

Abstract
BACKGROUND:
Mesenchymal stem cells (MSCs) have been shown to mitigate vascular permeability in hemorrhagic shock (HS) and trauma-induced brain and lung injury. Mechanistically, paracrine factors secreted from MSCs have been identified that can recapitulate many of the potent biologic effects of MSCs in animal models of disease. Interestingly, MSC-derived extracellular vesicles (EVs), contain many of these key soluble factors, and have therapeutic potential independent of the parent cells. In this study we sought to determine whether MSC-derived EVs (MSC EVs) could recapitulate the beneficial therapeutic effects of MSCs on lung vascular permeability induced by HS in mice.
METHODS:
Mesenchymal stem cell EVs were isolated from human bone marrow-derived MSCs by ultracentrifugation. A mouse model of fixed pressure HS was used to study the effects of shock, shock + MSCs and shock + MSC EVs on lung vascular endothelial permeability. Mice were administered MSCs, MSC EVs, or saline IV. Lung tissue was harvested and assayed for permeability, RhoA/Rac1 activation, and for differential phosphoprotein expression. In vitro, human lung microvascular cells junctional integrity was evaluated by immunocytochemistry and endothelial cell impedance assays.
RESULTS:
Hemorrhagic shock-induced lung vascular permeability was significantly decreased by both MSC and MSC EV infusion. Phosphoprotein profiling of lung tissue revealed differential activation of proteins and pathways related to cytoskeletal rearrangement and regulation of vascular permeability by MSCs and MSC EVs. Lung tissue from treatment groups demonstrated decreased activation of the cytoskeletal GTPase RhoA. In vitro, human lung microvascular cells, MSC CM but not MSC-EVs prevented thrombin-induced endothelial cell permeability as measured by electrical cell-substrate impedance sensing system and immunocytochemistry of VE-cadherin and actin.
CONCLUSION:
Mesenchymal stem cells and MSC EVs modulate cytoskeletal signaling and attenuate lung vascular permeability after HS. Mesenchymal stem cell EVs may potentially be used as a novel “stem cell free” therapeutic to treat HS-induced lung injury.
Keywords: Hemorrhagic shock, vascular permeability, rhoA signaling, MSC EVs, mesenchymal stem cells
Acute respiratory distress syndrome (ARDS) is a serious complication of traumatic injury.1,2 In a prospective randomized trial of trauma patients (PROPPR), the incidence of ARDS was 14%.3 Furthermore, in a recent review of trauma deaths at a Level I trauma center, approximately 33% of deaths were the result of a combination of infection, inflammation, microvascular compromise, and dysfunctional coagulation, all components which comprise the endotheliopathy of trauma (EOT).4 The EOT potentially contributes to the development of ARDS, multiple organ failure including acute kidney injury (AKI), and sepsis in trauma.5,6 To date, the greatest advances in the treatment of major trauma are primarily related to improved hemorrhage control and optimized resuscitation paradigms with blood products.4,7 Novel therapeutic interventions are needed to address the EOT following trauma and resuscitation, including trauma-induced ARDS. For trauma and non–trauma-related causes of ARDS, aside from supportive care with lung-protective ventilation, there is currently no effective therapeutic modality that reduces mortality. Development of a therapy that can significantly reduce the severity of ARDS, attenuate multiple organ failure, and decrease the number of secondary infections would be a significant advance for the management of trauma patients.
Over the past 15 years, there has been considerable progress in investigating the therapeutic application of stem cells in human disease.5 Mesenchymal stem cells (MSCs), the stromal progenitor cells isolated from bone marrow, have been demonstrated to have potent therapeutic effects that mitigate outcomes in diseases characterized by vascular dysfunction and inflammation.8 Mesenchymal stem cells may modulate coagulation as well as described in recent reports.9 These properties of MSCs have inspired a number of preclinical and clinical studies in broad disease areas including sepsis, traumatic brain injury (TBI), stroke, AKI, graft versus host disease, acute lung injury and ARDS, to name a few. Many of the noted beneficial effects are not dependent on the capacity of MSCs to differentiate into other cell types but are rather mediated through mechanisms involving the secretion of soluble factors and extracellular vesicles (EVs) by MSCs that mediate their therapeutic effects.10–12 Mesenchymal stem cells have been shown to mitigate inflammation, injury and lung edema in multiple rodent preclinical models of ARDS,13 ex vivo–perfused human lungs,14 and in sheep with infectious causes of ARDS.15 Mesenchymal stem cells were also effective in attenuating lung injury in a rodent model of hemorrhagic shock (HS)-induced lung injury16 where they reduced lung edema, decreased pulmonary inflammation and restored vascular endothelial cell (EC) tight and adherens junctions.16
Recent work has demonstrated that many of the therapeutic proteins secreted by MSCs are contained within EVs including specific DNA, RNA, miRNA, and lipids. Mesenchymal stem cell EVs enter neighboring cells locally or distally after which they have been shown to deliver their protein, lipid and nucleic acid cargo to receptive cells.17 Mesenchymal stem cell EVs have been shown to possess therapeutic properties similar to parent MSCs.18–22 They can recapitulate some of the therapeutic effects of MSCs in multiple disease models of lung injury, AKI and TBI.13,15,17–22 Zhu and colleagues23 demonstrated that EVs released by human bone marrow-derived MSCs are as effective as MSCs in restoring lung protein permeability and reducing inflammation in an Escherichia coli endotoxin-induced lung injury in C57BL/6 mice.
In this article, we hypothesized that MSC EVs could recapitulate the vasculoprotective effects of MSCs in a mouse model of HS and laparotomy-induced lung injury in a similar manner to MSCs.
METHODS
Primary Cells and Culture
Human lung microvascular EC cells (HLMVECs) were purchased from Lonza (Walkersville, MD). The HLMVECs were maintained in EC growth media 2 (Lonza Walkersville, MD) Human MSCs passage 1 were obtained from Rooster Bio Inc. (Frederick, MD). Both cell types were expanded on the Quantum cell expansion device (Terumo BCT, Lakewood, CO). For details of the expansion, culture and generation of EVs please see Supplemental Methods (http://links.lww.com/TA/B51).
Characterization of MSCs and EVs
Mesenchymal stem cells and MSC EVs were both characterized by flow cytometry. Mesenchymal stem cells were gated for CD90, CD44, CD105, and CD73. Mesenchymal stem cell EVs were gated with comparatively sized beads before being assayed for markers CD9, CD63, CD81 CD73, CD90, CD29, CD44, HLA-DR, CD45, and CD31. Mesenchymal stem cell EV particle size distribution was determined using ZetaView instrumentation. For more details see Supplemental Methods (http://links.lww.com/TA/B51).
