

Abstract
GABAA receptors (GABAARs) are pentameric ligand-gated ion channels that mediate synaptic inhibition throughout the central nervous system. The α1β2γ2 receptor is the major subtype in the brain; GABA binds at the β2(+)α1(−) interface. The structure of the homomeric β3 GABAAR, which is not activated by GABA, has been solved. Recently, four additional heteromeric structures were reported, highlighting key residues required for agonist binding. Here, we used a protein engineering method, taking advantage of knowledge of the key binding residues, to create a β3(+)α1(−) heteromeric interface in the homomeric human β3 GABAAR that enables GABA-mediated activation. Substitutions were made in the complementary side of the orthosteric binding site in loop D (Y87F and Q89R), loop E (G152T), and loop G (N66D and A70T). The Q89R and G152T combination enabled low-potency activation by GABA and potentiation by propofol but impaired direct activation by higher propofol concentrations. At higher concentrations, GABA inhibited gating of β3 GABAAR variants containing Y87F, Q89R, and G152T. Reversion of Phe87 to tyrosine abolished GABA's inhibitory effect and partially recovered direct activation by propofol. This tyrosine is conserved in homomeric GABAARs and in the Erwinia chrysanthemi ligand-gated ion channel and may be essential for the absence of an inhibitory effect of GABA on homomeric channels. This work demonstrated that only two substitutions, Q89R and G152T, in β3 GABAAR are sufficient to reconstitute GABA-mediated activation and suggests that Tyr87 prevents inhibitory effects of GABA.
Keywords: γ-aminobutyric acid (GABA), GABA receptor, Cys-loop receptor, mutagenesis in vitro, receptor structure-function, anesthetic, ion channel, ligand-binding protein, patch clamp, gating, benzodiazepine, general anaesthetic, propofol
Introduction
GABAARs2 are members of the pentameric ligand-gated ion channel family and mediate fast synaptic inhibition (1). Consequently, they are important pharmacological targets (2, 3).
GABAAR subunits are composed of three domains (4): 1) the extracellular domain (ECD), with 10 β-strands (β1–10), one α-helix, and the orthosteric binding site; 2) the transmembrane domain (TMD) comprising four helices (TM1–4), with the TM2 of each subunit forming the ion pore; and 3) the intracellular domain (ICD), between TM3 and TM4, which is a site for posttranslational modification that interacts with trafficking proteins (4–6).
The orthosteric binding site is located between the α and β subunits that comprise the complementary (−) and principal (+) components, respectively. The site contains seven noncontiguous binding loops (A–G): A–C belong to the principal side, whereas loops D–G belong to the complementary side (7–9).
There are 19 different GABAAR subunits that form at least 14 distinct combinations in vivo (10, 11), accounting for the physiological versatility and pharmacological selectivity of these channels (2). The major subtype in the central nervous system is the α1β2γ2 GABAAR. The β1, β3, and ρ subunits can form homomers when recombinantly expressed in vitro. Although the homomeric β3 has not been identified in vivo, it is of considerable interest as the first GABAAR to yield to high-resolution structural analysis (12) and for functional studies because histaminergic ligands and propofol activate the receptor (13–16). Recently, four heteromeric GABAAR structures were published, including the major subtype (17–19). These studies determined the important residues for GABA binding and suggest that variability on the complementary subunit influences ligand selectivity (19). The homomeric β3 cannot be activated by GABA (16, 20). This raises questions about which residues in the complementary side are required to reconstitute activation. The availability of the β3 structure provides an opportunity to locate candidate residues.
In the present study, we investigated whether substituting amino acids in the complementary side of the β3 GABAAR to corresponding residues in the α1 subunit would reconstitute activation by GABA. Four β3 mutants were designed and used for patch-clamp electrophysiology. We analyzed the activation by GABA and propofol, potentiation of GABA-evoked currents by propofol, and the kinetics of GABA-evoked currents. Comparative modeling and molecular docking calculations were used to predict the orientation of GABA at the orthosteric site of the mutant β3 GABAAR. Using these approaches, we demonstrated that Q89R and G152T substitutions reconstituted GABA activation of GABAAR β3 and potentiation by propofol. In addition, we found that the Y87F substitution caused GABA to inhibit receptor function.
Results
Designing the constructs
We modified the β3 GABAAR by replacing the ICD (residues 346–396) with the SQPARAA sequence to mimic the construct used to crystallize the β3 GABAAR (12), referred to from this point as β3-cryst (Table 1).
Table 1.
Protein constructs
| Construct name | ECD interface | Substitutions |
|---|---|---|
| GABAAR β3-cryst | β3(+)β3(−) | Substitute ICD (346–396) for SQPARAA |
| GABAAR β3 C1 | β3(+)α1(−) | β3-cryst + Y87F,Q89R,G152T |
| GABAAR β3 C1 N66D | β3(+)α1(−) | C1 + N66D |
| GABAAR β3 C1 A70T | β3(+)α1(−) | C1 + A70T |
| GABAAR β3 C1 F87Y | β3(+)α1(−) | β3-cryst + Q89R,G152T |
Docking GABA into the β3 orthosteric site
Docking calculations were performed between GABA and the β3-cryst model (Fig. 1A). As far as we are aware, there are no prior reports of docking GABA into the homomeric β3 receptor. The best GABA pose presented an energy of −38 kcal/mol, suggesting binding. Examination of residues within the orthosteric binding domain on the α1 subunit revealed amino acids that are not shared by β3 at key locations known to affect activation by GABA (Fig. 1B). Substitution of these residues into the β3-cryst model (GABAAR β3 C1) improved the binding energy of GABA as evidenced by docking calculations (−46 kcal/mol). The model suggests that the GABA amino group forms a salt bridge with Glu180 on the (+) interface of the β3 subunit (Fig. 1C) and that the GABA carboxyl makes a bidentate interaction with Arg89 and a hydrogen bond with the Thr152 hydroxyl group, substituted in the (−) interface. These interactions are in agreement with the cryo-EM structures of the human GABAAR α1β2γ2 and rat GABAAR α1β1γ2 (18, 19). In addition, they were described by other studies using docking calculations with human GABAAR α1β2γ2 (21) and insect GABAAR models (21, 22).
Figure 1.

Docking results for GABAAR constructs. The predicted GABA orientations (pink and yellow) in GABAAR β3-cryst (A) and GABAAR β3 C1 (C) show the carboxyl group facing the complementary side (green cartoon) and the amino group facing the principal side (cyan cartoon). Residues interacting with GABA are depicted as gray sticks, and polar interactions are depicted as black dashes. B, sequence alignment of the orthosteric site (loops A–G) based on structural comparisons of GABAAR β3 (PDB code 4COF) and the GABAAR α1 model (21), showing the substitutions (gray box). Secondary structure, loops (black line) and β-strands (yellow arrows), is depicted below the sequence. Residues are numbered according to the initiating Met1.
