

Abstract
β-Adrenergic stimulation of adipose tissue increases mitochondrial density and activity (browning) that are associated with improved whole-body metabolism. Whereas chronically elevated levels of reactive oxygen species (ROS) in adipose tissue contribute to insulin resistance, transient ROS elevation stimulates physiological processes such as adipogenesis. Here, using a combination of biochemical and cell and molecular biology–based approaches, we studied whether ROS or antioxidant treatment affects β3-adrenergic receptor (β3-AR) stimulation–induced adipose tissue browning. We found that β3-AR stimulation increases ROS levels in cultured adipocytes, but, unexpectedly, pretreatment with different antioxidants (N-acetylcysteine, vitamin E, or GSH ethyl ester) did not prevent this ROS increase. Using fluorescent probes, we discovered that the antioxidant treatments instead enhanced β3-AR stimulation–induced mitochondrial ROS production. This pro-oxidant effect of antioxidants was, even in the absence of β3-AR stimulation, associated with decreased oxygen consumption and increased lactate production in adipocytes. We observed similar antioxidant effects in WT mice: N-acetylcysteine blunted β3-AR stimulation–induced browning of white adipose tissue and reduced mitochondrial activity in brown adipose tissue even in the absence of β3-AR stimulation. Furthermore, N-acetylcysteine increased the levels of peroxiredoxin 3 and superoxide dismutase 2 in adipose tissue, indicating increased mitochondrial oxidative stress. We interpret this negative impact of antioxidants on oxygen consumption in vitro and adipose tissue browning in vivo as essential adaptations that prevent a further increase in mitochondrial ROS production. In summary, these results suggest that chronic antioxidant supplementation can produce a paradoxical increase in oxidative stress associated with mitochondrial dysfunction in adipocytes.
Keywords: reactive oxygen species (ROS), adipose tissue, adrenergic receptor, antioxidant, vitamin E, mitochondria, oxidative stress, adipocyte, metabolism, browning, glutathione, N-acetylcysteine
Introduction
Obese dysfunctional adipose tissue contributes to metabolic syndrome, whereas healthy adipose tissue, regardless of its size, is protective through e.g. its ability to remove excess nutrients from the blood stream. The discovery of new mechanisms underlying adipose tissue functionality is therefore important for increasing our knowledge about the pathogenesis and further treatment of obesity-related diseases. Several studies show that cold temperature or β-adrenergic receptor (β-AR)2 stimulation leads to browning of white and activation of brown adipose tissue, i.e. increased mitochondrial biogenesis associated with increased thermogenic activity and improved whole-body metabolic function. This phenomenon is also thought to involve formation of new beige/brite adipocytes within the white adipose depots (1), but the underlying molecular mechanism for this adipocyte browning process is not fully understood.
β3-AR activation of adipocytes potently induces lipolysis, which results in very high levels of free fatty acids. This increase in fatty acids is associated with a transient proinflammatory response and leads eventually to increased mitochondrial biogenesis and enhanced oxidative metabolism in adipose tissue (2, 3). Thus, we propose that browning should be interpreted as a favorable adaptation to metabolic stress.
Increased levels of reactive oxygen species (ROS) promote adipocyte differentiation (4), whereas treatment with the antioxidant N-acetyl-l-cysteine (NAC) reduces markers of differentiation in adipocytes in vitro (5). Furthermore, increased ROS levels have been shown to induce mitochondrial biogenesis (6), but the role of ROS and/or antioxidant treatment in adipose tissue browning has, to our knowledge, not been fully explored. Elevated fatty acid levels can cause an increase in ROS production from nonmitochondrial sources such as NADPH oxidases in adipocytes (7, 8). Furthermore, ROS is a by-product from oxidative metabolism, which also is expected to increase in response to β-adrenergic stimulation. Thus, we expect that the adipocyte ROS levels will increase at least transiently during the process of β3-AR agonist– or cold-induced browning. Based on the above-mentioned findings, we hypothesized that such increase in ROS plays an active role in adipose tissue browning and consequently that pretreatment with antioxidant will reduce the browning response. Here, we tested this hypothesis using CL316,246 (a selective β3-AR agonist)-treated cultured adipocytes and mice. In brief, we found that β3-AR stimulation increased the total ROS production in cultured adipocytes. However, we were unable to prevent this increase in ROS by 24-h antioxidant pretreatment. Neither NAC, vitamin E, nor GSH ethyl ester was effective in this regard despite leading to the expected increase in intracellular levels of reduced glutathione (GSH). Instead, these antioxidant treatment regimens led to increased mitochondrial ROS levels in β3-AR–stimulated adipocytes. Furthermore, this pro-oxidant effect of antioxidants in adipocytes was linked to reduced oxygen consumption, increased lactate production, and increased levels of endogenous mitochondrial antioxidant protein. Similar results were also seen in vivo: NAC treatment suppressed CL316,246-induced browning of white adipose tissue and mitochondrial activity of brown adipose tissue (BAT) in WT mice. In summary, this study shows that chronic antioxidant supplementation can lead to a paradoxical increase in oxidative stress associated with mitochondrial dysfunction in white and brown adipocytes. We interpret this negative impact of antioxidant on oxygen consumption in vitro and adipose tissue browning in vivo as essential adaptations to prevent further increase in mitochondrial ROS production. Finally, our data imply that antioxidant treatment may not be an appropriate approach to study the effect of ROS and ROS neutralization in (patho-)physiological processes.
Results
Antioxidant treatment increases the GSH content but does not decrease CL316,246-induced ROS production in cultured adipocytes
GSH can be found in both a reduced and an oxidized state, and combined, they form the most abundant protection system in the cell against oxidants or reducing agents with a normal ratio of 10:1 for the reduced versus the oxidized form in the cytosol (9). The synthesis of GSH is tightly regulated and highly dependent on the availability of cysteine (the sulfur amino acid precursor) (10). An effective strategy to increase the cellular GSH levels is therefore NAC treatment. Indeed, 48-h NAC treatment of cultured adipocytes increased the levels of GSH about 20%, whereas the level of oxidized GSH (GSSG) was unchanged, leading to an altered cellular redox balance (Fig. 1, A and B). Similarly, 48-h treatment with GSH ethyl ester that directly increases intracellular GSH led to a 10% increase of cellular GSH (Fig. 1A) without affecting GSSG levels (Fig. 1B). 48-h vitamin E treatment was equally effective as the GSH ethyl ester treatment with respect to the GSH increase (Fig. 1A), but unlike NAC and GSH ethyl ester, vitamin E also increased the levels of GSSG (Fig. 1B).
Figure 1.

β3-AR stimulation increases ROS production, and NAC pretreatment has no effect on this response. A, relative GSH levels in cultured adipocytes treated with or without NAC, GSH ethyl ester (Glut-est), and vitamin E (Vit E) for 48 h (n = 4/group; *, p < 0.05; **, p < 0.01; ****, p < 0.0001). B, relative GSSG levels in cultured adipocytes treated with or without NAC, GSH ethyl ester, and vitamin E for 48 h (n = 4/group; **, p < 0.01). C, representative ROS emission images (CM-H2DCFDA) of cultured adipocytes after 45-min (45′) incubation with 5 μm CL316,246. D, quantification of the ROS emission of cultured adipocytes pretreated with or without 1 mm NAC for 24 h after 60-min incubation with 1 μm CL316,246 (n = 3/group; experiment repeated three times). E and F, relative GSH (E) and GSSG (F) levels in cultured adipocytes in the presence or absence of 1 mm NAC for 48 h exposed to 1 μm CL316,246 or vehicle for 24 h (n = 4/group; *, p < 0.05). Error bars represent S.E. ns, not significant; RFU, relative fluorescence units; CTL, control.
