

Abstract
Reversible ubiquitination of G protein–coupled receptors regulates their trafficking and signaling; whether deubiquitinases regulate myocardial β1-adrenergic receptors (β1ARs) is unknown. We report that ubiquitin-specific protease 20 (USP20) deubiquitinates and attenuates lysosomal trafficking of the β1AR. β1AR-induced phosphorylation of USP20 Ser-333 by protein kinase A-α (PKAα) was required for optimal USP20-mediated regulation of β1AR lysosomal trafficking. Both phosphomimetic (S333D) and phosphorylation-impaired (S333A) USP20 possess intrinsic deubiquitinase activity equivalent to WT activity. However, unlike USP20 WT and S333D, the S333A mutant associated poorly with the β1AR and failed to deubiquitinate the β1AR. USP20–KO mice showed normal baseline systolic function but impaired β1AR-induced contractility and relaxation. Dobutamine stimulation did not increase cAMP in USP20–KO left ventricles (LVs), whereas NKH477-induced adenylyl cyclase activity was equivalent to WT. The USP20 homolog USP33, which shares redundant roles with USP20, had no effect on β1AR ubiquitination, but USP33 was up-regulated in USP20–KO hearts suggesting compensatory regulation. Myocardial β1AR expression in USP20–KO was drastically reduced, whereas β2AR expression was maintained as determined by radioligand binding in LV sarcolemmal membranes. Phospho-USP20 was significantly increased in LVs of wildtype (WT) mice after a 1-week catecholamine infusion and a 2-week chronic pressure overload induced by transverse aortic constriction (TAC). Phospho-USP20 was undetectable in β1AR KO mice subjected to TAC, suggesting a role for USP20 phosphorylation in cardiac response to pressure overload. We conclude that USP20 regulates β1AR signaling in vitro and in vivo. Additionally, β1AR-induced USP20 phosphorylation may serve as a feed-forward mechanism to stabilize β1AR expression and signaling during pathological insults to the myocardium.
Keywords: ubiquitylation (ubiquitination), protein kinase A (PKA), lysosome, G protein-coupled receptor (GPCR), endocytosis
Introduction
β-Adrenergic receptors (βARs)3 are G protein–coupled receptors (GPCRs) that respond to signals from the sympathetic nervous system and regulate cardiovascular functions (1, 2). Out of the three βAR subtypes expressed in the healthy heart, the β1AR and β2AR subtypes are the most abundant, representing 70–80% and 20–30% of total βARs, respectively (3). These receptors couple to the heterotrimeric G protein Gs upon catecholamine stimulation and activate adenylyl cyclase to promote cAMP production and protein kinase A (PKA) activation (1, 2). The β1AR is the main regulator of catecholamine-induced cardiac chronotropy and inotropy because mouse hearts lacking either β1AR or both β1- and β2AR function normally at the basal state but fail to respond to β-adrenergic agonists; in contrast, mice lacking only β2AR have preserved cardiac responsiveness to catecholamine stimulation (4–8).
Activated β1AR and β2AR are regulated via phosphorylation by GPCR kinases (GRKs) and binding of the multifunctional adaptor proteins called β-arrestins, which precede receptor endocytosis and post-endocytic sorting (1, 9). β1AR signaling is significantly attenuated in failing hearts due to receptor desensitization and down-regulation (10, 11). However, β2AR, which is mostly desensitized but not down-regulated in the failing heart, can promote anti-apoptotic signaling and is considered to be cardioprotective according to studies in mice and cardiomyocytes (12–14). β1AR down-regulation during heart failure may be compensatory to reduce cardiac metabolic demand and limit cardiotoxic signaling associated with chronic βAR stimulation. The down-regulation of β1AR in failing hearts is observed at both mRNA and protein levels; the latter process entails trafficking and lysosomal degradation of internalized receptors (10, 15–17). Another hallmark of failing hearts is the refractory nature of βARs due to sequestration of desensitized receptor protein in late endosomes (16). Recruitment of phosphoinositide 3-kinase (PI3K) activity to the β1AR complex promotes β1AR sequestration into late endosomes, and blockade of PI3K recruitment mobilizes the trafficking of β1AR from late endosomes to the cell surface (16, 17).
GPCR lysosomal trafficking and degradation can also be modulated by ubiquitination, a post-translational modification that appends monomers or polymeric chains of the 76-amino acid protein ubiquitin to substrate proteins (18, 19). Although the dynamics and mechanisms of β2AR ubiquitination have been broadly characterized (20–24), the mechanisms that regulate β1AR ubiquitination and associated vesicular trafficking are largely unknown. In this study, we discovered that ubiquitin-specific protease 20 (USP20) deubiquitinates β1AR and regulates β1AR trafficking. We also generated and characterized a novel USP20 knockout mouse model to determine the effect of this deubiquitinase (DUB) on βAR expression and cardiac response to catecholamine stimulation.
Results
USP20 functions as a cognate deubiquitinase for the β1AR
Agonist stimulation of FLAG-tagged β1AR expressed in HEK-293 cells with isoproterenol (Iso) resulted in a detectable increase in receptor ubiquitination, which peaked at 60 min after agonist treatment and decreased to basal levels beyond 6 h of agonist stimulation (Fig. 1, A and B). Agonist-induced lysosomal trafficking of the β1AR was initiated after 1 h of Iso stimulation as assessed by β1AR-LAMP1 colocalization. However, significant amounts of β1AR trafficked to lysosomes only after 6 h of agonist treatment (Fig. 1, C and D). Agonist-induced β1AR ubiquitination was detectable only when 10 mm N-ethylmaleimide (NEM, an inhibitor of deubiquitinating enzymes), but not when an inhibitor of 26S proteasomal activity such as MG132, was included in the lysis and immunoprecipitation buffers. This suggests that ubiquitination of β1ARs is rapidly reversed by deubiquitinase activity that associates with the β1AR. We previously demonstrated that the two homologous DUBs USP20 and USP33 function redundantly to deubiquitinate and regulate post-endocytic sorting of the β2AR to lysosomes (23). We therefore tested whether ubiquitination and trafficking of the β1AR are regulated by deubiquitinases in a similar manner as the β2AR.
Figure 1.

Agonist-induced ubiquitination and lysosomal trafficking of the β1AR. A, HEK-293 cells stably transfected with FLAG-β1AR were stimulated with 1 μm isoproterenol (Iso) for indicated times and subjected to FLAG IP followed by serial immunoblotting (IB) with anti-ubiquitin antibody (rabbit polyclonal, Bethyl Laboratories Inc.) and polyclonal FLAG antibody. The 1st lane shows the background signal obtained from HEK-293 cells transfected with vector. B, ubiquitin smears were quantitated and normalized to cognate FLAG-β1AR bands and plotted as % maximum signal (see “Experimental procedures”). The graph includes means ± S.E. from four independent experiments. *, p < 0.05 compared with 0 min, one-way ANOVA, and Bonferroni's test. C, HEK-293 cells with stable transfection of β1AR-CFP were stimulated with 1 μm Iso for the indicated times, and the distribution of β1AR (red) and LAMP1 (green) was visualized with LSM-510 confocal microscope. Representative images are shown, and quantification of colocalization from ≥13 z-stack images from three independent experiments are included in the bar graph in D. We used Imaris software to quantify volume of colocalization from images in a Z-stack. *, p < 0.05, versus 0 h, one-way ANOVA, and Bonferroni's test. Scale bars in C, 10 μm.
USP20 knockdown significantly increased ubiquitination of the β1AR in both unstimulated and agonist-stimulated cells when compared with cells transfected with control siRNA (Fig. 2, A and B). In contrast, depletion of USP33 had no significant effect on β1AR ubiquitination compared with control knockdown (Fig. 2C). For a number of GPCRs, ubiquitination serves as a sorting signal to promote their trafficking to late endosomes and lysosomes (19, 25). We therefore tested whether USP20-mediated deubiquitination of the β1AR affects lysosomal trafficking of internalized β1ARs. After 6 h Iso stimulation of cells transfected with control siRNA, we observed a significant increase in β1AR colocalization with the lysosomal marker protein LAMP1 (Fig. 2, D–F). Additionally, USP20 knockdown led to a statistically significant enhancement of β1AR/LAMP1 colocalization under basal and Iso-stimulated conditions as compared with quiescent cells with intact USP20 expression (Fig. 2, D–F). Accordingly, USP20 serves as a cognate DUB for the β1AR: USP20 deubiquitinates β1AR and attenuates its trafficking to lysosomes.
Figure 2.

USP20 serves as a cognate deubiquitinase for the β1AR. A, HEK-293 cells with stable FLAG-β1AR were transiently transfected with siRNAs targeting no mRNA (CTL) or USP20 for 48 h, serum-starved for 60 min, and stimulated ±1 μm Iso for 60 min. IP and immunoblotting (IB) are as in Fig. 1A. Lysate blots for USP20 and β-actin are shown in the lower two panels. Some batches of USP20 antisera cross-react with USP33, see under “Experimental procedures.” B, β1AR ubiquitination was quantitated as in Fig. 1B. Graph represents means ± S.E. from five independent experiments. *, p < 0.05, versus control-nonstimulated (NS) and USP20-Iso; **, p < 0.01 compared with all others, two-way ANOVA, Holm-Sidak's post-test. C, β1AR ubiquitination was determined as in A, but siRNA targeting USP33 was used. Blots shown are representative of three similar experiments performed. D, distribution of β1AR (red) and LAMP1 (green) in HEK-293 cells transfected with siRNA targeting either no mRNA (control, CTL) or USP20 ± Iso were visualized with LSM-710 confocal microscope. Representative images are shown, and quantification of colocalization from ≥20 z-stack images from three independent experiments are included in the bar graph in E. We used Imaris software to quantify volume of colocalization from images in a Z-stack. #, p < 0.05 versus CTL-NS; *, p < 0.01, versus all others, two-way ANOVA, Holm-Sidak's post-test. F, lysates serially immunoblotted for USP20 and β-actin show knockdown of USP20. Scale bars in D, 10 μm.