Three substitutions reconstituted GABA activation of homomeric β3 receptors
The Y87F, Q89R, and G152T substitutions were introduced into the β3-cryst construct using site-directed mutagenesis. This β3 C1 cDNA was transiently transfected into human embryonic kidney 293 (HEK293) cells for whole-cell electrophysiology recordings. Concentrations of GABA that are maximally efficacious at heteromeric GABAARs (1 mm) fail to activate homomeric β3 GABAARs (16, 20). We therefore applied higher concentrations of GABA (10 mm) to HEK293 cells expressing either β3-cryst or β3 C1. GABA (10 mm) evoked negligible currents mediated by β3-cryst with current densities of 2.3 ± 0.8 pA/pF (Fig. 2A). By contrast, GABA (10 mm)–evoked currents mediated by β3 C1 were larger with current densities of 15.7 ± 12.5 pA/pF (Fig. 2B). This was significantly different from β3-cryst (n = 7, p = 0.003, t test; Fig. 2C). These results indicate that the amino acid substitutions (Y87F, Q89R, and G152T) were sufficient to reconstitute activation by GABA.
Figure 2.

GABAAR β3 C1 is activated and inhibited by GABA. A and B, examples of currents recorded when GABA (10 mm) was applied to cells expressing GABAAR β3-cryst (A) and GABAAR β3 C1 (B) indicate that the latter is functional and activated by the neurotransmitter. C, mean ± S.D. current densities evoked by GABA (10 mm), with an asterisk indicating significant differences between the proteins (n = 7, p = 0.003, t test). D, examples of currents mediated by GABAAR β3 C1, evoked by increasing concentrations of GABA. Currents in gray are declining due to inhibition by GABA (>10 mm). The bar indicates GABA application (5 s). E, concentration-response relationships obtained using the percentage of the maximum amplitude recorded for each cell (n = 5). Logistic equations were fitted to the data points (see “Experimental procedures”). From the double-logistic fit, two distinct potencies were observed for activation (EC50 = 2.9 mm) and inhibition (IC50 = 50.5 mm). A summary of the data is in Table 2. F, graph of mean current 10–90% rise time. Activation rates are slowed somewhat by increasing the GABA concentration in β3 C1 (n = 6, F(4,25) = 42.2, one-way ANOVA post hoc Dunnett's, p = 0.04 comparing 10 with 1 mm GABA), whereas currents evoked by 100 mm GABA were activated faster (n = 6, p < 0.0001, F(4,25) = 42.2, one-way ANOVA post hoc Dunnett's, comparing 100 with 1 mm GABA). G, values for weighted τ of deactivation exhibited a similar trend with increasing GABA concentration (p = 0.04, one-way ANOVA, n = 6, F(4,19) = 3.2), although there was no significant difference comparing 1 mm with the other GABA concentrations tested. Detailed information about the components is in Table 3. Error bars represent S.D.
We subsequently determined the concentration-response relationship of β3 C1 to characterize the potency of GABA (Table 2). GABA was applied at increasing concentrations to cells expressing β3 C1. A representative example of these currents is shown in Fig. 2D. GABA-evoked current amplitudes were expressed as a percentage of the maximum and plotted as a concentration-response relationship (Fig. 2E). The data indicate that GABA exhibits a biphasic concentration-response relationship, which suggests two effects: activation and inhibition. We therefore fitted a two-component logistic function to the data (see “Experimental procedures”). GABA, up to 10 mm, activates β3 C1 with an EC50 of ∼3 mm (Table 2). Higher concentrations of GABA caused a reduction in current amplitude with an IC50 of ∼50 mm. This inhibitory effect has not been observed previously in GABAARs (9, 23–26) or in the bacterial pentameric ligand-gated ion channel ELIC (27), which, like β3 C1, also requires high concentrations of GABA for its activation (Fig. S1). We also observed a lack of inhibitory effect in heteromeric GABAARs formed from β3-cryst and β3 C1 subunits (Fig. S2).
Table 2.
Summary of Hill slope, EC50, and current density values obtained for GABA activation of GABAAR β3 C1 and F87Y
Mean ± S.D. Hill slope and EC50 values obtained from logistic function fit parameters of individual experiments and mean ± S.D. current densities evoked by peak concentrations of GABA are shown. No significant differences between GABAAR β3 C1 and F87Y were observed (t test; EC50 p = 0.2; current densities p = 0.6). n = number of experiments.
| Receptor | Hill slope | EC50 | Current density | n |
|---|---|---|---|---|
| mm | pA/pF | |||
| GABAAR β3 C1 | 1.3 ± 0.4 | 2.9 ± 2.1 | −15.7 ± 12.5 | 4 |
| GABAAR β3 C1 F87Y | 1.2 ± 0.3 | 1.3 ± 0.6 | −17.1 ± 11.9 | 4 |
Kinetics of β3 C1
In addition to the biphasic nature of the GABA concentration-response relationship, the representative currents shown in Fig. 2D also display unusual kinetics. Therefore, we analyzed the current activation and deactivation rates by measuring the 10–90% rise time and by fitting a two-component exponential function, respectively (see “Experimental procedures”). The mean values of rise times and weighted τ were plotted (Fig. 2, F and G). The individual components of the double-exponential fits for deactivation can be found in Table 3. Currents evoked by lower concentrations of GABA (0.1 and 0.3 mm) were excluded from the analysis due to their small amplitudes. Consistent with the concentration dependence of peak current activation (Fig. 2E), the concentration dependence of activation and deactivation also appears to be biphasic (Fig. 2, F and G).
Table 3.
Mean ± S.D. of the deactivation components in GABAAR β3 C1 and F87Y
No significant differences were observed between the mutants (n = 4, p = 0.1, F(9,34) = 1.8, one-way ANOVA post hoc Tukey's).
