

Abstract
Catecholamine neurotransmitter levels in the synapses of the brain shape human disposition—cognitive flexibility, aggression, depression, and reward seeking—and manipulating these levels is a major objective of the pharmaceutical industry. Certain neurotransmitters are extensively sulfonated and inactivated by human sulfotransferase 1A3 (SULT1A3). To our knowledge, sulfonation as a therapeutic means of regulating transmitter activity has not been explored. Here, we describe the discovery of a SULT1A3 allosteric site that can be used to inhibit the enzyme. The structure of the new site is determined using spin-label-triangulation NMR. The site forms a cleft at the edge of a conserved ∼30-residue active-site cap that must open and close during the catalytic cycle. Allosteres anchor into the site via π-stacking interactions with two residues that sandwich the planar core of the allostere and inhibit the enzyme through cap-stabilizing interactions with substituents attached to the core. Changes in cap free energy were calculated ab initio as a function of core substituents and used to design and synthesize a series of inhibitors intended to progressively stabilize the cap and slow turnover. The inhibitors bound tightly (34 nm to 7.4 μm) and exhibited progressive inhibition. The cap-stabilizing effects of the inhibitors were experimentally determined and agreed remarkably well with the theoretical predictions. These studies establish a reliable heuristic for the design of SULT1A3 allosteric inhibitors and demonstrate that the free-energy changes of a small, dynamic loop that is critical for SULT substrate selection and turnover can be calculated accurately.
Keywords: sulfotransferase, allosteric regulation, enzyme inhibitor, enzyme structure, neurotransmitter, catecholamine, dopamine, nuclear magnetic resonance (NMR), enzyme mechanism, enzyme kinetics, epinephrine, norepinephrine, serotonin, spin label, SULT1A3, allostery, mechanism, inhibition
Introduction
It is estimated that 1 in 5 individuals worldwide will suffer from major depression, a depressive episode lasting at least 2 weeks (1). Suicide is the tenth leading cause of death in the United States and occurs ∼20-fold more frequently during major depression episodes (2). Major depression is treated primarily with serotonin-reuptake and monoamine oxidase inhibitors (3), which increase synaptic levels of serotonin. More than 40% of patients fail to respond when taking these antidepressants alone but can respond well when they are taken in combination (4–6). The serotonin in brain micro-dialysates from living humans is nearly 100% oxidized by monoamine oxidase and ∼80% sulfonated (7). Thus, preventing sulfotransferase (SULT)3 activity in a monoamine oxidase–inhibited background can be expected to substantially enhance serotonin activity. Similarly compelling disease-relevant scenarios surround the sulfonation of other catecholamines, yet, to our knowledge, regulating sulfonation remains largely unexplored as a therapeutic strategy.
SULT1A3 shows a strong preference over other human SULT isoforms for catecholamine substrates (8, 9). The enzyme is unique to higher primates (10) and found at high levels in gut, platelets, and brain (11–13). Sulfonation prevents catecholamines from binding their receptors (14) and enhances their transport properties (15, 16), which decreases their terminal half-lives (17). SULT1A3 harbors two allosteric sites through which it communicates with catecholamine biosynthetic pathway metabolites (18, 19). While studying allosteres that bind these sites, we discovered an allostere that did not bind either site but inhibited with high affinity and isoform specificity. Here, we present the structure of that allostere bound to SULT1A3 and demonstrate that the allostere slows turnover by stabilizing the enzyme's active-site cap, a conserved stretch of ∼30 residues that is intimately involved in SULT substrate selection and turnover. A computational method that predicts the effects of inhibitors on cap stabilization is developed and used to design inhibitors with varying degrees of stabilization. The inhibitors were synthesized. These are the first man-made, isoform-specific allosteric SULT inhibitors; they bound tightly, and their inhibition characteristics correlated remarkably well with the predicted values. Thus, the behavior of this small, catalytically important active-site cap is predictable and can be reliably incorporated into SULT1A3 inhibitor designs.
Results and discussion
Discovering the site
SULTs harbor two physically distinct, noninteracting allosteric binding sites, the so-called catechin- and NSAID-binding sites (18, 21). The residues that line these sites differ in each SULT isoform, which begs the hypothesis that the sites evolved to enable isoforms to communicate with metabolites within their metabolic domains. This hypothesis was born out using the SULT1A3 isoform (18).
While testing the initial rate inhibition characteristics of compounds designed to inhibit SULT1A3 by binding at its catechin site, a high-affinity, high-specificity inhibitor that did not appear to bind either of the known sites was discovered: compound 8 (CMP8). The potency and specificity of CMP8 are demonstrated in Fig. 1. The Ki of CMP8 for SULT1A3 is 34 nm, and CMP8 does not detectably inhibit the other major SULT isoforms found in the liver and brain at concentrations as high as 2 μm. CMP8 proved to be a partial inhibitor; at saturation, its binding reduces SULT1A3 turnover to 46% of that in the absence of inhibitor. Partial inhibitors alleviate inhibition when used competitively against total inhibitors that bind at the same site. CMP8 did not alleviate inhibition by epigallochatechin gallate or mefenamic acid, which are near-total inhibitors that bind the catechin and NSAID sites, respectively (19, 20). Given these favorable characteristics, we reasoned that the CMP8-binding site offered a new target that could be used to control 1A3 activity. Consequently, the solution structure of the CMP8-binding site was determined using spin-label-triangulation NMR.
Figure 1.
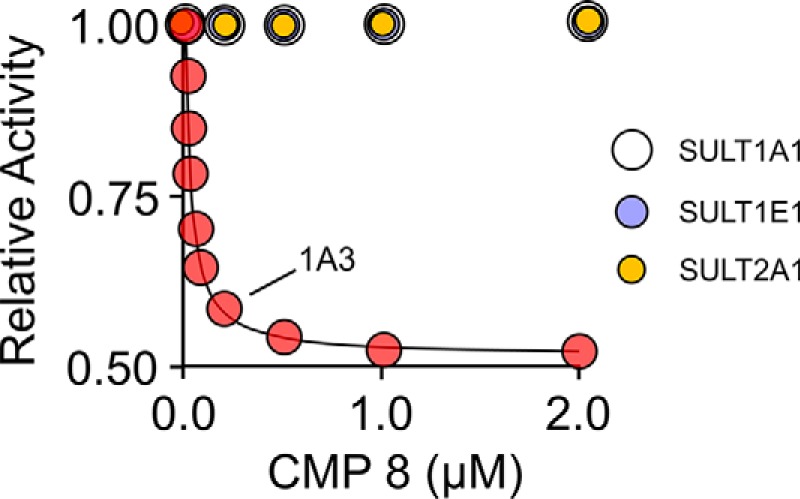
Inhibition of SULT isoforms by CMP8. The initial rates of SULT catalyzed 1-HP sulfonation are plotted as a function of inhibitor concentration and are normalized relative to [inhibitor] = 0. Enzyme activity was monitored by the sulfonation dependent change in 1-HP fluorescence (λex = 325 nm, λem = 370 nm). Less than 5% of the concentration-limiting substrate converted at the reaction end point was consumed during initial rate measurements. Each point is the average of three independent determinations. Conditions were as follows: SULT (20 nm, active sites), PAPS (0.50 mm, 17 × Km), 1-HP (5.0 μm, 61 × Km), KPO4 (50 mm), pH 7.5, 25 ± 2 °C.