β1AR activation promotes site-specific USP20 phosphorylation by PKAα
We previously identified that a serine residue in the consensus PKA phosphorylation motif (Arg–Lys–Phe–Ser) within the unique insertion domain of USP20 is phosphorylated by PKAα upon agonist activation of the β2AR (24). To determine whether β1AR activation also provokes USP20 phosphorylation on this serine residue (Ser-333 in human USP20, which is conserved across different species, including mouse), we stimulated HEK-293 cells expressing β1AR with increasing doses of Iso or dobutamine (Dob, a selective full β1AR agonist). As shown in Fig. 3, A–D, β1AR activation triggered robust USP20 phosphorylation within 5 min even at subsaturating doses of either agonist. Additionally, this β1AR-induced USP20 phosphorylation is mediated by the PKAα isoform expressed in HEK-293 cells, because PKAα knockdown significantly decreased phosphorylation of USP20 induced by 5 and 20 min of stimulation with Iso or Dob (Fig. 3, E and F). However, USP20 phosphorylation triggered by these agonists prevailed with control or PKAβ knockdown (Fig. 3, E and F). Accordingly, the PKAα isoform in HEK-293 cells phosphorylates USP20 Ser-333 downstream of both β1AR (Fig. 3) and β2AR (24) activation.
Figure 3.

β1AR activation promotes phosphorylation of USP20 on serine 333 by PKAα. A, HEK-293 cells stably expressing FLAG-β1AR were stimulated for 5 min with indicated amounts of Iso, and equivalent cell extracts were analyzed for USP20 phosphorylation (p-USP20) and expression (T-USP20). B, phospho-USP20 bands were normalized to total USP20 in each sample and plotted as % maximum signal showing means ± S.E. from n = 3 independent experiments. # denotes p < 0.01 compared with nonstimulated (NS) samples, one-way ANOVA, Bonferroni's post-test. C and D, experiments and analyses were as in A and B, except that the β1AR-selective agonist dobutamine was used. #, p < 0.05; *, p < 0.01 compared with nonstimulated. E, HEK-293 cells stably expressing FLAG-β1AR were transiently transfected with siRNAs targeting no mRNA (control), PKAα, or PKAβ for 48 h, serum-starved for 60 min, and then stimulated with 1 μm Dob or 1 μm Iso for 5 or 20 min. Cell lysates were immunoblotted for p-USP20, t-USP20, PKAα, PKAβ, and GAPDH. F, quantification of p-USP20 normalized to t-USP20. n = 3, *, p < 0.01; **, p < 0.001, ANOVA, Bonferroni's post-test.
Phospho-USP20 deubiquitinates β1AR and blocks β1AR trafficking to lysosomes
We next ascertained the role of USP20 phosphorylation in the regulation of β1AR trafficking by using two complementary approaches: by siRNA-mediated down-regulation of PKAα to stabilize de-phosphorylated USP20 in cells, and by overexpressing phosphorylation-impaired (S333A) and phosphomimetic (S333D) mutant constructs of USP20. Down-regulation of PKAα significantly increased the colocalization of internalized β1AR with LAMP1 both before and after 6 h of agonist stimulation as compared with quiescent cells with intact PKAα expression (Fig. 4, A and B). PKAα knockdown also preserved agonist-induced ubiquitination of the β1AR (Fig. 4C). We overexpressed recombinant adenoviruses encoding eGFP or USP20 WT, S333A, or S333D constructs in HEK-293 cells stably transfected with FLAG-β1AR and analyzed Iso-induced ubiquitination of the receptor (Fig. 5). In the absence of exogenous USP20, Iso stimulation produced robust β1AR ubiquitination. However, either WT USP20 or S333D overexpression led to significant deubiquitination of the receptor (Fig. 5, A and B). In contrast, S333A overexpression did not lead to receptor deubiquitination. These data suggest that USP20 Ser-333 phosphorylation by PKAα is required for its DUB activity toward the β1AR.
Figure 4.

Effects of PKAα knockdown on β1AR lysosomal trafficking and β1AR ubiquitination. A, distribution of β1AR (red) and LAMP1 (green) in HEK-293 cells transfected with siRNA targeting either no mRNA (control, CTL) or PKAα treated for 6 h ± Iso were visualized with LSM-710 confocal microscope. Images were acquired and analyzed as in Fig. 1. Scale bars, 10 μm. B, volume of colocalization from images in a Z-stack obtained from ≥15 images from four independent experiments. #, p < 0.05 versus CTL-NS and PKAα-Iso; *, p < 0.05, versus all others; two-way ANOVA, Holm-Sidak's post-test. NS, nonstimulated. C, HEK-293 cells with stable FLAG-β1AR were transiently transfected with siRNAs targeting no mRNA (CTL) or PKAα for 48 h, serum-starved for 60 min, and stimulated ± 1 μm Iso for 60 min. The receptor was immunoprecipitated (IP) with anti-FLAG affinity gel, and serially immunoblotted (IB) for ubiquitin and β1AR as in Fig. 1A. Lysate blots for PKAα and β-actin are shown. Blots shown are representative of three similar experiments.
Figure 5.

Ser-333 phosphorylation preserves DUB activity of USP20 facilitating β1AR deubiquitination. A, HEK-293 cells stably expressing FLAG-β1AR were infected at equal m.o.i. with recombinant adenoviruses encoding eGFP or HA-tagged USP20 WT, S333A (S/A), or S333D (S/D) constructs and stimulated with 1 μm Iso for 60 min. The receptors were isolated with M2 anti-FLAG affinity gel. The immunoprecipitates (IP) were separated by SDS-PAGE and immunoblotted (IB) with antibodies specific to ubiquitin (rabbit polyclonal, Bethyl Laboratories Inc.) and FLAG antibodies as in Fig. 1A. Bottom panels show the expression levels of USP20 WT or mutants in the lysates as detected by monoclonal anti-HA (12CA5) antibody and detection of β-actin. B, ubiquitin smears in each lane from the IP blot in A were quantified, normalized to β1AR signals, and plotted as bars. *, p < 0.05 versus GFP(NS), WT(NS and Iso), and S/D (NS and Iso); two-way ANOVA Holm-Sidak's post-test. NS, not stimulated. Data are means ± S.E. of four independent experiments.
These findings are paradoxical to our earlier findings with the β2AR; PKAα knockdown augmented β2AR deubiquitination and concomitantly blocked β2AR lysosomal trafficking (24). Our earlier work also showed that USP20 dissociates from the agonist-activated β2AR complex, and additionally, the phosphomimetic mutant S333D only weakly associated with the β2AR compared with the phospho-defective S333A mutant (23, 24). In contrast to the β2AR, which dissociates from USP20 upon activation (Fig. 6) (23), agonist-activated β1AR forms a stable complex with USP20, suggesting that phosphorylated USP20 associates robustly with the β1AR (Fig. 6). Additionally, although overexpressed USP20 WT and S333D constructs displayed equivalent binding with the β1AR, S333A binding was significantly reduced (Fig. 7, A and B). Despite the differences observed in the interaction with the β1-AR versus β2-AR, both phosphomimetic and phosphorylation-impaired forms of USP20 possessed similar intrinsic enzyme activity as assessed by active-site labeling and covalent binding of ubiquitin-vinyl methyl ester (Ub-VME) (Fig. 7, C and D). Ub-VME is an active-site–directed probe that has been widely used to assess enzyme activity of DUBs and can distinguish between active and inactive forms of DUBs (26–28). As expected, Ub-VME did not bind a catalytically inactive USP20 in which the active-site cysteine and histidine residues have been mutated (USP20-CH) (23, 29).
Figure 6.

Kinetics of agonist-induced binding of USP20 with β1AR and β2AR are dramatically different. A, HEK-293 cells stably expressing FLAG-β1AR were stimulated with 1 μm Iso for the indicated times. Receptors were immunoprecipitated (IP) with anti-FLAG affinity gel, followed by immunoblotting for USP20. HEK-293 cells without FLAG-β1AR were used as control cells (Mock). Center panel, the amount of receptors detected by FLAG antibody. Bottom panels, total cell lysate immunoblotted for USP20 and β-actin. B, USP20 band in each IP was normalized to the cognate β1AR band, and the mean values (±S.E.) from three independent experiments were plotted as % maximum signals denoting β1AR/USP20 association. C and D, same procedures were used as in A and B except that HEK-293 cells that stably express FLAG-β2AR were used. B and D, n = 3, *, p < 0.05 compared with 0 min (′); one-way ANOVA, Bonferroni's post-test.
Figure 7.

