

Abstract
Endogenous hydrogen sulfide (H2S) is a potent vasodilator and proangiogenic second messenger synthesized from L-cysteine by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH). Estrogens are potent vasodilators that stimulate H2S biosynthesis in uterine arteries (UA) in vivo; however, the underlying mechanisms are unknown. We hypothesized that estrogens stimulate H2S biosynthesis in UA endothelial cells (UAEC) via specific estrogen receptor (ER)-dependent mechanisms. In cultured primary UAEC, treatment with estradiol-17β (E2β) stimulated CBS and CTH mRNAs and proteins in a time- and concentration-dependent fashion. As little as 0.1 nM E2β was effective in increasing CBS and CTH expressions and these stimulatory effects maximized with 10–100 nM E2β at 48–72 h. E2β also activated CBS and CTH promoters in UAEC, leading to CBS and CTH expression. Treatment with E2β stimulated H2S production, which was blocked by specific inhibitors of either CBS or CTH and their combination and the ER antagonist ICI 182780. Treatment with either specific agonist of ERα or ERβ stimulated both CBS and CTH mRNA and protein expressions and H2S production to levels similar to that of E2β. Specific antagonist of either ERα or ERβ blocked E2β-stimulated CBS and CTH mRNA and protein expressions and H2S production. Combinations of either ERα or ERβ agonists or their antagonists had no additive effects. Thus, E2β stimulates H2S production by upregulating CBS and CTH mRNA and protein expressions through specific ERα or ERβ-dependent CBS and CTH transcription in UAEC in vitro.
Keywords: estrogen, estrogen receptors, hydrogen sulfide, endothelium, uterine artery, vasodilation
Estradiol-17β stimulates uterine artery endothelial cell hydrogen sulfide biosynthesis.
Introduction
Circulating estrogens are significantly elevated during the follicular phase of the ovarian cycle and pregnancy [1, 2]. Estrogens are potent vasoactive hormones that dilate selected vasculature beds in organs throughout the body with the greatest response in reproductive tissues, especially the uterus [3, 4]. The vasodilatory effect of estrogens is of major physiological significance during pregnancy because uterine vasodilation as measured by a rise in uterine blood flow (UBF) elevates more than 50- to 80-fold during pregnancy to facilitate the bidirectional mother–fetus exchanges of gases (i.e. O2 and CO2) and to provide the sole nutrient support to fetal development and survival [5]. Insufficient rise in UBF during pregnancy results in pregnancy disorders such as fetal growth restriction and preeclampsia [6], not only raising the risk of maternal and infant morbidity and mortality but also contributing to the susceptibility of both the mother and child to cardiovascular and other metabolic disorders later in life [7, 8].
Local uterine artery (UA) endothelium production of orchestrated vasodilators, including nitric oxide (NO) [9], endothelium-derived hyperpolarizing factor (EDHF) [10], and vascular endothelial growth factor (VEGFA) [11], etc., is crucial for mediating estrogen-induced and pregnancy-associated uterine vasodilation. Endothelial NO production via endothelial NO synthase (NOS3) activation and/or expression has been identified as a key player in uterine vasodilation as NOS3-derived NO is important for vascular remodeling, decreased vascular resistance, increased UBF, and pregnancy outcomes [12–15]. NOS3-derived NO seems to function as a focal mediator as it interacts with nearly all known vasodilators, including EDHF and VEGF [10, 11]. However, blockade of local NO production only inhibits approximately 65% estradiol-17β (E2β)-induced uterine vasodilation in nonpregnant ewes [9] and prolonged UA NOS inhibition only modestly reduces basal uteroplacental vasodilation in the last one third of ovine pregnancy [16], suggesting that mechanisms other than NO are present to maintain uterine hemodynamics.
Hydrogen sulfide (H2S) is a gaseous signaling molecule that belongs to the gasotransmitter family after NO and carbon monoxide [17]. Endogenous H2S in mammalian tissues is primarily synthesized via conversion of L-cysteine by two pyridoxal 5′-phosphate-dependent enzymes cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH, also called CSE) [18–20]. In mammals, H2S potently dilates various vasculatures via activating ATP-dependent potassium (KATP) channel [21] and relaxes smooth muscle via activating large conductance calcium-activated potassium (BKCa) channel [22]. We recently reported that a slow-releasing H2S donor GYY4137 dose-dependently relaxes UAs from nonpregnant and pregnant rats, with significantly greater potency in the pregnant state and vascular bed specificity [23]. Others have shown H2S to be a placental vasodilator [24, 25] and dysregulated CTH/H2S signaling in the placenta results in preeclampsia-like conditions [25]. Thus, H2S plays an important role in uterine and placental hemodynamic regulation.
We have recently reported that both CBS and CTH are expressed in UA endothelium and smooth muscle in sheep [26] and human [23] UAs and both contribute to baseline UA H2S biosynthesis. However, UA H2S biosynthesis and endothelium and smooth muscle CBS, but not CTH, upregulation are significantly stimulated by exogenous E2β in a sheep model of estrogen replacement therapy [26] and are associated elevated endogenous estrogens during the proliferative phase of the menstrual cycle and pregnancy in women [23]. These data show that CBS is the major enzymatic source of augmented UA H2S biosynthesis in response to both exogenous and endogenous estrogen stimulation. However, the underlying mechanism by which estrogens regulate CBS expression to stimulate H2S biosynthesis is unknown in UA vascular cells.
