, co-treatment with Akt inhibitor X/3-MA exhibited a stronger inhibitory effect on Cd-induced events (Fig. 7A and B). Consistently, the combination of Akt inhibition X with 3-MA also showed more potent inhibitory effects on Cd-elicited autophagosome expansion and apoptosis than Akt inhibitor X or 3-MA alone, as evidenced by MDC and DAPI staining (Fig. 7C and D). In addition, we also noted that Akt inhibitor X alone or combination with 3-MA, but not 3-MA alone, obviously reduced Cd-increased p62 expression (Fig. 7A and B). Taken together, the data support the idea that Cd activates Akt-mediated BECN1 phosphorylation promoting autophagosome-dependent neuronal apoptosis.
Fig. 7.

Pharmacological inhibition of Akt diminishes Cd-induced BECN1 phosphorylation and autophagosome-dependent neuronal apoptosis. PC12 cells and primary neurons were pretreated with/without Akt inhibitor X (20 μM) for 2 h and then 3-MA (4 mM) for 1 h, followed by exposure to Cd (10 μM) for 8 h (for Western blotting), 12 h (MDC staining) and 24 h (for DAPI staining). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-Akt (Thr308), p-BECN1 (Ser295), p62, cleaved-caspase-3 were semi-quantified. (C) The fluorescence intensity for MDC-labeled vacuoles in the cells was quantified. (D) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. For (B), (C) and (D), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, difference with 10 μM Cd group; $P < 0.05, difference with Cd/Akt inhibitor X group or Cd/3-MA group.
3.5. Expression of dominant negative Akt attenuates Cd-induced autophagic flux imparment, preventing autophagosome-dependent neuronal apoptosis
To corroborate the above finding, PC12 cells, infected with recombinant adenovirus encoding HA-tagged dominant negative Akt (Ad-dn-Akt) or GFP/LacZ (Ad-GFP/Ad-LacZ) (as control), were pretreated with/without 3-MA (4 mM) for 1 h, and then exposed to Cd (10 mM) for 8 h, 12 h or 24 h. As expected, the basal phosphorylation level of Akt was significantly inhibited by the infection with Ad-dn-Akt, compared to the control infection with Ad-GFP (Fig. 8A and B). Of note, ectopic expression of dn-Akt markedly attenuated Cd-induced phosphorylation of Akt and BECN1 as well as activation of caspase-3, and potentiated the inhibitory effect of 3-MA on cleaved-caspase-3 (Fig. 8A and B). The basal and Cd-increased p62 levels were attenuated by overexpressing dn-Akt, but were not potentiated by 3-MA (Fig. 8A and B). Overexpression of dn-Akt diminished the ratio of cells with large LC3 puncta in response to Cd, which was strengthened by 3-MA (Fig. 8C and D). Using live cell assay and DAPI staining, we also revealed that expression of dn-Akt alone partially prevented Cd-induced cell viability reduction (Fig. 8E) and apoptosis (Fig. 8F). Furthermore, addition of 3-MA rendered more significant resistance to Cd-induced cell death in Ad-dn-Akt-infected group than in Ad-GFP-infected group (Fig. 8E and F). Interestingly, using tandem mCherry-GFP-LC3 assay, we found that there existed fewer yellow puncta in Ad-dn-Akt-infected group than in Ad-GFP-infected group (Fig. 8G). The quantification data showed that overexpression of dn-Akt prevented Cd-induced decrease of GFP–/mCherry+-LC3 (red) puncta per cell (Fig. 8H), indicating that Cd-blocked degradation of mCherry-GFP-LC3 was attenuated in Ad-dn-Akt-infected cells. The results suggest that Cd-activated Akt is involved in mediating BECN1 phosphorylation and impairing autophagic flux, contributing to autophagosome-dependent apoptosis in neuronal cells.
Fig. 8.