Structure determination
We have developed a method (spin-label-triangulation NMR) that allows the solution structure of a SULT ligand-binding pocket to be determined without the use of high-resolution NMR or crystallography and regardless of the molecular mass of the protein (18–20). The method relies on the distance-dependent line broadening of peaks in the ligand's solution one-dimensional NMR spectrum, which occurs when a ligand docks within ∼25 Å of a spin label covalently attached to the protein. Using three spatially well-separated spin labels, it is possible to triangulate the position of each ligand proton with respect to the protein surface. Spin label insertion sites are identified by first screening for line-broadening effects using a previously defined set of six sites that when spin-labeled coat the entire surface of the SULT dimer in a paramagnetic field of sufficient strength to broaden the NMR line width of the ligand regardless of where it binds (18). Based on the findings of the screen, the spin label insertion sites can then be optimized as needed (20). It is critical that the initial rate and inhibition parameters of the spin-labeled protein closely match those of the native protein (see Table S1). Having positioned ligand protons on the protein surface, the structure is then refined using NMR distance-constrained molecular dynamics docking.
Distance measurements
The distance dependence of the magnetic interactions between an unpaired electron and a nuclear spin are well-understood (21–23). Given the effect of an unpaired electron spin on the line width of a ligand nucleus (R2), interspin distances can be calculated using the Solomon–Bloembergen equation (21). The 1H NMR line widths of protein-bound ligands are typically too broad to measure directly; however, the line-broadening effect of the electron can be obtained from a ligand's solution spectrum if the frequency of ligand exchange between bulk solution and the protein is comparable to or greater than the difference in the resonant frequencies of the free and bound ligand (21–23). In such cases, solution line widths provide observed transverse relaxation rates, R2obs, that depend linearly on the fraction of bound ligand, FB, according to Equation 1,
| (Eq. 1) |
where R2B and R2F are the transverse relaxation rates for bound and free protons, and R2ex is the chemical exchange contribution to the relaxation (21).
R2obs contains contributions from both protein nuclei and the unpaired-electron spins (18–20, 23). To isolate the contribution of the electron, a diamagnetic control is created for each spin-labeled construct in which the paramagnetic (PROXYL) moiety of the spin label is replaced with a diamagnetic homologue (i.e. a cyclohexyl moiety). The contribution of the unpaired electron is obtained by subtracting the slopes of the paramagnetic and diamagnetic R2obs versus FB plots.
A representative study characterizing the interaction of the C5 proton of CMP8 with the three spin-labeled constructs and diamagnetic control is presented in Fig. 2. The study was performed at saturating concentrations of CMP8 and PAP (see the legend to Fig. 2). The structure of CMP8 and its associated 1H NMR spectrum and peak assignments are shown in A. The line broadening of the C5 peak as a function of the percentage of bound CMP8 is shown in B. The construct used in the panel B study was spin-labeled at position 234. The line widths obtained from the spectra in B are plotted versus fraction-CMP8-bound in C (red dots). Similar studies were performed for the remaining two spin-labeled constructs (positions 198 and 116) and diamagnetic control (CTRL). As discussed above, the slope associated with the diamagnetic control was subtracted from those of the spin-labeled constructs to obtain the contribution of the spin label unpaired electron to the line broadening of the C5 proton, which was then used to calculate distances. Similar studies were performed on the C3, C3′, C4, C7, and C8 protons (see Fig. S1). The inter-spin distances from each proton to each of the three spin labels are compiled, along with the measurement errors, in Table S2.
Figure 2.
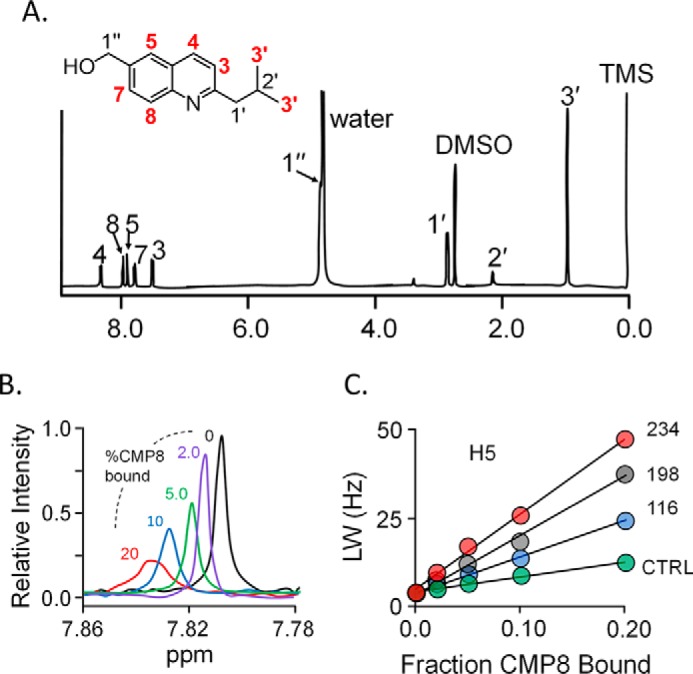
The NMR measurements. A, structure and 600-MHz 1H NMR spectrum of CMP8. The protons of CMP8 are labeled in the spectrum and structure. Red labels identify the proton positions used in NMR distance measurements. Conditions were as follows: CMP8 (1.9 mm), DMSO (0.50 mm), TMS (0.50 mm), D2O (>95%), 25 ± 1 °C. B, spin label effects on the H5 proton peak of CMP8. The solution 1H NMR spectrum (600 MHz) of the H5 peak of CMP8 is shown as a function of percentage of CMP8 bound to spin-labeled Cys-234-SULT1A3. Peak amplitudes were normalized to reflect 1.0 mm CMP8. Conditions were as follows: CMP8 (1.0 mm (purple), 400 μm (green), 200 μm (blue), 100 μm (red)), spin-labeled Cys-234-SULT1A3 (20 μm, active site), PAP (500 μm, 17 × Kd), KPO4 (50 mm), pD 7.4, 25 ± 1 °C. Conditions associated with the black and purple peaks were identical except that the black spectrum conditions lacked enzyme. CMP8 is saturating at all concentrations (i.e. ≥2300 Kd). C, line width versus fraction of inhibitor bound. The effects of paramagnetic spin labels (4-maleimido-PROXYL attached at Cys-234 (red), Cys-198 (black), or Cys-116 (blue)) and diamagnetic control (N-cyclohexylmaleimide attached at Cys-234 (green)) on the line width of the H5 proton peak are plotted as a function of fraction of enzyme-bound CMP8. Fractions of CMP8 bound were 0.02, 0.05, 0.10, and 0.20. Conditions were as described in B. Diamagnetic controls for Cys-198 and Cys-116 were indistinguishable from the Cys-234.