Animal Protocols
The animal studies were performed with approval of the Institutional Animal Care and Use Committee at ISIS Services LLC (San Carlos, CA). The experiments were conducted in compliance with the National Institutes of Health guidelines on the use of laboratory animals. All animals were housed in a room with access to food and water ad libitum, controlled temperature and 12:12-hour light-dark cycles.
In Vivo Lung Injury Induced by HS and Laparotomy
Male C57BL6 mice, 8 weeks to 12 weeks, were obtained from Harlan Laboratories (Livermore, CA). Under inhaled isoflurane anesthesia, animals were placed on a heating plank to maintain a body temperature between 35°C and 37°C. A 2.5-cm midline laparotomy was performed, and then the incision was sutured. Femoral arterial catheters were flushed with 1,000-U/mL heparin and then placed into the femoral arteries of both legs. No additional heparin was used. The left catheter was connected to the corresponding fluid reservoir and the right to a blood pressure monitor (PowerLab 8; ADInstruments, Dunedin, New Zealand). In this established model of HS and laparotomy, mice were bled to a mean arterial blood pressure of 35 mm Hg for 90 minutes except the sham group (n = 7).24,25 After the shock period, mice received fluid treatments of 200-μL phosphate-buffered saline (PBS) containing 10 × 106 MSCs (n = 7), 30 μg of MSC EVs (n = 6) or no treatment (shock group, n = 12). Mice were monitored for an additional 30 minutes after shock. Animals were allowed to ambulate freely for 60 minutes, then they were reanesthetized and administered 200 μL of 1-mg/mL Alexa Flour 680 dextran 10 kDa through the femoral cannula. After 1 hour, animals were perfused with 30 mL of ice-cold PBS. The lungs and heart were removed and then perfused with an additional 30-mL ice-cold PBS, ensuring that the lungs were inflated, and all blood was flushed from the lungs. The lungs were then harvested, inflated with freezing compound OCT Tissue Tek (Andwin Scientific, Schaumburg, IL). One lung was placed on an Odyssey scanner (LI-COR Biosciences, Lincoln, NE), and the fluorescence was read at 700 nm. With the use of image studio software (LI-COR Biosciences), an average fluorescence intensity per lung was quantitated to read lung permeability to 10-kDa proteins and averaged with others from the same treatment group after being normalized to the sham.
Phosphorylated Protein Expression Profiling and Gene Ontological Analysis
Four lung samples per treatment group were processed for protein analysis, and each sample was run on different 2D gel electrophoresis gels, fixed, then stained for proteins (green) and phosphoproteins (red). This work was performed by Applied Biomics (Hayward, CA). Gels were analyzed using DeCyder software, and 87 spots were selected that varied between the various treatment groups (sham, shock, shock + MSCs, and shock + MSC EVs). The quantitative changes in phosphorylation between groups were analyzed by DeCyder software. For each group of proteins altered between test groups, we applied a PANTHER (Version 11.1,26) statistical overrepresentation test and identified significant ( p<0.01) pathways or gene ontologies in the areas of biological processes, cellular components and molecular functions. (See Supplemental Methods, http://links.lww.com/TA/B51).
Rho and Rac Activity Assay
RhoA or Rac1 activity were assayed by using the luminescence based, enzyme linked capture assay, G-LISA (Cytoskeleton; Denver, Colorado) as per manufacturer’s protocols. (See Supplemental Methods, http://links.lww.com/TA/B51).
Endothelial Tansendothelial Resistance Assay
Endothelial cell permeability was assessed as a function of transendothelial electrical resistance (TEER), which was measured by using an electrical cell-substrate impedance sensing system (ECIS) (Applied Biophysics; Troy, NY) as described previously.27,28 (see Supplemental Methods, http://links.lww.com/TA/B51).
Immunocytochemistry
The HLMVECs, grown on 12-well plates, were treated with MSC EVs (15 μg/mL) or conditioned media (CM) for 6 hours. After treatment, cells were stained with Texas-Red phalloidin and an antibody to VE-cadherin (see Supplemental Methods, http://links.lww.com/TA/B51).
Statistics
All values are presented as ± standard deviation. Between group comparisons were conducted via one-way analysis of variance (ANOVA) analysis followed by Tukey post hoc tests performed using GraphPad Software (La Jolla, CA). Data were considered significant if p value is less than 0.05. For statistical analysis of the phosphorylated-protein expression, see Supplemental Methods (http://links.lww.com/TA/B51).
RESULTS
Characterization of MSCs and MSC EVs
To characterize the MSCs and the MSC EVs we used flow cytometry for markers specific to MSC EVs (exosomes and microvesicles) and MSCs. Exosomes are the smaller component of EVs with sizes ranging between 20 nm and 0.2 microns. MVs typically range from 0.2 to 2.0 microns. Collectively, MVs and exosomes form the EV component derived from MSCs. The MSCs used for this study were characterized via flow cytometry and expressed MSC markers (CD105, CD90, and CD73) and were negative for (CD45, CD34, CD11b, CD19, HLA-DR) (see Supplemental Figure 1E, http://links.lww.com/TA/B51).29 The EVs were also analyzed for surface markers via flow cytometry. Using beads of defined sizes: 0.2, 0.5, 2.0, and 3.0 um, we found 97% of the EVs (see Supplemental Figure 1A, http://links.lww.com/TA/B51) to be in the 0.2-μm to 0.5-μm range. 1.1% of the EVs were between 0.5 μm and 1 μm and the remaining were found to be between 1.0 μm and 2.0 μm. Less than 10% of the EVs expressed exosomal markers (CD9, CD63, and CD81) and MSC markers (CD44, CD73, and CD90) (see Supplemental Figure 1B and 1C, http://links.lww.com/TA/B51), and most of the EVs expressing MSC markers were exosomes as determined in Supplemental Figure 1D (http://links.lww.com/TA/B51) demonstrating coexpression of exosomal and MSC surface markers, CD44 and CD73.
However, flow cytometry has limits in quantitating the size of small particles, so we performed a quantitative analysis of MSC-derived EVs by ZetaView (see Supplemental Figure 1F, http://links.lww.com/TA/B51), which revealed the EV particle sizes range between 5 nm and 375 nm with greater than 95% of EVs in the 45-nm to 250-nm range with a peak at ~100 nm. Furthermore, ZetaView analysis indicates that the aliquots of 30 μg of protein, used in our in vivo studies, have approximately 8.9 × 1010 EV particles within each dose (see below). Taken together these data indicate that the EVs analyzed are a mix of exosomes and microvesicles, where a majority of the EVs are exosomal in origin. Supplementary Table 1 (http://links.lww.com/TA/B51) confirms the presence of key therapeutic mRNAs within the MSC EVs (TIMP3, ANG-1, and TSG-6) determined by qPCR of the MSC EV RNA content.