Expression of dominant negative Akt attenuates Cd-blocked autophagic flux and autophagosome-dependent neuronal apoptosis. PC12 cells, infected with Ad-dn-Akt or Ad-GFP (as control) and/or infected with Ad-mCherry-GFP-LC3, respectively, were exposed to Cd (10 μM) for 12 h (for mCherry-GFP-LC3 assay), or pretreated with/without 3-MA (4 mM) for 1 h and then exposed to Cd (10 μM) for 8 h (for Western blotting), 12 h (MDC staining) and 24 h (for DAPI staining). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-Akt (Thr308), p-BECN1 (Ser295), p62, cleaved-caspase-3 were semi-quantified. (C and D) Overlay of large LC3 puncta (in green) was shown (C) and the ratio of cells with large LC3 punctate structures was counted and calculated (D). Scale bar: 2 μm. (E) Live cells were detected by counting viable cells using trypan blue exclusion. (F) Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. (G) Co-localization of both GFP and mCherry fluorescence for GFP+/mCherry+-LC3 (yellow) and GFP–/mCherry+-LC3 (red) puncta was shown in the cells. Scale bar: 2 μm. (H) GFP–/mCherry+-LC3 puncta per cell was quantified. For (B), (D), (E), (F) and (H), all data were expressed as means ± SE (n = 3–5). *P < 0.05, difference with control group; #P < 0.05, difference with 10 μM Cd group; †P < 0.05, Ad-dn-Akt group versus Ad-LacZ or Ad-GFP group.
4. Discussion
The present study shows that Cd treatment increased autophagosome formation with a concomitant elevation of LC3-II and p62 protein levels, leading to caspase-dependent apoptosis in PC12 cells and primary cortical neurons. Further, we found that Cd-induced autophagosome formation involved both LC3 and BECN1. Moreover, Cd-activated Akt mediated BECN1 activation and impaired autophagic flux, resulting in autophagosome-dependent apoptosis in neuronal cells. The data suggest that Cd elicited a special noncanonical pathway of autophagy with impairment of autophagic flux, thus contributing to neuronal apoptosis.
Many studies have documented that Cd neurotoxicity contributes to the development of neurodegenerative diseases, such as PD, AD, HD and ALS [11; 12; 13; 14; 15]. In the context of neurodegenerative disorders, autophagosome is abundant in neurons, which renders neuroprotection or fosters neuronal cell death, and has been recognized as an arbiter that decides neuronal survival and death [2; 4; 7; 47]. It has been described that Cd induces autophagy, which can result in either cytoprotection or cytotoxicity/cell death in various cells [16; 17; 18; 19; 20]. In the current study, we demonstrated that Cd induced defective autophagy triggering neuronal apoptosis. This is supported by the findings that Cd induced increases in autophagosomes, large LC3 puncta and apoptosis with a concomitant elevation of LC3-II/p62 and cleavages of caspase-3/PARP, which was attenuated by 3-MA, an autophagy inhibitor via suppressing class III PI3K [48] in PC12 cells and primary neurons induced by Cd exposure (Fig. 2). Our results are in contrast to the report that Cd-induced autophagy is cytoprotective in PC12 cells [16]. Whether this is related to different experimental conditions used remains to be determined. However, our findings, to some extent, are in line with the previous observations that Cd elicits autophagy or overactivation of mitophagy, leading to cytotoxicity or cell death in a variety of non-neuronal cells, including mesangial cells [17], epidermal cells [18], mouse kidney cells [19], as well as brain neurons [20]. Of note, the abovementioned studies [17; 18; 19; 20] have described that Cd induces autophagosome formation (LC3 puncta and/or LC3-II expression) and cell death, which can be prevented by 3-MA, but have not shown whether Cd increases or decreases the protein level of p62, an indicator of executed autophagy. It would be interesting to clarify whether Cd indeed induces autophagy/mitophagy in those cells. Taken together, the findings in this study and others imply that the effect of Cd-induced autophagosomes/autophagy on cell fate may be dependent on cell type or experimental conditions.