To measure transient changes in the adipocyte's ROS production directly in real time (as opposed to indirectly through analysis of ROS-related damages), we used the probe 2′,7′-dichlorofluorescein diacetate (CM-H2DCFDA). This probe becomes fluorescent when reacting with ROS (H2O2, ROO−, and ONOO−) and is passively and homogeneously distributed in the cell and can thus be found in both cytosol and mitochondria (11–13). The ROS production was followed for 60 min post-β3-AR stimulation. Indeed, CL316,246-treated adipocytes had about 3-fold higher ROS levels than control adipocytes (Fig. 1, C and D), but NAC pretreatment had no significant effect (Fig. 1D). Similar results were obtained with GSH ethyl ester and vitamin E pretreatment (data not shown). Thus, in line with our hypothesis, β3-AR stimulation increased the general ROS production in cultured adipocytes, but to our surprise, we were unable to prevent this ROS increase by antioxidant pretreatment.
CL316,246 stimulation alone had no effect on either GSH or GSSG levels in cultured 3T3-L1 adipocytes (Fig. 1, E and F). This indicates either that healthy adipocytes are able to restore their GSH levels or that the possible decrease in GSH levels (due to the elevated ROS production) may have been too small to be detected. Surprisingly, when adipocytes were treated with both NAC and CL316,246, the GSH levels were similar to control cells, implying a very fast depletion of the increased GSH pool. Nevertheless, this GSH depletion was not sufficient to reduce the total ROS levels as judged by the H2DCFDA fluorescence shown in Fig. 1D. GSSG levels were not affected by single or combined treatment (Fig. 1F).
Antioxidant treatment increases the mitochondrial ROS production and reduces the oxygen consumption in cultured adipocytes
To, in real time, elucidate the impact of antioxidants specifically on mitochondrial ROS production, we used the fluorescent probe MitoSOX as described previously (8). CL316,246 treatment alone did not significantly affect mitochondrial superoxide production at early time points (the first 15 min post-β3-AR stimulation), indicating that the endogenous antioxidant system effectively neutralizes superoxide generated from the anticipated immediate increase in mitochondrial metabolism. At later time points, there was, however, an increase in mitochondrial superoxide levels in response to β3-AR stimulation, and paradoxically this increase was further enhanced by NAC pretreatment (Fig. 2A). The mitochondrial localization of MitoSOX depends on the mitochondrial membrane potential (extended information about this probe is provided under “Experimental procedures”). We therefore evaluated whether our NAC treatment regimen uncouples the mitochondria, but no such effect was seen (Fig. S1C). This indicates that the observed difference in MitoSOX staining is driven primarily by a difference in mitochondrial ROS production. Furthermore, a similar effect on CL316,246-induced mitochondrial superoxide levels was also detected in GSH ethyl ester– or vitamin E–treated adipocytes (Fig. 2, B and C), although vitamin E was less potent than the other antioxidants in this regard.
Figure 2.

NAC increases the mitochondrial ROS production and reduces oxygen consumption in cultured adipocytes. A, area under the curve (AUC) quantification of MitoSOX Red fluorescence of cultured adipocytes pretreated with or without 1 mm NAC for 24 h, 30 min after incubation with 5 μm CL316,246 (n = 3/group; **, p < 0.01). B, AUC quantification of MitoSOX Red fluorescence of cultured adipocytes pretreated with or without 5 mm GSH ethyl ester for 48 h, 30 min after incubation with 5 μm CL316,246 (n = 3/group; **, p < 0.01). C, AUC quantification of MitoSOX Red fluorescence of cultured adipocytes pretreated with or without 50 μm vitamin E for 48 h, 90 min after incubation with 5 μm CL316,246 (n = 3/group; *, p < 0.05). D, lactate levels in medium from cultured adipocytes incubated for 24 h with or without 1 mm NAC in combination with CL316,246 (n = 3/group; *, p < 0.05). E, oxygen consumption rate (OCR) of cultured adipocytes treated in the presence or absence of 1 mm NAC for 48 h and thereafter exposed to 1 μm CL316,246 or vehicle (Veh) for 20 min (n = 12/group; ****, p < 0.0001). F, oxygen consumption rate of cultured adipocytes treated with 1 mm NAC or 10 μm vitamin E (Vit E) (n = 12/group; *, p < 0.05; ****, p < 0.0001). G, oxygen consumption rates of cultured adipocytes after 30-min NAC treatment at the indicated concentrations (n = 10/group; ****, p < 0.0001). Error bars represent S.E. ns, not significant; RFU, relative fluorescence units; CTL, control.
Increased mitochondrial superoxide levels may lead to mitophagy and even apoptosis (14), but we did not detect any signs of increased cell death based on morphological appearance of the adipocytes. Therefore, we hypothesized that the mitochondrion protects itself from exceedingly high superoxide levels by reducing its oxidative phosphorylation, thus increasing the cell's reliance on glycolysis for sufficient cellular ATP production. To investigate this hypothesis, we analyzed mitochondrial function and lactate production in cultured adipocytes. β3-AR agonist treatment increased both oxygen consumption and lactate levels, whereas 48-h NAC treatment reduced oxygen consumption (Fig. 2E and Fig. S1B) and increased both basal and β3-AR agonist–induced lactate production (Fig. 2D) in cultured adipocytes. There was a trend for reduced basal oxygen consumption already after 24-h NAC treatment, and this effect became more pronounced after extended (48-h) treatment (Fig. 2F). Long-term vitamin E treatment (48 h) in cultured adipocytes caused a similar, albeit less potent, negative effect on basal oxygen consumption (Fig. 2F). In contrast but well in line with a previous study (11), short-term (30-min) NAC treatment caused a small dose-dependent increase in basal oxygen consumption (Fig. 2G). Furthermore, 48-h NAC treatment led to an ∼20% reduction of ATP production–linked respiration (Fig. S1D), whereas there was a strong tendency for increased maximal respiration rate and a significant ∼50% increase of the spare respiratory capacity (Fig. S1, E and F). Collectively, these data suggest that chronic NAC treatment of cultured adipocytes reduces the mitochondrial metabolism even though the capacity for respiration is increased.
Antioxidant treatment blunts β3-AR agonist–induced browning of white adipose tissue
To investigate whether the observed effect of antioxidants on mitochondrial function in cultured adipocytes also is relevant in the context of adipose tissue browning in vivo, we treated WT mice with either regular or NAC-supplemented drinking water. After 1 week, we induced browning by daily injections with CL316,246 for 10 days. Control animals received vehicle (PBS). No significant differences in total body weight or weight gain were found among the four groups (Fig. 3A). The inguinal and gonadal white adipose tissue (IWAT and GWAT) weights (normalized to body weight) were, however, increased in NAC-treated vehicle animals compared with regular water–drinking controls (Fig. 3B). Phosphoenolpyruvate carboxykinase (PEPCK), the rate-limiting enzyme for glyceroneogenesis and essential for re-esterification of fatty acids in adipose tissue, was increased 3.5-fold by NAC treatment in IWAT (Fig. S2A). This increase in PEPCK may explain the increased fat pad weight in NAC-treated vehicle animals. Furthermore, NAC reduced the CL316,246-induced mRNA and protein expression of several browning markers in IWAT (Fig. 3, C–E), and cytochrome c oxidase subunit 4 (COX4) protein expression was significantly reduced in NAC-treated mice independently of β3-AR activation. In line with these results, NAC blunted both the CL316,246-induced reduction in lipid density and the increase in mitochondrial density/activity (measured as percentage of cell area) as judged by combined coherent anti-Stokes Raman spectroscopy (CARS) and two-photon fluorescence microscopy (rhodamine 123), respectively, of freshly dissected IWAT (Fig. 4, A–C). CL316,246-induced browning of IWAT is typically associated with a reduction in lipid droplet size combined with an increase in the total number of droplets per cell. Both these effects could be seen in CL316,246-treated animals, but NAC preconditioning blunted this reduction in lipid density (Fig. 4, A and B). The CL316,246-induced difference in mitochondrial density/activity was almost significant between groups (p = 0.054) also when mitochondrial density and activity were analyzed as absolute areas, whereas the lipid density area was similar between groups, arguing that reduced mitochondrial density/activity is the primary mechanism underlying the blunted IWAT browning in NAC-treated animals (Fig. S3, A and B). The average adipocyte size did not differ between groups, but there is considerable variability in cell size, leading to larger variability in lipid density and mitochondrial density/activity when these parameters are analyzed as absolute area as opposed to percentage of cell area (Fig. S3E). No changes in morphology of lipid droplets could be observed in cells treated with NAC alone, either with regard to total density or number of droplets per cell (data not shown). It should, however, be noted that we found no effect of NAC pretreatment on the expression of browning markers in IWAT 3 and 24 h post-CL316,246 treatment (Fig. S3, B and C). This suggests that NAC treatment does not directly interfere with the initial browning response to β3-AR stimulation but rather affects components necessary to accomplish a complete browning response.