The Effects of MSCs and MSC EVs on HS-Induced Lung Permeability
We compared the effects of MSCs and MSC EVs using an established 3-hour mouse model of laparotomy24,25 followed by fixed pressure HS (Fig. 1A). Hemorrhagic shock mice were treated with one dose of MSCs (1.0 × 106 cells) or MSC EVs (30 μg protein). Supplementary Figure 2 (http://links.lww.com/TA/B51) demonstrates our dose selection of 1 × 106 MSCs to be active in attenuating lung permeability. Figure 1B and1C demonstrates that MSCs and MSC EVs equivalently attenuate pulmonary vascular permeability induced by HS. Both treatment groups (MSC, 1119 ± 202.7; MSC EVs, 1290 ± 337.2) as well as sham (973.7 ± 233.2) were significantly lower than HS alone (2010 ± 101) (see Fig. 1B). Mean arterial pressures (MAPs) were also recorded (Fig. 1D) and demonstrate no significant differences in the MAPs between the groups.
Figure 1.

Both MSC and MSC EV treatment attenuate pulmonary vascular permeability after hemorrhagic shock. (A) Schematic of 90 minute hemorrhagic shock model experiment. (B) Average fluorescence intensity of 10 kDa dextran conjugated to Alexafluor 680 in the left lobes of the mouse lungs after harvest. Bars indicate significant differences by one-way ANOVA Tukey test (p< 0.05). (C) Representative images of the left lobes of mouse lungs at 700 nm after sacrifice. (D) Resuscitation MAPs for shock mice are shown. Error bars indicate mean ± SD.
Phosphorylated-Protein Expression Profiling Reveals Differential Pathways Activated in the Lungs of HS Mice by MSCs and MSC EVs
To better understand the molecular mechanisms underlying the changes in lung permeability induced by HS in the four groups of mice, we conducted phosphoprotein expression profiling of lung tissue. We sought to globally screen for signaling pathways that are altered by HS and MSCs or MSC EV treatment. Using 2D gel electrophoresis (2D-DIGE) and phosphorprotein identification by mass spectrometry (Fig. 2A–2C), we were able to assess early signaling pathways that are activated or altered by shock and MSC or MSC EV treatment.
Figure 2.

Treating HS with MSCs or MSC EV triggers a change in the phosphorylation state of cytoskeletal proteins. (A) Workflow schematic of 2D gel phosphorylation screen. Four lungs from each treatment group were randomly selected (sham, shock, shock + MSCs, and shock + MSC EVs) for analysis. They are separated by size and pH. The gels are stained for protein (green) and phosphorylation (red). Since the experiment endpoint was acute we focused on phosphorylation changes. DeCyder software identified 87 spots on the DIGE gels that changed between the treatment groups. Each of those spots was removed from the gels and the proteins identified by mass spectrometry. These protein lists then allowed us to run a PANTHER analysis to find over represented pathways, run gene ontologies and find proteins of interest. Representative 2D gels stained for phosphorylated proteins for (B) sham and (C) shock groups, the 87 spots where differences were found in phosphorylation content by DeCyber analysis are circled and labeled. (D) Heat map of phosphorylation changes with comparing shock to the treatment groups on the left and sham to the treatment groups on the right. The blue lines map spots 10 and 11 from the gel to the heat map. Green indicates a positive change and red indicates a negative change relative the comparisons group. Note that the shock + MV group is has less changes compared to sham than the shock + MSC group.
The data were analyzed and expressed as fold change in phosphorylated proteins between the following treatment groups: (1) shock versus sham, (2) shock versus shock+MSCs, (3) shock versus shock+MSC EVs, (4) shock + MSC EVs versus shock + MSCs (see Figs. 2D for heat map comparisons). Shock clearly induced phosphoprotein changes compared with sham mice. Both treatments with MSCs and MSC EV-induced changes in protein phosphorylation patterns compared with shock mice, but not all changes were the same (Fig. 2D). Furthermore, when comparing the heat maps (protein phosphorylation changes) of shams to the treatment groups (Fig. 2D), shock + MSC EVs appear to be more similar to sham (uninjured mice) than to the MSC-treated mice, suggesting that the state of protein signaling in the shock animals treated with MSC EVs is closer to the uninjured baseline. Taken together, these data indicate that the proteins and signaling pathways activated by MSC versus MSC EV treatment in shock mice may differ at the molecular level from each other.
To identify exactly which proteins and signaling pathways were altered in phosphorylation profiles between the four groups, the 87 protein spots that differed between mouse groups were extracted from each gel and subjected to mass spectrometry (Fig. 2A). The MASCOT search (see Methods) identified up to 10 proteins present within each spot (see Supplemental Table 2, http://links.lww.com/TA/B51). Analysis of the protein list revealed differences primarily related to lung cytoskeleton changes and signaling proteins, such as the Rho kinase signaling pathway.
PANTHER analysis of the protein phosphorylation changes produced gene ontologies for changes in the areas of molecular signaling pathways, cellular components and molecular functions (Fig. 3B and Supplemental Figures 3 and 4, http://links.lww.com/TA/B51). In all the ontology groups, a predominance of activation and alteration of cytoskeletal signaling pathways related to the Rho GTPase pathway is identified. Additional signaling pathways altered between shock and treated groups included inf lammatory cytokine/chemokine signaling pathways, the cadherin pathways, and integrin signaling pathways.
Figure 3.

Proteins related to cytoskeleton reorganization are enriched in the phosphor-protein screen. (A) Proteins of interest found in the screen. (B) Gene ontological donut plots of molecular pathways found by PANTHER analysis. Pathways related to cytoskeletal reorganization and vascular permeability are highlighted in green. Pathways are presented in with the name, then the number of genes found, followed by the percentage of those genes in the gene ontology.
RhoA GTPase Activity Is Modulated by Both MSC and MSC EV Treatment in the Lungs of HS Mice
The phosphoprotein screen found changes between treatment groups in cytoskeletal organization pathways and the Rho GTPase pathway (Fig. 4A). Specifically, the screen identified actin, myosin light chain, RhoA, and Rab GDP dissociation inhibitor alpha (GDI1) as significantly changed (Fig. 3A and 4A).
Figure 4.