Estrogens elicit diverse biological functions in target cells/tissues that possess specific ERs (i.e. ERα and ERβ) [27, 28], including vascular endothelial cells (ECs) such as uterine artery ECs (UAEC) [29]. By binding to specific transcription factor ERs, estrogens initiate the transcription of target genes that possess estrogen-responsive elements in their promoters in the nucleus [30]. Estrogen-induced uterine vasodilation is at least partially mediated by specific estrogen receptors since both exogenous and endogenous estrogen-induced rises in UBF can be inhibited by ICI 182780 (ICI) [31]. We hypothesize herein that estrogens stimulate H2S biosynthesis by specific ERα and/or ERβ-dependent upregulation of CBS transcription. The objectives of this study were to determine in cultured UAEC whether (1) E2β stimulates H2S production with enhanced mRNA and protein expression of CBS and/or CTH; (2) E2β stimulation of CBS and/or CTH expression is mediated by ER-dependent transcription; and (3) ERα and ERβ plays different roles in E2β-stimulated H2S biosynthesis.
Materials and methods
Chemicals and antibodies
E2β, hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), fatty acid free bovine serum albumin (BSA), O-(carboxymethyl)hydroxylamine hemihydrochloride (CHH), sodium dodecyl sulfate (SDS), and all other chemicals unless specified were from Sigma (St. Louis, MO). ICI 182780 (ICI), 4,4,4-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT), diarylpropionitrile (DPN), 1,3-Bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP), 4-[2-Phenyl-5,7-bis (trifluo-romethyl)pyrazolo[1,5-a]pyrimidin-3yl]phenol (PHTPP) were from Tocris (Ellisville, MO). β-cyano-L-alanine (BCA) was from Cayman Chemical (Ann Arbor, MI). Anti-ACTH monoclonal antibody was from Ambion (Austin, TX). Monoclonal antibodies of CBS and CTH were from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Anti-biotin antibody was from Cell Signaling Technology (Beverly, MA). Cell culture media MCDB131 and M199, prolong gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI), and Alexa488 goat anti-mouse IgG were from Invitrogen (Carlsbad, CA).
Cell culture and treatments
Primary UAEC were isolated by collagenase digestion from late pregnant ewes (120–130 days of gestation, normal term ≈ 145 days) as previously described [13, 32]. The Animal Care and Use Committee from the University of California approved the animal use protocol. Frozen UAEC aliquots (passage 2) were thawed and seeded in MCDB131 containing 10% fetal bovine serum (Lonza, Walkersville, MD) and 1% antibiotics for experimental use at passages 4–5. Briefly, cells at approximately 70% confluence were cultured in serum/phenol red-free M-199 medium containing 0.1% fatty acid-free BSA, 0.5% charcoal stripped FBS, 1% penicillin/streptomycin, and 25 mM HEPES overnight for approximately 16 h. Following equilibration in fresh serum-free M-199, cells were treated with E2β, ER agonists, or E2β with or without ER antagonists as previously described [32]. Ethanol was the vehicle used to dissolve E2β and ER agonists and antagonists. Final ethanol concentrations used were less than 0.5% and did not alter cellular responses surveyed in this study.
Methylene blue assay
UAEC (1 × 106/treatment in duplicate) were homogenized in 50 mM ice-cold potassium phosphate buffer pH 8.0. Using the methylene blue assay, H2S production was determined as previously described [23, 26]. The H2S concentration was calculated based on a calibration curve generated from NaHS solutions. CHH or BCA at a final concentration of 2 mM was added to the reaction mixtures prior to initiating the assay for determining specific CBS and CTH activities, respectively.
RNA extraction, polymerase chain reaction, and quantitative real-time RT-PCR
RNA extraction, polymerase chain reaction (PCR), and quantitative real-time RT-PCR was performed as previously described [23, 26]. Comparative CT method (ΔΔCT method) was used to calculate mRNA expression with L19 as the internal reference control.
SDS-PAGE and immunoblotting
SDS-PAGE and immunoblotting with specific antibodies listed in supplemental table S1 were performed as previously described [23, 26].
Immunofluorescence microscopy
UAEC were grown on glass coverslips to reach approximately 80% confluence and treated as described above. Following treatments, cells were washed in PBS and fixed in 4% paraformaldehyde for 20 min at room temperature. Cells were permeablized by incubating in 0.2% Triton-X in PBS for 15 min at room temperature. Autofluorescence was quenched by washing with 300 mM glycine in PBS, and nonspecific binding was blocked by incubation with PBS containing 1% BSA, 0.125% saponin, and 1% gelatin. Cells were incubated with 1 μg/mL of anti-CBS or anti-CTH. Following incubation with Alexa488-labeled secondary antibody (1:1000), coverslips were mounted onto slides with Prolong Gold antifade reagent (Invitrogen) containing DAPI. Slides were examined under a Leica fluorescence microscope (Leica Corporation, Deerfield, IL). Digital images were acquired using a CCD camera and SimplePCI image analysis software (Hamamatsu Corporation, Sewickley, PA) and used for determining relative CBS and CTH proteins by quantifying mean green fluorescence intensity (15 cells/image and 5 images/animal) using SimplePCI. Normalized CBS and CTH protein levels were presented as fold change in the average fluorescence intensity of vehicle control treated cells.