Figure 3.
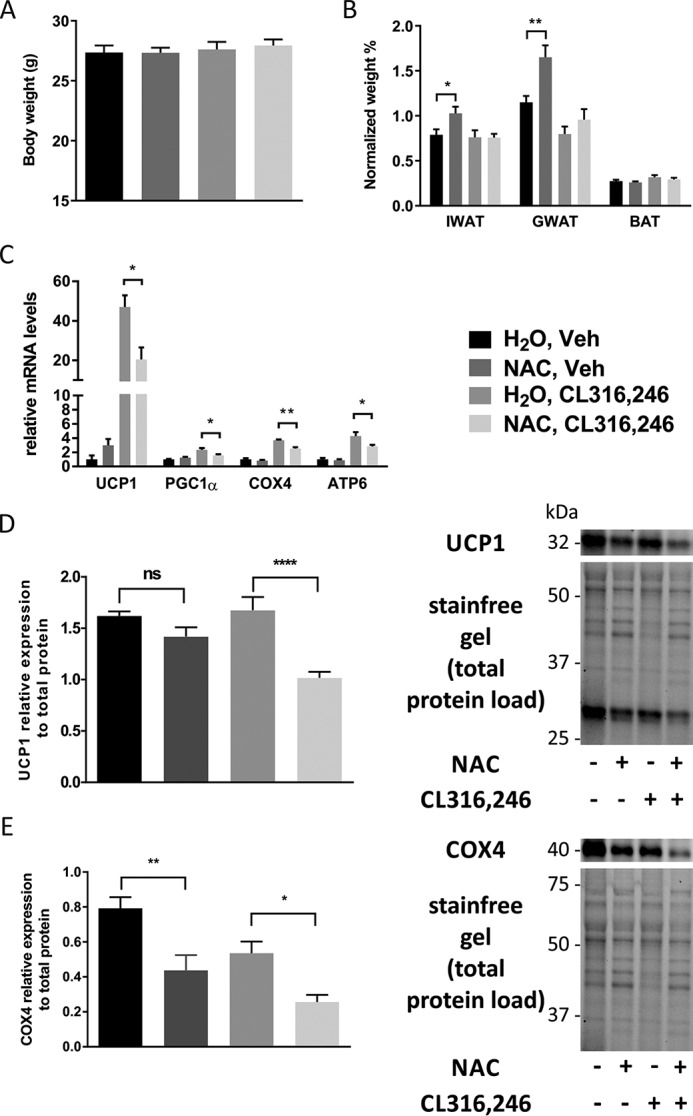
NAC pretreatment reduces browning in white adipose tissue in response to β3-AR stimulation. A, total body weight of C57/Bl6 mice pretreated for 1 week with either water or NAC (1 g/liter solution) followed by 10 days of injection with either vehicle or 1 μg of CL316,246/g of body weight. B and C, IWAT, GWAT, and BAT weight (normalized to total body weight) in response to the different treatment regimens in C57/Bl6 mice (B) and relative mRNA expression of UCP1, PGC1α, COX4, and ATP6 in IWAT (C) (n = 10/group; *, p < 0.05; **, p < 0.01). D and E, relative UCP1 (D) and COX4 (E) protein expression and representative blots in IWAT (n = 5/group; *, p < 0.05; **, p < 0.01; ****, p < 0.0001). Error bars represent S.E. ns, not significant; Veh, vehicle.
Figure 4.

NAC pretreatment reduces mitochondrial activity in white and brown adipose tissue in response to β3-AR stimulation. A, representative images of fresh pieces of IWAT of C57/Bl6 mice pretreated for 1 week with either water or NAC (1 g/liter solution) followed by 10 days of injection with either vehicle or 1 μg of CL316,246/g of body weight as judged by CARS (lipids; in purple) and two-photon fluorescence microscopy (mitochondria; in green). B and C, quantification of lipid content (B) and mitochondrial density/activity (C) in experiments depicted in A (n = 5/group; *, p < 0.05; **, p < 0.01). D, representative images of fresh pieces of BAT of C57/Bl6 mice pretreated for 1 week with either water or NAC (1 g/liter solution) followed by 10 days of injection with either vehicle or 1 μg of CL316,246/g of body weight as judged by CARS (lipids; in purple) and two-photon fluorescence microscopy (mitochondria; in green). E and F, quantification of lipid content (E) and mitochondrial density/activity (F) in experiments shown in D (n = 5/group; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). Error bars represent S.E.
To test whether this effect of NAC on browning in the context of chronic β3-AR stimulation could be generalized also to other antioxidants, we investigated the effect of high-dose vitamin E treatment on CL316,246-induced browning in mice. Vitamin E–treated mice did not show an increase of fat pad weights (Fig. 5A) but displayed a reduction in CL316,246-induced expression of the mitochondrial markers ATP synthase Fo subunit 6 (ATP6) and COX4 (Fig. 5B). Vitamin E treatment had, however, no significant effect on uncoupling protein 1 (UCP1) and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α) mRNA expression. Thus, both antioxidants interfered with CL316,246-induced browning, but NAC had a more potent effect than vitamin E (at least in the dosages tested).
Figure 5.
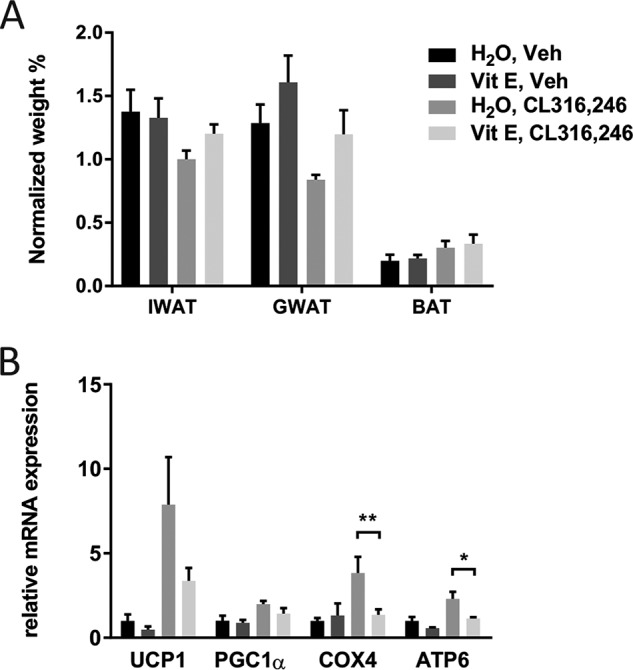
Vitamin E pretreatment reduces mitochondrial markers in white adipose tissue in response to β3-AR stimulation. A, IWAT, GWAT, and BAT weight (normalized to total body weight) in response to the different treatment regimens in C57/Bl6 mice (n = 10/group). B, relative mRNA expression of UCP1, PGC1α, COX4, and ATP6 in IWAT, post-chronic CL316,246 treatment (n = 10/group; *, p < 0.05; **, p < 0,01). Error bars represent S.E. Veh, vehicle; Vit E, vitamin E.