Autophagy is a dynamic process [21; 24]. Autophagy adaptor p62 is known to bind to ubiquitinated substrates and LC3, and is degraded along with its cargo [25]. The accumulation of LC3-II or autophagosomes, event previously interpreted as induction of autophagy, is in fact a consequence of an impaired autophagic flux [49]. The combination of elevated p62 and punctuate LC3-II could be reflecting an inhibition of autophagosome degradation [49]. In this study, we found that Cd evoked a robust expression of p62 protein dose- and time-dependently in PC12 cells and primary neurons (Fig. 1E–H), suggesting that Cd might impair autophagic flux. To confirm the Cd-impaired autophagic flux and its relationship to neuronal apoptosis, we next monitored the manifestation of autophagic flux using tandem mCherry-GFP-LC3 assay, which can generate a LC3 construct tandem tagged with mCherry and GFP probes [45]. In line with the immunoblotting results, PC12 cells and primary neurons treated with Cd for 8–24 h exhibited a decline in autophagic flux, as shown by the decrease in the number of red LC3 puncta (autolysosomes) without the concomitant decrease in the number of yellow puncta (autophagosomes) per cell (Fig. 3A and B). Subsequently, we showed that administration of chloroquine (CQ), an agent that leads to impairment of the function of lysosome’s degradation or inhibition of fusion of autophagosome with lysosome [24; 48], potentiated the basic and Cd-elevated LC3-II and p62 levels, autophagosome accumulation and cell apoptosis (Fig. 3E–H). Taken together, these results support the idea that Cd may inhibit the fusion of autophagosomes with lysosomes and thus impair autophagic flux, causing accumulation of LC3-II/autophagosomes and consequential cytotoxicity in neuronal cells.
Rapamycin, a known autophagy inducer, has been shown to promote autophagy flux and accelerate autophagosome-lysosome fusion [42; 43; 44; 45]. In this study, we also demonstrated that rapamycin alone elicited a significant high LC3-II alteration and a marked low p62 in PC12 cells and primary neurons (Fig. 4A and B). Of note, however, rapamycin substantially attenuated Cd-elicited increases of LC3-II and p62 proteins, which were in line with Cd-induced cleaved-caspase-3 and apoptosis in the cells (Fig. 4A–C). Using LysoTracker/GFP-LC3 assay, we clearly observed that there existed the large majority of the LC3-decored vesicles (green) in PC12 cells and primary neurons exposed to Cd alone, whereas acceleration of autophagosome-lysosome fusion by rapamycin showed autolysosome vesicles (yellow), as evidenced by the images and quantification co-labeled by LysoTracker/GFP-LC3 (Fig. 4D and E). Collectively, these findings further highlight that Cd impairs autophagic flux by inhibiting autophagosome-lysosome fusion, which contributes to accumulation of autophagosomes leading to apoptosis in neuronal cells.
It is known that Akt is an important regulator of neuronal cell survival [28]. BECN1, an essential and core component in autophagy, can be phosphorylated by different protein kinases, and different phosphorylation sites of BECN1 are involved in induction or inhibition of autophagy [29; 50; 51]. It has been reported that Akt inhibits autophagy,[46] which is associated with its phosphorylation of BECN1 (Ser295) [29]. Our recent studies have shown that Cd induces neuronal apoptosis by activating Akt/mTOR signaling pathway [31]. In this