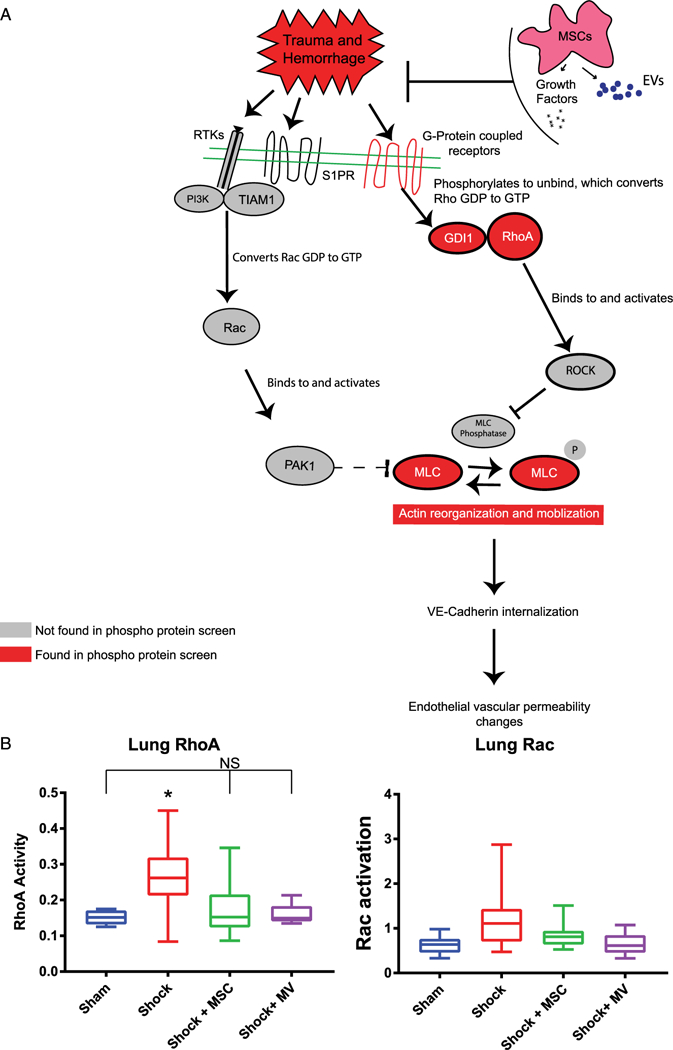
Hemorrhagic shock increases RhoA activity. (A) Model for MSC-mediated endothelial barrier function via Rho/Rac GTP production and signaling. The in vivo conditions of MSC or MSC EV treatment promote an inhibition of RhoA activation by MSCs, leading to a protection against vascular leakage. Changes in phosphorylation of GDI1, RhoA, and MLC (all shown in red) were observed in a phosphoprotein screen of lung tissue from our mouse model. B) GLISA results of Rho and Rac activity of mouse lungs. *Indicates significant difference from all other groups by one-way ANOVA Tukey test (p < 0.05). Mice treated with either MSCs of MSC MVs show no elevation of RhoA activity. Rac activity showed no significant differences.
To understand the mechanisms of action of MSCs and MSC EVs on RhoA GTPase pathways, lungs of HS mice from the four groups were assayed for RhoA and Rac1 activity by an enzyme-linked immunoabsorbent assay-based kit (GLISA) (Fig. 4B). The lungs in shock mice revealed increased expression of activated RhoA (0.27 ± 0.10) as compared with sham (0.151 ± 0.020) without a change in Rac1 expression (sham, ± 0.22; shock, 1.3 ± 0.80). The increase in expression of active RhoA induced by shock is significantly down regulated to sham levels by both MSCs and MSC EVs (RhoA: sham, 0.1511 ± 0.020; shock + MSCs, 0.17 ± 0.074; shock + MSC EVs, 0.16 ± 0.31.) Both MSC and MSC EV treatment groups are not significantly different from the sham mice. Activated Rac1 expression levels did not change in any of the groups (see Fig. 4B). These data suggest that MSCs and MSC EVs inhibit HS-induced RhoA GTPase activity, which may contribute to the demonstrated inhibition of lung vascular permeability by both treatments (Fig. 1). There are no significant effects of any of the test groups on Rac1, suggesting that Rac1 may not be involved in HS-mediated effects in vivo.
In Vitro MSCs But Not MSC EVs Are Protective Against Thrombin-Induced Permeability
We measured the TEER (equating to assessment of para-cellular permeability) of HLMVECs in response to thrombin. Mesenchymal stem cell CM was used in vitro since the CM has all of the protective factors secreted by MSCs that inhibit barrier permeability. Increased TEER is representative of decreased permeability of EC monolayers. The resulting TEER traces (Fig. 5A) and calculation of area under the curve (AUC) for each of the groups (Fig. 5B), demonstrate that MSC CM prevents thrombin-induced permeability (decreases in TEER); however, the MSC EVs do not prevent thrombin-induced permeability in vitro. Area under the curve for MSC CM (7.01 ± 0.122) was significantly higher than thrombin-treated control HLMVECs (6.03 ± 0.245) (Fig. 5B) and similar to the HLVMEC media control without thrombin (7.00 ± 0.0934). Additionally, MSC control media, which had not been exposed to MSCs, had no protective effect (5.60 ± 0.252). In contrast to MSC CM, the cells treated with MSC EVs (5.72 ± 0.334) showed no protective effect compared to thrombin-treated controls. Cells treated with recombinant ANG1, a positive control and another MSC-secreted protein, demonstrated significant protective (inhibitory) effects on permeability as expected. These results overall indicate a divergence in effect of MSCs and MSC EVs in vitro, suggesting that MSC EVs may be acting through different cells, other than ECs, or pathways in vivo to regulate pulmonary vascular permeability in HS.
Figure 5.
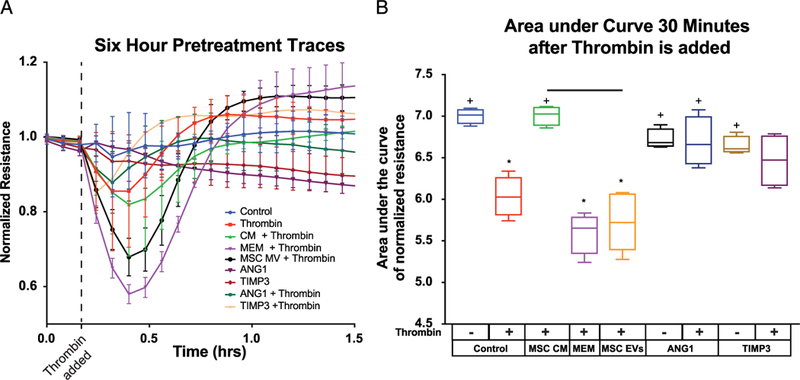
MSC CM and MSC EVs show differential effects in vitro. (A) TEER ECIS traces after six hours of treatment. ANG1 and TIMP3 were used as a positive control. MEM is the MSC media with serum that has not been exposed to MSCs. (B) Area under the curve quantitation of the first 30 minutes after Thrombin exposure. *Indicates significant difference from control while + indicates a significant difference from thrombin alone. All tests determined by one-way ANOVA followed by post hoc Tukey test analysis (p < 0.05).