Binding properties and DUB activity of USP20 phospho-mutants. A, HEK-293 cells stably expressing FLAG-β1AR were infected at equal m.o.i. with recombinant adenoviruses encoding eGFP or HA-tagged USP20 WT, S333A, or S333D constructs, and the receptors were isolated with M2 anti-FLAG affinity gel. The immunoprecipitates (IP) were separated by SDS-PAGE and immunoblotted (IB) with anti-HA (rabbit polyclonal, Cell Signaling Technology) and anti-FLAG antibodies. Lysates were serially immunoblotted for HA and β-actin. B, quantification of USP20 in receptor IPs from four independent experiments is plotted as USP20/β1AR ratio. *, p < 0.05; versus WT and S/D, one-way ANOVA, Bonferroni's test. C, expression of HA-tagged USP20 WT, S333A, S333D, or Cys-His were induced in HEK-293 cells by respective recombinant adenoviral infections. Lysates that contain ∼ 4 pmol of each USP20 (see “Experimental procedures”) were incubated with buffer or 20 pmol of ubiquitin-VME in vitro at 37 °C. Unmodified USP20 and USP20 that is covalently linked with ubiquitin-VME were detected by immunoblotting with rabbit polyclonal anti-HA antibody (Cell Signaling Technology). D, ratios of USP20-Ub and unmodified USP20 for each construct are plotted. No significant difference between WT, S/A, and S/D (one-way ANOVA, Bonferroni's test).
To determine the effects of USP20 phosphorylation on β1AR trafficking, we overexpressed WT, S333A, S333D, and USP20-CH constructs in HEK-293 cells stably transfected with CFP-tagged β1AR and analyzed receptor colocalization with LAMP1 at 6 h of agonist stimulation. As shown in Fig. 8, both WT USP20 and phosphomimetic S333D overexpression resulted in almost no agonist-induced increase in β1AR colocalization with LAMP1 compared with vector. In contrast, both phospho-defective USP20 S333A and USP20-CH mutants significantly increased β1AR trafficking to lysosomes when compared with vector-transfected cells (Fig. 8). These data suggest that USP20 activity and its phosphorylation status play a critical role in attenuating the trafficking of internalized β1AR to lysosomes.
Figure 8.

Phospho-USP20 inhibits lysosomal trafficking of the β1AR. A and B, HEK-293 cells stably expressing β1AR-CFP were transiently transfected with vector pcDNA3, USP20 WT, S333A (S/A), S333D (S/D), and catalytic site mutant (Cys–His, CH), starved for 60 min, and stimulated with Iso for 6 h. Cells were fixed, permeabilized, and immunostained for LAMP1. Confocal images are shown with β1AR-CFP in red, LAMP1 in green (Alexa Fluor 488), and USP20 in cyan (Alexa Fluor 633). C, quantification of volume of β1AR colocalized with LAMP1 from ≥20 images from three independent experiments. *, p < 0.05, compared with pc3 Iso, S/A Iso, CH Iso; #, p < 0.05 compared with pc3 Iso. Two-way ANOVA, Holm-Sidak's post-test. Scale bars, 10 μm.
Baseline characteristics and function of USP20–KO mice hearts
Because USP20 regulates β1AR, which is the predominant βAR subtype for cardiac contractility, we hypothesized that disruption of USP20 in vivo could impact β1AR function in the myocardium. To determine the role of USP20 in vivo, we used USP20 gene trap mice (USP20–KO) in which we confirmed the absence of USP20 protein expression by Western blotting (Fig. 9A). We next compared USP20–KO and WT mice for overall cardiac morphology, histopathology, and function (Fig. 9, B–E, and Table 1). USP20–KO mice are similar to WT controls in terms of body and heart weights (Fig. 9). Similar to WT mice, USP20–KO mice have normal heart morphology and no fibrosis in cross-sections assessed by Masson's trichrome staining (Fig. 9). However, conscious, nonanesthetized USP20–KO mice presented a faster heart rate than WT during noninvasive echocardiography, a response that can be attributed to greater excitement or stress in these animals during the procedure (Table 1). Although the LV wall dimensions of USP20–KO mice were comparable with WT, left ventricle volumes and internal dimensions were significantly greater in the USP20–KO mice compared with WT mice during both systole and diastole as assessed by M-mode echocardiography. Nevertheless, basal systolic function was normal in these mice and comparable with WT as determined by the percentage of LV fractional shortening (%FS, 53.5 ± 7.2, n = 16 for USP20–KO versus %FS, 57.5 ± 6.5, n = 15 for WT). These echocardiography findings are further complemented with invasive hemodynamic studies on anesthetized WT and USP20–KO mice, which demonstrated a similar heart rate as well as equivalent load-dependent and load-independent cardiac function but with a modest difference in diastolic function (dP/dtmin) and no significant differences in systolic function and cardiac output (Tables 2 and 3). Accordingly, USP20 gene deletion does not have a major adverse effect on cardiac development and on overall baseline cardiac performance in mice.
Figure 9.

Cardiac morphology, histopathology, and morphometry of WT and USP20–KO mice. A, immunoblot of cardiac lysates from 3- to 4-month-old WT and USP20–KO mice with USP20 and GAPDH antibodies. Blots are representative of similar results obtained from >10 mice of each genotype. B, representative whole hearts from 3- to 4-month-old WT and USP20–KO mice (n = 3, scale bar = 1 mm). C, heart weight to body weight ratio from 3- to 4-month-old WT (n = 12) and USP20–KO (n = 12) mice. Error bars indicate average ± S.E. D, representative Masson Trichrome staining of cardiac sections from 3- to 4-month-old WT and USP20–KO (n = 5, scale bar, 0.5 mm). E, representative baseline echocardiograms via M-mode, short axis imaging, of WT and USP20–KO mice (see Table 1 for ECHO analyses).
Table 1.
Echocardiographic measurements in WT and USP20-KO
Values are expressed as mean ± S.D. The following abbreviations are used: LV, left ventricle; s, systole; d, diastole; IVS, intraventricular septum; LVID, left ventricle internal dimension; LVPW, left ventricular posterior wall; EF, ejection fraction; FS, fractional shortening of LV; Vol, volume; BPM, beats/min; AET, aortic ejection time; mVcf, mean velocity of circumferential fiber shortening; mVcfc, mean velocity of circumferential fiber shortening corrected for heart rate; cir/s, circumferences/s. Statistical significance was determined by Student's t test.
| WT, n = 15 | USP20-KO, n = 16 | |
|---|---|---|
| LVID; d (mm) | 2.96 ± 0.34 | 3.27 ± 0.40a |
| LVID; s (mm) | 1.27 ± 0.32 | 1.54 ± 0.38a |
| LVPW; d (mm) | 1.01 ± 0.18 | 0.95 ± 0.16 |
| IVS; d (mm) | 1.05 ± 0.13 | 0.99 ± 0.12 |
| EF (%) | 88.18 ± 4.82 | 84.36 ± 5.89 |
| FS (%) | 57.50 ± 6.46 | 53.58 ± 7.20 |
| LV mass (corrected) (mg) | 84.88 ± 14.13 | 90.03 ± 20.49 |
| LV Vol; d (μl) | 34.68 ± 10.32 | 44.17 ± 13.35a |
| LV Vol; s (μl) | 4.41 ± 3.23 | 7.47 ± 4.23a |
| Heart rate (BPM) | 652 ± 51 | 710 ± 57a |
| AET (ms) | 38.82 ± 4.93 | 36.45 ± 2.89 |
| mVcf (cir/s) | 15.07 ± 2.79 | 14.87 ± 2.81 |
| mVcfc (cir/s) | 4.57 ± 0.80 | 4.31 ± 0.73 |
a p < 0.05 versus WT.
Table 2.
Baseline load-dependent hemodynamics
The following abbreviations are used: LVESP, left ventricle end systolic pressure; LVEDP, left ventricle end diastolic pressure; dP/dtmax, maximum first derivative of left ventricular pressure; dP/dtmin, minimum first derivative of left ventricular pressure; τ, left ventricular diastolic time constant. Values are expressed as mean ± S.D.
| WT, n = 11 | USP20-KO, n = 14 | |
|---|---|---|
| Heart rate (BPM) | 453 ± 34 | 428 ± 55 |
| LVESP (mm Hg) | 130.4 ± 16.6 | 121.2 ± 26.4 |
| LVEDP (mm Hg) | 9.4 ± 4.9 | 10.4 ± 8.6 |
| Arterial elastance (mm Hg/μl) | 6.5 ± 2.2 | 6.7 ± 2.7 |
| Systolic function parameters | ||
| Stroke volume (μl) | 21.4 ± 5.2 | 19.4 ± 5.1 |
| Ejection fraction (%) | 62.4 ± 19.1 | 51.9 ± 20.8 |
| Cardiac output (μl/min) | 9590.1 ± 1941.3 | 8263.2 ± 2197.7 |
| Stroke work (mm Hg·μl) | 2066.3 ± 593.7 | 1787.1 ± 561.6 |
| dP/dtmax (mm Hg/s) | 10,237.4 ± 1305.5 | 9048.4 ± 1834.3 |
| Diastolic function parameters | ||
| dP/dtmin (mm Hg/s) | −8946.4 ± 1063.2 | −7596.5 ± 1286.4a |
| τ (ms) (Weiss) | 7.9 ± 1.3 | 8.6 ± 1.8 |
| τ (ms) (Glantz) | 12.1 ± 2.6 | 13.4 ± 3.4 |
a p < 0.05 compared with WT, Student's t test.