Cell transfection and luciferase assay
RenSP luciferase-reporter constructs containing the promoters of human CBS (S711027), CTH (S712215), β-actin (S717678), and all transfection and luciferase reagents were purchased from Switchgear Genomics (Carlsbad, CA). The RenSP luciferase plasmid DNA and cypridina luciferase TK control construct were co-transfected into UAEC by using FuGENE HD transfection reagents (1:3, μl/ng) for 24 h at 37°C. UAEC transfected with an empty vector and β-actin promoter vector were served as negative and positive transfection controls, respectively. After transfection, cells were allowed to recover for 18–20 h in DMEM containing 10% FBS. Cells were serum-starved overnight and treated with vehicle or E2β (10 nM) for 24 h with or without ICI 182780. The RenSP luciferase activity in both cells and supernatant was measured and normalized to cypridina luciferase activity as previously described [33].
Statistical analysis
Each experiment was repeated at least three times with cells derived from different pregnant ewes. Data are presented as means ± SEM and analyzed by one-way or two-way analysis of variance (ANOVA), followed by the Bonferroni test for multiple comparisons using SigmaPlot (Systat Software Inc.). Student paired t-test was used for comparison of data between two groups. Significance was defined as P < 0.05, unless higher statistical power is indicated in the figure legends.
Results
E2β stimulates H2S production: role of CBS, CTH, and ER
Compared to vehicle-treated controls, treatment with E2β (10 nM) for 48 h stimulated a 2.43 ± 0.21-fold (P < 0.01) increase in H2S production in UAEC in vitro. The stimulation was blocked by pretreatment with 1 μM ICI; incubation with ICI alone had no effect (Figure 1A). E2β-stimulated H2S production was significantly inhibited by the specific CBS inhibitor CHH or CSE inhibitor BCA alone; the combination of CHH and BCA completely blocked E2β-stimulated H2S production (Figure 1B).
Figure 1.
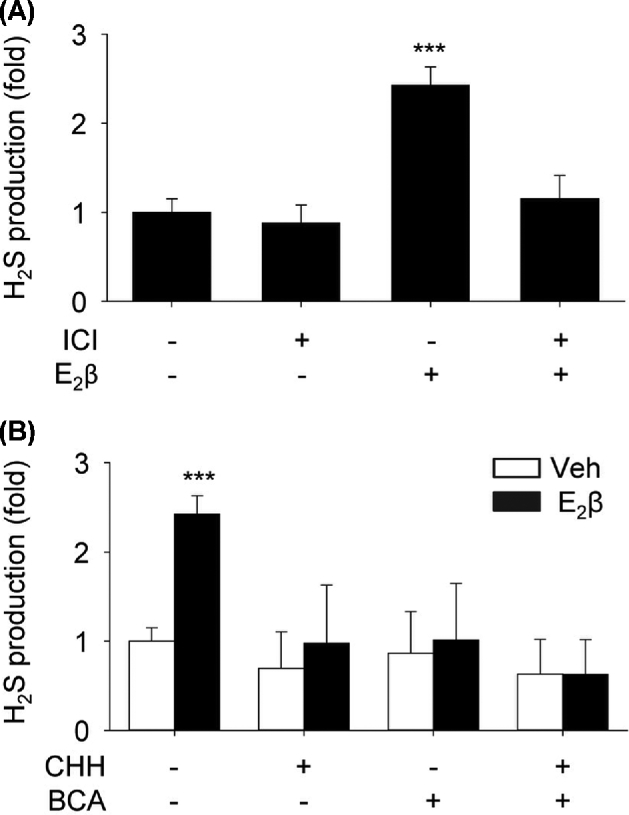
Effects of E2β on H2S production—role of CBS, CTH, and ER. (A) Primary uterine artery endothelial cells (UAEC) were treated with vehicle or estradiol-17β (E2β, 10 nM) with or without the estrogen receptor (ER) antagonist ICI 182780 (ICI, 1 μM) for 48 h. Protein extracts (1 × 106 cells) were used to determine hydrogen sulfide (H2S) production. (B) UAEC were treated with vehicle or 10 nM E2β for 48 h, and protein extracts were used to determine H2S production in the presence or absence of an inhibitor of cystathionine β-synthase (CBS, cystathionine γ-lyase (CTH), or CHH and BCA, respectively. Data (means ± SEM) were collected from different cell preparations cells prepared from three to five ewes. *** P < 0.001 vs control.
E2β stimulates CBS and CTH mRNA/protein expressions in a time- and concentration-dependent manner
E2β significantly stimulated CBS and CTH mRNAs in a time-dependent manner in UAEC in vitro. Following treatment with 10 nM E2β, both CBS and CTH mRNA levels began to increase significantly at 24 h, maximized at 48 h (CBS: 2.61 ± 0.27-fold vs control; P < 0.01; CTH: 3.24 ± 0.41 fold vs control, P < 0.01), and plateaued at 72 h (Figure 2A). E2β also significantly stimulated CBS and CTH proteins in a time-dependent manner. Following treatment with 10 nM E2β, both CBS and CTH protein levels began to significantly increase at 24 h and the stimulation continued up to 3 days (Figure 2B).