VE-Cadherin and Actin Staining Display Differential Effects in HMLVECS Treated With of MSCs or MSC EVs
To examine the effects MSCs and MSC EVs on EC junctional stability, we analyzed EC cell expression of VE-cadherin and actin. These data qualitatively and quantitatively demonstrate that MSC CM treatment thickened the width and prominence of VE-cadherin junctions between cells compared with media alone (control, 3.1 ± 0.10 μm; MSC CM, 3.4 ± 0.20 μm) and did not noticeably alter the actin fibers (Fig. 6A and C). This is consistent with our results in Figure 5 demonstrating the MSC CM decreases HLMVEC permeability and increases TEER. The MSC EVs did not thicken VE-cadherin junctions (MSC EVs, 3.1 ± 0.15 μm) and increased the amount of actin stress fibers in agreement with our finding that HLMVEC permeability to thrombin is not attenuated in vitro by MSC EVs. Our positive control, Ang1, increased VE-Cadherin junction thickness (Ang1, 3.8 ± 0.12 μm) without reorganizing the actin fibers in a noticeable manner as expected (Fig. 6B). Overall, these data suggest that in vitro, there is a difference between the effects of MSCs and MSC EVs on EC integrity, cytoskeletal reorganization, and EC permeability. This diverges from our findings in vivo (Fig. 1A), where both treatments attenuate lung permeability in HS similarly. These results suggest that there are potentially other effects of MSC EVs in vivo, possibly on different cell types that result in the decreased lung permeability.
Figure 6.
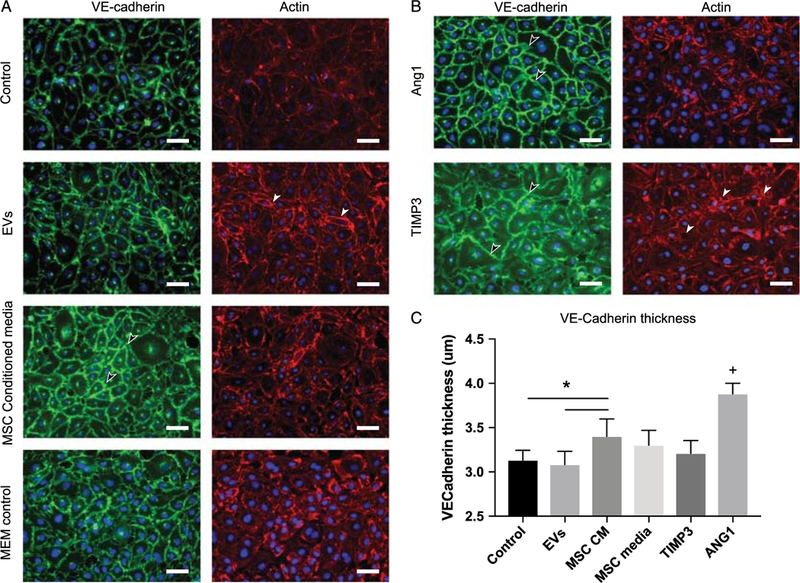
MSC CM increases the deposition of VE-Cadherin junctions and MSC EVs increases the presence of stress fibers. Representative images of HLMVEC cells stained for VE-cadherin (green) and actin (red) after treatment with (A) PBS control, MSC MVs, MSC CM, MEM control media, and positive controls (B) recombinant angiopoietin 1 (dose) and TIMP3 (dose). Qualitatively, MVs slightly increase the width of VE-cadherin junctions at the cell–cell interface (see arrow) but also increase the amount of stress fibers present in the cytoplasm of the cell (white arrows). In contrast, MSC CM, which is barrier protective, increases the width of the VE-cadherin junction at the cell-cell interface (black arrows) but has no visible effects on cytoskeletal stress fiber formation. TIMP3 and Ang1, positive controls, both effect the adherens junctions but TIMP3 also upregulates actin stress fiber formation. The VE-Cadherin thickness is measured. (C) *MSC CM was significantly different from both EV and control groups (p < 0.005), + the ANG1-treated cells had thicker VE-Cadherin staining than all other group (p < 0.0001) via one-way ANOVA followed by post hoc Tukey test analysis (p < 0.05).
DISCUSSION
Our data demonstrate that HS-induced lung vascular permeability is significantly decreased by both MSC and MSC EV infusion in our mouse model of HS+ laparotomy. Our previous studies, have shown that MSCs decrease endothelial permeability in multiple preclinical models of traumatic injury including TBI and HS-induced acute lung injury.11,16,30,31 These effects were found to be dependent on increased VE-cadherin expression and enhanced beta-catenin/VE-cadherin interactions at the EC surface.31 Adherens junction proteins are linked to the actin cytoskeleton and therefore regulated by pathways that control it, such as the Rho Kinase pathway.32 Our data from the lung phosphoprotein profiling screen suggest that the major early pathways activated in HS lungs and modulated by MSCs and MSC EVs are related to cytoskeletal signaling pathways that regulate EC permeability, cell motility, cell proliferation, and cell contraction. More specifically, the protein profile data implicated several members of the RhoA GTPase pathway, which is mechanistically closely linked to cell contraction and EC permeability (Fig. 5).32,33 Indeed, in the Rho pathway, Rho kinase (ROCK) has been shown to be activated in sepsis-induced lung injury,34 however the role of this pathway in HS-induced lung injury has not been elucidated. In animal models of sepsis, inhibition of the pathway via the ROCK inhibitor Y-27632, attenuated the infiltration of inflammatory cells and leakage of proteins into the bronchoalveolar lavage fluid.34 Our data suggest that the Rho Kinase pathway is involved in vivo in modulating the effects of both the MSCs and MSC EVs as noted by the GLISA results (Fig. 4), demonstrating decreased RhoA activity in lungs treated with both MSC EVs and MSCs.
Interestingly, in in vitro HLMVECs, we find that MSC EVs increased stress fiber formation, did not change adherens junctions and failed to suppress EC permeability induced by thrombin as noted by the ECIS impedance assay results (Figs. 5B and 6). These data demonstrate divergent effects of the MSC EVs in vitro and in vivo. One potential explanation for this finding is that there are other cells that modulate EC permeability that are potentially affected by MSC EVs in the lungs, such as pulmonary epithelial cells, inflammatory cells, smooth muscle cells, monocytes, and macrophages. This milieu of cells is not reproduced in vitro in the HLMVEC cultures, which may be the reason for the divergent effects.