NAC reduces the mitochondrial activity in BAT
NAC treatment had no effect on BAT weight (Fig. 3B), and in line with several other studies, chronic CL316,246 treatment (10 days) did not affect the mRNA expression of the browning markers UCP1, PGC1α, and COX4 (Fig. 6A). BAT expression of ATP6 was, however, significantly increased compared with control, and expression of both COX4 and ATP6 was significantly reduced by NAC treatment (Fig. 6A). Interestingly, BAT protein expression of UCP1 was significantly up-regulated in CL316,246-treated mice, and this up-regulation was slightly blunted in NAC-supplemented animals (Fig. 6B). COX4 protein levels were elevated by β3-AR activation, but NAC had no effect (Fig. 6C). Mitochondrial density/activity (measured as percent area/cell) was unaffected by CL316,246 treatment as judged by two-photon fluorescence microscopy (rhodamine 123), but lipid density was reduced from 53 to 36% as determined from the CARS signal of freshly harvested BAT (Fig. 4, D–F). NAC preconditioning completely blocked this effect on lipid density of CL316,246 treatment, and much to our surprise, NAC treatment caused a 40% reduction in mitochondrial density and/or activity in brown adipocytes even in the absence of β3-AR stimulation (Fig. 4F). The difference in mitochondrial density/activity between NAC and controls remained significant even when measured as absolute area, and there was a similar trend in CL316,246-treated mice (not reaching significance due to slightly more heterogeneous cell size in these animals) (Fig. S3D). Lipid density measured as absolute area/cell and average brown adipocyte size did not differ between groups (Fig. S3, C and F), suggesting that the NAC-induced difference in mitochondrial density/activity is the primary driver of the observed BAT phenotype. Moreover, measurements of the relative protein levels of four OXPHOS complexes in BAT mitochondria showed no differences across the four groups, thus arguing that it is the mitochondrial activity rather than the density that is affected in BAT by the treatments (Fig. S4, A and B). In line with these assumptions, 48-h NAC treatment of cultured brown adipocytes reduced basal respiration, uncoupled respiration, ATP production–linked respiration, and spare respiratory capacity. With the exception of maximal respiration and spare respiratory capacity, these differences remained significant also in the CL316,246-treated brown adipocytes (Fig. S5, A–F). Lipid droplet morphology and size were similar between groups (data not shown). Thus, the negative effect of 48-h NAC treatment on respiration was even stronger in cultured brown adipocytes than in white 3T3-L1 adipocytes.
Figure 6.

NAC reduces mitochondrial activity in brown adipose tissue. A, relative mRNA expression of UCP1, PGC1α, COX4, and ATP6 in BAT of mice pretreated for 1 week with either water or NAC (1 g/liter solution) followed by 10 days of injection with either vehicle or 1 μg of CL316,246/g of body weight (n = 5/group; *, p < 0.05; **, p < 0.01). B and C, relative UCP1 (B) and COX4 (C) protein expression and representative blots in BAT (n = 5/group; *, p < 0.05) D, AUC quantification of the glycerol level in serum for a 60-min time course after β3-AR activation (n = 5/group). E, AUC quantification of FFA levels in serum for a 60-min time course after β3-AR activation (n = 5/group; ****, p < 0.0001; ≠, p < 0.0001). Error bars represent S.E. ns, not significant; Veh, vehicle.
To get a better understanding of these results and the impact of antioxidants on adipose tissue, we further investigated the effect of chronic NAC treatment on the acute lipolytic response to β3-AR agonist treatment. There was no difference in serum glycerol levels between control and NAC-treated animals either at baseline or in response to β3-AR stimulation (Fig. 6D), indicating that lipolysis is not affected by chronic NAC treatment. In contrast, free fatty acid (FFA) levels reached significantly higher levels in NAC-treated animals, both in vehicle control and in the β3-AR–stimulated group (Fig. 6E). Healthy adipocytes are able to take up a significant amount of released fatty acids during lipolysis, but these data suggest that this is not occurring to the same extent in NAC-treated animals compared with controls. Although we cannot determine which cells and tissues are responsible for the reduced fatty acid clearance under this condition, the facts that NAC treatment was associated with increased IWAT and GWAT size (Fig. 3B) and increased IWAT PEPCK expression (Fig. S2A) and that BAT is regarded as the major plasma lipid–clearing organ in rodents (15) indicate that BAT indeed is dysfunctional in NAC-treated mice. Collectively, we believe all these observations suggest that NAC treatment alone has a negative impact on mitochondrial activity in brown adipocytes.
Chronic NAC treatment leads to oxidative stress
Increased mitochondrial ROS production and reduced oxygen consumption along with the reduction of mitochondrial activity in brown adipocytes (even in the absence of β3-AR stimulation) in response to chronic antioxidant treatment were, to us, unexpected findings. We speculated that these results might result either from reduced activity of the endogenous mitochondrial antioxidant system due to exogenous delivery of antioxidants or from NAC and, to some extent, vitamin E acting as pro-oxidants under our experimental conditions.
Increased cellular ROS levels stabilize the transcription factor nuclear factor (erythroid-derived 2)–like 2 (NRF2) that protects cells against oxidative damage by stimulating the expression of antioxidant proteins such as peroxiredoxins (PRDXs) (16) or the mitochondrial superoxide dismutase 2 (SOD2) (17, 18). Protein levels of PRDX3 in IWAT were significantly up-regulated in animals treated with NAC, independently of the β3-AR activation (Fig. 7A), indicating increased oxidative stress in the mitochondria. Furthermore, SOD2 mRNA expression was up-regulated in IWAT in NAC-pretreated animals when treated chronically with CL316,246 (Fig. 7B), and IWAT SOD2 protein levels were elevated in NAC-treated animals, independently of β3-AR activation (Fig. 7C). To further confirm the effect of NAC pretreatment on the redox status of adipocytes, cultured adipocytes were treated with NAC for 24 and 48 h. At both time points, there was a significant up-regulation of NRF2 protein levels (Fig. 7D). It is important to highlight that PRDX2, abundant in the cytosol, was not up-regulated in any of the studied conditions (data not shown).
Figure 7.
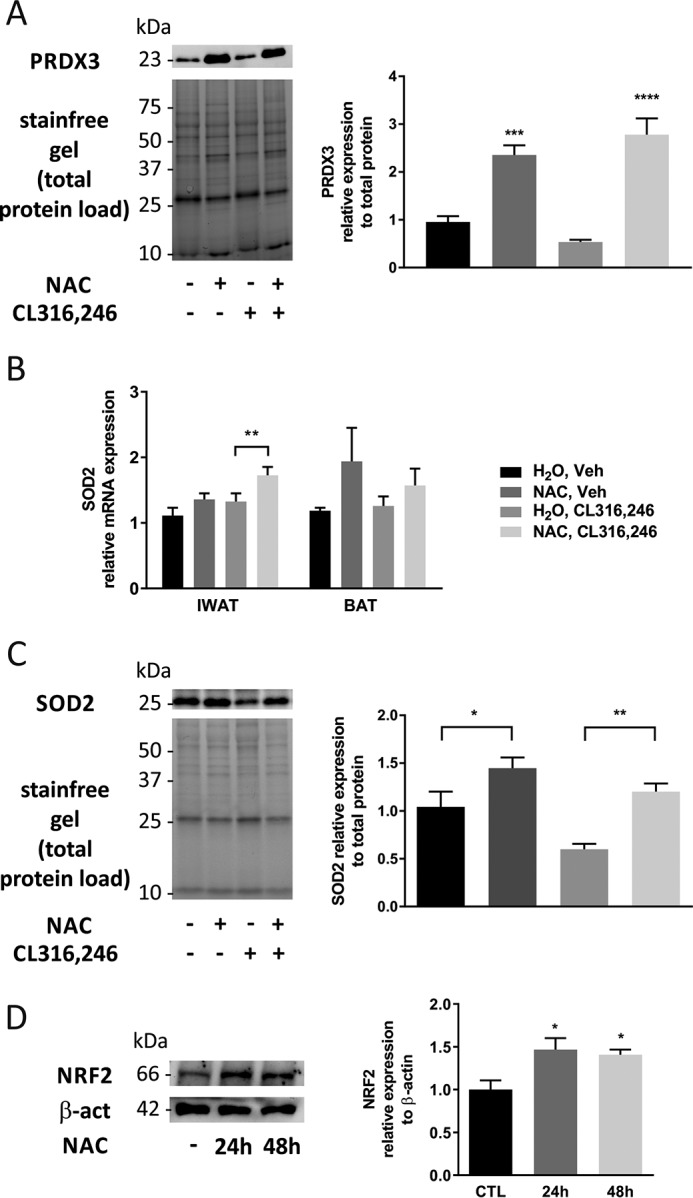
NAC changes the oxygen consumption profile and leads to oxidative stress in adipose tissue and cultured adipocytes. A, PRDX3 protein levels in IWAT after 10-day treatment with CL316,246 in C57/Bl6 mice on either NAC-supplemented or regular water (n = 5/group; ***, p < 0.001; ****, p < 0.0001). B, IWAT and BAT SOD2 mRNA expression after 10-day treatment with CL316,246 in C57/Bl6 mice on either NAC-supplemented or regular water (n = 5/group; **, p < 0.01). C, SOD2 protein levels in IWAT after 10-day treatment with CL316,246 in C57/Bl6 mice on either NAC-supplemented or regular water (n = 5/group; *, p < 0.05; **, p < 0.01). D, NRF2 protein levels in cultured adipocytes treated with 1 mm NAC at the indicated time points (n = 4/group; *, p < 0.05). Error bars represent S.E. CTL, control; Veh, vehicle; β-act, β-actin.