Figure 2.

Time course and concentration response of E2β on mRNA and protein expressions of CBS and CTH. Primary uterine artery endothelial cells (UAEC) were treated with 10 nM estradiol-17β (E2β) for up to 3 days to assess cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH) mRNA (A) and protein (B), or with increasing concentrations of E2β (0–1 μM) for 48 h to assess CBS and CTH mRNA (C) and protein (D). Data (means ± SEM) were collected from different cell preparations cells prepared from three to five ewes. Bars with different letters differ significantly (P < 0.05); capital and lower case letters pertain to CBS and CTH, respectively.
E2β also significantly stimulated CBS and CTH mRNAs in a concentration-dependent manner in UAEC in vitro. Treatment with 0.1 nM E2β for 48 h effectively stimulated both CBS and CTH mRNA expressions. Within the E2β concentrations tested, CBS mRNA continued to increase and maximized at 1 μM (3.02 ± 0.32-fold vs control, P < 0.01). CTH mRNA maximized by treatment with 10 nM E2β (3.25 ± 0.41-fold vs control, P < 0.01) and plateaued with 1 μM E2β (Figure 2C). Treatment with 0.1 nM E2β for 48 h effectively stimulated both CBS and CTH proteins. The responses increased with increasing concentrations of E2β, and maximized with 100 nM E2β (CBS: 3.04 ± 0.16-fold vs control, P < 0.01; CTH: 3.17 ± 0.63 fold vs control, P < 0.01) (Figure 2D).
E2β stimulation of CBS and CTH expressions is mediated by specific ERs
In the presence of the specific ER antagonist ICI (1 μM), E2β-stimulated CBS and CTH mRNAs (Figure 3A) were completely abrogated. Treatment with 10 nM E2β for 48 h also significantly stimulated CBS (2.46 ± 0.17-fold vs control, P < 0.01) and CTH (2.39 ± 0.10-fold vs control, P < 0.01) protein expressions, which were completely blocked by ICI (Figure 3B). Immunofluorescence analysis with specific anti-CBS and anti-CTH antibodies showed relative low baseline CBS and high CTH proteins in UAEC and confirmed ICI blockade of E2β stimulation of CBS and CTH protein expressions (Figure 3C). Treatments with vehicle or ICI alone did not alter CBS and CTH mRNA and protein expressions.
Figure 3.

ER-dependency of E2β effects on mRNA and protein expressions of CBS and CTH. Primary uterine artery endothelial cells (UAECs) were treated with vehicle, 10 nM estradiol-17β (E2β), 1 μM ICI 182 780 (ICI), or both for 48 h. Cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH) mRNA (A) and protein (B) were determined. (C) Immunofluorescence labeling of CBS and CTH proteins. Nuclei were stained with DAPI (blue). Protein expression was determined by relative green fluorescence intensity as fold change of control. Data (means ± SEM) were collected from different cell preparations from three to five ewes. *** P < 0.001 vs. Control. Scale bar = 50 μm.
Either ERα or ERβ is sufficient to mediate E2β stimulation of CBS and CTH expressions
UAEC expresses both ERα and ERβ [29] and they may play common or even opposite roles in mediating estrogen regulation of gene expression in a cell [34]; we determined the specific roles of ERα and ERβ in estrogen stimulation of CBS and CTH expressions by using ER isoform-specific agonists or antagonists. Compared to vehicle-treated controls, treatment with the ERα agonist PPT alone induced 2.55 ± 0.26-fold (P < 0.01) and 2.98 ± 0.91-fold (P < 0.01) increases in CBS and CTH mRNAs, respectively. Treatment with the ERβ agonist DPN alone induced 2.45 ± 0.15-fold (P < 0.01) and 2.39 ± 0.41-fold (P < 0.01) increases in CBS and CTH mRNAs, respectively (Figure 4A). These increases were comparable to that of E2β-stimulated fold increases in CBS (2.61 ± 0.27 vs vehicle, P < 0.01) and CTH (3.25 ± 0.41 vs vehicle, P < 0.01) mRNAs, respectively. PPT alone induced 2.02 ± 0.12-fold (P < 0.01) and 2.70 ± 0.39-fold (P < 0.01) increases in CBS and CTH proteins, respectively; DPN alone induced 2.69 ± 0.12-fold (P < 0.01) and 2.80 ± 0.17-fold (P < 0.01) increases in CBS and CTH proteins, respectively (Figure 4B). These changes were also comparable to that of E2β-stimulated fold increases in CBS (2.46 ± 0.17 vs vehicle, P < 0.01) and CTH (2.39 ± 0.10 vs vehicle, P < 0.01) proteins, respectively. The combination of PPT and DPN had no additive effects on either CBS or CTH mRNA and protein expressions (Figure 4). Treatment with either ERα antagonist MPP or ERβ antagonist PHTPP alone or their combination was sufficient to completely attenuate E2β-stimulated CBS and CTH mRNA (Figure 4C) and protein (Figure 4D) expressions.