study, having observed that Cd-induced autophagosome accumulation is related to neuronal apoptosis, we further asked whether Cd activation of Akt links to BECN1 mediation of the events. As expected, we observed that Cd evoked phosphorylation of Akt (Ser473) and BECN1 (Ser295) dose- and time-dependently in PC12 cells and primary neurons (Fig. 5). Further, BECN1, especially phosphorylation of the BECN1 Ser295 residue, appeared to play a key role in Cd-induced autophagosome-dependent neuronal apoptosis, as BECN1 knockdown or mutant BECN1 (S295A) deletion attenuated or silenced the basal and Cd-induced of BECN1 (Ser295) phosphorylation, and downregulation of BECN1 or BECN1 (S295A) substantially diminished LC3-II expression, MDC fluorescence intensity, the ratio of cells with large LC3 puncta, cleaved-caspase-3, cell viability reduction and/or apoptosis in PC12 cells in response to Cd (Fig. 6). Next, to pinpoint whether Cd-activated Akt mediates BECN1 phosphorylation, leading to autophagosome-dependent neuronal apoptosis, pharmacological or genetic inhibition experiments for Akt were conducted. We found that pharmacological inhibition of Akt with Akt inhibitor X or ectopic expression of dn-Akt in the presence or absence of 3-MA significantly alleviated Cd-triggered phosphorylation of Akt and BECN1, autophagosomes/large LC3 puncta, and cell apoptosis (Fig. 7 and 8). Basal and Cd-increased p62 were attenuated by pretreating with Akt inhibitor X or overexpressing dn-Akt, but not potentiated by 3-MA (Fig. 7 and 8). Moreover, our tandem mCherry-GFP-LC3 assay revealed that Cd activation of Akt functioned in impairing autophagic flux (Fig. 8G and H). Collectively, these observations support such concept that Cd-activated Akt mediates BECN1 activation and impairs autophagic flux, leading to accumulated autophagosome-dependent apoptosis in neuronal cells.
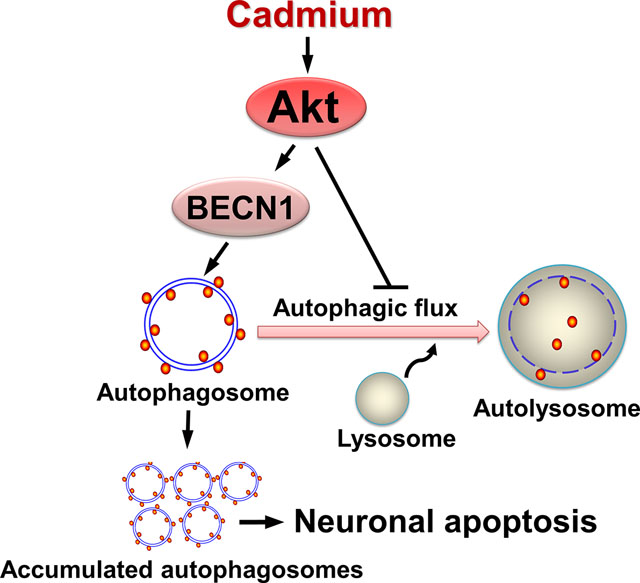
In conclusion, we have shown that Cd induces autophagosome formation and impairs autophagic flux, contributing to neuronal apoptosis. Our results suggest that Cd activates Akt, which not only mediates autophagosome formation by phosphorylating BECN1, but also functions in impairing autophagic flux, thereby resulting in accumulation of autophagosomes, which eventually triggers neuronal apoptosis (Fig. 9). Our findings underline that inhibition of Akt is a promising intervention against Cd-induced neurotoxicity and neurodegeneration.
Fig. 9.

Diagram illustrating how Cd induces accumulation of autophagosomes contributing to neuronal apoptosis. Cd activates Akt, which not only mediates autophagosome formation by phosphorylating BECN1, but also functions in imaparing autophagic flux, thereby resulting in accumulation of autophagosomes, which finally triggers neuronal apoptosis.
Highlights.
Cd induces accumulation of autophagosomes contributing to apoptosis in neuronal cells.
Cd impairs autophagy flux promoting accumulation of autophagosomes leading to neuronal apoptosis.
Cd-activated Akt evokes autophagosome-dependent neuronal apoptosis via phosphorylating BECN1.
Cd-activated Akt impairs autophagic flux causing autophagosome-dependent neuronal apoptosis.
Acknowledgements
This work was supported in part by the grants from National Natural Science Foundation of China (No. 81873781; LC), NIH (CA115414; SH), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010; LC), and American Cancer Society (RSG-08-135-01-CNE; SH).