Overall, these data imply that NAC can act as a mitochondrial pro-oxidant, exacerbating the oxidative stress, leading to activation of the NRF2/Kelch-like ECH-associated protein 1 (KEAP1) pathway rather than reducing the endogenous antioxidant defense. The difference between short- and long-term effects of NAC on oxygen consumption could hence be explained on the basis of NAC acting both as an antioxidant and as a pro-oxidant depending on the condition. Acute NAC exposure (minutes) may effectively reduce oxidative stress, leading to increased oxygen consumption, whereas extensive NAC exposure (days) may instead engender the mitochondrial pro-oxidant effect (11).
Discussion
In the present study, we found that β3-AR stimulation increases the ROS levels in cultured adipocytes and that antioxidant treatment is ineffective in preventing this increase in ROS due to mitochondrial pro-oxidant activity under the studied experimental conditions. This increase in mitochondrial ROS levels was associated with increased lactate production, reduced oxygen consumption, and increased levels of NRF2 in cultured adipocytes. Thus, NAC increases the mitochondrial ROS levels, and this likely triggers the activation of the protective NRF2 pathway, the increase in glycolysis, the reduction in respiration, and the reduced β3-AR agonist–induced browning response. We also observed an increase in fat pad size along with elevated expression of the PEPCK (essential for re-esterification of fatty acids) together with increased expression of SOD2 and PRDX3 in IWAT of NAC-treated animals. Collectively, these effects of NAC in IWAT may be regarded as secondary adaptations to reduce the risk of lipotoxic side effects from increased fatty acid levels and to limit excessive mitochondrial ROS generation.
Short-term NAC treatment associated with reduced ROS levels has been shown to reduce lipolysis in cultured human adipocytes (19), but the effect of chronic NAC treatment on lipolysis has to our knowledge not previously been tested. We found that the acute lipolytic response to β3-AR stimulation as judged by the glycerol levels was unchanged in NAC-treated animals. This is well in line with our notion that long-term NAC treatment does not decrease the ROS levels in adipocytes. Interestingly, the circulating FFAs reached higher levels in NAC-treated animals compared with controls, suggesting reduced fatty acid clearance. Given that BAT is regarded as the major plasma lipid–clearing organ in rodents (15), we believe our data indicate that our chronic NAC treatment regimen leads to BAT dysfunction.
Clearly, we have a situation in the adipocytes where antioxidants act as mitochondrial pro-oxidants. Although such pro-oxidant effect of antioxidants may appear surprising, it is well in line with a previous myoblast study (20). We propose that this mitochondrial pro-oxidant effect of antioxidant treatment in cultured adipocytes results from increased levels of GSH. Similar to our study, it has been reported that NAC treatment, especially at low ROS levels, triggers a reductive GSH redox potential associated with pro-oxidative consequences in mitochondria in myoblasts (9). This alteration in GSH homeostasis can lead to a too reduced state that increases the mitochondrial ROS production through reduction of oxygen to superoxide anion (21), which has been referred as reductive stress in opposition to oxidative stress (22). Our data confirm the occurrence of such an undesired redox reaction where increased GSH levels in β3-AR–stimulated adipocytes may increase both the reduction of hydrogen peroxide to water in the cytosol and the reduction of molecular oxygen to superoxide in the mitochondria (Fig. 8). Reductive stress has also been connected to the unfolded protein response in the endoplasmic reticulum (23), but we did not find any evidence of such an effect under our experimental conditions (data not shown).
Figure 8.
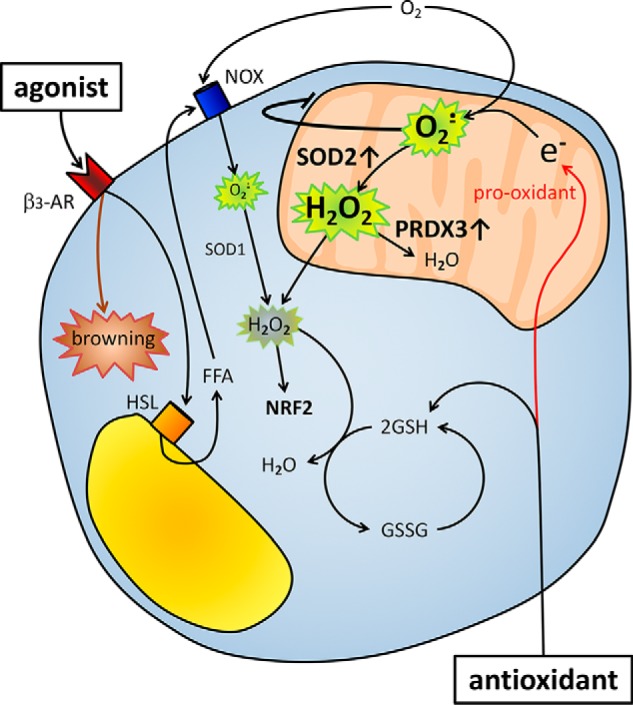
Model of NAC and β3-AR agonist effects on ROS and mitochondrial activity in adipocytes. β3-AR agonist treatment induces lipolysis, leading to an increase in FFAs, which in turn trigger an increase in adipocyte ROS levels, presumably via NOX4 (7, 8). NAC pretreatment increases GSH levels, leading to increased neutralization of hydrogen peroxide to water in the cytosol. Chronic antioxidant treatment leads to an increase in chemical reduction of molecular oxygen to superoxide in the mitochondrion. The adipocytes adapt to this pro-oxidant effect of chronic antioxidant treatment by reducing their oxidative metabolism and increasing their mitochondrial antioxidant proteins such as PRDX3 and SOD2. Consequently, chronic antioxidant treatment will blunt the β3-AR agonist–induced browning response and reduce BAT activity. NOX, NADPH oxidase; HSL, hormone-sensitive lipase.
Our results may seem difficult to reconcile with previous findings where antioxidant treatment has been shown to e.g. reduce lipolysis and stimulate oxidative metabolism in adipocytes as well as increase oxygen consumption in high-fat diet–fed mice (11, 19). Those studies have, however, focused on either short-term effects (hours) of antioxidants on adipocytes or effects of antioxidant treatment in high-fat diet–induced obese mice. Thus, we propose that the divergent results in our study compared with previous studies can be explained by a difference in duration of antioxidant exposure as well as by the prevailing redox state of the cell. Scavenging ROS with exogenous antioxidants in an acute setting and/or in a situation where the endogenous antioxidant system is already severely challenged by chronically increased ROS levels (24) may well have beneficial effects. Moreover, one may also consider the existence of an alleged hormesis effect where the mitochondrial pro-oxidant effect of NAC leads to protective NRF2 signaling and increased PRDX3 and SOD2 expression (as demonstrated in this study) that over time will overrule the negative impact of increased mitochondrial superoxide. Such a hormesis effect with NAC pretreatment has been previously reported in myoblasts (20). Finally, different responses to antioxidants in different cell types may well be explained by differences in mitochondrial concentration and activity combined with the different extents of treatment.