Figure 4.

Specific role of ERα or ERβ in mediating E2β stimulation of mRNA and protein expressions of CBS and CTH. (A, B) ERα or ERβ activation: primary uterine artery endothelial cells (UAECs) were treated with vehicle, 10 nM of estradiol-17β (E2β), PPT (ERα agonist), DPN (ERβ agonist), or PPT + DPN for 48 h. (C, D) ERα or ERβ inhibition: primary uterine artery endothelial cells (UAECs) were treated with vehicle or estradiol-17β (E2β) (10 nM) with or without 1 μM MPP (ERα antagonist), PHTPP (ERβ antagonist), or MPP + PHTPP. Cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH) mRNA (A, C) and protein (B, D) were determined. Data (means ± SEM) were collected from different cell preparations from three to five ewes. Bars with different letters differ significantly; capital and lower case letters pertain to CBS and CTH, respectively. *** P < 0.001 vs Control.
E2β stimulates ER-dependent activation of CBS and CTH promoters
Compared to vehicle-treated controls, treatment with 10 nM E2β for 24 h significantly stimulated the promoter activities of CBS (3.71 ± 0.65-fold, P < 0.001) and CTH (3.77 ± 0.31-fold, P < 0.001) in UAEC. Co-treatment with ICI completely attenuated E2β-stimulated activation of CBS and CTH promoters (Figure 5).
Figure 5.
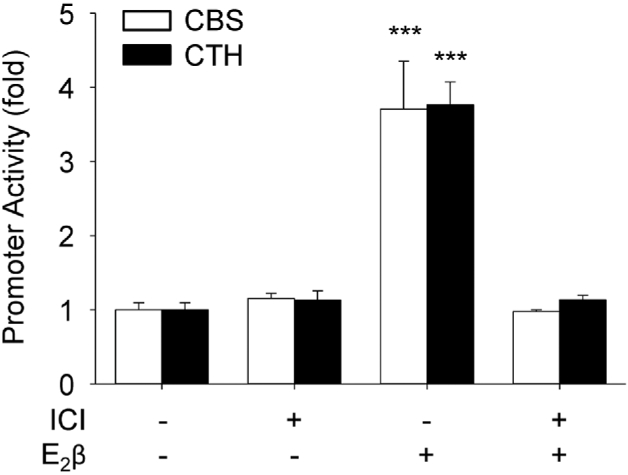
ER-dependency of E2β activation of CBS and CTH promoters. Human cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH) promoter luciferase-reporter constructs were transfected into primary uterine artery endothelial cells (UAECs). Twenty-four hours later, the cells were treated with vehicle or estradiol-17β (E2β, 10 nM) with or without estrogen receptor (ER) antagonist ICI 182780 (ICI, 1 μM) for 24 h. Luciferase activity was determined as an index for promoter action. Data (means ± SEM) were collected from cells prepared from three to five ewes. *** P < 0.001 vs Control.
Discussion
Consistent with our most recent in vivo findings that UA H2S production and endothelium and smooth muscle CBS but not CSE expressions are significantly greater in the proliferative phase and pregnancy, positively linked to elevated endogenous estrogens in women [23] and that exogenous estrogens significantly stimulate UA H2S biosynthesis via selective upregulation of endothelium and smooth muscle CBS, but not CTH, mRNA, and protein expressions in OVX ewes [26], we now report that E2β stimulates UAEC H2S production in association with upregulation of not only CBS but also CSE expression in vitro. The stimulatory effect of E2β on CTH mRNA and protein expression in UAEC in vitro is unexpected; however, the increased CBS and CTH protein expressions contribute to the increased H2S production upon E2β stimulation in UAEC in vitro because UAEC H2S production can be significantly inhibited by the specific inhibitor of either CBS (CHH) or CTH (BCA) alone and completely by their combination.
Our current data shown that E2β stimulates CBS mRNA and protein expressions in a time- and concentration-dependent manner, with as little as 0.1 nM E2β being effective to stimulate both CBS mRNA and protein expressions in UAEC. This low-dose E2β is physiologically relevant because 0.1–10 nM E2β is in the range of circulating estrogen levels in nonpregnant and pregnant states [1, 35, 36]. Together with our recent in vivo findings [23, 26], these observations further strengthened CBS as an estrogen-responsive gene in ECs. E2β stimulates both mRNA and protein expressions of CBS in UAEC in vitro, suggesting that the stimulation occurs mainly at the level of transcription. Indeed, E2β activates the CBS promoter in UAEC in vitro. ICI completely attenuates E2β stimulation of CBS mRNA and protein expressions as well as its promoter activation, indicating the involvement of specific ERs.