NMR distance–constrained molecular dynamics docking
Each NMR-determined distance represents a vector between the time-averaged position of the spin label oxygen (calculated using GROMACS) and the given proton. Three such vectors are associated with each proton, each originates at the oxygen of a different spin label, and all three intersect at the proton. The errors associated with these three measurements constitute an error ellipsoid that centers on the proton and whose principal axis magnitudes are given by the S.E. values (±1 σ) of the NMR measurements. Docking is constrained by applying a 50-kJ mol−1 Å−1 restoring force (using distance_restraints, GROMACS (24, 25)) that drives the proton toward the ellipsoid center if any part of its van der Waals surface lies outside the ellipsoid; the restoring force inside the ellipsoid is zero. As is appropriate for NMR spin–spin interaction measurements (21, 26, 27), distance_restraints was parameterized to utilize time-averaged (1/r6) weighted restraints. The motions of all six CMP8 protons (H3, H3′, H4, H5, H7, and H8) were constrained simultaneously during docking. Docking was repeated 100 times; identical structures were achieved 92 times, and the structures did not change once the distance constraints were removed.
The refined structure
The energy-minimized, NMR distance–constrained structure of the CMP8·E·PAPS·dopamine complex is presented in Fig. 3 (A and B) and is available at www.modelarchive.org,4 archive number ma-qtj80. Fig. 3A provides an at-a-distance view of the complex and reveals that CMP8 binds in a crux of the protein where the cap helix connects to the base. Whereas the majority of residues that are in direct contact with CMP8 reside in the base, CMP8 directly contacts the cap at residue 235. The residues linearly aligned along the left-facing edge of the cap helix (Fig. 3A) either directly contact CMP8 or layer immediately over direct-contact residues; these interactions likely contribute to the CMP8-induced cap stabilization. Fig. 3B offers a more detailed view of the CMP8 interactions. The quininoid base of CMP8 is engaged in π-stacking interactions with planar residues (Phe-222 and His-226) on both of its faces. A hydrophobic interaction with Leu-67 anchors the CMP8 isobutyl moiety and is geometrically positioned such that CMP8 can hydrogen-bond with Gln-225.
Figure 3.
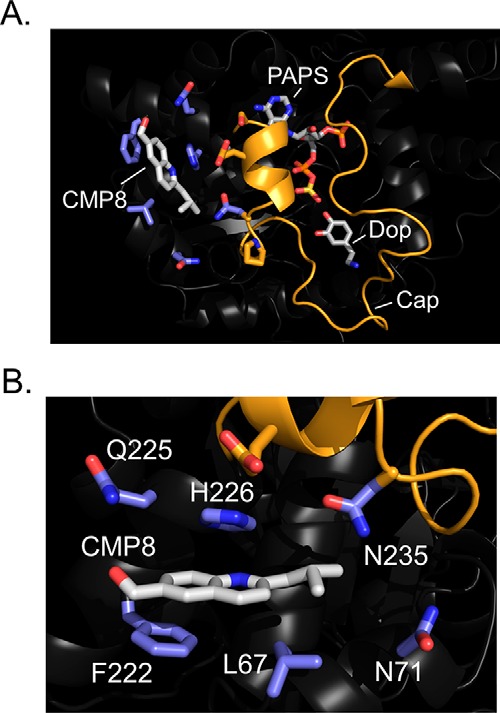
Structure of the CMP8·SULT1A3·PAPS·dopamine complex. A, structure at a distance. CMP8 is labeled and shown in white. Carbon atoms of residues in direct contact with CMP8 are blue. The cap (orange) is shown in the closed conformation and sitting above the substrates, PAPS and dopamine (Dop). B, CMP8-binding pocket. CMP8 is shown interacting with six direct-contact residues, shown in blue. The refined structure (shown) was generated by molecular dynamics energy minimization of the NMR distance–constrained structure (see “Results and discussion”).
Confirming the structure
The structure was validated by testing its ability to predict the consequences of mutations on the initial rate parameters of the enzyme. The effects of binding-site mutations on the initial rate parameters of SULT1A3 are compiled in Table 1. Consistent with the structure, replacing the isobutyl moiety of Leu-67 with a proton (L67G) results in a significant (5.3-fold) increase in KiCMP8, and replacing the planar moieties at positions 226 and 222 with methyl groups (F222A/H226A) causes a significantly greater (60-fold) increase in KiCMP8. With one exception (N71H), the SULT1A3 residues that directly contact CMP8 are identical to those of its evolutionary precursor, SULT1A1 (9). Reconstructing the SULT1A1 pocket in the SULT1A3 scaffold, by replacing Asn-71 with His caused KiCMP8 to increase beyond the detectible limit (i.e. no inhibition detected at 100 μm CMP8); thus, this single residue swap appears to determine the CMP8 specificity of the isoform. In conclusion, the mutagenesis data fully support the structure.
Table 1.
Initial rate parameters for WT and mutant SULT1A3
| Enzyme | Ki CMP8 | kcat inha | kcatb | Km 1-HP |
|---|---|---|---|---|
| μm | s−1 | s−1 | nm | |
| WT | 0.034 (0.003)c | 1.1 (0.1) | 2.1 (0.3) | 82 (2) |
| L67G | 0.18 (0.01) | 0.95 (0.05) | 1.9 (0.2) | 80 (1) |
| F222A H226A | 2.0 (0.2) | 1.0 (0.1) | 2.0 (0.2) | 85 (2) |
| N71H | NDd | ND | 2.0 (0.3) | 82 (3) |
a kcat inh, turnover at saturating inhibitor (20 × Ki).
b kcat, turnover at [inhibitor] = 0.
c Values in parentheses indicate one S.D. unit.
d ND, not determined.
It is notable that whereas the allosteric site mutations affect KiCMP8, they do not alter other initial rate parameters, including turnover at saturating inhibitor (kcat inh). Thus, the mutations weaken CMP8's affinity but do not affect the linkages that couple CMP8 binding to turnover; these linkages must lie elsewhere in the cavity.
The mechanism of inhibition
SULTs harbor an ∼30-residue active-site cap that must isomerize between open and closed states during the catalytic cycle (28–32). The cap isomerization equilibrium constant (Kiso) is defined as the ratio of the closed-to-open forms of the cap and is a function of bound ligand (29–33). Nucleotide binding causes the cap to move from a largely open (Kiso ≤ 0.1) to largely closed (Kiso ∼ 20) configuration. Cap stabilization by PAP or PAPS is virtually identical, and neither acceptors nor their sulfates detectably influence cap closure regardless of whether nucleotide is bound. Cap closure encapsulates the nucleotide, which can release only from a cap-open form (28, 29). For all isoforms studied thus far (18, 31–33), nucleotide release is rate-determining (34, 35), and the release rate depends on Kiso, which determines the fraction of enzyme-bound PAP poised for escape.