The findings presented here may also have implications for other studies where antioxidant treatment regimens are evaluated for their potential effect on disease progression. For instance, our data may explain why attempts to target the ROS balance in the treatment of type 2 diabetes have failed to prevent or improve the metabolic condition (25). It is also feasible that previously shown negative effects of antioxidants on e.g. tumor progression (26) and exercise (27) do not chiefly depend on excessive ROS neutralization but are rather due to an increase in pro-oxidant activity of the given antioxidant.
In conclusion, this study highlights how chronic antioxidant supplementation can lead to a paradoxical increase in oxidative stress associated with mitochondrial dysfunction in white and brown adipocytes. In the long run, chronic antioxidant treatment may thus impair adipose tissue functionality, leading to increased risk for developing whole-body metabolic dysfunction.
Experimental procedures
Cell culture of adipocytes
3T3-L1 cells (Zen-Bio, Research Triangle Park, NC) were maintained as subconfluent cultures in Dulbecco's modified Eagle's medium (DMEM) (high glucose, 4.5 g/liter; Life Technologies) with 10% calf serum and 1% penicillin-streptomycin. Differentiation was carried out as described previously (28). Studies were performed in completely differentiated adipocytes (based on occurrence of large lipid droplets) between days 8 and 9 from start of differentiation; only cultures in which >90% of cells displayed adipocyte morphology were used. Cells were washed twice with PBS and incubated with serum-free DMEM with or without N-acetyl-l-cysteine, vitamin E, or GSH ethyl ester for the specified time. β3-AR stimulation was performed using CL316,243 (Tocris, UK). All cell culture reagents were purchased from Sigma-Aldrich unless otherwise specified.
Isolation and culture of brown adipocytes
Brown adipose tissue from four mice (aged 3 weeks old) was carefully dissected, minced, and digested for 40 min in buffer containing 123 mm NaCl, 5 mm KCl, 5 mm CaCl2, 5 mm glucose, 100 mm HEPES, 4% BSA, and 1.5 mg/ml collagenase A. The filtered digest was spun at 1500 × g for 5 min, and the pellet was resuspended in high-glucose DMEM with 20% newborn calf serum and 20 mm HEPES and seeded in the corresponding plate homogeneously distributed in all wells. Culture medium (DMEM containing 10% fetal bovine serum, 1% penicillin-streptomycin, 4 nm insulin, 25 μg/ml sodium ascorbate, 10 mm HEPES, 1 mm sodium pyruvate, and 4 mm glutamine) was changed every other day until complete differentiation. Studies were performed in completely differentiated adipocytes (based on occurrence of multilocular lipid droplets) between days 8 and 9 from start of differentiation; only cultures in which >90% of cells displayed adipocyte morphology were used. Cells were washed twice with PBS and incubated with serum-free DMEM with or without N-acetyl-l-cysteine. β3-AR stimulation was performed using CL316,243 (Tocris). All cell culture reagents were purchased from Sigma-Aldrich unless otherwise specified.
Lactate release from cultured adipocytes
Medium from cultured adipocytes was centrifuged at 1000 × g for 5min. Lactate content was determined in the supernatant using the EnzyChrom l-Lactate assay kit (BioAssay Systems, Hayward, CA).
Measurement of GSH and GSSG in cultured adipocytes
Cultured adipocytes were washed twice with PBS and thereafter lysed with a 0.5% Triton X-100 (AMRESCO, Cleveland, OH) solution in PBS. Total GSH and GSH levels were measured in protein lysates using the GSH/GSSG ratio Detection Assay kit (Abcam, UK). The GSSG levels were calculated from the total GSH and GSH levels (GSSG = (Total GSH − GSH)/2).
Intracellular ROS generation in cultured adipocytes
Cultured adipocytes grown and differentiated in 12-well glass-bottom black plates (In Vitro Scientific, Mountain View, CA) or optical-ready culture glass Petri-Dishes (MatTek Corp., Ashland, MA) were washed twice with PBS and incubated with phenol red– and serum-free DMEM with or without NAC. After 24 h, membrane-permeable CM-H2DCFDA (Life Technologies) or MitoSOX (Life Technologies) was added, and cells were incubated for 30 min at 37 °C. Two washes with PBS were performed, and phenol red– and serum-free DMEM was added. Stained cells were analyzed using fluorometry (SpectraMax i3 fluorometer, Molecular Devices, San Jose, CA) or laser scanning confocal microscopy (LSM 700 laser scanning confocal microscope, Zeiss, Germany). To quantify the effect of β3-AR stimulation, fluorescence measurements or images were obtained at baseline followed by sequential measurements after addition of CL316,246. The total fluorescence level at every time point was quantified by calculating averages of 12-point readouts per well. Reduced mitochondrial membrane potential may decrease the mitochondrial specificity of MitoSOX (29). Our control experiments showed, however, that the nuclear MitoSOX stain, as judged by overlap between MitoSOX and Hoechst stains, was similar between groups, indicating that this error is unlikely to contribute to the observed differences in MitoSOX staining intensity.
Animal experiments
Five-week-old male WT C57/Bl6J mice were obtained from Harlan Netherlands B.V. (Horst, The Netherlands) and housed under standard conditions at the animal facility of the Laboratory for Experimental Biomedicine at the University of Gothenburg, Sweden. Water and food were provided ad libitum. All experiments were approved in advance by the ethics committee for animal care at the University of Gothenburg and were performed in accordance with all the relevant guidelines and regulations.
In vivo antioxidant and CL316,246 treatment
At the age of 7 weeks, the mice were randomized and divided into control and antioxidant groups. The antioxidant groups either received NAC through their drinking water (1 g/liter) or vitamin E–enriched food (0.5 g/kg of food; Lantmännen, Stockholm, Sweden). After 1 week of antioxidant preconditioning, mice were injected intraperitoneally daily with either vehicle or CL316,246 (1 μg/g of body weight) for 10 days. Thereafter, animals were euthanized, blood was collected, and tissues were rapidly dissected out, weighed, and snap frozen in liquid nitrogen. Serum and tissue samples were kept at −80 °C until further analysis.
Gene expression analysis
Total RNA from subcutaneous IWAT and BAT (intrascapular) was extracted with Isol-RNA Lysis Reagent (5 PRIME, Germany) with TissueLyser II (Qiagen Nordic, Sweden) and 5-mm stainless steel beads followed by isolation using the ReliaPrep RNA Miniprep System (Promega, Madison, WI). RNA was reverse transcribed into cDNA using the qScript kit (Quantabio), and SYBR Green Master Mix (Applied Biosystems, Beverly, MA) was used in the quantitative real-time PCRs. Primer sequences used were as follows: UCP1: forward, 5′-GTGAAGGTCAGAATGCAAGC-3′; reverse, 5′-AGGGCCCCCTTCATGAGGTC-3′; PGC1α: forward, 5′-ACAGCTTTCTGGGTGGATTG-3; reverse, 5′-TGAGGACCGCTAGCAAGTTT-3′; COX4: forward, 5′-CATACTTTCGATCGTGACTGGGTG-3; reverse, 5′-TTCTTGTCATAGTCCCACTTGGCG-3′; ATP6: forward, 5′-ACTTGCCCACTTCCTTCCACA-3; reverse, 5′-TAAGCCGGACTGCTAATGCCA-3′; PEPCK: forward, 5′-CTGCATAACGGTCTGGACTTC-3; reverse, 5′-CAGCAACTGCCCGTACTCC-3′; SOD2: forward, 5′-CCAGTGCAGGACCTCATTTT-3; reverse, 5′-CACCTTTGCCCAAGTCATCT-3′; and β-actin: forward, 5′-GACCCAGATCATGTTTGAGA-3′; reverse, 5′-GAGCATAGCCCTCGTAGAT-3′. A melting curve analysis was performed in each experiment for all genes to confirm specificity of single-target amplification. All samples were amplified in duplicate, and all measurements were performed under nuclease-free conditions. Gene expression levels were calculated using the 2−ΔΔCt method with β-actin as endogenous control (30).