We have previously shown that UAEC are direct targets of estrogens as both ERα and ERβ are expressed in UAEC [29, 37]. In target cells expressing ERα and ERβ, ligated receptors function as transcription factors to regulate gene expression via interaction with estrogen response elements (ERE) in the promoter of target genes [28]. In this mode, estrogens also regulate the expression of genes without classical EREs via crosstalk between ligated ERs with other ERE-interacting transcription factors such as AP-1 or Sp1 [27, 38]. The genomic effects of estrogens regulate the expression of various genes that are key enzymes such as NOS3 [39] and prostaglandin synthase [40] for synthesizing NO and prostacyclin, respectively. In addition, ERα and ERβ may play different and even opposite roles in regulating cellular responses to estrogens [34]. We show herein that either PPT or DPN stimulates CBS mRNA and protein expressions to levels comparable to that of E2β-stimulated. Co-treatment with either MPP or PHTPP attenuates E2β-stimulated CBS mRNA and protein expressions and neither the combination of the agonists nor the antagonists have additive effects. Thus, activation of either ERα or ERβ is sufficient to mediate E2β stimulation of CBS expression in UAEC in vitro.
E2β also stimulates CTH mRNA and protein expressions in UAEC in vitro, similar to its effects on CBS expression. Also similar is that E2β stimulation of CTH mRNA and protein expressions is mediated by ER-dependent CTH transcription, which can be activated by either ERα or ERβ because E2β stimulates CTH mRNA and protein expressions as well as CTH promoter activation and all responses can be blocked by ICI. Thus, these data demonstrate that CTH is also an estrogen-responsive gene in UAEC in vitro. Nonetheless, estrogen stimulation of CTH expression is unexpected because it contradicts not only to our in vivo studies showing that exogenous or endogenous E2β does not alter CTH mRNA and protein in UA endothelium in women [23] and sheep [26], but also to other studies showing that E2β does not stimulate CTH expression in mouse mesenteric smooth muscle cells in vitro [41]. The mechanism underlying the discrepancy between the effect of E2β on CTH expression in UAEC in vitro and in vivo is currently unknown. However, it is not uncommon that in vitro experiments do not always agree with in vivo findings. Nonetheless, this discrepancy is at least, in part, due to loss of cell–cell interactions and the microenvironment that ECs reside in in vivo. Regardless, caution should be exercised when interpreting in vitro findings as they pertain to in vivo conditions because in vitro and in vivo studies can sometimes produce different outcomes.
Although our current study shows that E2β activates CBS and CTH transcription that is linked to ER activation in UAEC, it does not provide direct evidence as to how ER interacts with the regulatory cis-elements in CBS and CTH promoters. The human CBS promoter spans over >4K bp [42] and the CTH promoter is approximately 1000 bp in length [43]. Both contain multiple putative cis-elements for binding various transcription factors, including ERE, Sp1, AP-1, and AP-3 [43, 44]. AP-1 and Sp1 have been implicated in stimulating CTH expression in vascular smooth muscle cells [43] as well as CTH [45] and CBS [42] expression in hepatocellular carcinoma cells. Since E2β stimulation of CBS and CTH expression in UAEC and trans-activation of their promoters by E2β is blocked by ICI, it is reasonable to infer that estrogen stimulation of CBS and CTH expression is mediated mainly by ER (ERα and/or ERβ) interaction with ERE. However, others transcription factors, such as Sp1 and AP1, cannot be excluded because E2β can stimulate gene expression through interactions of ER with other transcription factors [38].
Of note, in addition to ERα and ERβ, estrogens also signal via G protein-coupled estrogen receptor 1 (GPER) located on the plasma membrane [46, 47]. In this “extranuclear” nongenomic mode of estrogen signaling, binding to membrane ERα, ERβ, or GPER activates multiple protein kinase cascades within seconds to minutes to activate downstream target proteins to elicit biological functions or nuclear transcription factors to regulate latent gene expression [47]. ERα and ERβ [29] and GPER [48] are expressed in UA. The nongenomic action of estrogens is also of critical importance to the vascular effects of estrogens. For instance, activation of extracellular signal-activated protein kinases [13] and protein kinase B/Akt1 [49] via membrane ER-mediated mechanisms mediates rapid activation of NOS3 to produce NO by E2β in ECs, which contributes greatly to the endothelium-dependent mechanisms for the vascular effects of estrogens [50], including estrogen-induced uterine vasodilation [48]. To this end, whether estrogens activate the H2S system via nongenomic pathway awaits to be determined.
In summary, our present study demonstrates that E2β stimulates H2S production by upregulating CBS and CTH mRNA and protein expressions through specific ER-dependent CBS and CTH transcription in UAEC in vitro. Estrogen stimulation of H2S biosynthesis in UAEC in vivo [23, 26] and in vitro (current study) obviously raises a question as to what role H2S plays in the UA. We believe that our findings underline a physiological role of H2S in mediating estrogen-induced and pregnancy-associated rises in UBF due to the potent vasodilatory properties of H2S [21]. This idea is supported by observations that exogenous H2S donor dilates rat UA more effectively in P vs NP states [23], although a role of endogenous H2S in mediating uterine vasodilation is waiting to be determined.
Supplementary data
Supplementary Table S1. Antibody Table.
Supplementary Material
Notes
Edited by Dr. Romana Nowak, PhD, University of Illinois Urbana-Champaign
Footnotes
Grant Support: This study was supported in part by National Institutes of Health (NIH) grants RO1 HL70562 and R21 HL98746 to DBC and PO1 HD38843 and RO1 HL117341 to RRM. TJL was an American Heart Association (AHA) Pre-doctoral Fellow (AHA14PRE18570033). The content is solely the responsibility of the authors and does not necessarily the official views of NIH and AHA.