Table 3.
Load-independent hemodynamic measures
Values are expressed as mean ± S.D. The following abbreviations are used: EDPVR, end diastolic pressure volume relationship; ESPVR, end systolic pressure volume relationship; PRSW, preload recruitable stroke work; EDV, end diastolic volume; Emax, maximal elastance.
| WT, n = 10 | USP20-KO, n = 13 | |
|---|---|---|
| EDPVR (mm Hg/μl) | 0.23 ± 0.1 | 0.3 ± 0.2 |
| ESPVR (mm Hg/μl) | 4.33 ± 1.7 | 3.5 ± 1.3 |
| PRSW (mm Hg) | 63.6 ± 20.8 | 48.8 ± 17.5 |
| dP/dt-EDV (mm Hg/s/μl) | 244.7 ± 95.2 | 195.01 ± 75.9 |
| Emax (mm Hg/μl) | 6.7 ± 2.2 | 5.6 ± 2.2 |
β1AR signaling and expression are down-regulated in USP20–KO mouse hearts
To ascertain whether the absence of USP20 affects myocardial β1ARs, we first repeated invasive hemodynamic measurements with Iso stimulation. Compared with WT controls, we observed a marked blunting of Iso-induced cardiac contractility (dP/dtmax, Fig. 10A) and relaxation (dP/dtmin, Fig. 10B) in USP20–KO. The heart rates between the WT and USP20–KO measured before Iso infusion were not significantly different: mean beats/min ± S.D.; WT 397 ± 78, n = 10; KO 339 ± 52, n = 10. However, the increase in heart rate induced by Iso stimulation was significantly blunted in the USP20–KO mice compared with the WT mice (Fig. 10C). Additionally, as shown in Fig. 10D, Dob infusion did not induce cAMP increase in USP20–KO LVs, whereas direct activation of adenylyl cyclase with the water-soluble forskolin analog NKH477 (30) resulted in equivalent cAMP production in both WT and USP20–KO LVs (n = 5–7 mice of each genotype, mean cAMP pmol/mg: WT-saline, 75 ± 41; WT-dobutamine, 270 ± 43; WT-NKH477, 242 ± 57; KO-saline, 69 ± 18; KO-dobutamine 40 ± 18; KO-NKH477, 300 ± 44).
Figure 10.
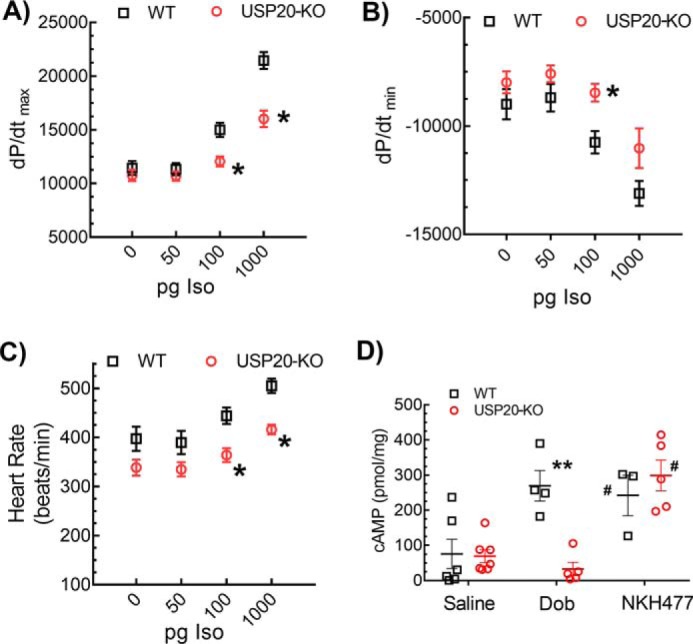
β1AR-induced cardiac contractility and cAMP response are impaired in USP20–KO mice. Hemodynamic parameters measured in anesthetized mice after infusion with increasing doses of isoproterenol are shown in A, maximal first derivative of LV pressure, and in B, minimal first derivative of LV pressure. Data shown are average values ± S.E. *, p < 0.01, two-way ANOVA, Holm-Sidak's post-test. n = 10 (WT) and n = 10 (KO). C, heart rate of anesthetized WT (n = 10) and USP20–KO (n = 10) mice in response to increasing doses of isoproterenol. Error bars indicate average ± S.E. *, p < 0.05, two-way ANOVA, Holm-Sidak's post-test. D, cAMP production in 3-month-old WT and USP20–KO hearts, following acute saline, dobutamine (Dob) or NKH477 infusion plotted as means ± S.E. **, p < 0.01, versus saline groups and KO-Dob; #, p < 0.05 compared with saline groups; two-way ANOVA, Holm-Sidak's post-test.
To determine whether the impaired βAR activity was due to a reduction in receptor density, we measured the expression levels of β1 and β2 ARs in LV membranes by radioligand binding (Fig. 11, A and B). Interestingly, although the total βAR levels were not significantly different between WT and USP20–KO mice, β1AR levels were significantly decreased, and β2AR levels were reciprocally increased in USP20–KO LVs. These data suggest that USP20 functions as the cognate DUB to regulate trafficking and prevent degradation of the β1AR in vivo, and as such, the β1ARs are tonically down-regulated in USP20–KO. Because previous studies have shown a redundant regulation of the β2AR by USP20 and USP33 (23), we also ascertained whether the expression level of USP33 was altered in USP20–KO hearts. Indeed, we detected 2-fold more USP33 in USP20–KO LV compared with WT LV extracts (Fig. 11, C and D). These findings suggest that although the β1AR is primarily regulated by USP20, cardiac β2AR expression and trafficking might be regulated by USP33 in addition to USP20 in vivo.
Figure 11.

β1AR is down-regulated, and β2AR and USP33 are up-regulated in USP20–KO LVs. A, βAR subtypes in LV of WT and USP20–KO mice were measured by radioligand binding with 125I-cyanopindolol and using the β1AR subtype-selective antagonist CGP20712A as reported before (72, 74). Nonspecific binding was defined by the nonselective antagonist propranolol. Experiments were performed in triplicate. n = 21 for WT and n = 17 for USP20–KO. Error bars indicate average ± S.E. *, p < 0.05, ANOVA Bonferroni's post-test. B, bar graphs show relative proportion of β1 and β2 AR in USP20–KO and WT LVs calculated from the binding values shown in A. C, immunoblot (IB) of cardiac lysates from 3- to 4-month-old WT and USP20–KO mice probed with USP20, USP33, and GAPDH antibodies. D, band intensities of USP20 and USP33 normalized to GAPDH in each sample are shown and represented as means ± S.E. *, p < 0.05, versus USP33 in WT hearts, ANOVA, Bonferroni's post-test.
USP20 Ser-333 phosphorylation is triggered in mouse hearts following chronic catecholamine stress and pressure overload
Excessive or chronic catecholamine stimulation of cardiac β1ARs is deleterious to heart function and leads to adverse cardiac remodeling, perhaps by promoting pathological signaling via CaMKII signaling or via PI3K/AKT/GSK3 signaling (1, 8, 31, 32). β1ARs also transactivate epidermal growth factor receptors in a β-arrestin–dependent manner and mitigate apoptosis induced by chronic Iso stimulation (33). To determine whether chronic catecholamine stimulation affects cardiac USP20 and/or activity, which in turn might affect β1AR trafficking and signaling, we subjected WT mice to chronic saline or catecholamine exposure for 1 week, and we tested the levels of USP20 and phospho-USP20 in extracts prepared from left ventricles. The experimental mice (C57BL/6, 8–10 weeks old) had normal heart function at baseline as measured by echocardiography (Table 4). A 1-week infusion of saline did not alter cardiac function. However, chronic Iso (3 mg/kg/day for 7 days) led to a marked increase in LV chamber size and a decrease in fractional shortening (Table 4), consistent with previous studies (33). Although saline-treated mice showed weak phospho-USP20, Iso-infused mice showed a 2–4-fold increase in USP20 phosphorylation (Fig. 12). However, both saline- and Iso-treated hearts had identical levels of total USP20. The sustained phospho-USP20 signals suggest the interesting possibility of preserved expression and signaling via the β1AR in nonlysosomal compartments with chronic Iso stimulation.
Table 4.