Given that CMP8 interacts with the SULT1A3 cap, we reasoned that it might inhibit by stabilizing the cap-closed form(s) of the enzyme. To test this hypothesis, the microscopic rate constants for nucleotide binding and release were determined in the presence and absence of saturating CMP8. The binding reactions were monitored via intrinsic fluorescence changes associated with ligand binding (30–32) (see “Methods”). The resulting kobs versus [PAP] plots are presented in Fig. 4. kon and koff are obtained from the slopes and intercepts of the plots, respectively (30, 31) (Table 2). As can be seen, saturation with CMP8 does not detectibly alter the slopes but decreases the intercepts by a factor of 0.41 ± 0.05. Thus, CMP8 does indeed slow nucleotide release by increasing Kiso and stabilizing the closed form of the cap. Notably, the effect of CMP8 on the ratio of the nucleotide off-rate constants (koff(+CMP8)/koff(−CMP8) = 0.41 ± 0.05) is comparable with its effect on the kcat ratio (kcat(+CMP8)/kcat(−CMP8) = 0.46 ± 0.08), a coincidence that strongly suggests that nucleotide release is rate-determining and that turnover is controlled by Kiso.
Figure 4.
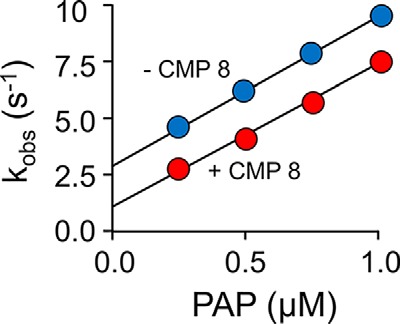
Mechanism of inhibition. The effect of CMP8 on PAP binding to SULT1A3 was monitored using a stopped-flow spectrofluorimeter (λex = 290 nm, λem ≥ 330 nm (cutoff filter)). Reactions were initiated by rapidly mixing (1:1, v/v) a solution containing SULT1A3 (25 nm, dimer), CMP8 (0 (blue) or 1.7 μm, 50 × Kd (red)), KPO4 (50 mm), pH 7.5, 25 ± 2 °C, with a solution that was identical except that it lacked SULT1A3 and contained PAP at twice the indicated concentrations. kobs values were obtained by fitting the average of 6–9 progress curves to a single exponential. Each kobs value was determined in triplicate, and the averaged values are shown. kon and koff are given, respectively, by the slopes and intercepts obtained from linear least-squares fitting of the kobs versus [PAP] plots.
Table 2.
Rate constants governing nucleotide binding to SULT1A3
| Nucleotide | Without CMP8 |
With CMP8 |
||
|---|---|---|---|---|
| kon | koff | kon | koff | |
| μm−1 s−1 | s−1 | μm−1 s−1 | s−1 | |
| PAP | 6.6 (0.2)a | 2.7 (0.1) | 6.3 (0.3) | 1.1 (0.1) |
a Values in parentheses indicate S.E.
Building better inhibitors
Given that 1A3 turnover is determined by the fraction of enzyme in the cap-open form, we reasoned that further inhibition could be achieved if CMP8 were modified in ways that enhance cap closure. Toward this end, CMP8 derivatives were designed via visual inspection of GROMACS-docked structures and computational assessments of cap stabilization. The compounds selected for synthesis and further studies are presented in Table 3 along with their experimentally determined Ki and percentage inhibition at saturation values. The synthesis and characterization of these new allosteric inhibitors are described in the supporting material.
Table 3.
Structures and characterization of allosteric inhibitors

akcat at saturating inhibitor is calculated as follows: 120 × [1 − (% inhibition at saturation/100)]. kcat in the absence of inhibitor is 120 min−1.
b ND, inhibition was not detected at 30 μm compound.
c Numbers in parentheses indicate one standard deviation unit.
To computationally assess the cap-stabilizing effects of the inhibitors, the free energies of the closed caps were calculated with and without inhibitor bound to the E·PAPS·dopamine complex. The cap free energy was calculated, using g_energy (24, 25, 37), by summing the Gibbs free energy of all atoms in the system (including those of solvent) and subtracting from it the free energy of all noncap atom interactions. The free energies are time-averaged values calculated using equilibrated models (see “Experimental procedures”) and are a measure of the free energy of interaction of the closed cap with its milieu. To ensure that the time intervals over which the free energies were calculated are sufficiently long to average the fluctuations that occur around equilibrium, the free energy was calculated for each complex over 1-, 2-, 4-, 6-, 8-, and 10-ns intervals (5 ps/frame), and the values were compared. The values agreed to within ±1% in all cases.
Inhibitor-induced changes in the free energy of the closed cap were both calculated and determined experimentally, and the values were correlated to assess the fitness of the cap-stabilization model for predicting the effects of inhibitors on turnover and further improving inhibitor design. The factor by which any two allosteric inhibitors differ in their extent of inhibition at saturation is given by the ratio of their Kiso values, which can be calculated from the Gibbs equation.
| (Eq. 2) |
Subscripts 1 and 2 refer to different enzyme forms (i.e. different inhibitors bound). The Kiso ratio can be obtained from ΔGcap0 values calculated as described above or from kcat at saturating inhibitor, as follows,
| (Eq. 3) |
where Etot represents total enzyme concentration, EO and EC represent concentrations of the cap-open cap-closed forms of the enzyme, respectively, and krel is the rate constant governing release of nucleotide from the cap-open enzyme. Substituting Equation 4 into Equation 3 and rearranging yields Equation 5,
| (Eq. 4) |
| (Eq. 5) |
which can be written for any two cap-closed enzyme forms. Assuming that inhibitors do not affect krel, any two such equations can be set equal to one another and rearranged to yield the following.
| (Eq. 6) |
The kcat values associated with inhibitor complexes were calculated from the data compiled in Table 3.
As is seen in Equation 6, Kiso and kcat ratios approach equivalence as Kiso becomes ≫1. To assess the extent to which this equivalence holds for SULT1A3, Kiso values were determined as described previously (29–33). In short, as nucleotide binds and the cap closes, a pore forms at the acceptor-binding site. The affinity of an acceptor that is too large to pass through the pore is diminished by the presence of nucleotide, which fosters cap closure. At saturating nucleotide, the apparent affinity, Kapp, of such acceptors for the fully cap-closed enzyme is given by Equation 7 (29–33), where Kd is the affinity for the cap-open (nucleotide-free) form of the enzyme.
| (Eq. 7) |
Kapp and Kd were obtained by fitting titrations that monitor changes in SULT1A3 intrinsic fluorescence as a function of the concentration of 4-hydroxytamoxifen, a large acceptor (29) (see Fig. S2). Titrations were performed at 0 and saturating PAP (0.50 mm, 17 × Kd). Kd and Kapp are 0.41 ± 0.03 and 5.4 ± 0.3 μm, respectively, and predict that Kiso = 13 ± 1.7. Notably, adding inhibitors will increase Kiso; consequently, the Equation 6 approximation should hold well for studies that use inhibitors.