| [GABA] | τf | %f | τs | %s | Weighted τ |
|---|---|---|---|---|---|
| mm | |||||
| β3 C1 | |||||
| 1 | 34 ± 3 | 59 ± 5 | 253 ± 3 | 41 ± 5 | 142 ± 54 |
| 3 | 56 ± 32 | 48 ± 14 | 347 ± 139 | 52 ± 14 | 181 ± 97 |
| 10 | 58 ± 45 | 47 ± 2 | 342 ± 137 | 72 ± 26 | 243 ± 130 |
| 30 | 39 ± 10 | 37 ± 10 | 282 ± 98 | 72 ± 20 | 260 ± 124 |
| 100 | 37 ± 23 | 63 ± 33 | 261 ± 125 | 37 ± 33 | 84 ± 29 |
| β3 C1 F87Y | |||||
| 1 | 40 ± 9 | 64 ± 13 | 424 ± 142 | 36 ± 13 | 176 ± 56 |
| 3 | 51 ± 19 | 66 ± 11 | 345 ± 139 | 34 ± 11 | 151 ± 76 |
| 10 | 67 ± 30 | 56 ± 13 | 337 ± 79 | 44 ± 14 | 194 ± 66 |
| 30 | 79 ± 49 | 52 ± 15 | 511 ± 173 | 48 ± 15 | 287 ± 114 |
| 100 | 63 ± 7 | 77 ± 17 | 518 ± 232 | 23 ± 17 | 198 ± 171 |
GABA does not cause a voltage-dependent channel block
The inhibitory effect of GABA at higher concentrations could be due to binding at a lower-affinity site, which blocks the channel pore. We therefore examined whether GABA causes a voltage-dependent block of β3 C1 by comparing the current–voltage (I–V) relationships of currents evoked by 1 and 100 mm GABA. These concentrations were chosen because the inhibitory effect was observed at 100 mm but not at 1 mm GABA. Representative examples of the currents evoked by GABA at voltages ranging from −60 to 60 mV are shown in Fig. 3A. GABA (1 mm) produced an outwardly rectifying I–V relationship, consistent with previous observations of currents mediated by β3 and α1β3 GABAARs (16), as did 100 mm GABA. We quantified outward rectification by expressing the current amplitudes as a ratio of those evoked at −60 mV (Fig. 3B). The rectification indexes calculated (I60 mV/I−60 mV) were 3.5 ± 0.7 and 3.6 ± 0.5 (n = 4) for 1 and 100 mm GABA, respectively (p = 0.8, t test, n = 4). These results suggest that the inhibitory effect of 100 mm GABA was not caused by voltage-dependent channel block.
Figure 3.
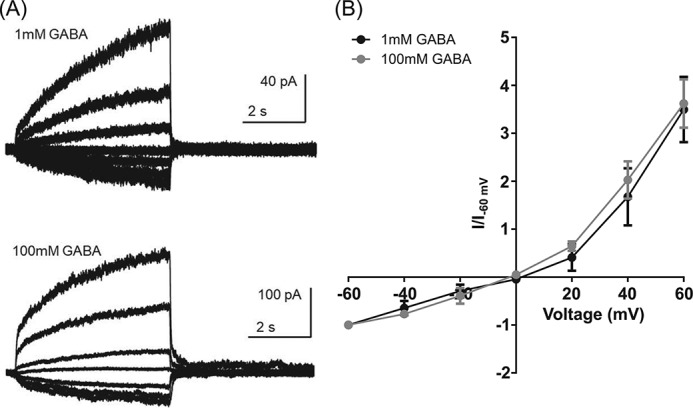
GABA does not block the channel by a voltage-dependent process. A, representative examples of the currents evoked by GABA (1 and 100 mm) recorded at voltages ranging from −60 to 60 mV. B, the amplitude of the currents was expressed as a ratio of those evoked at −60 mV (I/I−60 mV) and plotted against the voltage, indicating similar outward rectification for both concentrations. Error bars represent S.D.
Substitutions in loop G do not affect the activation of β3 C1 by GABA
Mutagenesis studies in GABAAR α1β2γ2 indicate that the identities of α1 loop G residues at positions 71 and 75 influence gating and, thereby, the apparent potency of GABA (9, 23, 25, 28). In an attempt to increase the apparent potency of GABA, substitutions in loop G were made, introducing α1 residues into β3 C1 N66D and β3 C1 A70T GABAARs (residues equivalent to those at α1 positions 71 and 75, respectively). Neither the potency nor the efficacy of GABA was affected (one-way ANOVA post hoc Dunnett's, p = 0.8, F(2,12) = 0.28). This is perhaps not surprising due to the conservative nature of the N66D and A40T substitutions (Fig. S3 and Table S1).
A loop D Tyr conserved in homomeric receptors prevents block by GABA
An amino acid sequence alignment of the pentameric ligand-gated ion channel subunits that form homomeric GABA-activated receptors, including ELIC, reveals conservation of the Tyr at the position equivalent to β3 amino acid 87 (Fig. S4A). We investigated whether replacement of ELIC Tyr38 with Phe affects activation by GABA. Interestingly, GABA failed to evoke currents mediated by ELIC Y38F despite the conservative nature of this substitution (Fig. S4C). These data suggest that the Phe is detrimental to ELIC function. Because ELIC, GABAA ρ, and GABAA β all contain a Tyr, this residue may be necessary for preventing block of homomeric receptors by GABA. We tested the hypothesis that the Tyr is required in β3 receptors to prevent inhibitory effects of GABA at high concentrations by creating the β3 C1 F87Y in which the Phe87 was reverted back to the tyrosine found in WT β3.
Cells expressing β3 C1 F87Y were voltage-clamped at −60 mV, and GABA-evoked currents were recorded. Representative examples are shown in Fig. 4A. The current amplitudes were expressed as a percentage of maximum and plotted as a concentration-response relationship, which was fitted with a single-component logistic function (Fig. 4B). The potency of activation by GABA was similar when compared with β3 C1 (p = 0.2, n = 4, t test; Table 2) as was the maximum current density (p = 0.6, n = 4, t test; Table 2). However, the inhibition by 100 mm GABA was absent in β3 C1 F87Y with a significant difference in the current amplitude evoked by 100 mm GABA compared with that mediated by β3 C1 (p = 0.003, n = 4, t test). Similarly, higher concentrations of GABA (300 mm) did not reduce GABA-mediated current amplitude (Fig. S5), indicating that the inhibitory component was abolished with the F87Y substitution. Furthermore, the rate of activation of β3 C1 F87Y increased with GABA concentration and was not biphasic (Fig. 4C). There was also no apparent influence of GABA concentration on deactivation (Fig. 4D), consistent with the previous data for GABAARs (29) and ELIC (Fig. S1, C and D).
Figure 4.

The C1 F87Y substitution abolished the biphasic nature of concentration-response relationship. A, examples of currents mediated by GABAAR β3 C1 F87Y, evoked by increasing concentrations of GABA. B, concentration-response relationship obtained using the percentage of the maximum amplitude recorded for each cell. The inhibition caused by 100 mm GABA in β3 C1 was abolished by the F87Y substitution. A summary of the data is in Table 2. C, mean 10–90% rise times showed no significant change with increasing GABA concentrations in β3 C1 F87Y except comparing 100 with 1 mm (p = 0.008, n = 4, F(4,15) = 5.2, one-way ANOVA post hoc Dunnett's). D, mean deactivation weighted τ was also independent of GABA concentrations (p = 0.5, one-way ANOVA, F(4,15) = 0.96). Detailed information about the components is in Table 3. Error bars represent S.D.
Effects of propofol on β3 mutants
Consistent with a previous report of β3 receptor activation (16), propofol (30 μm) evoked inward currents when applied locally to HEK293 cells expressing β3-cryst recorded under voltage clamp at −60 mV (Fig. 5A); however, no response was observed in cells expressing β3 C1 (Fig. 5B). A partial recovery of propofol direct activation was observed in β3 C1 F87Y (Fig. 5C) as evidenced by the significant difference in current densities between β3-cryst and β3 C1 (n = 10, t test, p < 0.0001) and between β3 C1 and β3 C1 F87Y (n = 10, t test, p = 0.007; Fig. 5D).