Sources of funding
TJL was an American Heart Association (AHA) Pre-doctoral Fellow (AHA14PRE18570033). This study was supported in part by National Institutes of Health (NIH) grants RO1HL70562 and R21HL98746 to DBC and PO1HD38843, RO1HL87144, and RO1HL117341 to RRM. The content is solely the responsibility of the authors and does not necessarily the official views of NIH and AHA.
Conflict of Interest/Disclosure Statement: The authors have no financial interests to disclose.
References
- 1. O’Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem 1991; 37:667–672. [PubMed] [Google Scholar]
- 2. Albrecht ED, Pepe GJ. Placental steroid hormone biosynthesis in primate pregnancy. Endocr Rev 1990; 11:124–150. [DOI] [PubMed] [Google Scholar]
- 3. Rosenfeld CR. Responses of reproductive and nonreproductive tissues to 17 beta-estradiol during ovine puerperium. Am J Physiol 1980; 239:E333–E339. [DOI] [PubMed] [Google Scholar]
- 4. Magness RR, Rosenfeld CR. Local and systemic estradiol-17 beta: effects on uterine and systemic vasodilation. Am J Physiol 1989; 256:E536–E542. [DOI] [PubMed] [Google Scholar]
- 5. Magness RR, Rosenfeld CR. The role of steroid hormones in the control of uterine blood flow. In: The Uterine Circulation. Ithaca, NY: Perinatology Press; 1989; 10:239–271. [Google Scholar]
- 6. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 2009; 24:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barker DJ. Intrauterine programming of adult disease. Mol Med Today 1995; 1:418–423. [DOI] [PubMed] [Google Scholar]
- 8. Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science 2014; 345:760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosenfeld CR, Cox BE, Roy T, Magness RR. Nitric oxide contributes to estrogen-induced vasodilation of the ovine uterine circulation. J Clin Invest 1996; 98:2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gokina NI, Kuzina OY, Vance AM. Augmented EDHF signaling in rat uteroplacental vasculature during late pregnancy. Am J Physiol Heart Circ Physiol 2010; 299:H1642–H1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ni Y, May V, Braas K, Osol G. Pregnancy augments uteroplacental vascular endothelial growth factor gene expression and vasodilator effects. Am J Physiol 1997; 273:H938–H944. [DOI] [PubMed] [Google Scholar]
- 12. Chen DB, Jia S, King AG, Barker A, Li SM, Mata-Greenwood E, Zheng J, Magness RR. Global protein expression profiling underlines reciprocal regulation of caveolin 1 and endothelial nitric oxide synthase expression in ovariectomized sheep uterine artery by estrogen/progesterone replacement therapy. Biol Reprod 2006; 74:832–838. [DOI] [PubMed] [Google Scholar]
- 13. Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology 2004; 145:113–125. [DOI] [PubMed] [Google Scholar]
- 14. van der Heijden OW, Essers YP, Fazzi G, Peeters LL, De Mey JG, van Eys GJ. Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase-deficient mice. Biol Reprod 2005; 72:1161–1168. [DOI] [PubMed] [Google Scholar]
- 15. Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA, Adamson SL. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension 2012; 60:231–238. [DOI] [PubMed] [Google Scholar]
- 16. Rosenfeld CR, Roy T. Prolonged uterine artery nitric oxide synthase inhibition modestly alters basal uteroplacental vasodilation in the last third of ovine pregnancy. Am J Physiol Heart Circ Physiol 2014; 307:H1196–H1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 2012; 92:791–896. [DOI] [PubMed] [Google Scholar]
- 18. Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL. Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol 2011; 300:H440–H447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatia M. Hydrogen sulfide as a vasodilator. IUBMB Life 2005; 57:603–606. [DOI] [PubMed] [Google Scholar]
- 20. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008; 322:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 2001; 20:6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li Y, Zang Y, Fu S, Zhang H, Gao L, Li J. H2S relaxes vas deferens smooth muscle by modulating the large conductance Ca2+ -activated K+ (BKCa) channels via a redox mechanism. J Sex Med 2012; 9:2806–2813. [DOI] [PubMed] [Google Scholar]
- 23. Sheibani L, Lechuga TJ, Zhang HH, Hameed A, Wing DA, Kumar S, Rosenfeld CR, Chen DB. Augmented H2S production via CBS upregulation plays a role in pregnancy-associated uterine vasodilation. Biol Reprod 2017; 96:664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol 2013; 182:1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E et al. . Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013; 127:2514–2522. [DOI] [PubMed] [Google Scholar]
- 26. Lechuga TJ, Zhang HH, Sheibani L, Karim M, Jia J, Magness RR, Rosenfeld CR, Chen DB. Estrogen replacement therapy in ovariectomized nonpregnant ewes stimulates uterine artery hydrogen sulfide biosynthesis by selectively up-regulating cystathionine beta-synthase expression. Endocrinology 2015; 156:2288–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors alpha and beta. J Biol Chem 2005; 280:347–354. [DOI] [PubMed] [Google Scholar]
- 28. Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am J Cardiol 2002; 90:F3–F6. [DOI] [PubMed] [Google Scholar]