Plotting the calculated and experimentally determined free energies against one another (Fig. 5) reveals that the data sets are highly linearly correlated (slope = 1.2 ± 0.1). Thus, the ab initio calculations are capable of reliably predicting small changes in the free energy of a dynamic, 30-residue loop of the enzyme that is intimately involved in SULT substrate selection and turnover. Given the method's reliability, GROMACS-docked structures of inhibitor-bound complexes should provide plausible molecular explanations for the structure–activity relationships seen in the data. For example, the length of the aliphatic spacer that connects the propyl moiety to the quinoline ring system markedly affects the inhibition, as is seen when comparing compound 1, compound 2, and CMP8, which are otherwise identical. Although Ki values for 1 and 2 could not be determined, because they do not inhibit, their Kd values, determined by fluorescence titration, are 160 ± 10 and 87 ± 6 nm, respectively. Thus, whereas all three compounds bind tightly, only one inhibits. Structures suggest that the key to the inhibition is optimizing hydrogen bonding to Gln-225, which is situated in a loop whose conformation seems to be coupled to that of the cap. Further, compound 11 is identical to CMP8 except for the substitution of the amido-group for the hydroxyl at C1″, which appears to engage in essentially perfect hydrogen bonding with Gln-255. Finally, relative to CMP8, compound 11 shifts toward Gln-255 and thus allows Asn-235 and Asn-71 to form optimal hydrogen-bonding interactions. These and related insights will be incorporated into future ligand designs.
Figure 5.
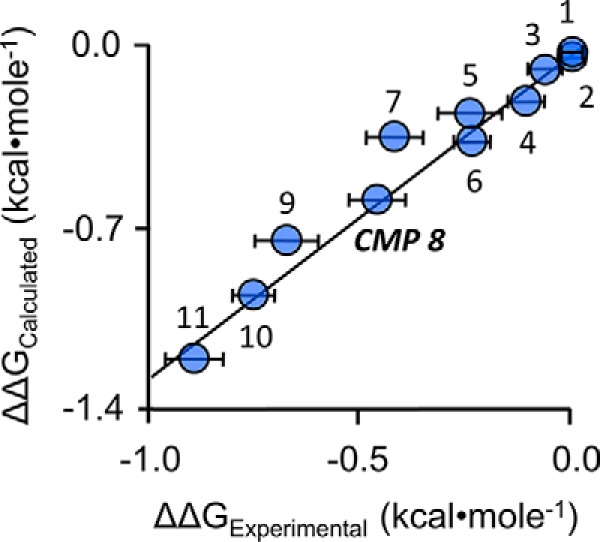
Free energy correlation plot. ΔΔG values were either calculated ab initio, using molecular dynamics simulations, or obtained from experimentally determined kcat values (see “Results and discussion”). The data show strong linear correlation, slope = 1.2 ± 0.1. Numbers associated with data points correspond to the compound numbering in Table 3. Error bars, ± 1 standard unit. Error in the calculated (y axis) values is vanishingly small.
Conclusions
An allosteric pocket has been discovered in SULT1A3. Given the pocket's potential to be used to regulate catecholamine neurotransmitter activity, the solution structure of the pocket and the mechanism of allosteric inhibition were determined. Allosteres inhibit by stabilizing the closed form of the SULT1A3 active-site cap, which must open to release nucleotide. The effects of inhibitors on the closed-cap free energy were calculated using a GROMACS-based method. The calculations were used to design a series of synthetically tractable inhibitors intended to both bind the pocket and stabilize the cap. The inhibitors were synthesized, and their SULT1A3 binding and inhibition characteristics were determined. All of the inhibitors bound well (34 nm to 7.4 μm), and the predictions regarding cap stabilization and percent inhibition at saturating allosteres (0–80%) proved remarkably accurate. These findings demonstrate that it is possible to accurately calculate small changes in the free energy of a dynamic stretch of ∼30 residues that is critically engaged in SULT substrate selection and turnover. Efforts under way will use these methods to develop even more effective allosteric inhibitors that will be used to explore the consequences of preventing catecholamine sulfonation in living systems.
Experimental procedures
Materials
Chemicals and media
The materials and sources used in this study are as follows: 5,5′-dithiobis-2-nitrobenzoic acid, DTT, EDTA, l-GSH (reduced), 1-hydroxypyrene (1-HP), imidazole, isopropyl-thio-β-d-galactopyranoside, lysozyme, 4-hydroxyltamoxifen, lysozyme, 3-maleimido-PROXYL, pepstatin A, and potassium phosphate were the highest grade available from Sigma. Ampicillin, KOH, lysogeny broth medium, MgCl2, NaCl, phenylmethylsulfonyl fluoride, and tetramethylsilane (TMS) were purchased from Fisher. GSH- and nickel-chelating resins were obtained from GE Healthcare. Competent Escherichia coli (BL21(DE3)) was purchased from Novagen. The syntheses and purification of PAPS and PAP were described previously (38), and the purity of the nucleotides was determined by anion-exchange HPLC to be ≥99%.
Computer and software
Molecular dynamics simulations were performed using a Parallel Quantum Solutions QS32-2670C-XS8 computer. PQS Molecular Builder was purchased from Parallel Quantum Solutions (39). The source code for GROningen MAchine for Chemical Simulation (GROMACS) version 4.5 was downloaded from http://www.GROMACS.org (24, 25)4 under the GROMCAS General Public License (GPL). Antechamber was acquired as part of AmberTools, under the GPL. A Genetically Optimized Ligand Docking (GOLD) license was obtained from the Cambridge Crystallographic Data Center.
Methods
SULT1A3 DNA constructs
WT and mutant SULT1A3 coding regions were inserted into a pGEX-6P expression vector that fuses a triple-tag (N-His/GST/MBP) PreScission protease–cleavable protein to the SULT1A3 N terminus (18). Mutated coding regions were constructed using PCR mutagenesis and confirmed by DNA sequencing.