Figure 5.
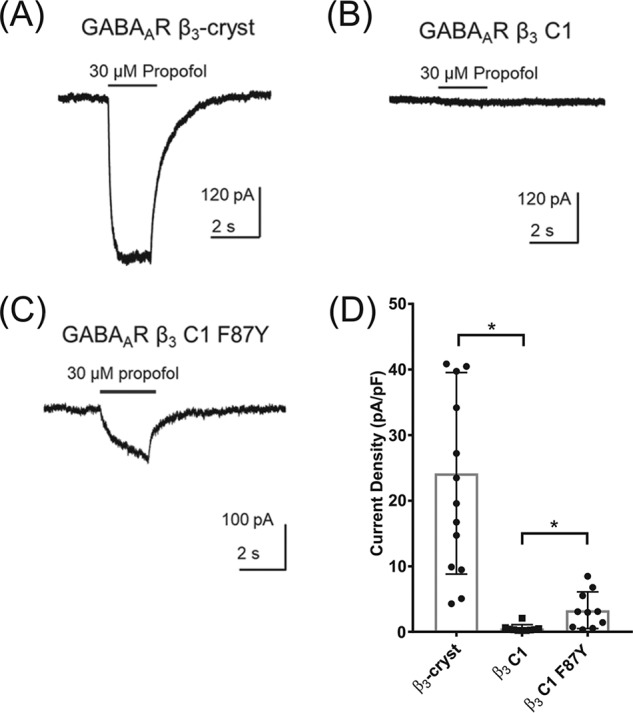
Propofol does not activate β3 C1. Examples of currents recorded in the presence of propofol from HEK293 cells expressing β3-cryst (A), β3 C1 (B), and β3 C1 F87Y (C) are shown. D, mean ± S.D. current densities evoked by propofol (30 μm) demonstrate that the function of β3 C1 is impaired, with values indicated by asterisks significantly different from the β3-cryst (n = 10, t test, β3 C1 p < 0.0001). However, propofol direct activation was partially restored in β3 C1 F87Y with values significantly different from β3 C1 (n = 10, p = 0.007, t test). Error bars represent S.D.
Potentiation, activation, and blockade of GABAARs occur at different propofol concentrations, consistent with the possibility of distinct sites with differing affinities (30–32). The substitutions introduced in β3 C1 and β3 C1 F87Y may have affected the gating mechanism or induced structural rearrangements that disrupt the binding site of propofol responsible for the direct activation of the receptor.
We investigated whether propofol can potentiate GABA-induced currents mediated by β3 C1 and β3 C1 F87Y. Cells were stepped from GABA (1 mm) to a solution of GABA (1 mm) plus propofol (10 or 30 μm) and back to GABA (1 mm). This concentration of GABA corresponds to EC25 according to the concentration-response relationship, allowing ample scope for enhancement (Fig. 2E).
As observed previously (Fig. 2A), GABA failed to evoke currents when applied to cells expressing β3-cryst (Fig. 6A). The current observed when GABA was applied with propofol (30 μm) to cells expressing β3-cryst was equivalent to that observed when propofol was applied alone (Fig. 5), indicating a lack of interaction with GABA (Fig. 6A). By contrast, in cells expressing GABAAR β3 C1, propofol (10 and 30 μm) enhanced GABA-evoked currents (Fig. 6, B and C) by 404 ± 183 (n = 3) and 405 ± 176% (n = 7), respectively (Fig. 6F). When applied alone, propofol did not evoke a current. Therefore, the enhancement by propofol of GABA-evoked currents mediated by β3 C1 is caused by potentiation rather than additive activation. Propofol (10 and 30 μm) also enhanced GABA-induced currents mediated by β3 C1 F87Y (Fig. 6, D and E) by 242 ± 140 (n = 5) and 663 ± 233% (n = 8), respectively (Fig. 6F). The significant increase (one-way ANOVA post hoc Tukey's, p = 0.008, F(3,19) = 5.3) in current enhancement by propofol (30 μm) is due in this case to the additive effect of direct activation rather than increased potentiation. However, the observation that propofol (10 μm) alone failed to activate a current in the absence of GABA indicates that, similar to GABAAR β3 C1, β3 C1 F87Y also supports propofol-evoked potentiation.
Figure 6.

Potentiation of GABA-evoked currents by propofol was unaffected in the β3 mutants. A, an exemplar current evoked by propofol mediated by β3-cryst. GABA had no effect, and the current amplitude evoked by propofol is similar to that seen in the absence of GABA. B and C, examples of GABA (1 mm)–evoked currents mediated by β3 C1 enhanced in the presence of 10 μm propofol (B) and 30 μm propofol (C). D and E, examples of GABA (1 mm)–evoked currents mediated by β3 C1 F87Y enhanced in the presence of 10 μm propofol (D) and 30 μm propofol (E). F, the percentages of potentiation by propofol at 10 and 30 μm for β3 C1 and β3 C1 F87Y were plotted, with an asterisk indicating a significant difference for β3 C1 F87Y between 10 and 30 μm propofol (n = 4, p = 0.008, F(3,19) = 5.3, one-way ANOVA post hoc Tukey's). This difference can be explained by the additive effect of propofol (30 μm) activation of β3 C1 F87Y (Fig. 5C). The bars indicate application of GABA (1 mm) or propofol (30 and 10 μm) with GABA (1 mm). Error bars represent S.D.
Discussion
This study demonstrates that the replacement of two key residues in the orthosteric binding site of the β3 subunit (Gln89 and Gly152) by the equivalent ECD loci in the α subunit, Arg and Thr, respectively, enables gating of β3 receptors by GABA. Docking to the β3 C1 model, which includes these substitutions plus the additional F87Y substitution, confirmed the interaction of GABA with all three of these binding residues. The favored GABA-binding pose was similar to that of heteromeric GABAAR structures (18, 19) and to observations in previous docking studies using the mammalian heteromeric and the insect homomeric GABAARs (21, 22) and in general agreement with the literature (21, 33, 34). The GABA carboxyl makes a bidentate interaction with Arg89 and a hydrogen bond with the Thr152 hydroxyl group in β3 C1. The same interactions were reported in heteromeric GABAAR structures solved in the presence of the agonist (18, 19). In addition, site-directed mutagenesis studies demonstrate that substitution of these residues in the α subunit affects GABA potency in GABAAR α1β2γ2 and GABAAR α1β2 (24, 35, 36). Taken together, the results of docking and functional analysis are consistent with the idea that the introduction of Q89R and G152T substitutions into β3 generates a heteromeric β3(+)α1(−)-like interface capable of activation by GABA albeit at high concentrations (>300 μm).