Two-photon fluorescence and CARS microscopy
About 100 mg of adipose tissue were collected from the same anatomical region in each animal and thereafter cut in <1-mm3 pieces for analysis with nonlinear microscopy: two-photon fluorescence and CARS microscopy of mitochondria and lipid store densities, respectively. Immediately, living mitochondria were stained with 50 μm rhodamine 123 (Life Technologies) in PBS for 10 min at 37 °C. Tissues were rinsed twice with PBS, submerged in PBS in a glass-bottom dish (In Vitro Scientific), and kept in an on-stage incubator mounted on the microscope.
The custom-built nonlinear microscope setup and analysis method for intracellular lipid storages has been described in detail elsewhere (31). In brief, the specific vibration at 2845 cm−1 of carbon–hydrogen bonds in the alkylic chains of triglycerides were probed in a CARS process by overlapping two picosecond-pulsed laser beams at wavelengths 817 and 1064 nm (7 ps, 76 MHz), respectively, in the focal plane of an inverted optical microscope (Eclipse TE2000-E with C2 confocal scanning head, Nikon). The 1064 nm beam was generated together with a 532 nm beam by a diode-pumped solid-state laser (neodymium vanadate, PicoTrain, HighQ Laser/Spectra-Physics, Hohenems, Austria). The 532 nm beam pumped an optical parametric oscillator (Levante Emerald OPO, APE Angewandte Physik and Elektronik GmbH) to generate the 817 nm beam. The OPO output was spatially and temporally overlapped with the 1064 nm beam from the pump laser and tightly focused onto the sample using an oil immersion objective (40×; numerical aperture, 1.30). By means of dichroic mirrors and high-optical density filters, the forward-scattered CARS signal and backscattered two-photon fluorescence signal (excited by the 817 nm beam; collected in the 495–530 nm regime) were recorded using single-photon counting detector technology (Becker and Hickl GmbH).
Multiple planes, each with 512 × 512 pixels, were recorded at 5.04-μs pixel dwell time and summed over eight acquisitions resulting in 3D images of the tissues. Three different areas were recorded per sample. Images were analyzed with ImageJ offline using an adapted version of the quantitative voxel analysis as described previously (32). From thresholded two-photon fluorescence and CARS signal tissue images, cell size and lipid and mitochondrial contents were quantified by manually extracting these areas.
Protein expression quantification
3T3-L1 adipocytes were washed with PBS and thereafter lysed in lysis buffer (50 mm Tris, 0.25 m sucrose, 1 mm DTT, Complete, PhosSTOP, 1% Triton X-100, 1 mm EGTA, and 1 mm EDTA, pH 7.4), scraped, collected, and sonicated for 5 min to completely disrupt membranes. The resulting lysates were centrifuged at 13,000 × g for 5 min to remove cell debris. After protein quantification assay (Pierce BCA assay, Thermo Fisher), supernatants were further diluted with lysis buffer, mixed with 4× loading buffer with β-mercaptoethanol, and boiled for 5 min. Samples were loaded into an AnykD Mini or Midi-PROTEAN TGX Precast Protein Stain-Free Gel (Bio-Rad) and transferred to a blot using a Trans-Blot Turbo nitrocellulose or polyvinylidene difluoride Transfer Pack (Bio-Rad). Anti-UCP1 (1:500 dilution; ab10983, Abcam), Total OXPHOS Rodent WB Antibody Mixture (1:250; ab110413, Abcam), anti-NRF2 (1:500 dilution; MAB3926, R&D Systems, Minneapolis, MN), anti-PRDX3 (1:500 dilution; ab73349, Abcam), anti-rabbit-HRP (1:5000; 31461, Thermo Fisher), anti-mouse-HRP (1:1000; ab6789, Abcam), anti-PRDX3 (1:500 dilution; ab73349, Abcam), anti-SOD2 (1:5000; ab13533, Abcam), and anti-β-actin-HRP (1:1000 dilution; ab20272, Abcam) were used as antibodies, and the blot pictures were acquired and analyzed with the ChemiDoc Touch imager (Bio-Rad). Total protein in samples was quantified through stain-free technology (33).
IWAT and BAT proteins were extracted with lysis buffer using 5-mm stainless steel beads and a TissueLyser II. After tissue disruption (5 min, 30 Hz), the resulting homogenates were centrifuged at 10,000 × g for 10 min. The aqueous phase was moved into new tubes and centrifuged at 4000 × g for 10 min. Thereafter, the same protocol as described above was followed.
Oxygen consumption rate
3T3-L1 cells or stromal vascular cells isolated from brown adipose tissue were seeded into XF96 microplates (Seahorse Bioscience, Agilent Technologies, Santa Clara, CA) at a density of 5000 cells/well. Cells were differentiated into mature adipocytes following standard protocols. Mature adipocytes were washed three times with assay medium (nonbuffered DMEM, Seahorse Biosciences) supplemented with 25 mm glucose and 1 mm sodium pyruvate and incubated for 1 h in a non-CO2 incubator at 37 °C. Basal respiration was determined as oxygen consumption rate in the presence of substrate alone. OXPHOS parameters were calculated following Agilent Technologies instructions, and conditions are described for every experiment. Results were normalized to the number of cells (quantified as described below) in each well.
Cell counting and imaging of adipocytes
After each experiment with the XF96 Seahorse, 8 μl of NucRed Live 647 (Thermo Fisher) were added to each well. Cells were incubated for 20 min at 37 °C and 5% CO2 and washed twice with PBS prior to addition of phenol red–free DMEM with 10% fetal bovine serum. Thereafter, cells were imaged and counted using a SpectraMax i3x.
Serum glycerol and FFA analysis
Serum glycerol and FFA levels were determined using, respectively, the Free Glycerol Determination kit (Sigma Aldrich) and the acyl-CoA synthetase–acyl-CoA-oxidase method using the HR Series NEFA-HR(2) Determination kit (FUJIFILM Wako Chemicals, Neuss, Germany) according to the manufacturers' protocols.
Statistical analysis
GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for statistical analysis. Values are shown as mean ± S.E. Comparisons were performed using one-way analysis of variance, two-way analysis of variance, or two-tailed t test; data were log-transformed as necessary to achieve normal distributions; and p < 0.05 was considered significant.
Data availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author contributions
E. P. and I. W. A. conceptualization; E. P., P. M., A. P., and C. S. O. data curation; E. P. and A. P. formal analysis; E. P. validation; E. P., P. M., A. P., and V. P. investigation; E. P. and A. P. visualization; E. P., A. P., and I. W. A. writing-original draft; E. P., C. S. O., and I. W. A. writing-review and editing; A. P., A. E., and M. B.-T. methodology; V. P., A. E., and I. W. A. resources; M. B.-T. and I. W. A. supervision; I. W. A. funding acquisition; I. W. A. project administration.
Supplementary Material
Acknowledgments
We thank Birgit Linder and Ann-Marie Alborn (Department of Metabolic Physiology, University of Gothenburg) for excellent assistance with cell culturing and Professor Henrik Hagberg (Department of Obstetrics and Gynecology, University of Gothenburg) who generously allowed the use of the Seahorse instrument.
This work was supported by Swedish Research Council Grants 2012-1601, 2013-7107, and 2017-00792; a Novo Nordisk Foundation Excellence project grant; VINNOVA (Swedish Governmental Agency for Innovation Systems); the Swedish Diabetes Foundation; the Diabetes Wellness Research Foundation; the Åke Wiberg Foundation; the IngaBritt and Arne Lundberg Foundation; the Magnus Bergvall Foundation; and the European Union's Seventh Framework Programme (FP7/2007–2013, Grant 607842). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5.
- β-AR
- β-adrenergic receptor
- ATP6
- ATP synthase Fo subunit 6
- BAT
- brown adipose tissue
- CARS
- coherent anti-Stokes Raman spectroscopy
- CM-H2DCFDA
- 2′,7′-dichlorofluorescein diacetate
- COX4
- cytochrome c oxidase subunit 4
- GWAT
- gonadal white adipose tissue
- IWAT
- inguinal subcutaneous white adipose tissue
- NAC
- N-acetyl-l-cysteine
- NRF2
- nuclear factor (erythroid-derived 2)–like 2
- PEPCK
- phosphoenolpyruvate carboxykinase
- PGC1α
- peroxisome proliferator-activated receptor γ coactivator 1-α
- PRDX
- peroxiredoxin
- ROS
- reactive oxygen species
- SOD2
- superoxide dismutase 2
- UCP1
- uncoupling protein 1
- OXPHOS
- oxidative phosphorylation
- FFA
- free fatty acid
- DMEM
- Dulbecco's modified Eagle's medium
- OPO
- optical parametric oscillator
- HRP
- horseradish peroxidase
- AUC
- area under the curve.