SULT1A3 expression and purification
E. coli (BL21(DE3)) containing the human SULT1A3 expression plasmid were grown at 37 °C in lysogeny broth medium (18). At A600 ∼ 0.6, the culture was temperature-shifted to 17 °C in an ice-water bath. Upon reaching 17 °C, isopropyl-thio-β-d-galactopyranoside was added (0.30 mm), and the culture was incubated at 17 °C for 18 h. Cells were then pelleted and resuspended in lysis buffer (phenylmethylsulfonyl fluoride (290 μm), pepstatin A (1.5 μm), lysozyme (0.10 mg/ml), EDTA (2.0 mm), KCl (400 mm), K2PO4 (50 mm), pH 7.5). The suspension was sonicated and then centrifuged (10,000 × g, 1.0 h, 4 °C). MgCl2 (5.0 mm) was added to chelate EDTA before passing the solution through a chelating Sepharose Fast Flow column charged with Ni2+. The column was washed (imidazole (10 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5), and enzyme was eluted (imidazole (250 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5) and loaded directly onto a GSH-Sepharose column. The GST column was washed (DTT (2.0 mm), KCl (400 mm), and KPO4 (50 mm), pH 7.5) before eluting the tagged enzyme (reduced GSH (10 mm), DTT (2.0 mm), KCl (400 mm), and Tris (100 mm), pH 8.0). The fusion protein was digested overnight at 4 °C using PreScission Protease and passed through a GST column to remove the tag. The protein was ≥95% pure as judged by SDS-PAGE, and its concentration was determined by UV absorbance (ϵ280 = 53.9 mm−1 cm−1). The extinction coefficient was calculated using the ExPASy ProtParam tool (40). SULT1A3 concentrations determined by absorbance at 280 nm agreed with those obtained by the Bradford method (41). The pure protein was concentrated, flash-frozen, and stored at −80 °C.
Covalent tagging of SULT1A3 Cys constructs
Labeling of the SULT1A3 Cys constructs was performed as described previously (18–20). Briefly, 3-maleimido-PROXYL (spin label) or N-cyclohexylmaleimide (diamagnetic label) was added to an enzyme-containing solution at 20-fold excess over reactive Cys. At 3 h, and every hour thereafter until the reaction was complete, 50-μl aliquots from each reaction were tested for unreacted Cys using 5,5′-dithiobis-(2-nitrobenzoic acid) (18–20). The reactions were considered complete when >98% of the Cys was labeled. Reaction conditions were as follows: SULT1A3 (50 μm, monomer), 3-maleimido-PROXYL or N-cyclohexylmaleimide (1.0 mm), PAP (0.50 mm), KPO4 (50 mm), pH 7.5, 4 ± 2 °C. To prepare samples for NMR, reaction mixtures were dialyzed three times against 40 volumes of PAP (0.50 mm), KPO4 (50 mm), pD 7.4, D2O (> 95%), 4 ± 2 °C. Following dialysis, labeled enzyme was assayed to ensure that the initial-rate parameters (kcat, Km, and Ki) were not substantially altered by Cys insertion and labeling (see Table S1).
Allostere synthesis
The synthesis and characterization of the 11 allosteres compiled in Table 3 are described in the supporting material.
NMR studies
NMR experiments were performed using a Bruker 600-MHz spectrometer equipped with a TCI H/F-cryogenic probe at 298 K. CMP8 peak assignments were determined from one-dimensional proton and carbon spectra using 1H-13C heteronuclear single-quantum coherence (42) and heteronuclear multiple-bond correlation (42). Sample composition was as follows: CMP8 (1.9 mm), TMS (0.5 mm), DMSO (0.5 mm), D2O ≥ 99%, and temperature 25 ± 1 °C. Sample composition in the line-broadening studies was as follows: labeled SULT1A3 (20 μm active sites), CMP8 (100, 200, 400, or 1000 μm), PAP (500 μm, 17 × Kd), KPO4 (50 mm), pD 7.4, 25 ± 1 °C. Proton line widths were fit to a Lorentzian distribution using NMRdraw (43).
NMR distance–constrained molecular dynamics modeling
A ligand-free homology model of SULT1A3 was constructed from the SULT1A3·PAP·dopamine structure (Protein Data Bank code 2A3R (8)) using SWISS-MODEL. The model was protonated at pH 7.4 and energy-minimized using GROMACS. Generalized AMBER force field energy parameter files were constructed using Antechamber (44–46) for CMP8, PAPS, and a spin-labeled cysteine analogue in which the nitroxyl moiety was replaced by a hydroxyl group, as described previously (18).
The cysteine analogue parameter file was added to the AMBER energy file as a noncanonical amino acid and inserted into SULT1A3 by replacing residues Gln-116, Glu-198, and Lys-234. PAPS was positioned in the active site of the ligand-less enzyme using GOLD. The system was equilibrated (298 K, NaCl (50 mm), pH 7.4) in 100-ps increments using GROMACS. Once equilibrated, CMP8 was positioned randomly in a simulated box of water (52 × 52 × 52 Å) containing the spin-labeled SULT1A3·PAPS construct and docked using GROMACS. Docking was constrained using NMR-determined, spin label/CMP8-proton distances, as described under “NMR distance–constrained molecular dynamics docking.” The docking simulations were repeated 100 times; 92 of the docking experiments yielded the structure described in the text, and the remaining eight structures showed a reversed orientation.
Molecular dynamics docking
Docking of CMP8 and its analogues was performed using a previously described equilibrated model of the SULT1A3·PAPS·dopamine complex without spin label (18). Briefly, compounds were positioned in the NMR-determined allosteric binding site using GOLD, minimized with GROMACS (AMBER energy field), and equilibrated in 100-ps increments at 298 K, NaCl (50 mm), pH 7.4. Once the root mean square deviation of the system had stabilized, indicating that equilibrium had been reached, equilibrium was confirmed by ensuring that the root mean square deviation remained stable over an additional 10 ns.
Initial rate studies
Initial rate parameters for the labeled or mutant SULT1A3 constructs were determined using a previously described 1-HP assay (47). Briefly, reactions were initiated by the addition of PAPS (0.50 mm, 17 × Km) to a solution containing enzyme (20 nm, active sites), 1-HP (2.0 μm, ∼24 × Km), and KPO4 (50 mm), pH 7.5, 25 ± 2 °C. Reactions were monitored via fluorescence change associated with the conversion of 1-HP to 1-HP-S (λex = 325 nm, λem = 370 nm). Keq for the 1-HP sulfonation reaction is ∼250. Given the reaction conditions outlined above, ≥99% of 1-HP is converted to 1-HP-S, and ∼0.4% of PAPS is converted to PAP at equilibrium. The affinities of PAPS and PAP for SULT1A3 are comparable, and the affinity of 1-HP-S is quite low (Kd = 240 ± 30 μm); consequently, the conditions result in quantitative conversion of 1-HP to 1-HP-S with negligible product inhibition. Reaction progress curves were analyzed to obtain initial-rate parameters as described previously (48, 49).
Inhibition studies
Inhibition parameters were determined under conditions identical to those described above (see “Initial rate studies”) except that [1-HP] was increased to 5.0 μm (∼61 × Km), and inhibitor concentration was varied between 0.20 and 20 × Ki. Ki was obtained using a least-squares fit to the partial inhibition equation (36, 47), V/Vmax = (Ki + α·[I])/(Ki + [I]), where α is the fraction of enzyme turnover remaining at saturating substrates and allosteric inhibitor.