In addition, our GLISA data on HS lungs demonstrate that the Rac1 pathway, which is typically closely linked to RhoA signaling, do not seem to be involved in the effects of the MSCs and MSC EVs on lung permeability. It is possible that the Rac1 pathway is separate from RhoA-mediated effects on permeability in the case of MSCs and MSC EVs. Divergent effects on these two pathways have been reported in the case of chemotaxis, where RhoA is involved independently of Rac1.35
It is of interest to note in the phosphoprotein profiles of treated HS lungs (Figs. 2 and 3) and in vitro studies suggest that there are differences between the molecular effects of MSCs and MSC EVs. These differences indicate that the two therapies potentially overlap in some of their therapeutic effects; however, they are not mechanistically equivalent as many groups have suggested.13,19,23 It is feasible that there are particular clinical applications that MSCs or MSC EVs may be optimally suited for and further studies are warranted.
The primary limitation of the study is that lung tissue obtained at the time of sacrifice, 3 hours post-HS, may be too late to capture the initial early effects of the treatments such as MSC-mediated effects on Rac 1. Further investigation of timing and dose studies on MSCs and MSC EVs are warranted both in vivo and in vitro models of vascular compromise. This also raises the question of whether the biological effects of MSCs and MSC EVs are sustained in vivo and for how long. Our past studies have demonstrated that MSCs can be detected in the lungs up to 72 hours after treatment, with lasting biological effects that can be detected weeks after treatment.36 Similar studies are warranted for MSC EVs, which may provide sustained and different therapeutic effects through delivery of RNA, DNA and nucleic acid content to recipient cells.
The clinical therapeutic potential of MSCs in ARDS is still unknown. Recently a phase 1 dose escalation and Phase 2a randomized controlled trial of bone marrow-derived human MSCs were completed for non–trauma-associated ARDS.37 No safety issues have been reported in these trials. It is still unknown if MSCs will be safe and efficacious in trauma-induced ARDS. To begin answering this question, a multicenter Department of Defense–funded randomized placebo controlled trial for MSCs in trauma-related ARDS is planned to start in 2018.38 Interestingly, EVs from other cell types have also been tested in clinical applications, such as cancer; however, none have involved MSC EVs.39 Whether cellular and EV-based therapies are truly equivalent warrants further investigation.
Taken together, our data demonstrate that MSCs and MSC EVs modulate some components of the EOT in HS.24,40 Although our in vivo data and studies by other groups support the premise that MSC EVs may potentially be used in lieu of MSCs as a novel “stem cell free” therapy to treat HS-induced lung vascular permeability, it is possible that the mechanisms of action are similar but not necessarily equivalent too MSCs as noted by the divergence in our in vitro and in vivo findings. Interestingly, targeting cytoskeletal changes may be a viable therapeutic target for novel therapies to treat trauma-induced organ failure.
Supplementary Material
FUNDING
This work was supported by NIH grant: 7R01GM111899-03.
DISCUSSION
Dr. David H. Livingston (Newark, New Jersey): Dr. Pati, President Coimbra, Recorder Spain, members and guests. I really want to thank the AAST for the opportunity to discuss this really very elegant and very complicated cutting-edge study.
For those of you in the audience whose eyelids go down when the gene maps go up, I think you may need to actually read this one and pay attention when it gets published. If you didn’t get enough coffee or fogged out during the presentation I’ll try to put it in a little bit of perspective.
Clearly, over the past decades we’ve made great strides in resuscitation and our ability to control hemorrhage and overall critical care management. We are saving many more patients now than we did when I trained in the “dark ages.” But organ failure, persistent inflammation, immune dysfunction, infection, still is a very vexing problem. And while it may lead to fewer deaths, it leads to a lot of disability in our surviving patients. These patients really come out of the ICU half the people they used to be with many lifelong changes.
Searching for and identifying mechanisms and solutions to this problem really has been our Holy Grail of this. In the 1990s it was not only the decade of rap and grunge rock but it was the decade of cytokines. We tried to identify and block a whole bunch of them. None of them worked. The mechanisms were probably too distal in the immune cascade and we had numerous and sometimes spectacular failures of very promising experimental therapies. It was here that big pharma bet big and lost bigger and now they’re risk-adverse and they’re not really in the game very much. As newer models and molecular techniques evolved we got more proximal, hoping to sort of find our way through this.
In addition, totally blocking a biologic system may not be exactly what we really want. We, as surgeons, know it’s much easier to cut something out than to adjust something to make is just right. Now entered this era of cell-based therapy. As Dr. Pati alluded to in an off-handed thankful way, the Bush ban on embryonal stem cells made us very clever. We now have moved into, really, the MSCs or mesenchymal stem cells have now emerged as the next “sliced bread.”
As she also mentioned, a quick internet search demonstrates about 500 clinical studies treating a myriad of diseases, really varied diseases; and these are clinical studies, not pigs, not mice, not rats.
But one problem with cellular products is they’re still cells. Makes some people nervous. It’s really hard to commercialize them to give them right off the shelf or in the battlefield. In the classic story of one man’s trash is another man’s treasure, the detritus which we used to spin and throw away, have now emerged as biologically important, these microvesicles, exosomes, nanoparticles.
In a recent study by our president, Dr. Coimbra, it was likely that the exosomes are real bad actors in all those mesenteric lymph papers that we had to listen to; not proteins, not lipids but the exosomes. Now we may have to give exosomes from other cells to counteract the bad exosomes.
So are we done? No, hardly. Despite the excellent presentation of this elegant work, it really opens up a lot of questions. I’m not going to recapitulate her data, just ask a couple of questions.
I want to congratulate you on the attempt to characterize the extracellular vesicles. But they are still a really pretty “dirty” group of reagents. There is a lot of different kind of bits in that. It’s kind of like granola. There are nuts; there are raisins; there are craisins; there is everything in it. So can you maybe hone it down a little bit. What do you think is driving your results in the “soup?”
In addition, some of your data demonstrates that the whole cells are much better than the extracellular vesicles. But do you think the extracellular vesicles are good enough?
Most people use bone marrow-derived MSCs. I’ve used bone marrow-derived MSCs in my own studies. But as you say, they tend to go right to lung and maybe they are better on preventing ARDS than they are, for example, AKI. Have you tried similar studies with endothelial, adipose-derived MSCs? Do you think a cocktail of MSCs or MSC vesicles are really going to eventually be the way to go in multisystem trauma?
The other thing is although your model is fairly standard if you put those little mice back into their cages, they all survive. Our patients don’t do that. IACUC won’t allow us to treat mice and rats as badly as we treat our patients so is your model really clinically-relevant?
Lastly, the only molecular question I have for you is you spent a lot of time in the paper talking about TIMP3 but your TIMP3 data failed here. So is it the cell and type different? Is it really that finicky? Is the pathway really biologically important? How do you reconcile your previous work with this new stuff?
Once again, I want to thank the AAST for the privilege of discussing this really elegant and ground-breaking work.