References
- 1. Wu J., Cohen P., and Spiegelman B. M. (2013) Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 27, 234–250 10.1101/gad.211649.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li P., Zhu Z., Lu Y., and Granneman J. G. (2005) Metabolic and cellular plasticity in white adipose tissue II: role of peroxisome proliferator-activated receptor-α. Am. J. Physiol. Endocrinol. Metab. 289, E617–E626 10.1152/ajpendo.00010.2005 [DOI] [PubMed] [Google Scholar]
- 3. Wernstedt Asterholm I., Tao C., Morley T. S., Wang Q. A., Delgado-Lopez F., Wang Z. V., and Scherer P. E. (2014) Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20, 103–118 10.1016/j.cmet.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee H., Lee Y. J., Choi H., Ko E. H., and Kim J.-W. (2009) Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 284, 10601–10609 10.1074/jbc.M808742200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calzadilla P., Sapochnik D., Cosentino S., Diz V., Dicelio L., Calvo J. C., and Guerra L. N. (2011) N-Acetylcysteine reduces markers of differentiation in 3T3-L1 adipocytes. Int. J. Mol. Sci. 12, 6936–6951 10.3390/ijms12106936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chevtzoff C., Yoboue E. D., Galinier A., Casteilla L., Daignan-Fornier B., Rigoulet M., and Devin A. (2010) Reactive oxygen species-mediated regulation of mitochondrial biogenesis in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 285, 1733–1742 10.1074/jbc.M109.019570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., and Shimomura I. (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 114, 1752–1761 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han C. Y., Umemoto T., Omer M., Den Hartigh L. J., Chiba T., LeBoeuf R., Buller C. L., Sweet I. R., Pennathur S., Abel E. D., and Chait A. (2012) NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 287, 10379–10393 10.1074/jbc.M111.304998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang H., Limphong P., Pieper J., Liu Q., Rodesch C. K., Christians E., and Benjamin I. J. (2012) Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 26, 1442–1451 10.1096/fj.11-199869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu S. C. (2009) Regulation of glutathione synthesis. Mol. Aspects Med. 30, 42–59 10.1016/j.mam.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang T., Si Y., Shirihai O. S., Si H., Schultz V., Corkey R. F., Hu L., Deeney J. T., Guo W., and Corkey B. E. (2010) Respiration in adipocytes is inhibited by reactive oxygen species. Obesity 18, 1493–1502 10.1038/oby.2009.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oparka M., Walczak J., Malinska D., van Oppen L. M. P. E., Szczepanowska J., Koopman W. J. H., and Wieckowski M. R. (2016) Quantifying ROS levels using CM-H2DCFDA and HyPer. Methods 109, 3–11 10.1016/j.ymeth.2016.06.008 [DOI] [PubMed] [Google Scholar]
- 13. Eruslanov E., and Kusmartsev S. (2010) Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 594, 57–72 10.1007/978-1-60761-411-1_4 [DOI] [PubMed] [Google Scholar]
- 14. Frank M., Duvezin-Caubet S., Koob S., Occhipinti A., Jagasia R., Petcherski A., Ruonala M. O., Priault M., Salin B., and Reichert A. S. (2012) Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 1823, 2297–2310 10.1016/j.bbamcr.2012.08.007 [DOI] [PubMed] [Google Scholar]
- 15. Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K., Kaul M. G., Tromsdorf U. I., Weller H., Waurisch C., Eychmüller A., Gordts P. L., Rinninger F., Bruegelmann K., Freund B., et al. (2011) Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17, 200–205 10.1038/nm.2297 [DOI] [PubMed] [Google Scholar]
- 16. Ma Q. (2013) Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426 10.1146/annurev-pharmtox-011112-140320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bryan H. K., Olayanju A., Goldring C. E., and Park B. K. (2013) The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 85, 705–717 10.1016/j.bcp.2012.11.016 [DOI] [PubMed] [Google Scholar]
- 18. Hamanaka R. B., and Chandel N. S. (2010) Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513 10.1016/j.tibs.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krawczyk S. A., Haller J. F., Ferrante T., Zoeller R. A., and Corkey B. E. (2012) Reactive oxygen species facilitate translocation of hormone sensitive lipase to the lipid droplet during lipolysis in human differentiated adipocytes. PLoS One 7, e34904 10.1371/journal.pone.0034904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh F., Charles A.-L., Schlagowski A.-I., Bouitbir J., Bonifacio A., Piquard F., Krähenbühl S., Geny B., and Zoll J. (2015) Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim. Biophys. Acta 1853, 1574–1585 10.1016/j.bbamcr.2015.03.006 [DOI] [PubMed] [Google Scholar]
- 21. Korge P., Calmettes G., and Weiss J. N. (2015) Increased reactive oxygen species production during reductive stress: the roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 1847, 514–525 10.1016/j.bbabio.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schafer F. Q., and Buettner G. R. (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212 10.1016/S0891-5849(01)00480-4 [DOI] [PubMed] [Google Scholar]
- 23. Maity S., Rajkumar A., Matai L., Bhat A., Ghosh A., Agam G., Kaur S., Bhatt N. R., Mukhopadhyay A., Sengupta S., and Chakraborty K. (2016) Oxidative homeostasis regulates the response to reductive endoplasmic reticulum stress through translation control. Cell Rep. 16, 851–865 10.1016/j.celrep.2016.06.025 [DOI] [PubMed] [Google Scholar]
- 24. Matsuzawa-Nagata N., Takamura T., Ando H., Nakamura S., Kurita S., Misu H., Ota T., Yokoyama M., Honda M., Miyamoto K., and Kaneko S. (2008) Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 57, 1071–1077 10.1016/j.metabol.2008.03.010 [DOI] [PubMed] [Google Scholar]
- 25. Castro J. P., Grune T., and Speckmann B. (2016) The two faces of ROS in adipocyte function and dysfunction. Biol. Chem. 397, 709–724 10.1515/hsz-2015-0305 [DOI] [PubMed] [Google Scholar]
- 26. Sayin V. I., Ibrahim M. X., Larsson E., Nilsson J. A., Lindahl P., and Bergo M. O. (2014) Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6, 221ra15 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- 27. Ristow M., Zarse K., Oberbach A., Klöting N., Birringer M., Kiehntopf M., Stumvoll M., Kahn C. R., and Blüher M. (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8665–8670 10.1073/pnas.0903485106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arsenijevic T., Grégoire F., Delforge V., Delporte C., and Perret J. (2012) Murine 3T3-L1 adipocyte cell differentiation model: validated reference genes for qPCR gene expression analysis. PLoS One 7, e37517 10.1371/journal.pone.0037517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Polster B. M., Nicholls D. G., Ge S. X., and Roelofs B. A. (2014) Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol. 547, 225–250 10.1016/B978-0-12-801415-8.00013-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 31. Enejder A., Brackmann C., and Svedberg F. (2010) Coherent anti-Stokes Raman scattering microscopy of cellular lipid storage. IEEE J. Sel. Top. Quantum Electron. 16, 506–515 10.1109/JSTQE.2009.2032512 [DOI] [Google Scholar]
- 32. Brännmark C., Paul A., Ribeiro D., Magnusson B., Brolén G., Enejder A., and Forslöw A. (2014) Increased adipogenesis of human adipose-derived stem cells on polycaprolactone fiber matrices. PLoS One 9, e113620 10.1371/journal.pone.0113620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gürtler A., Kunz N., Gomolka M., Hornhardt S., Friedl A. A., McDonald K., Kohn J. E., and Posch A. (2013) Stain-free technology as a normalization tool in Western blot analysis. Anal. Biochem. 433, 105–111 10.1016/j.ab.2012.10.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.