Pre-steady-state binding studies
The pre-steady-state binding of PAP to SULT1A3 was monitored via ligand-induced enzyme fluorescence changes in SULT1A3 using an Applied Photophysics SX20 stopped-flow spectrofluorimeter (29). Fluorescence was measured at λex = 290 nm and λem ≥ 330 nm (using a cutoff filter). kon and koff of PAP binding to SULT1A3 were obtained by rapidly mixing (1:1, v/v) a solution containing SULT1A3 (25 nm, dimer), CMP8 (0 or 1.7 μm, 50 × Kd), KPO4 (50 mm), pH 7.5, with a solution that was identical except that it contained PAP (0.5, 1.0, 1.5, or 2.0 μm) and did not contain enzyme. All reactions were pseudo-first-order in [PAP]. The observed rate constant (kobs) at a given [PAP] was taken as the average of three independent progress curves, each obtained from a single-exponential fit of 6–9 averaged, binding progress curves. The rate constants, kon and koff, were obtained from the slopes and intercepts predicted by linear least-squares analysis of four-point kobs versus [PAP] plots.
Author contributions
K. D., T. W., I. C., M. C., and T. S. L. formal analysis; I. C. software; A. D. and T. S. L. supervision; A. D. and T. S. L. funding acquisition; A. D. and T. S. L. project administration; T. S. L. conceptualization; T. S. L. resources; T. S. L. validation; T. S. L. methodology; T. S. L. writing-original draft.
Supplementary Material
This work was supported by National Institutes of Health Grants GM112728 (to A. D.) and GM121849 and GM127144 (to T. S. L.). A provisional patent covering the compounds described in this paper has been submitted. All authors listed are co-inventors on the patent. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1 and S2.
The energy-minimized, NMR distance–constrained structure of the CMP8·E·PAPS·dopamine complex is available in the Model Archive database under archive number ma-qtj80.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- SULT
- sulfotransferase
- CMP
- compound 8
- 1-HP
- 1-hydroxypyrene
- PAP
- 3′-phosphoadenosine 5′-phosphate
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- TMS
- tetramethylsilane
- GPL
- General Public License
- GOLD
- Genetically Optimized Ligand Docking.
References
- 1. Merikangas K. R., He J. P., Burstein M., Swanson S. A., Avenevoli S., Cui L., Benjet C., Georgiades K., and Swendsen J. (2010) Lifetime prevalence of mental disorders in U.S. adolescents: results from the National Comorbidity Survey Replication–Adolescent Supplement (NCS-A). J. Am. Acad. Child Adolesc. Psychiatry 49, 980–989 10.1016/j.jaac.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chesney E., Goodwin G. M., and Fazel S. (2014) Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry 13, 153–160 10.1002/wps.20128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berton O., and Nestler E. J. (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151 10.1038/nrn1846 [DOI] [PubMed] [Google Scholar]
- 4. Souery D., Amsterdam J., de Montigny C., Lecrubier Y., Montgomery S., Lipp O., Racagni G., Zohar J., and Mendlewicz J. (1999) Treatment resistant depression: methodological overview and operational criteria. Eur. Neuropsychopharmacol. 9, 83–91 10.1016/S0924-977X(98)00004-2 [DOI] [PubMed] [Google Scholar]
- 5. Olgiati P., Serretti A., Souery D., Dold M., Kasper S., Montgomery S., Zohar J., and Mendlewicz J. (2018) Early improvement and response to antidepressant medications in adults with major depressive disorder: meta-analysis and study of a sample with treatment-resistant depression. J. Affect. Disord. 227, 777–786 10.1016/j.jad.2017.11.004 [DOI] [PubMed] [Google Scholar]
- 6. Thomas S. J., Shin M., McInnis M. G., and Bostwick J. R. (2015) Combination therapy with monoamine oxidase inhibitors and other antidepressants or stimulants: strategies for the management of treatment-resistant depression. Pharmacotherapy 35, 433–449 10.1002/phar.1576 [DOI] [PubMed] [Google Scholar]
- 7. Suominen T., Uutela P., Ketola R. A., Bergquist J., Hillered L., Finel M., Zhang H., Laakso A., and Kostiainen R. (2013) Determination of serotonin and dopamine metabolites in human brain microdialysis and cerebrospinal fluid samples by UPLC-MS/MS: discovery of intact glucuronide and sulfate conjugates. PLoS One 8, e68007 10.1371/journal.pone.0068007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu J. H., Li H. T., Liu M. C., Zhang J. P., Li M., An X. M., and Chang W. R. (2005) Crystal structure of human sulfotransferase SULT1A3 in complex with dopamine and 3′-phosphoadenosine 5′-phosphate. Biochem. Biophys. Res. Commun. 335, 417–423 10.1016/j.bbrc.2005.07.091 [DOI] [PubMed] [Google Scholar]
- 9. Dajani R., Hood A. M., and Coughtrie M. W. (1998) A single amino acid, glu146, governs the substrate specificity of a human dopamine sulfotransferase, SULT1A3. Mol. Pharmacol. 54, 942–948 10.1124/mol.54.6.942 [DOI] [PubMed] [Google Scholar]
- 10. Hildebrandt M. A., Salavaggione O. E., Martin Y. N., Flynn H. C., Jalal S., Wieben E. D., and Weinshilboum R. M. (2004) Human SULT1A3 pharmacogenetics: gene duplication and functional genomic studies. Biochem. Biophys. Res. Commun. 321, 870–878 10.1016/j.bbrc.2004.07.038 [DOI] [PubMed] [Google Scholar]
- 11. Riches Z., Stanley E. L., Bloomer J. C., and Coughtrie M. W. (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab. Dispos. 37, 2255–2261 10.1124/dmd.109.028399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salman E. D., Kadlubar S. A., and Falany C. N. (2009) Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain. Drug Metab. Dispos. 37, 706–709 10.1124/dmd.108.025767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heroux J. A., and Roth J. A. (1988) Physical characterization of a monoamine-sulfating form of phenol sulfotransferase from human platelets. Mol. Pharmacol. 34, 194–199 [PubMed] [Google Scholar]
- 14. Goldstein D. S., Swoboda K. J., Miles J. M., Coppack S. W., Aneman A., Holmes C., Lamensdorf I., and Eisenhofer G. (1999) Sources and physiological significance of plasma dopamine sulfate. J. Clin. Endocrinol. Metab. 84, 2523–2531 10.1210/jcem.84.7.5864 [DOI] [PubMed] [Google Scholar]
- 15. Strott C. A. (2002) Sulfonation and molecular action. Endocr. Rev. 23, 703–732 10.1210/er.2001-0040 [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto T., Yamatodani A., Nishimura M., and Wada H. (1985) Determination of dopamine-3- and 4-O-sulphate in human plasma and urine by anion-exchange high-performance liquid chromatography with fluorimetric detection. J. Chromatogr. 342, 261–267 10.1016/S0378-4347(00)84516-8 [DOI] [PubMed] [Google Scholar]