Echocardiographic measurements in WT mice after chronic Iso
Values are expressed as mean ± S.D. The following abbreviations are used: LV, left ventricle; s, systole; d, diastole; IVS, intraventricular septum; LVD, left ventricle internal dimension; LVPW, left ventricular posterior wall; ET, ejection time; BPM, beats/min; FS, fractional shortening of LV; LVm, LV mass; mVcf, mean velocity of circumferential fiber shortening; mVcfc, mean velocity of circumferential fiber shortening corrected for heart rate; cir/s, circumferences/s. Statistical significance was determined by two-way ANOVA with post hoc Sidak analysis.
| Saline |
Iso |
|||
|---|---|---|---|---|
| Pre | Post | Pre | Post | |
| N | 6 | 6 | 5 | 5 |
| LVDd (mm) | 3.0 ± 0.2 | 3.0 ± 0.2 | 2.8 ± 0.2 | 3.9 ± 0.4a |
| LVDs (mm) | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | 2.4 ± 0.4a |
| IVSW (mm) | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 0.9 ± 0.1 |
| PW (mm) | 1.1 ± 0.2 | 1.1 ± 0.2 | 1.2 ± 0.1 | 1.1 ± 0.1 |
| ET (ms) | 37.4 ± 3.2 | 36.8 ± 2.1 | 36.6 ± 3.1 | 42.2 ± 9.2 |
| HR (BPM) | 692 ± 31 | 723 ± 107 | 701 ± 10 | 620 ± 94 |
| FS (%) | 60.4 ± 2.6 | 60.3 ± 3.7 | 62.1 ± 3.4 | 38.3 ± 5.6a |
| LVm (mg) | 85.2 ± 19.9 | 88.7 ± 16.4 | 93.5 ± 9.3 | 117.6 ± 10.5a |
| mVcf (cir/s) | 16.3 ± 1.7 | 16.5 ± 1.6 | 17.0 ± 1.8 | 9.4 ± 2.5a |
| mVcfc (cir/s) | 4.8 ± 0.5 | 4.7 ± 0.1 | 5.0 ± 0.5 | 2.9 ± 0.6a |
a p < 0.05 versus pre-saline.
Figure 12.

βAR stimulation induces USP20 phosphorylation in vivo. Soluble extracts from left ventricles of WT mice exposed chronically to saline or Iso were immunoblotted for p-USP20 and total USP20. Quantification of p-USP20 normalized to USP20, saline n = 6; Iso n = 5, *, p = 0.01, unpaired, two-tailed t test.
β1AR and β2AR also contribute to pressure-overload hypertrophy induced by transverse aortic constriction (TAC) (34, 35). Previous studies have also shown that with moderate pressure gradients, only β1ARs are responsible for left ventricular hypertrophy (35). We tested whether USP20 expression or phosphorylation are affected during hypertrophy, as this might regulate expression and desensitization of βARs. Two weeks of chronic pressure overload was induced by TAC in WT, β1AR KO, and β1AR/β2AR double KO mice as described previously (34, 36–39). At 2 weeks, the blood pressure gradient was measured to assess the efficacy of surgery. Pre- and post-TAC echocardiography was also performed on conscious mice. The sham-operated mice for WT, β1AR KO, and β1AR/β2AR double KO displayed no pre- versus post-surgery change in fractional shortening, and moreover, Western blottings of heart extracts showed weak to no phospho-USP20 signals (Fig. 13 and Table 5). The WT 2-week TAC group but not the β1AR KO nor β1AR/β2AR double KO displayed robust phosphorylation of USP20 as well as deterioration of heart function as evaluated by ECHO (% FS, WT: pre-sham 57.8 ± 4.8 and post-sham 55.8 ± 5.5; pre-TAC 57.7 ± 3.3 and post-TAC 43.1 ± 13.6, see Table 5). The WT TAC group but neither β1AR KO nor β1AR/β2AR double KO showed significant hypertrophy (heart weight/body weight WT-sham 4.6 ± 0.5 and WT-TAC 6.5 ± 1.0, see Table 5). According to these data, an increase in USP20 phosphorylation induced by TAC requires expression of β1ARs. Collectively, these in vivo data show a correlation of USP20 phosphorylation in two models of cardiac dysfunction (chronic Iso and TAC) and suggest that βAR expression and signaling in the heart is regulated by USP20 and its phosphorylation. In addition, it appears that β1AR regulates and preserves its own trafficking itinerary as well as signaling by triggering downstream phosphorylation of USP20. Thus β1AR-dependent USP20 Ser-333 phosphorylation is a feed-forward mechanism in vivo that might facilitate and sustain pathological cardiotoxic β1AR signaling.
Figure 13.
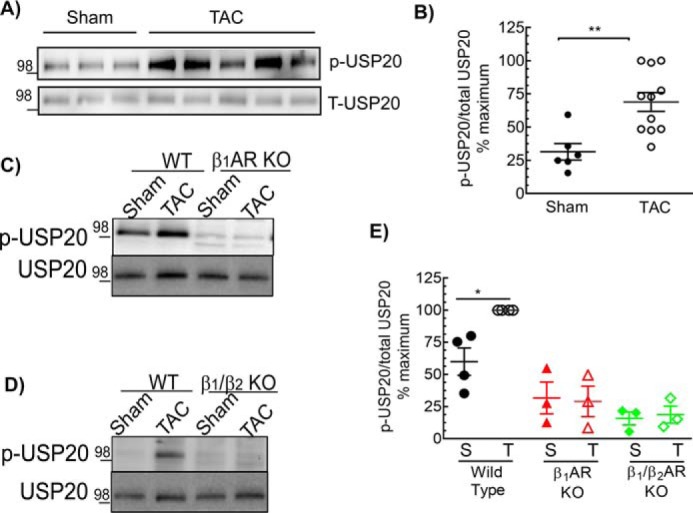
Cardiac pressure overload induces USP20 phosphorylation in a β1AR-dependent manner. A, soluble extracts from left ventricles of WT mice with sham or TAC surgery were immunoblotted for p-USP20 and total USP20. B, quantification of p-USP20 normalized to USP20, sham n = 5, and TAC n = 11, **, p = 0.003, unpaired, two-tailed t test. The maximum p-USP20/USP20 ratio in the experimental cohort was taken as 100%. C and D, soluble extracts from left ventricles of mice that underwent sham or TAC surgery of indicated genotypes were serially immunoblotted for p-USP20 and total USP20. E, graph shows quantification of p-USP20 normalized to USP20 plotted as in B. *, p < 0.05, ANOVA, Bonferroni's post-test.
Table 5.
Echocardiographic measurements following trans-aortic constriction
Values are expressed as mean ± SD. LV, left ventricle; s, systole; d, diastole; IVS, intra-ventricular septum; LVD, left ventricle internal dimension; LVPW, left ventricular posterior wall; ET, ejection time; BPM, beats per minute; FS, fractional shortening of LV; LVm, LV mass; mVcf, mean velocity of circumferential fiber shortening; mVcfc, mean velocity of circumferential fiber shortening corrected for heart rate; cir/sec, circumferences per second; HW, heart weight; BW, body weight. Statistical significance was determined by two-way ANOVA with post hoc Sidak analysis.

* p < 0.05 versus WT Sham, PRE, is shown.
# p < 0.05 versus all groups.
Discussion
We have identified USP20 as a novel deubiquitinase that regulates β1AR expression, trafficking, and signaling in cultured cells and in mouse hearts (Fig. 14). Our findings reveal βAR-induced phosphorylation of USP20 as a critical layer of regulation with unique reciprocal effects on the lysosomal trafficking of β1AR and β2AR subtypes (Fig. 14). USP20 phosphorylation is induced by βAR stimulation and by cardiac pressure overload through the β1AR; phospho-USP20 deubiquitinates β1AR and blocks lysosomal trafficking of the β1AR, whereas phospho-USP20 promotes both ubiquitination and lysosomal degradation of the β2AR (24). Furthermore, conditions that stabilize unphosphorylated USP20 are more favorable for β1AR lysosomal trafficking, whereas unphosphorylated USP20 favors stabilization of β2AR expression and signaling. Thus, phospho-USP20 presents as a molecular signature that engenders trafficking bias in the lysosomal sorting of the two major βAR subtypes.
Figure 14.

Effects of USP20 and its phosphorylation on β1AR signaling. A, activation of the β1AR induces USP20 phosphorylation on Ser-333 catalyzed by PKAα. Phosphorylated USP20 deubiquitinates β1AR and attenuates lysosomal trafficking, thus facilitating β1AR signaling. B, PKAα down-regulation by RNAi or overexpression of weakly interacting USP20 S333A mutant leads to increased lysosomal trafficking of ubiquitinated β1ARs. C, in USP20–KO mice, myocardial β1AR levels are significantly decreased, whereas levels of β2AR and USP33 (homolog of USP20) are increased. Accordingly, although USP20 and USP33 serve as redundant DUBs for the β2AR, USP20 functions as the predominant DUB to regulate β1AR expression and trafficking in the myocardium.
We previously identified USP20, and its homolog USP33, as interacting partners of the β2AR and as mediators of β2AR ubiquitination and recycling (23). USP20 and USP33 share 70% identity and play redundant roles in the regulation of β2AR ubiquitination and trafficking (23). In fact, significant lysosomal degradation of the β2AR occurs in HEK-293 cells only after down-regulation of both deubiquitinases, whereas receptor recycling is maintained when expression of either one of these DUBs is preserved (23). In contrast, we found that only USP20 and not USP33 protects β1AR from lysosomal degradation even though β1AR and β2AR share 52% identity (40). Ablation of USP20 in the heart coincides with a reduction in density of the β1AR even though cardiac USP33 is up-regulated in the USP20-deficient heart. Thus, although USP33 up-regulation may preserve β2AR expression in the heart, it is ineffective in regulating cardiac β1AR expression that is mainly modulated by USP20. Accordingly, the deubiquitinases USP20 and USP33 may play a prominent role in defining which βAR subtype signaling is sustained in normal, failing, or pressure-overloaded hearts.