GABA concentrations above 10 mm caused a blocking effect in β3 C1. This has not been observed in other physiologically relevant heteromeric GABAARs (9, 23–26) or in ELIC (27). The effect was abolished when the phenylalanine in β3 C1 was reverted back to tyrosine, F87Y. Interestingly, this effect was also abolished in heteromeric GABAARs formed from β3 C1 (where position 87 is a Phe) and β3-cryst subunits (where position 87 is a Tyr). The apparent potency of GABA-mediated activation is not altered in these heteromeric GABAARs. Although the stoichiometries of heteromeric GABAARs formed from β3 C1 and β3-cryst subunits are not known, our data suggest that the incorporation of one or more Tyr87 is sufficient to prevent GABA-mediated blockade while preserving GABA-mediated activation, highlighting the importance of this residue in GABAAR function.
The kinetics of GABA-evoked currents mediated by β3 C1 GABAARs were also unusual. Activation and deactivation became slower and then faster with increasing concentrations of GABA, whereas the kinetics in β3 C1 F87Y GABAARs were more consistent with those of heteromeric GABAARs (29) and ELIC (Fig. S1). Interestingly, activation and deactivation rates of GABA-evoked currents mediated by β3 C1 GABAARs appear similar to those described for GABAARs activated in the presence of modulators, such as propofol (37) and benzodiazepines (38). In addition to its role as an agonist and an inhibitor of β3 C1 GABAARs, GABA may also act as a positive allosteric modulator. In the homomeric β3 C1 GABAARs, GABA may bind to all five subunit interfaces, and the Hill slope of 1.3 suggests cooperativity between at least two of these sites. It is possible that binding to additional orthosteric sites may result in potentiation, similar to the effect of benzodiazepines (38). However, our data with β3 C1 and β3-cryst heteromeric GABAARs suggest that GABA-mediated activation does not require GABA binding to all interfaces.
Moreover, bell-shaped concentration-response curves have been described for allosteric activators and modulators of GABAARs, such as propofol (16), valerenic acid (39), and pentobarbital (40–42). Pentobarbital, at low concentrations (low micromolar), can potentiate GABAAR currents by increasing the mean open duration. Higher concentrations (high micromolar) of pentobarbital can activate GABAARs, and millimolar concentrations can inhibit the channel, slowing deactivation (42). Similarly, GABA may act as an agonist, modulator, and inhibitor of β3 C1. However, the inhibition is not through a voltage-dependent channel block. Instead, there may be a lower-affinity inhibitory site for GABA. A similar mechanism has been proposed for the inhibitory effect observed with high concentrations of propofol (32).
Although the potentiation of GABA-evoked currents was unaffected, propofol's direct activation of β3 C1 was impaired compared with β3-cryst. There was partial recovery of propofol-activated current medicated by β3 C1 F87Y GABAARs. It is clear that substitutions in the orthosteric site can influence direct activation by propofol despite its binding site being in the TM region. In keeping with a need for conformational rearrangement in the orthosteric binding site during gating by propofol, the activation is also inhibited by bicuculline (43). Furthermore, we recently demonstrated faster deactivation of propofol-evoked currents with α1 loop D (F64C) and loop G (T47R) substitutions in GABAAR α1β2γ2, which adds additional support for a role of residues in or near the orthosteric binding site in the efficacy of gating by an allosteric agonist (26). Several studies suggest that gating by both orthosteric and allosteric agonists involves an interaction of the loops in the ECD with the TMD, particularly the loops between the β1-β2 strands and TM2-TM3 helices (44–47) and between β6-β7 strands and TM2-TM3 helices (12). It is important to note that loop G is located in β1 strand, loop D is in β2, and loop E is in β6. The substitutions in GABAAR β3 C1 are located in loops D and E; therefore, they may affect a concerted gating mechanism.
It is not yet clear why the substitution Y87F causes GABA to act as an inhibitor of β3 C1 GABAARs at high concentrations and impair propofol direct activation. The substitution may affect channel gating, consistent with previous mutagenesis studies of homologous residues in GABAAR ρ1 that produced spontaneous opening and affected GABA, trans-4-aminocrotonic acid, and imidazole-4-acetic acid potencies (48) and in GABAAR α1β1,2γ2 that affected GABA potency and kinetics (9, 49).
The tyrosine is found in all GABAAR β and ρ subunits and in ELIC. The latter two form homomers that can be activated by GABA (20, 27, 50). Tyrosine may prevent an inhibitory effect of GABA in homomeric receptors, and its substitution to phenylalanine may enable GABA to bind at another lower-affinity site and inhibit gating.
In summary, this study demonstrated that only two substitutions (Q89R and G152T) were required to reconstitute activation by GABA in homomeric β3 constructs. The potency of GABA was 2 orders of magnitude lower compared with heteromeric GABAARs. Similar to heteromeric GABAARs, propofol potentiated submaximal GABA-evoked currents and caused direct activation of β3 C1 F87Y receptors. Surprisingly, the conservative replacement of Tyr87 by phenylalanine abolished gating by propofol and caused GABA to have inhibitory effects at high concentrations.
These findings identify structural requirements for the reconstitution of a functional GABA-binding site in β3 homomeric receptors by transplanting key residues of the α subunit at the heteromeric interface. This approach provides a novel method for developing a better understanding of the structural requirements for gating.
Experimental procedures
Constructs
The GABAAR constructs were designed based on the published GABAAR β3 structure, i.e. substituting the ICD for the amino acid sequence SQPARAA (12) and using the human GABAAR β3 sequence (UniProt accession number P28472). The ELIC WT construct (UniProt accession number P0C7B7) was modified for expression in HEK293 cells, adding a Kozak sequence before the cDNA and using the human 5-HT3A subunit signal peptide as described previously (51).
Mutagenesis of GABAAR β3 subunit
Genes encoding the human GABAAR β3 WT, human GABAAR β3 C1, and Erwinia chrysanthemi ELIC WT were ordered from GeneWiz and cloned into pRK5 and pcDNA3.1 vectors. Single point mutations were performed by overlap extension PCR (52). The QuikChange® tool (Agilent) was utilized to design the primers. Multiple template-based sequential PCRs were used to obtain the 5-HT3A signal peptide–ELIC WT chimera (53).
PCR products, mutagenesis reactions, and ligations were verified using agarose gel electrophoresis and DNA sequencing (DNA Sequencing and Services, University of Dundee). The PCR and cloning reagents were bought from Agilent and Thermo Fisher, respectively.
The genes cloned into their respective vectors were used to transform Escherichia coli DH5α cells and grow cultures (500 ml of lysogeny broth medium with 50 μg/ml carbenicillin) at 37 °C overnight. The cells were harvested (6000 × g, 4 °C, 20 min) and used for Maxiprep (Qiagen) to obtain a higher yield of the plasmid.