- 29. Liao WX, Magness RR, Chen DB. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol Reprod 2005; 72:530–537. [DOI] [PubMed] [Google Scholar]
- 30. Martini PG, Katzenellenbogen BS. Modulation of estrogen receptor activity by selective coregulators. J Steroid Biochem Mol Biol 2003; 85:117–122. [DOI] [PubMed] [Google Scholar]
- 31. Magness RR, Phernetton TM, Gibson TC, Chen DB. Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol-17beta-treated, intact follicular and pregnant sheep. J Physiol 2005; 565:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang HH, Feng L, Wang W, Magness RR, Chen DB. Estrogen-responsive nitroso-proteome in uterine artery endothelial cells: role of endothelial nitric oxide synthase and estrogen receptor-beta. J Cell Physiol 2012; 227:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mata-Greenwood E, Liao WX, Wang W, Zheng J, Chen DB. Activation of AP-1 transcription factors differentiates FGF2 and vascular endothelial growth factor regulation of endothelial nitric-oxide synthase expression in placental artery endothelial cells. J Biol Chem 2010; 285:17348–17358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hurst AG, Goad DW, Mohan M, Malayer JR. Independent downstream gene expression profiles in the presence of estrogen receptor alpha or beta. Biol Reprod 2004; 71:1252–1261. [DOI] [PubMed] [Google Scholar]
- 35. Gibson TC, Phernetton TM, Wiltbank MC, Magness RR. Development and use of an ovarian synchronization model to study the effects of endogenous estrogen and nitric oxide on uterine blood flow during ovarian cycles in sheep. Biol Reprod 2004; 70:1886–1894. [DOI] [PubMed] [Google Scholar]
- 36. Dawood MY, Ratnam SS. Serum unconjugated estradiol-17 beta in normal pregnancy measured by radioimmunoassay. Obstet Gynecol 1974; 44:194–199. [PubMed] [Google Scholar]
- 37. Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen DB, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: ovarian steroid and pregnancy control of ERalpha and ERbeta levels. J Physiol 2005; 565:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem 2010; 285:2676–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Si H, Yu J, Jiang H, Lum H, Liu D. Phytoestrogen genistein up-regulates endothelial nitric oxide synthase expression via activation of cAMP response element-binding protein in human aortic endothelial cells. Endocrinology 2012; 153:3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su EJ, Lin ZH, Zeine R, Yin P, Reierstad S, Innes JE, Bulun SE. Estrogen receptor-beta mediates cyclooxygenase-2 expression and vascular prostanoid levels in human placental villous endothelial cells. Am J Obstet Gynecol 2009; 200:427.e1–427.e8e421-428. [DOI] [PubMed] [Google Scholar]
- 41. Li H, Mani S, Cao W, Yang G, Lai C, Wu L, Wang R. Interaction of hydrogen sulfide and estrogen on the proliferation of vascular smooth muscle cells. PLoS One 2012; 7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ge Y, Konrad MA, Matherly LH, Taub JW. Transcriptional regulation of the human cystathionine beta-synthase -1b basal promoter: synergistic transactivation by transcription factors NF-Y and Sp1/Sp3. Biochem J 2001; 357:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang G, Pei Y, Teng H, Cao Q, Wang R. Specificity protein-1 as a critical regulator of human cystathionine gamma-lyase in smooth muscle cells. J Biol Chem 2011; 286:26450–26460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kraus JP, Oliveriusova J, Sokolova J, Kraus E, Vlcek C, de Franchis R, Maclean KN, Bao L, Bukovsk, Patterson D, Paces V, Ansorge W et al. . The human cystathionine beta-synthase (CBS) gene: complete sequence, alternative splicing, and polymorphisms. Genomics 1998; 52:312–324. [DOI] [PubMed] [Google Scholar]
- 45. Yin P, Zhao C, Li Z, Mei C, Yao W, Liu Y, Li N, Qi J, Wang L, Shi Y, Qiu S, Fan J et al. . Sp1 is involved in regulation of cystathionine gamma-lyase gene expression and biological function by PI3K/Akt pathway in human hepatocellular carcinoma cell lines. Cell Signal 2012; 24:1229–1240. [DOI] [PubMed] [Google Scholar]
- 46. Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vasc Pharmacol 2011; 55:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pedram A, Razandi M, Aitkenhead M, Hughes CC, Levin ER. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J Biol Chem 2002; 277:50768–50775. [DOI] [PubMed] [Google Scholar]
- 48. Tropea T, De Francesco EM, Rigiracciolo D, Maggiolini M, Wareing M, Osol G, Mandala M. Pregnancy augments G protein estrogen receptor (GPER) induced vasodilation in rat uterine arteries via the nitric oxide - cGMP signaling pathway. PLoS One 2015; 10:e0141997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haynes MP, Sinha D, Russell KS, Collinge M, Fulton D, Morales-Ruiz M, Sessa WC, Bender JR. Membrane estrogen receptor engagement activates endothelial nitric oxide synthase via the PI3-kinase-Akt pathway in human endothelial cells. Circ Res 2000; 87:677–682. [DOI] [PubMed] [Google Scholar]
- 50. Murphy E. Estrogen signaling and cardiovascular disease. Circ Res 2011; 109:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.