Although USP20 phosphorylation is triggered by agonists, it is reversed or blocked by β-blockers or antagonists (24). Possibly, β-blockers by inhibiting cAMP production can inhibit PKA-mediated phosphorylation of USP20, thus differentially affecting the trafficking and lysosomal degradation of βAR subtypes in the failing heart. The β-blockers also show differential effects on the expression level of βAR subtypes in the failing heart (41–43). Although β-blocker therapy improves cardiac function by antagonizing catecholamine activity, βAR density can also be increased by some β-blockers (for example metoprolol), which contribute to the improved cardiac performance observed in heart failure patients (44, 45). However, other β-blockers such as carvedilol do not up-regulate or stabilize βAR expression in failing hearts, but nonetheless, they decrease morbidity and mortality of patients with heart failure (43, 46–51). Although carvedilol has been shown to block USP20 interaction with the β2AR and inhibit β2AR recycling (52), future work should reveal the exact effects of various β1AR subtype-selective and nonselective blockers on β1AR-induced USP20 phosphorylation and interaction of USP20 with the β1AR.
DUBs exist in cells as fully processed enzymes; however, they require conformational activation induced by binding of substrates (53, 54). The binding of the activity-based Ub-VME probe suggests that Ser-333 phosphorylation does not affect the intrinsic DUB activity of USP20. However, Ser-333 phosphorylation by PKAα dictates USP20's specific association and affinity for each βAR subtype, which further defines deubiquitination and effects on post-endocytic sorting to lysosomes of each βAR subtype. Accordingly, phospho-USP20 deubiquitinates β1AR and blocks lysosomal trafficking of the β1AR, whereas it promotes both ubiquitination and lysosomal degradation of the β2AR. In contrast, conditions that stabilize unphosphorylated USP20, are more favorable for β1AR lysosomal trafficking and for stabilization of β2AR expression and signaling. Although pathological insults provoke USP20 phosphorylation in vivo, the effects of phospho-USP20 on the development of cardiac dysfunction and heart failure remain to be defined when USP20 S333A and S333D knockin mouse models become available.
We also identified the physiological effects of USP20 ablation in the heart of our newly characterized USP20–KO mice. The basal systolic function in USP20–KO and WT are comparable, but USP20–KO manifests modest load–dependent relaxation impairment determined by invasive hemodynamic studies on anesthetized mice. Nevertheless, overall cardiac performance was normal as cardiac output in USP20–KO mice was similar to WT. Importantly, USP20–KO mice show markedly impaired cardiac responsiveness to β-agonist stimulation. This impaired cardiac contractility induced by adrenergic stimulation is ascribed to the inability of the heart to promote cAMP production after stimulation with the β1AR-selective agonist dobutamine. However, USP20 ablation did not affect the signaling machinery downstream of β1AR as NKH477-induced adenylyl cyclase activation was preserved. The impaired β-adrenergic signaling in USP20 KO mouse was associated with lower density of β1AR. The inability of the remaining β1AR to activate cAMP in the USP20–KO LVs may be linked to their ubiquitination status, which promotes their compartmentalization in lysosomes or induces conformational changes that do not favor G protein activation. Under chronic or intermittent stress of pressure overload, the densities of both β1- and β2-ARs are significantly reduced (55). Future studies should reveal the exact role of USP20 in regulating β1AR expression and signaling in the heart subjected to pressure overload.
The lessened Iso-induced cardiac contractility in USP20–KO mice is consistent with β1AR deficiency because mice lacking β1AR also present normal basal cardiac function and impaired cardiac responsiveness to β-adrenergic stimulation (5, 7). However, the exact underlying mechanism that leads to modest reduction in relaxation in USP20–KO mice remains to be defined. Moreover, preserved expression of β2AR in cardiac membranes of USP20–KO was associated with up-regulation of USP33. Whether USP20 regulates additional GPCRs and proteins in the heart and whether USP20 modulates alternative signaling mechanisms in pressure overloaded ventricles and heart failure also remain to be defined.
In conclusion, we have determined a novel role of USP20 in the regulation of myocardial β1AR density and cardiac responsiveness after catecholamine stimulation. Although catecholamines stimulate βAR signaling that is critical for inducing powerful contractions of the heart muscle, catecholamines also trigger GRK2-mediated signal desensitization that is followed by receptor endocytosis and degradation (56–60). These events are maladaptive in the long term as they either cause βARs to be degraded or to be refractory to stimulus and thus reduce cardiac performance. In this scenario, β1ARs also promote noncanonical signaling pathways that promote apoptosis or cardiac hypertrophy (61). Accordingly, restoring βAR activity without enabling endocytosis or directly inhibiting the endocytic machinery has been found to be cardioprotective (16, 61–69). Thus, preserving βAR levels and activity by preventing lysosomal trafficking of βARs is a valuable therapeutic strategy for heart failure; in this context, exploiting deubiquitinases and their activity might prove beneficial.
Experimental procedures
Reagents
N-Ethylmaleimide, anti-FLAG M2 affinity gel, CGP20712A, propranolol, isoproterenol, dobutamine, and NKH477 were purchased from Sigma. The IgGs obtained from the following sources were used at 1:1000 dilution unless indicated otherwise: mouse monoclonal anti-β-actin (catalog no. A5441, 1:10,000) and rabbit polyclonal anti-FLAG (catalog no. F7425) Sigma; rabbit polyclonal anti-USP20 (catalog no. A301-189A, 1:2000), anti-USP33 (catalog no. A300925A), and anti-ubiquitin (catalog no. A300-317A) Bethyl Laboratories, Inc.; anti-LAMP1 (catalog no. sc-18821), rabbit polyclonal anti-PKAα (catalog no. sc-903), anti-PKAβ (catalog no. sc-904), anti-β-tubulin (catalog no. sc-5274), anti-β2AR H20 (catalog no. sc-569), and anti-β1AR V-19 (catalog no. sc-568), Santa Cruz Biotechnology; and rabbit polyclonal anti-GAPDH (catalog no. 3683) Cell Signaling Technology. Custom antibody specific for mouse USP33 and USP20 phosphorylated at serine 333 was produced by GenScript and described previously (24, 70). Horseradish peroxidase-conjugated secondary antibodies were from Rockland Immunochemicals, Cell Signaling Technology, and Bethyl Laboratories, Inc., and used at a dilution of 1:3000. Alexa Fluor 488-, 594-, and 633-conjugated secondary antibodies were purchased from Invitrogen and used at a dilution of 1:300 for immunofluorescence labeling.
Experimental animals
Genetically engineered β1AR-KO and β1AR/β2AR-KO mice were described previously (5, 7). The USP20 gene trap heterozygous mouse line (Usp20tm1a(EUCOMM)Hmgu) was purchased from the International Knockout Mouse Consortium. The USP20 gene trap heterozygous mice were mated to produce homozygous gene trap and WT mice that were used as experimental cohorts. Additionally, we generated USP20 gene trap KO with C57BL/6N ES cell lines (HEPD0524_2_C09 and HEPD0524_2_H10, EUCOMM), with the help of Duke Transgenic Core. All three USP20 gene trap KO lines (designated as USP20–KO) were studied and were assigned into age- and gender-matched experimental cohorts for cardiac phenotyping. Genotypes of all breeding and experimental animals were confirmed by PCR-based and Western blotting assays. All in vivo measurements were made by observers blinded to specimen identity. All animals were handled according to the approved protocols and animal welfare regulations of the Duke University Institutional Animal Care and Use Committee.
Cell lines
Human embryonic kidney (HEK-293) cells were purchased from the American Type Culture Collection and grown in minimal essential medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Plasmid transfections in these cells were performed at 40–50% confluency in the presence of Lipofectamine 2000TM (ThermoFisher Scientific) and following manufacturer's protocol as described before (52). Stable cell lines expressing FLAG-tagged β1AR, CFP-tagged β1AR, or FLAG-tagged β2AR were selected by supplementing growth medium with 1 mg/ml G418, and upon passaging they were maintained in growth medium containing 400 μg/ml G418, as described previously (71).
Cardiac morphological and histological examinations
Morphological and histological cardiac assessments were performed as described previously (72). Hearts were excised and briefly washed in PBS before photographing with a Nikon SMZ 800 stereomicroscope and a DS-Fi1 camera. Histological heart specimens were fixed in 10% formaldehyde solution, sectioned into 5-μm thick slices, and stained with Masson trichrome before visualization at ×20 magnification under a Zeiss Axio Imager widefield fluorescence microscope. ZEN software version 2.3 (Carl Zeiss Microscopy) was used for histological image processing and stitching, and NIH ImageJ was used for quantification and analysis.
Transthoracic echocardiography
All cardiovascular phenotyping was performed at the Duke Cardiovascular Physiology Core. Echocardiography was performed on 2–4-month-old awake mice using a Vevo 2100 high-resolution imaging system (VisualSonics, Toronto, Ontario, Canada) as described previously (72). Left ventricle structure and dimension were assessed via parasternal short axis view in M-mode, whereas aortic ejection time was measured by pulsed-wave Doppler. All data and images were analyzed with the Vevo 2100 Imaging System by an examiner blinded to mouse identity.
Hemodynamic evaluation in intact anesthetized mice
Mice were anesthetized with a mixture of ketamine (100 mg/kg i.p.) and xylazine (2.5 mg/kg i.p.) and connected to a rodent ventilator via endotracheal intubation (8, 34). Intraventricular pressure was measured in real-time with a 1.4F high-fidelity micromanometer catheter (Millar Instruments) inserted into the left ventricle via the right carotid artery. Isoproterenol was administered through a polyethylene catheter (PE10) inserted into the right external jugular vein. Ventricular contractility was assessed at baseline and 45 s after injection of increasing doses of isoproterenol.