- 17. Le Corre P., Malledant Y., Tanguy M., and Le Verge R. (1993) Steady-state pharmacokinetics of dopamine in adult patients. Crit. Care Med. 21, 1652–1657 10.1097/00003246-199311000-00013 [DOI] [PubMed] [Google Scholar]
- 18. Cook I., Wang T., and Leyh T. S. (2017) Tetrahydrobiopterin regulates monoamine neurotransmitter sulfonation. Proc. Natl. Acad. Sci. U.S.A. 114, E5317–E5324 10.1073/pnas.1704500114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang T., Cook I., and Leyh T. S. (2017) The NSAID allosteric site of human cytosolic sulfotransferases. J. Biol. Chem. 292, 20305–20312 10.1074/jbc.M117.817387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cook I., Wang T., Girvin M., and Leyh T. S. (2016) The structure of the catechin-binding site of human sulfotransferase 1A1. Proc. Natl. Acad. Sci. U.S.A. 113, 14312–14317 10.1073/pnas.1613913113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Solomon I. (1955) Relaxation processes in a system of two spins. Phys. Rev. 99, 559–566 10.1103/PhysRev.99.559 [DOI] [Google Scholar]
- 22. Girvin M. E., and Fillingame R. H. (1995) Determination of local protein structure by spin label difference 2D NMR: the region neighboring Asp61 of subunit c of the F1F0 ATP synthase. Biochemistry 34, 1635–1645 10.1021/bi00005a020 [DOI] [PubMed] [Google Scholar]
- 23. Gochin M., Zhou G., and Phillips A. H. (2011) Paramagnetic relaxation assisted docking of a small indole compound in the HIV-1 gp41 hydrophobic pocket. ACS Chem. Biol. 6, 267–274 10.1021/cb100368d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 25. Berendsen H. J. C., Vanderspoel D., and Vandrunen R. (1995) Gromacs: a message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 91, 43–56 10.1016/0010-4655(95)00042-E [DOI] [Google Scholar]
- 26. Schmitz U., Ulyanov N. B., Kumar A., and James T. L. (1993) Molecular dynamics with weighted time-averaged restraints for a DNA octamer: dynamic interpretation of nuclear magnetic resonance data. J. Mol. Biol. 234, 373–389 10.1006/jmbi.1993.1593 [DOI] [PubMed] [Google Scholar]
- 27. Battiste J. L., and Wagner G. (2000) Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear Overhauser effect data. Biochemistry 39, 5355–5365 10.1021/bi000060h [DOI] [PubMed] [Google Scholar]
- 28. Wang T., Cook I., and Leyh T. S. (2016) Isozyme specific allosteric regulation of human sulfotransferase 1A1. Biochemistry 55, 4036–4046 10.1021/acs.biochem.6b00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang T., Cook I., and Leyh T. S. (2014) 3′-Phosphoadenosine 5′-phosphosulfate allosterically regulates sulfotransferase turnover. Biochemistry 53, 6893–6900 10.1021/bi501120p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cook I., Wang T., Almo S. C., Kim J., Falany C. N., and Leyh T. S. (2013) Testing the sulfotransferase molecular pore hypothesis. J. Biol. Chem. 288, 8619–8626 10.1074/jbc.M112.445015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cook I., Wang T., Almo S. C., Kim J., Falany C. N., and Leyh T. S. (2013) The gate that governs sulfotransferase selectivity. Biochemistry 52, 415–424 10.1021/bi301492j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cook I., Wang T., Falany C. N., and Leyh T. S. (2012) A nucleotide-gated molecular pore selects sulfotransferase substrates. Biochemistry 51, 5674–5683 10.1021/bi300631g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cook I., Wang T., Wang W., Kopp F., Wu P., and Leyh T. S. (2016) Controlling sulfuryl-transfer biology. Cell Chem. Biol. 23, 579–586 10.1016/j.chembiol.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang T., Cook I., and Leyh T. (2016) The design and interpretation of human SULT1A1 assays. Drug Metab. Dispos. 44, 481–484 10.1124/dmd.115.068205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang T., Cook I., Falany C. N., and Leyh T. S. (2014) Paradigms of sulfotransferase catalysis: the mechanism of SULT2A1. J. Biol. Chem. 289, 26474–26480 10.1074/jbc.M114.573501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whiteley C. G. (1999) Enzyme kinetics: partial and complete non-competitive inhibition. Biochem. Educ. 27, 15–18 10.1016/S0307-4412(98)00265-9 [DOI] [PubMed] [Google Scholar]
- 37. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun M., and Leyh T. S. (2010) The human estrogen sulfotransferase: a half-site reactive enzyme. Biochemistry 49, 4779–4785 10.1021/bi902190r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baker J., Wolinski K., Malagoli M., Kinghorn D., Wolinski P., Magyarfalvi G., Saebo S., Janowski T., and Pulay P. (2009) Quantum chemistry in parallel with PQS. J. Comput. Chem. 30, 317–335 10.1002/jcc.21052 [DOI] [PubMed] [Google Scholar]
- 40. Artimo P., Jonnalagedda M., Arnold K., Baratin D., Csardi G., de Castro E., Duvaud S., Flegel V., Fortier A., Gasteiger E., Grosdidier A., Hernandez C., Ioannidis V., Kuznetsov D., Liechti R., et al. (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603 10.1093/nar/gks400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 42. Castañar L., and Parella T. (2015) Recent advances in small molecule NMR: improved HSQC and HSQMBC experiments. Annu. Rep. NMR Spectrosc. 84, 163–232 10.1016/bs.arnmr.2014.10.004 [DOI] [Google Scholar]
- 43. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 44. Case D. A., Babin V., Berryman J. T., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E., Darden T., Duke R., Gohlke H., Götz A. W., Gusarov S., Homeyer N., Janowski P., Kaus J., et al. (2014) AMBER 14, University of California, San Francisco [Google Scholar]
- 45. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 46. Wang B., and Merz K. M. (2006) A fast QM/MM (quantum mechanical/molecular mechanical) approach to calculate nuclear magnetic resonance chemical shifts for macromolecules. J. Chem. Theory Comput. 2, 209–215 10.1021/ct050212s [DOI] [PubMed] [Google Scholar]
- 47. Cook I., Wang T., Falany C. N., and Leyh T. S. (2015) The allosteric binding sites of sulfotransferase 1A1. Drug Metab. Dispos. 43, 418–423 10.1124/dmd.114.061887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cook I., Wang T., Falany C. N., and Leyh T. S. (2013) High accuracy in silico sulfotransferase models. J. Biol. Chem. 288, 34494–34501 10.1074/jbc.M113.510974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang Q., and Leyh T. S. (2010) Precise, facile initial rate measurements. J. Phys. Chem. B 114, 16131–16136 10.1021/jp1055528 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.