Abbreviations
- 3-MA
3-methyladenine
- AD
Alzheimer disease
- PKB
Akt protein kinase B
- ATG
autophagy-related
- ALS
amyotrophic lateral sclerosis
- BECN1
beclin 1
- Cd
cadmium
- CQ
chloroquine diphosphate
- DAPI
4′6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
fetal bovine serum
- HD
Huntington’s disease
- LC3
microtuble-associated protein 1 light chain 3 MDCmonodansylcadaverine
- mTOR
mammalian target of rapamycin
- PBS
phosphate buffered saline
- PD
Parkinson disease
- PDL
poly-D-lysine
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Feng Y, Yao Z, Klionsky DJ, How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell. Biol 25 (2015) 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Los MJ, Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol 112 (2014) 24–49. [DOI] [PubMed] [Google Scholar]
- [3].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC, Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev 90 (2011) 1383–1435. [DOI] [PubMed] [Google Scholar]
- [4].Zhang Z, Miah M, Culbreth M, Aschner M, Autophagy in neurodegenerative diseases and metal neurotoxicity. Neurochem. Res 41 (2016) 409–422. [DOI] [PubMed] [Google Scholar]
- [5].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K, Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441 (2006) 880–884. [DOI] [PubMed] [Google Scholar]
- [6].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N, Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441 (2006) 885–889. [DOI] [PubMed] [Google Scholar]
- [7].Martinez-Vicente M, Autophagy in neurodegenerative diseases: From pathogenic dysfunction to therapeutic modulation. Semin. Cell Dev. Biol 40 (2015) 115–126. [DOI] [PubMed] [Google Scholar]
- [8].Mizushima N, Levine B, Cuervo AM, Klionsky DJ, Autophagy fights disease through cellular self-digestion. Nature 451 (2008) 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP, Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br. J. Pharmacol 138 (2003) 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen S, Ren Q, Zhang J, Ye Y, Zhang Z, Xu Y, Guo M, Ji H, Xu C, Gu C, Gao W, Huang S, Chen L, N-acetyl-L-cysteine protects against cadmium-induced neuronal apoptosis by inhibiting ROS-dependent activation of Akt/mTOR pathway in mouse brain. Neuropathol. Appl. Neurobiol 40 (2014) 759–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang B, Du Y, Cadmium and its neurotoxic effects. Oxid. Med. Cell. Longev 2013 (2013) 898034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Okuda B, Iwamoto Y, Tachibana H, Sugita M, Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg 99 (1997) 263–265. [DOI] [PubMed] [Google Scholar]
- [13].Jomova K, Valko M, Advances in metal-induced oxidative stress and human disease. Toxicology 283 (2011) 65–87. [DOI] [PubMed] [Google Scholar]
- [14].Panayi AE, Spyrou NM, Iversen BS, White MA, Part P, Determination of cadmium and zinc in Alzheimer’s brain tissue using inductively coupled plasma mass spectrometry. J. Neurol. Sci 195 (2002) 1–10. [DOI] [PubMed] [Google Scholar]
- [15].Bar-Sela S, Reingold S, Richter ED, Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. Int. J. Occup. Environ. Health 7 (2001) 109–112. [DOI] [PubMed] [Google Scholar]
- [16].Wang Q, Zhu J, Zhang K, Jiang C, Wang Y, Yuan Y, Bian J, Liu X, Gu J, Liu Z, Induction of cytoprotective autophagy in PC-12 cells by cadmium. Biochem. Biophys. Res. Commun 438 (2013) 186–192. [DOI] [PubMed] [Google Scholar]
- [17].Wang SH, Shih YL, Ko WC, Wei YH, Shih CM, Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. Life Sci 65 (2008) 3640–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Son YO, Wang X, Hitron JA, Zhang Z, Cheng S, Budhraja A, Ding S, Lee JC, Shi X, Cadmium induces autophagy through ROS-dependent activation of the LKB1-AMPK signaling in skin epidermal cells. Toxicol. Appl. Pharmacol 255 (2011) 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wei X, Qi Y, Zhang X, Qiu Q, Gu X, Tao C, Huang D, Zhang Y, Cadmium induces mitophagy through ROS-mediated PINK1/Parkin pathway. Toxicol. Mech. Methods 24 (2014) 504–511. [DOI] [PubMed] [Google Scholar]
- [20].Wei X, Qi Y, Zhang X, Gu X, Cai H, Yang J, Zhang Y, ROS act as an upstream signal to mediate cadmium-induced mitophagy in mouse brain. Neurotoxicology 46 (2015) 19–24. [DOI] [PubMed] [Google Scholar]
- [21].Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX, Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 7 (2011) 188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ohsumi Y, Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol 2 (2001) 211–216. [DOI] [PubMed] [Google Scholar]