Cell culture and transfection
HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 μg/ml penicillin, and 100 units/ml streptomycin at 37 °C and 5% CO2. Cells were seeded at low density in 35-mm dishes for electrophysiology. Transfections were performed by calcium phosphate precipitation using 1 μg of total cDNA per dish as described previously (26). The cDNAs encoding GABAAR β3 WT and the mutants were cloned into the pRK5 mammalian expression vector. The cDNA encoding ELIC WT was cloned into the pcDNA3.1 vector. The cDNA that encodes enhanced green fluorescent protein (0.1 μg; in pEGFP vector) was included to identify successfully transfected cells using fluorescence microscopy. Cells were washed with medium 16 h after transfection and used for voltage-clamp electrophysiology after 48–72 h. The tissue culture reagents were obtained from Invitrogen.
Electrophysiology
The whole-cell configuration of the patch-clamp technique was used to record propofol- or GABA-evoked currents from HEK293 cells transiently expressing GABAAR β3 WT, GABAAR β3 mutants, and ELIC WT. Recording electrodes were fabricated from borosilicate glass capillaries with resistances of 1.2–3.5 megaohms when filled with intracellular solution, which contained 140 mm CsCl, 2 mm MgCl2, 1.1 mm EGTA, 3 mm Mg-ATP, and 10 mm HEPES (pH 7.4 with CsOH). The extracellular solution contained 140 mm NaCl, 4.7 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 10 mm HEPES, and 10 mm glucose (pH 7.4 with NaOH). The solutions for ELIC WT were different. The intracellular solution contained 140 mm NaCl, 0.5 mm CaCl2, 5 mm EGTA, and 10 mm HEPES (pH 7.4 with NaOH). The extracellular solution contained 140 mm NaCl, 4.7 mm KCl, 1.2 mm MgCl2, 0.2 mm CaCl2, 10 mm HEPES, and 10 mm glucose (pH 7.4 with NaOH).
Cells were voltage-clamped at an electrode potential of −60 mV unless otherwise stated. Currents were evoked by rapid application of GABA or propofol using the three-pipe Perfusion Fast Step system (Warner Instruments) as described previously (26).
The data were recorded using an Axopatch 200B amplifier (Axon Instruments), low pass–filtered at 2 kHz, digitized at 10 kHz using a Digidata 1320 A interface (Molecular Devices), and acquired using pCLAMP8 software (Molecular Devices).
Data analyses
The analyses were carried out using Clampfit 10 (Molecular Devices), Excel 2011 (Microsoft), and Prism 5 (GraphPad). Peak amplitudes were measured using averaged traces from at least three currents. GABA-evoked current amplitudes were expressed as a percentage of the maximum and plotted as a concentration-response relationship. The following logistic (Equation 1) and bell-shaped equations (Equations 2 and 3) were fitted to the data points to determine the Hill slopes (nH) and the EC50.
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
Peak current densities were calculated by normalizing the peak current amplitude to the cell capacitance. The potentiation effect of propofol was calculated using the following formula,
| (Eq. 4) |
where Ipot and IGABA represent the potentiated and control peak current amplitudes, respectively. Activation rates were measured using 10–90% rise time of the GABA-evoked current. Deactivation rate was calculated by fitting a double-exponential function to the decay phase of the GABA-evoked current as follows,
| (Eq. 5) |
where τn are time constants and An represent the proportion of the particular τ. The best-fit number of exponential terms was determined using an F-test with confidence at the 95% level. Deactivation rates were provided as weighted τ values using the following equation.
| (Eq. 6) |
Statistical analyses
Data are presented as mean ± S.D. Differences of three or more groups were compared using one-way ANOVA. Subsequent multiple pairwise comparisons were performed using the Dunnett's or Tukey's correction. Student's t test was used for other pairwise comparisons. In all cases, p < 0.05 was considered statistically significant. Statistical analyses were performed in Prism 5.
Comparative modeling
The model for GABAAR β3 C1 was generated in Modeller v9.13 (54) using the GABAAR β3 structure (Protein Data Bank (PDB) code 4COF) (12) as a template. The proteins share 99% sequence identity according to MUSCLE sequence alignment (55) and thus are suitable for comparative modeling. The best model according to energy, spatial restraints, and stereochemistry was chosen using the Discrete Optimized Protein Energy (DOPE) score (56) and Ramachandran plot (57).
Molecular docking
Molsoft ICM v.3.8-3 (58) was used to perform docking calculations of GABA into the GABAAR β3 WT structure (PDB code 4COF) and the GABAAR β3 C1 model. The preparation of the receptor and ligand models involved adding hydrogens, calculating charges at pH 7.0, deleting waters, and treating the receptor as rigid and the ligand as flexible. The whole receptor or potentially important residues of the binding site were selected (principal side, Asp95–Leu99, Leu152–Thr161, and Asn197–Arg207; complementary side, Asn41–Ala45, Met61–Tyr66, Asn113–Leu118, Leu125–Ala135, and Ala174–Val178), and a box was created around the selection with a 3-Å distance between the residues and the edges. The results were ranked according to the ICM score, which takes into consideration the quality of the complex based on van der Waals interactions and the internal force-field energy of the ligand (58).
Author contributions
C. G. C. and D. T. B.-H. data curation; C. G. C. and D. T. B.-H. software; C. G. C. and D. T. B.-H. formal analysis; C. G. C. and D. T. B.-H. validation; C. G. C. and D. T. B.-H. investigation; C. G. C. and D. T. B.-H. visualization; C. G. C. and D. T. B.-H. methodology; C. G. C. and T. G. H. writing-original draft; D. T. B.-H., W. N. H., and T. G. H. supervision; D. T. B.-H., W. N. H., and T. G. H. writing-review and editing; W. N. H. and T. G. H. conceptualization; W. N. H. and T. G. H. resources; W. N. H. funding acquisition; W. N. H. and T. G. H. project administration.
Supplementary Material
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Science without Borders Scheme Grant BEX 0321/13-3 (to C. G. C.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Table S1.
- GABAAR
- GABA type A receptor
- ECD
- extracellular domain
- ELIC
- E. chrysanthemi ligand-gated ion channel
- HEK293
- human embryonic kidney 293
- ICD
- intracellular domain
- TM
- transmembrane
- TMD
- transmembrane domain
- pF
- picofarad
- I–V
- current–voltage
- ANOVA
- analysis of variance.