Dr. Mitchell J. Cohen (Denver, Colorado): Shibani, beautiful work. I’m interested in your thoughts about the actions of cells versus the microparticles or conditinoned media.
It seems the idea of going after microparticles or something “in the soup” seems antithetical to the whole proposed benefit of using cells which is they should go to the area of injury and do what they know how to do and modulate their actions as needed locally.
And it seems a little reductionist, like we’re going back toward the idea of individually blocking cytokines which failed spectacularily in the past. I know we have talked about that in the past but I’d like your current thinking on this, please.
Dr. Matthew C. Hernandez (Rochester, Minnesota): Matt Hernandez, Mayo Clinic. So was it the duration of hypotension or the degree of hypotension that influenced your results? And then how close does this lead to a liquid biopsy for patients?
Dr. Shibani Pati (San Francisco, California): Thank you very much for those very thoughtful questions. So the question that Dr. Livingston posed about whether this is a “dirty” agent, and have we attempted to isolate which component of the extra-cellular vesicles is active, is a good question and is still unknown. To be honest in many ways a cell therapy is a “dirty agent” and we don’t know exactly what it is secreting in what quantities and how these factors synergize or antagonize each other. About 90 percent of the EV fraction of MSCs we tested are exosomes. We are in the process of characterizing the content of the EVs by RNA deep sequencing and also proteomics based studies. The work of others on MSCs EVs has demonstrated that about 50% of the content remains the same between MSC donors but there is variability on the remaining 50%. This is an area of active investigation.
The second question that you asked – and this is something that pertains to what Mitch also asked relates to whether the EVs are equivalent to the MSCs and can we substitute EVs for the cells? Our data suggests that the MSC EVs do indeed recapitulate a number of the activities of the MSCs in HS induced lung injury, but is appears that the mechanisms of action may differ based on our divergence in findings in vitro versus in vivo. The full spectrum of therapeutic benefits conferred by MSCs is likely not fully recapitulated by EVs. This is actually the focus of a second paper we are working on currently.
In terms of MSC EV activity in other models of disease, there have been a number of studies, in multiple models of disease that demonstrate the effectiveness of MSC EVs in comparison to MSCs. One interesting study conducted by Darwin Prockop’s group in a mouse model TBI, showed clearly that the MSC exosomal component is the most active component in attenuating brain injury and neurocognitive dysfunction and is comparable to the parent MSCs themselves. Logistically, there are a number of reasons why you would want to use EVs in lieu of cells. Cells come with issues with storage, cell viability, potential immune rejection and tumorigenicity that are circumvented by EVs. But the truth is that EVs are not actually the cells themselves. They only contain a very small fraction and participate in a fraction of the activities the cells do after infusion. EVs are self-limiting. It’s a one-shot therapy. Once they get into the cell they don’t replicate. I think there are going to be very specific applications for their extracellular vesicles, perhaps as an early bridge to prevent organ failure before a patient can actually receive cells in hospital such as for far forward care in the military or in remote and austere locations in civilian settings.
Can you make them more cell-like is another question that Dr. Livingston asked. Interestingly, yes, you can. What we’ve been able to do is to actually gene modify the cells that are producing the extracellular vesicles. We can transfect genes of interest into the MSCs and they then subsequently “package” the RNA and proteins of interest which then are transmitted/unloaded into to cells they come in contact with. So you pretty much can deliver nucleic acid, proteins and lipids of choice to other cells by using EVs and reconstitute some of the factors that would typically be delivered by the cells themselves. EVs in many ways are a vehicle for nature’s own gene therapy.
In terms of the question about sources of MSCs, there are multiple sources. And, no, they are not the same. So the attractive thing that you will hear in the literature is taking somebody’s fat and turning them into MSCs and then treating that person with autologous cells. Mind you, those adipose-derived MSCs are not equivalent to bone marrow-derived MSCs. They have different phenotypic and therapeutic effects.
Andre Cap’s group at the Army ISR has actually looked at the coagulation potential/contribution of two different kinds of MSCs, bone marrow versus adipose tissue. The adipose tissue MSCs were actually pro-coagulant and the bone marrow tissue MSCs were not.
An MSC from one tissue is not an MSC from another tissue, hence indicating that these cells will likely be tailored to the particular application they are intended to treat.
Footnotes
This was a podium talk presented at the 76th annual meeting of AAST on September 13, 2017 in Baltimore, Maryland.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal’s Web site (www.jtrauma.com).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353(16):1685–1693. [DOI] [PubMed] [Google Scholar]
- 2.Park PK, Cannon JW, Ye W, Blackbourne LH, Holcomb JB, Beninati W, Napolitano LM. Transfusion strategies and development of acute respiratory distress syndrome in combat casualty care. J Trauma Acute Care Surg 2013; 75(2 Suppl 2):S238–S246. [DOI] [PubMed] [Google Scholar]
- 3.Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, del Junco DJ, Brasel KJ, Bulger EM, Callcut RA, et al. Transfusion of Plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313(5):471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyeniyi BT, Fox EE, Scerbo M, Tomasek JS, Wade CE, Holcomb JB. Trends in 1029 trauma deaths at a level 1 trauma center: impact of a bleeding control bundle of care. Injury 2017;48(1):5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pati S, Pilia M, Grimsley JM, Karanikas AT, Oyeniyi B, Holcomb JB, Cap AP, Rasmussen TE. Cellular therapies in trauma and critical care medicine: forging new frontiers. Shock 2015;44(6):505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holcomb JB, Fox EE, Wade CE. PROPPR Study Group. Mortality and ratio of blood products used in patients with severe trauma—reply. JAMA 2015; 313(20):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannon JW, Khan MA, Raja AS, Cohen MJ, Como JJ, Cotton BA, Dubose JJ, Fox EE, Inaba K, Rodriguez CJ, et al. Damage control resuscitation in patients with severe traumatic hemorrhage: a practice management guideline from the Eastern Association for the Surgery of Trauma. J Trauma Acute Care Surg 2017;82:605–617. [DOI] [PubMed] [Google Scholar]
- 8.Matthay MA, Pati S, Lee JW. Concise review: mesenchymal stem (stromal) cells: biology and preclinical evidence for therapeutic potential for organ dysfunction following trauma or sepsis. Stem Cells 2017;35(2):316–324. [DOI] [PubMed] [Google Scholar]