- [23].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T, LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19 (2000) 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mizushima N, Yoshimori T, Levine B, Methods in mammalian autophagy research. Cell 140 (2010) 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yin Y, Sun G, Li E, Kiselyov K, Sun D, ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev 34 (2017) 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang XJ, Chen S, Huang KX, Le WD, Why should autophagic flux be assessed? Acta Pharmacol. Sin 34 (2013) 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sharifi MN, Mowers EE, Drake LE, Macleod KF, Measuring autophagy in stressed cells. Methods Mol. Biol 1292 (2015) 129–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME, Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275 (1997) 661–665. [DOI] [PubMed] [Google Scholar]
- [29].Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B, Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338 (2012) 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Levine B, Kroemer G, Autophagy in the pathogenesis of disease. Cell 132 (2008) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu C, Liu C, Liu L, Zhang R, Zhang H, Chen S, Luo Y, Chen L, Huang S, Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology 97 (2015) 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S, Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab. Invest 90 (2010) 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, Zhou H, Han X, Huang S, Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J. Biol. Chem 285 (2010) 38362–38373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K, Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19 (2005) 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S, Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene 25 (2006) 7029–7040. [DOI] [PubMed] [Google Scholar]
- [36].Wang W, Malcolm BA, Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 26 (1999) 680–682. [DOI] [PubMed] [Google Scholar]
- [37].Biederbick A, Kern HF, Elsasser HP, Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol 66 (1995) 3–14. [PubMed] [Google Scholar]
- [38].Munafo DB, Colombo MI, A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci 114 (2001) 3619–3629. [DOI] [PubMed] [Google Scholar]
- [39].Chen L, Liu L, Luo Y, Huang S, MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J. Neurochem 105 (2008) 251–261. [DOI] [PubMed] [Google Scholar]
- [40].Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD, Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol 33 (2013) 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA Jr., Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, et al. , Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4 (2008) 151–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang Y, Han Q, You S, Cao Y, Zhang X, Liu H, Hu L, Liu CF, Rapamycin Promotes the Autophagic Degradation of Oxidized Low-Density Lipoprotein in Human Umbilical Vein Endothelial Cells. J. Vasc. Res 52 (2015) 210–219. [DOI] [PubMed] [Google Scholar]
- [43].Yi H, Wang K, Du B, He L, Ho H, Qiu M, Zou Y, Li Q, Jin J, Zhan Y, Zhao Z, Liu X, Aleuritolic acid impaired autophagic flux and induced apoptosis in hepatocellular carcinoma HepG2 cells. Molecules 23 (2018) e1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Qiu W, Su M, Xie F, Ai J, Ren Y, Zhang J, Guan R, He W, Gong Y, Guo Y, Tetrandrine blocks autophagic flux and induces apoptosis via energetic impairment in cancer cells. Cell Death Dis 5 (2014) e1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A, Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 125 (2012) 3170–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K, Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol 183 (2008) 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Banerjee R, Beal MF, Thomas B, Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci 33 (2010) 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM, Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem 285 (2010) 10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillon J, Lo Iacono O, Corazzari M, Fimia GM, Piacentini M, Muntane J, Bosca L, Garcia-Monzon C, Martin-Sanz P, Valverde AM, Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 5 (2014) e1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A, DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 10 (2009) 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL, ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol 15 (2013) 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]