References
- 1. Sigel E., and Steinmann M. E. (2012) Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231 10.1074/jbc.R112.386664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnston G. A. (2005) GABAA receptor channel pharmacology. Curr. Pharm. Des. 11, 1867–1885 10.2174/1381612054021024 [DOI] [PubMed] [Google Scholar]
- 3. Zhang J., Xue F., Liu Y., Yang H., and Wang X. (2013) The structural mechanism of the Cys-loop receptor desensitization. Mol. Neurobiol. 48, 97–108 10.1007/s12035-013-8420-z [DOI] [PubMed] [Google Scholar]
- 4. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346, 967–989 10.1016/j.jmb.2004.12.031 [DOI] [PubMed] [Google Scholar]
- 5. Kelley S. P., Dunlop J. I., Kirkness E. F., Lambert J. J., and Peters J. A. (2003) A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature 424, 321–324 10.1038/nature01788 [DOI] [PubMed] [Google Scholar]
- 6. Baptista-Hon D. T., Deeb T. Z., Lambert J. J., Peters J. A., and Hales T. G. (2013) The minimum M3-M4 loop length of neurotransmitter-activated pentameric receptors is critical for the structural integrity of cytoplasmic portals. J. Biol. Chem. 288, 21558–21568 10.1074/jbc.M113.481689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nys M., Kesters D., and Ulens C. (2013) Structural insights into Cys-loop receptor function and ligand recognition. Biochem. Pharmacol. 86, 1042–1053 10.1016/j.bcp.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 8. Hibbs R. E., and Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 10.1038/nature10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baptista-Hon D. T., Krah A., Zachariae U., and Hales T. G. (2016) A role for loop G in the β1 strand in GABAA receptor activation. J. Physiol. 594, 5555–5571 10.1113/JP272463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whiting P. J. (2003) GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery? Drug Discov. Today 8, 445–450 10.1016/S1359-6446(03)02703-X [DOI] [PubMed] [Google Scholar]
- 11. Dawson G. R., Collinson N., and Atack J. R. (2005) Development of subtype selective GABAA modulators. CNS Spectr. 10, 21–27 10.1017/S1092852900009871 [DOI] [PubMed] [Google Scholar]
- 12. Miller P. S., and Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 10.1038/nature13293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saras A., Gisselmann G., Vogt-Eisele A. K., Erlkamp K. S., Kletke O., Pusch H., and Hatt H. (2008) Histamine action on vertebrate GABAA receptors: direct channel gating and potentiation of GABA responses. J. Biol. Chem. 283, 10470–10475 10.1074/jbc.M709993200 [DOI] [PubMed] [Google Scholar]
- 14. Seeger C., Christopeit T., Fuchs K., Grote K., Sieghart W., and Danielson U. H. (2012) Histaminergic pharmacology of homo-oligomeric β3 γ-aminobutyric acid type A receptors characterized by surface plasmon resonance biosensor technology. Biochem. Pharmacol. 84, 341–351 10.1016/j.bcp.2012.04.008 [DOI] [PubMed] [Google Scholar]
- 15. Kumar M., and Dillon G. H. (2016) Assessment of direct gating and allosteric modulatory effects of meprobamate in recombinant GABAA receptors. Eur. J. Pharmacol. 775, 149–158 10.1016/j.ejphar.2016.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies P. A., Kirkness E. F., and Hales T. G. (1997) Modulation by general anaesthetics of rat GABAA receptors comprised of α1β3 and β3 subunits expressed in human embryonic kidney 293 cells. Br. J. Pharmacol. 120, 899–909 10.1038/sj.bjp.0700987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miller P., Masiulis S., Malinauskas T., Kotecha A., Rao S., Chavali S., De Colibus L., Pardon E., Hannan S., Scott S., Sun Z., Frenz B., Klesse G., Li S., Diprose J., et al. (2018) Heteromeric GABAA receptor structures in positively-modulated active states. bioRxiv 10.1101/338343 [DOI] [Google Scholar]
- 18. Phulera S., Zhu H., Yu J., Claxton D. P., Yoder N., Yoshioka C., and Gouaux E. (2018) Cryo-EM structure of the benzodiazepine-sensitive α1β1γ2S tri-heteromeric GABAA receptor in complex with GABA. Elife 7, e39383 10.7554/eLife.39383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu S., Noviello C. M., Teng J., Walsh R. M. Jr., Kim J. J., and Hibbs R. E. (2018) Structure of a human synaptic GABAA receptor. Nature 559, 67–72 10.1038/s41586-018-0255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wooltorton J. R., Moss S. J., and Smart T. G. (1997) Pharmacological and physiological characterization of murine homomeric β3 GABAA receptors. Eur. J. Neurosci. 9, 2225–2235 10.1111/j.1460-9568.1997.tb01641.x [DOI] [PubMed] [Google Scholar]
- 21. Bergmann R., Kongsbak K., Sørensen P. L., Sander T., and Balle T. (2013) A unified model of the GABAA receptor comprising agonist and benzodiazepine binding sites. PLoS One 8, e52323 10.1371/journal.pone.0052323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ashby J. A., McGonigle I. V., Price K. L., Cohen N., Comitani F., Dougherty D. A., Molteni C., and Lummis S. C. (2012) GABA binding to an insect GABA receptor: a molecular dynamics and mutagenesis study. Biophys. J. 103, 2071–2081 10.1016/j.bpj.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jones M. V., Sahara Y., and Dzubay J. A., and Westbrook G. L. (1998) Defining affinity with the GABAA receptor. J. Neurosci. 18, 8590–8604 10.1523/JNEUROSCI.18-21-08590.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kloda J. H., and Czajkowski C. (2007) Agonist-, antagonist-, and benzodiazepine-induced structural changes in the α1 Met113-Leu132 region of the GABAA receptor. Mol. Pharmacol. 71, 483–493 10.1124/mol.106.028662 [DOI] [PubMed] [Google Scholar]
- 25. Hollands E. C., Dale T. J., Baxter A. W., Meadows H. J., Powell A. J., Clare J. J., and Trezise D. J. (2009) Population patch-clamp electrophysiology analysis of recombinant GABAA α1β3γ2 channels expressed in HEK-293 cells. J. Biomol. Screen. 14, 769–780 10.1177/1087057109335675 [DOI] [PubMed] [Google Scholar]