Left ventricular pressure–volume analysis
After bilateral vagotomy, cardiac catheterization was performed with a 1.4F high-fidelity micromanometer catheter (Millar Instruments) on anesthetized mice as described for the hemodynamic experiments and as described previously (8, 34). Left ventricle volume and intraventricular pressure during systole and diastole were measured in real-time and recorded using a P-V conductance system (PowerLab, AD Instruments). Various preload conditions were elicited by transient inferior vena cava occlusion. Parallel conductance (Vp) of the blood pool was determined by a 10-μl injection of 15% saline into the right jugular vein and used for correction of P-V loop data. Data were analyzed with P-V analysis software (PVAN data analysis software version 3.3; Millar Instruments) by an observer blinded to mouse genotype.
Determination of cardiac cAMP levels
For catecholamine-induced cAMP, 3–4-month-old mice were injected with a single bolus of a β1AR-selective agonist of dobutamine (10 μg/g body weight) or an equivalent volume of vehicle (0.9% NaCl) given intraperitoneally. For direct adenylyl cyclase activation, mice were treated with 1 μg/g body weight of the water-soluble forskolin analog NKH477 (30). 2 min after drug injection, the mice were rapidly euthanized, and their hearts were excised, flash-frozen in liquid nitrogen, and stored at −80 °C until assays. Total cAMP levels in mouse ventricles were measured with Direct cAMP ELISA kit (catalog no. ADI-900-066; Enzo Life Sciences Inc) according to the manufacturer's instructions. Optical density associated with cAMP levels was measured at 405 nm with Synergy Neo2 Hybrid Multi-Mode Reader (Biotek).
Cardiac membrane isolation
Left ventricles isolated from 3 to 4 months old WT and USP20–KO mice were lysed in ice-cold homogenization buffer containing 25 mm Tris-Cl (pH 7.4), 5 mm EDTA (pH 8.0), 5 μg/ml leupeptin, and 10 μg/ml aprotinin as described previously. After low-speed centrifugation (500 × g for 5 min at 4 °C) to remove debris, the supernatants were centrifuged at high-speed (35,000 × g for 30 min at 4 °C) to precipitate membranes. Membrane fractions were then suspended in buffer containing 75 mm Tris-Cl (pH 7.4), 2 mm EDTA (pH 8.0), 12.5 mm MgCl2, 5 μg/ml leupeptin, and 10 μg/ml aprotinin and stored at −80 °C until radioligand binding assays.
Radioligand-binding assays
Radioligand-binding assays were performed as described previously (72–74). Briefly, membranes isolated from LV of WT and USP20–KO mice were diluted to a concentration of 0.5 mg/ml in ice-cold binding assay buffer containing 50 mm Tris-Cl (pH 7.4), 2 mm EDTA (pH 8.0), 12.5 mm MgCl2, and 180 μg/ml l-ascorbic acid. 500 pm 125I- cyanopindolol was added to all membranes, and inhibition of specific 125I-cyanopindolol binding was determined by adding 20 μm nonselective antagonist propranolol or β1AR-selective antagonist CGP20712A. Samples were prepared in triplicate in polypropylene 96-well plates (Costar, Cambridge, MA), and at least three independent experiments were performed. After 90 min of incubation at room temperature, membrane-bound radioligand was harvested onto Whatman GF/B glass fiber filters using a Brandel cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with ice-cold washing buffer (50 mm Tris-Cl (pH 7.4), 2 mm EDTA (pH 8.0), 12.5 mm MgCl2), and bound radioactivity were measured with a Packard Cobra gamma counter.
RNA interference
Control siRNA targeting no mRNA and siRNA targeting USP20, USP33, PKAα, or PKAβ were purchased from Dharmacon GE Healthcare as described previously (23, 24). Transfections were performed in serum-free medium in 40–50% confluent cells using 20 μg of siRNA in the presence of Lipofectamine 2000TM and following the manufacturer's protocol. 4 h after transfection, serum-containing medium was added to the cells and maintained for 48 h until experiment was performed. Cells in which knockdown of target protein attained >80% efficiency were used for experimental analyses.
Immunoprecipitation and immunoblotting
For cardiac samples, freshly isolated mouse ventricles were solubilized in an ice-cold lysis buffer containing 20 mm Tris-Cl (pH 7.4), 137 mm NaCl, 20% (v/v) glycerol, and 1% (v/v) IGEPAL CA-630, supplemented with phosphatase and protease inhibitors (1 mm sodium orthovanadate, 10 mm sodium fluoride, 10 mm phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml phosphatase inhibitor mixture 2 (P5726), and 1 μg/ml phosphatase inhibitor mixture 3 (P0044); all were from Sigma. Cardiac homogenates were centrifuged at 35,000 × g for 30 min at 4 °C, and the supernatant was used for experiments. For immunoprecipitation assays, HEK-293 cells stably expressing FLAG-tagged β1- or β2-AR were solubilized in ice-cold lysis buffer containing 50 mm HEPES (pH 7.5), 2 mm EDTA (pH 8.0), 250 mm NaCl, 10% (v/v) glycerol, and 0.5% (v/v) IGEPAL CA-630 supplemented with the aforementioned phosphatase and protease inhibitors and also with 10 mm N-ethylmaleimide to inhibit cellular deubiquitinase activities and preserve receptor ubiquitination. Lysates from these cells were centrifuged at 13,000 rpm for 20 min at 4 °C, and 1–2 mg of supernatant whole-cell extracts were immunoprecipitated using anti-FLAG M2 resin. After overnight end-over-end rotation at 4 °C, immune complexes were washed three times with lysis buffer and eluted in 2× SDS-PAGE sample buffer. Immune complexes and 20 μg of corresponding lysates were resolved on 4–20% gradient gels and transferred onto nitrocellulose membranes. Membrane blocking and antibody incubation were in 5% (w/v) dried skim milk powder dissolved in TTBS (0.2% (v/v) Tween 20, 10 mm Tris-Cl (pH 8.0), and 150 mm NaCl), whereas in-between washes were performed in TTBS. Note that many batches of commercial anti-USP20 IgG also detect endogenous USP33, but anti-USP33 IgG does not detect endogenous USP20; optimal separation of the two proteins is achieved on 4–20 or 4–12% Tris glycine gradient gels. Immunoblotted proteins were detected with enhanced chemiluminescence (SuperSignal West Pico reagent, Pierce). Signals were detected and acquired with a charge-coupled device camera system (Bio-Rad Chemidoc-XRS) and analyzed with ImageLab software (Bio-Rad).
Quantification of β1AR ubiquitination
The FLAG-β1AR IPs were serially immunoblotted for ubiquitin and FLAG tag. Band densities detected by the ubiquitin antibody between 70 and 300 kDa were determined in all lanes, and those values from the lane that had no FLAG-β1AR was used as the negative control to subtract nonspecific signals. Similar densitometry was performed for the FLAG blot to evaluate the signals of prominent β1AR bands between 45 and 70 kDa. The specific values obtained for ubiquitinated species in each lane were then divided by the corresponding β1AR value, which was used in producing bar graphs. In Fig. 1B, maximal ubiquitin signal in each experiment is taken as 100%. In Figs. 2B, and 5B, the signals from control + agonist is fixed as 100% in each experiment.
Adenovirus-mediated transduction
HA-tagged adenovirus expressing WT, phosphomimetic, phospho-deficient, or catalytically inactive USP20 mutants were generated with the AdEasy system (Agilent Technologies) according to published protocols (75, 76), and their identity was confirmed by sequencing.
For conjugation assays with Ub-VME (U-203; R&D Systems) and for co-immunoprecipitation experiments, cells were transduced with adenovirus for 2 h and harvested 12–14 h post-transduction in co-immunoprecipitation buffer deprived of NEM. For Ub-VME conjugation assays, the level of expression of each USP20 construct was estimated by densitometric comparison of known amounts of purified HA-USP20 (76) as detected by immunoblotting with polyclonal anti-HA antibody. The final volume of lysate used in Ub-VME binding was adjusted to obtain 4 pmol of USP20 in each reaction. 20 pmol of Ub-VME was added to the reaction. In these assays, the maximal shift for USP20 WT required a 30-min incubation of the lysate + Ub-VME mixture at 37 °C. We used this condition to compare the reactivity of S333A, S333D, and USP20-CH constructs. The reactions were terminated by addition of SDS-PAGE sample buffer, and further analysis was carried out by Western blotting with a polyclonal anti-HA antibody (Cell Signaling Technology).