- 9.Christy BA, Herzig MC, Montgomery RK, Delavan C, Bynum JA, Reddoch KM, Cap AP. Procoagulant activity of human mesenchymal stem cells. J Trauma Acute Care Surg 2017;83(Suppl 1):S164–S169. [DOI] [PubMed] [Google Scholar]
- 10.Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, Krasnodembskaya AD. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med 2017;196:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menge T, Zhao Y, Zhao J, Wataha K, Gerber M, Zhang J, Letourneau P, Redell J, Shen L, Wang J, et al. Mesenchymal stem cells regulate blood-brain barrier integrity through TIMP3 release after traumatic brain injury. Sci Transl Med 2012;4(161):161ra150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibb SL, Zhao Y, Potter D, Hylin MJ, Bruhn R, Baimukanova G, Zhao J, Xue H, Abdel-Mohsen M, Pillai SK, et al. TIMP3 attenuates the loss of neural stem cells, mature neurons and neurocognitive dysfunction in traumatic brain injury. Stem Cells 2015;33(12):3530–3544. [DOI] [PubMed] [Google Scholar]
- 13.Monsel A, Zhu Y-G, Gennai S, Hao Q, Hu S, Rouby J-J, Rosenzwajg M, Matthay MA, Lee JW. Therapeutic effects of human mesenchymal stem cell-derived microvesicles in severe pneumonia in mice. Am J Respir Crit Care Med 2015;192(3):324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mordant P, Nakajima D, Kalaf R, Iskender I, Maahs L, Behrens P, Coutinho R, Iyer RK, Davies JE, Cypel M, et al. Mesenchymal stem cell treatment is associated with decreased perfusate concentration of interleukin-8 during ex vivo perfusion of donor lungs after 18-hour preservation. J Heart Lung Transplant 2016;35(10):1245–1254. [DOI] [PubMed] [Google Scholar]
- 15.Asmussen S, Ito H, Traber DL, Lee JW, Cox RA, Hawkins HK, McAuley DF, McKenna DH, Traber LD, Zhuo H, et al. Human mesenchymal stem cells reduce the severity of acute lung injury in a sheep model of bacterial pneumonia. Thorax 2014;69(9):819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pati S, Gerber MH, Menge TD, Wataha KA, Zhao Y, Baumgartner JA, Zhao J, Letourneau PA, Huby MP, Baer LA, et al. Bone marrow derived mesenchymal stem cells inhibit inflammation and preserve vascular endothelial integrity in the lungs after hemorrhagic shock. Schulz C, editor. PLoS ONE 2011; 6(9):e25171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lener T, Gimona M, Aigner L, Börger V, Buzas E, Camussi G, Chaput N, Chatterjee D, Court FA, del Portillo HA, et al. Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J Extracell Vesicles 2015;4(1):30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, Camussi G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant 2011;26(5):1474–1483. [DOI] [PubMed] [Google Scholar]
- 19.Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, Sordi A, Biancone L, Tetta C, Camussi G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int 2012;82(4):412–427. [DOI] [PubMed] [Google Scholar]
- 20.Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol 2009;20(5):1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gennai S, Monsel A, Hao Q, Park J, Matthay MA, Lee JW. Microvesicles derived from human mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. Am J Transplant 2015;15(9):2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson CL, Soeder Y, Dahlke MH. Concise review: mesenchymal stromal cell-based approaches for the treatment of acute respiratory distress and sepsis syndromes. Stem Cells Transl Med 2017;6(4):1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y-G, Feng X-M, Abbott J, Fang X-H, Hao Q, Monsel A, Qu J-M, Matthay MA, Lee JW. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells 2014;32(1):116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pati S, Potter DR, Baimukanova G, Farrel DH, Holcomb JB, Schreiber MA. Modulating the endotheliopathy of trauma: Factor concentrate versus fresh frozen plasma. J Trauma Acute Care Surg 2016;80(4):576–84; discussion 584–5. [DOI] [PubMed] [Google Scholar]
- 25.Potter DR, Baimukanova G, Keating SM, Deng X, Chu JA, Gibb SL, Peng Z, Muench MO, Fomin ME, Spinella PC, et al. Fresh frozen plasma and spray-dried plasma mitigate pulmonary vascular permeability and inflammation in hemorrhagic shock. J Trauma Acute Care Surg 2015;78:S7–S17. [DOI] [PubMed] [Google Scholar]
- 26.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45(D1):D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baimukanova G, Miyazawa B, Potter DR, Muench MO, Bruhn R, Gibb SL, Spinella PC, Cap AP, Cohen MJ, Pati S. Platelets regulate vascular endothelial stability: assessing the storage lesion and donor variability of apheresis platelets. Transfusion 2016;56(S1):S65–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baimukanova G, Miyazawa B, Potter DR, Gibb SL, Keating S, Danesh A, Beyer A, Dayter Y, Bruhn R, Muench MO, et al. The effects of 22°C and 4°C storage of platelets on vascular endothelial integrity and function. Transfusion 2016;56(S1):S52–S64. [DOI] [PubMed] [Google Scholar]
- 29.Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, Stolz DB, Watkins SC, Di YP, Leikauf GD, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Comms 2015;6:8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker PA, Harting MT, Jimenez F, Shah SK, Pati S, Dash PK, Cox CS. Direct intrathecal implantation of mesenchymal stromal cells leads to enhanced neuroprotection via an NFkappaB-mediated increase in interleukin-6 production. Stem Cells Dev 2010;19(6):867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menge T, Gerber M, Wataha K, Reid W, Guha S, Cox CS, Dash P, Reitz MS, Khakoo AY, Pati S. Human mesenchymal stem cells inhibit endothelial proliferation and angiogenesis via cell-cell contact through modulation of the VE-cadherin/β-catenin signaling pathway. Stem Cells Dev 2013;22:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ 2014;4(4):535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duluc L, Wojciak-Stothard B. Rho GTPases in the regulation of pulmonary vascular barrier function. Cell Tissue Res 2014;355(3):675–685. [DOI] [PubMed] [Google Scholar]
- 34.Cinel I, Ark M, Dellinger P, Karabacak T, Tamer L, Cinel L, Michael P, Hussein S, Parrillo JE, Kumar A, et al. Involvement of Rho kinase (ROCK) in sepsis-induced acute lung injury. J Thorac Dis 2012;4(1):30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, Roccaro AM, Ngo HT, Melhem MR, Sacco A, et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood 2009; 114(3):619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, Nguyen AT, Malide D, Combs CA, Hall G, et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med 2006;203(5):1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, Cosgrove K, Vojnik R, Calfee CS, Lee JW, et al. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med 2015; 3(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pati S, Rasmussen TE. Cellular therapies in trauma and critical care medicine: looking towards the future. PLoS Med 2017;14(7). e1002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andaloussi SE, Mäger I, Breakefield XO, Wood MJA. Extracellular vesicles: biology and emerging therapeutic opportunities. Nature Publishing Group 2013;12(5):347–357. [DOI] [PubMed] [Google Scholar]
- 40.Holcomb JB, Pati S. Optimal trauma resuscitation with plasma as the primary resuscitative fluid: the surgeon’s perspective. Hematology Am Soc Hematol Educ Program 2013;2013(1):656–659. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.