- 26. Baptista-Hon D. T., Gulbinaite S., and Hales T. G. (2017) Loop G in the GABAA receptor α1 subunit influences gating efficacy. J. Physiol. 595, 1725–1741 10.1113/JP273752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spurny R., Ramerstorfer J., Price K., Brams M., Ernst M., Nury H., Verheij M., Legrand P., Bertrand D., Bertrand S., Dougherty D. A., de Esch I. J., Corringer P.-J., Sieghart W., Lummis S. C., et al. (2012) Pentameric ligand-gated ion channel ELIC is activated by GABA and modulated by benzodiazepines. Proc. Natl. Acad. Sci. U.S.A. 109, E3028–E3034 10.1073/pnas.1208208109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Colquhoun D. (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists acid of the effects of mutating receptors. Br. J. Pharmacol. 125, 923–947 10.1038/sj.bjp.0702164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lavoie A. M., Tingey J. J., Harrison N. L., Pritchett D. B., and Twyman R. E. (1997) Activation and deactivation rates of recombinant GABAA receptor channels are dependent on α-subunit isoform. Biophys. J. 73, 2518–2526 10.1016/S0006-3495(97)78280-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hales T. G., and Lambert J. J. (1991) The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br. J. Pharmacol. 104, 619–628 10.1111/j.1476-5381.1991.tb12479.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orser B. A., Wang L. Y., Pennefather P. S., and MacDonald J. F. (1994) Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J. Neurosci. 14, 7747–7760 10.1523/JNEUROSCI.14-12-07747.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adodra S., and Hales T. G. (1995) Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1-7 neurones by propofol. Br. J. Pharmacol. 115, 953–960 10.1111/j.1476-5381.1995.tb15903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Defeudis F. V. (1986) Muscimol and central nervous system γ-aminobutyric acid receptors: studies with ligand-binding techniques, in The Receptors (Conn P. M., ed) pp. 135–152, Academic Press, Cambridge, Massachusetts [Google Scholar]
- 34. Rognan D., Boulanger T., Hoffmann R., Vercauteren D. P., Andre J. M., Durant F., and Wermuth C. G. (1992) Structure and molecular modeling of GABAA receptor antagonists. J. Med. Chem. 35, 1969–1977 10.1021/jm00089a005 [DOI] [PubMed] [Google Scholar]
- 35. Boileau A. J., Newell J. G., and Czajkowski C. (2002) GABAA receptor β2 Tyr97 and Leu99 line the GABA-binding site. Insights into mechanisms of agonist and antagonist actions. J. Biol. Chem. 277, 2931–2937 10.1074/jbc.M109334200 [DOI] [PubMed] [Google Scholar]
- 36. Holden J. H., and Czajkowski C. (2002) Different residues in the GABAA receptor α1T60-α1K70 region mediate GABA and SR-95531 actions. J. Biol. Chem. 277, 18785–18792 10.1074/jbc.M111778200 [DOI] [PubMed] [Google Scholar]
- 37. Bai D., Pennefather P. S., MacDonald J. F., and Orser B. A. (1999) The general anesthetic propofol slows deactivation and desensitization of GABAA receptors. J. Neurosci. 19, 10635–10646 10.1523/JNEUROSCI.19-24-10635.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mellor J. R., and Randall A. D. (1997) Frequency-dependent actions of benzodiazepines on GABAA receptors in cultured murine cerebellar granule cells. J. Physiol. 503, 353–369 10.1111/j.1469-7793.1997.353bh.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khom S., Baburin I., Timin E., Hohaus A., Trauner G., Kopp B., and Hering S. (2007) Valerenic acid potentiates and inhibits GABAA receptors: molecular mechanism and subunit specificity. Neuropharmacology 53, 178–187 10.1016/j.neuropharm.2007.04.018 [DOI] [PubMed] [Google Scholar]
- 40. Schwartz R. D., Suzdak P. D., and Paul S. M. (1986) γ-Aminobutyric acid (GABA)- and barbiturate-mediated 36Cl- uptake in rat brain synaptoneurosomes: evidence for rapid desensitization of the GABA receptor-coupled chloride ion channel. Mol. Pharmacol. 30, 419–426 [PubMed] [Google Scholar]
- 41. Thompson S. A., Whiting P. J., and Wafford K. A. (1996) Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br. J. Pharmacol. 117, 521–527 10.1111/j.1476-5381.1996.tb15221.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feng H. J., Bianchi M. T., and Macdonald R. L. (2004) Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABAA receptor currents. Mol. Pharmacol. 66, 988–1003 10.1124/mol.104.002543 [DOI] [PubMed] [Google Scholar]
- 43. McCartney M. R., Deeb T. Z., Henderson T. N., and Hales T. G. (2007) Tonically active GABAA receptors in hippocampal pyramidal neurons exhibit constitutive GABA-independent gating. Mol. Pharmacol. 71, 539–548 10.1124/mol.106.028597 [DOI] [PubMed] [Google Scholar]
- 44. Kash T. L., Jenkins A., Kelley J. C., Trudell J. R., and Harrison N. L. (2003) Coupling of agonist binding to channel gating in the GABAA receptor. Nature 421, 272–275 10.1038/nature01280 [DOI] [PubMed] [Google Scholar]
- 45. Hales T. G., Deeb T. Z., Tang H., Bollan K. A., King D. P., Johnson S. J., and Connolly C. N. (2006) An asymmetric contribution to γ-aminobutyric type A receptor function of a conserved lysine within TM2–3 of α1, β2, and γ2 subunits. J. Biol. Chem. 281, 17034–17043 10.1074/jbc.M603599200 [DOI] [PubMed] [Google Scholar]
- 46. Calimet N., Simoes M., Changeux J.-P., Karplus M., Taly A., and Cecchini M. (2013) A gating mechanism of pentameric ligand-gated ion channels. Proc. Natl. Acad. Sci. U.S.A. 110, E3987–E396 10.1073/pnas.1313785110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Althoff T., Hibbs R. E., Banerjee S., and Gouaux E. (2014) X-ray structures of GluCl in apo states reveal a gating mechanism of Cys-loop receptors. Nature 512, 333–337 10.1038/nature13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Torres V. I., and Weiss D. S. (2002) Identification of a tyrosine in the agonist binding site of the homomeric ρ1 γ-aminobutyric acid (GABA) receptor that, when mutated, produces spontaneous opening. J. Biol. Chem. 277, 43741–43748 10.1074/jbc.M202007200 [DOI] [PubMed] [Google Scholar]
- 49. Szczot M., Kisiel M., Czyzewska M. M., and Mozrzymas J. W. (2014) α1F64 residue at GABAA receptor binding site is involved in gating by influencing the receptor flipping transitions. J. Neurosci. 34, 3193–3209 10.1523/JNEUROSCI.2533-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harrison N. J., and Lummis S. C. (2006) Locating the carboxylate group of GABA in the homomeric rho GABAA receptor ligand-binding pocket. J. Biol. Chem. 281, 24455–24461 10.1074/jbc.M601775200 [DOI] [PubMed] [Google Scholar]
- 51. Trumper P., Hunter W. N., and Hales T. G. (2014) Development of a High Throughput Ligand Screening Method and Structural Studies of Pentameric Ligand Gated Ion Channels. Ph.D. thesis, University of Dundee, Dundee, UK [Google Scholar]
- 52. Heckman K. L., and Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 10.1038/nprot.2007.132 [DOI] [PubMed] [Google Scholar]
- 53. Shan Q., and Lynch J. W. (2010) Chimera construction using multiple-template-based sequential PCRs. J. Neurosci. Methods 193, 86–89 10.1016/j.jneumeth.2010.08.033 [DOI] [PubMed] [Google Scholar]
- 54. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.32 10.1002/0471250953.bi0506s47 [DOI] [PubMed] [Google Scholar]
- 55. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shen M. Y., and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 10.1110/ps.062416606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 58. Neves M. A., Totrov M., and Abagyan R. (2012) Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput. Aided. Mol. Des. 26, 675–686 10.1007/s10822-012-9547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.