Immunofluorescence staining and confocal imaging
Immunofluorescence staining and confocal imaging were performed as described previously (23, 24). HEK-293 cells stably expressing exogenous β1AR-CFP were transfected with siRNA or plasmids encoding pcDNA3-HA and HA-tagged USP20 WT or mutants. 24 h post-transfection, the cells were seeded on poly-d-lysine–coated 35-mm glass bottom plates (MatTek Corp, Ashland, MA), serum-starved for 1 h the next day, stimulated with 1 μm Iso, and fixed with 5% formaldehyde diluted in calcium/magnesium-containing Dulbecco's PBS (DPBS). 15 min after fixation, cells were permeabilized for 20–30 min with 0.1% Triton X-100, incubated with primary antibody overnight at 4 °C, and with cognate secondary antibody for 1 h at room temperature. Washes after cell fixation and antibody incubations were performed with DPBS, whereas DPBS containing 2% BSA was used for permeabilizing solution and antibody dilutions. Cells were visualized with Zeiss LSM-710 or LSM-510 META confocal microscope with filter settings for respective fluorophores: excitation nm are 435 CFP, 488 (Alexa 488), 568 (Alexa 594), and 633 (Alexa 633); emission nm are 460–500 CFP, 515–540 Alexa fluor 488, 585–615 Alexa fluor 594, and 650 (Alexa Fluor 633). Images acquired by the LSM operating software (ZEISS ZEN imaging software) were analyzed with Imaris software (Bitplane, South Windsor, CT) to quantify fluorophore colocalization. A total of 13–20 z-stack images from three independent experiments were used for analyses.
Micro-osmotic pump implantation
Micro-osmotic pump implantation for chronic isoproterenol administration was performed as described previously (77, 78). Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (2.5 mg/kg). Saline- or isoproterenol-filled micro-osmotic pumps (Alzet model 1007D, Durect Corp., Cupertino, CA) were subcutaneously implanted in 3–4-month-old WT mice and adjusted to a dosage of 3 mg/kg/day for 7 days. After treatment, mice were rapidly euthanized, and their hearts were processed for biochemical analyses.
TAC
Transverse aortic constriction was performed in 8–12-week-old mice anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (2.5 mg/kg) as described previously (34). After thoracotomy, the aortic arch was ligated with a 6.0 nylon suture to the width of a 27-gauge needle, inducing a 60–80% constriction. The sham control mice underwent a similar procedure but without aortic ligation. After needle removal and chest/skin closure, animals were maintained for 2 weeks and then euthanized for molecular analyses of cardiac tissues.
Statistical analysis
Data from at least three independent experiments were averaged and represented as means ± S.E. Two-tailed Student's t test was used for basal assessments of WT and USP20–KO mice. Studies involving more than two groups of mice, as well as time-course and dose-response experiments were analyzed with ANOVA followed by post hoc correction for multiple comparisons. Statistical analyses were performed using GraphPad Prism 7 (GraphPad, Inc.), and significance was established for p < 0.05.
Author contributions
S. M.-W. Y., P.-Y. J.-C., H. A. R., and S. K. S. conceptualization; S. M.-W. Y., P.-Y. J.-C., D. M. A., S. K., and C. G. formal analysis; S. M.-W. Y., S. K., and L. M. investigation; S. M.-W. Y., P.-Y. J.-C., D. M. A., S. K., C. G., and L. M. methodology; S. M.-W. Y., P.-Y. J.-C., and S. K. S. writing-original draft; P.-Y. J.-C. and S. K. S. data curation; D. M. A., H. A. R., and S. K. S. supervision; D. M. A., H. A. R., and S. K. S. writing-review and editing; H. A. R. and S. K. S. funding acquisition; S. K. S. resources; S. K. S. validation; S. K. S. project administration.
This work was supported by National Institutes of Health Grants HL118369 (to S. K. S.), HL125905 (to D. A.), and HL56687 and P01 HL75443 (to H. A. R.), American Heart Association Grant 15GRNT25550051(to S. K. S.), the Mandel Center for Hypertension and Atherosclerosis Research, and Duke O'Brien Center for Kidney Research National Institutes of Health/NIDDK Award P30-DK096493. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- βAR
- β-adrenergic receptor
- β1AR
- β1-adrenergic receptor
- PKA
- protein kinase A
- LV
- left ventricle
- TAC
- transverse aortic constriction
- GPCR
- G protein–coupled receptor
- GRK
- GPCR kinase
- DUB
- deubiquitinase
- NEM
- N-ethylmaleimide
- Iso
- isoproterenol
- Dob
- dobutamine
- Ub-VME
- ubiquitin vinyl methyl ester
- m.o.i.
- multiplicity of infection
- ANOVA
- analysis of variance
- PI3K
- phosphoinositide 3-kinase
- FS
- fractional shortening
- DPBS
- Dulbecco's PBS
- IP
- immunoprecipitation
- eGFP
- enhanced green fluorescent protein
- CFP
- cyan fluorescent protein
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Rockman H. A., Koch W. J., and Lefkowitz R. J. (2002) Seven-transmembrane-spanning receptors and heart function. Nature 415, 206–212 10.1038/415206a [DOI] [PubMed] [Google Scholar]
- 2. Rohrer D. K., and Kobilka B. K. (1998) Insights from in vivo modification of adrenergic receptor gene expression. Annu. Rev. Pharmacol. Toxicol. 38, 351–373 10.1146/annurev.pharmtox.38.1.351 [DOI] [PubMed] [Google Scholar]
- 3. Bristow M. R., Ginsburg R., Umans V., Fowler M., Minobe W., Rasmussen R., Zera P., Menlove R., Shah P., and Jamieson S. (1986) β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res. 59, 297–309 10.1161/01.RES.59.3.297 [DOI] [PubMed] [Google Scholar]
- 4. Chruscinski A. J., Rohrer D. K., Schauble E., Desai K. H., Bernstein D., and Kobilka B. K. (1999) Targeted disruption of the β2 adrenergic receptor gene. J. Biol. Chem. 274, 16694–16700 10.1074/jbc.274.24.16694 [DOI] [PubMed] [Google Scholar]
- 5. Rohrer D. K., Chruscinski A., Schauble E. H., Bernstein D., and Kobilka B. K. (1999) Cardiovascular and metabolic alterations in mice lacking both β1- and β2-adrenergic receptors. J. Biol. Chem. 274, 16701–16708 10.1074/jbc.274.24.16701 [DOI] [PubMed] [Google Scholar]
- 6. Rohrer D. K., Schauble E. H., Desai K. H., Kobilka B. K., and Bernstein D. (1998) Alterations in dynamic heart rate control in the β1-adrenergic receptor knockout mouse. Am. J. Physiol. 274, H1184–H1193 [DOI] [PubMed] [Google Scholar]
- 7. Rohrer D. K., Desai K. H., Jasper J. R., Stevens M. E., Regula D. P. Jr, Barsh G. S., Bernstein D., and Kobilka B. K. (1996) Targeted disruption of the mouse β1-adrenergic receptor gene: developmental and cardiovascular effects. Proc. Natl. Acad. Sci. U.S.A. 93, 7375–7380 10.1073/pnas.93.14.7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoo B., Lemaire A., Mangmool S., Wolf M. J., Curcio A., Mao L., and Rockman H. A. (2009) β1-Adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 297, H1377–H1386 10.1152/ajpheart.00504.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jean-Charles P. Y., Kaur S., and Shenoy S. K. (2017) G protein-coupled receptor signaling through β-arrestin-dependent mechanisms. J. Cardiovasc. Pharmacol. 70, 142–158 10.1097/FJC.0000000000000482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ungerer M., Böhm M., Elce J. S., Erdmann E., and Lohse M. J. (1993) Altered expression of β-adrenergic receptor kinase and β1-adrenergic receptors in the failing human heart. Circulation 87, 454–463 10.1161/01.CIR.87.2.454 [DOI] [PubMed] [Google Scholar]
- 11. Ginsburg R., Bristow M. R., Billingham M. E., Stinson E. B., Schroeder J. S., and Harrison D. C. (1983) Study of the normal and failing isolated human heart: decreased response of failing heart to isoproterenol. Am. Heart J. 106, 535–540 10.1016/0002-8703(83)90698-1 [DOI] [PubMed] [Google Scholar]
- 12. Bernstein D., Fajardo G., Zhao M., Urashima T., Powers J., Berry G., and Kobilka B. K. (2005) Differential cardioprotective/cardiotoxic effects mediated by β-adrenergic receptor subtypes. Am. J. Physiol. Heart Circ. Physiol. 289, H2441–H2449 10.1152/ajpheart.00005.2005 [DOI] [PubMed] [Google Scholar]
- 13. Devic E., Xiang Y., Gould D., and Kobilka B. (2001) β-Adrenergic receptor subtype-specific signaling in cardiac myocytes from β(1) and β(2) adrenoceptor knockout mice. Mol. Pharmacol. 60, 577–583 [PubMed] [Google Scholar]
- 14. Xiang Y., and Kobilka B. K. (2003) Myocyte adrenoceptor signaling pathways. Science 300, 1530–1532 10.1126/science.1079206 [DOI] [PubMed] [Google Scholar]
- 15. Engelhardt S., Böhm M., Erdmann E., and Lohse M. J. (1996) Analysis of β-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of β1-adrenergic receptor mRNA in heart failure. J. Am. Coll. Cardiol. 27, 146–154 10.1016/0735-1097(95)00425-4 [DOI] [PubMed] [Google Scholar]
- 16. Perrino C., Naga Prasad S. V., Schroder J. N., Hata J. A., Milano C., and Rockman H. A. (2005) Restoration of β-adrenergic receptor signaling and contractile function in heart failure by disruption of the βARK1/phosphoinositide 3-kinase complex. Circulation 111, 2579–2587 10.1161/CIRCULATIONAHA.104.508796 [DOI] [PubMed] [Google Scholar]
- 17. Perrino C., Schroder J. N., Lima B., Villamizar N., Nienaber J. J., Milano C. A., and Naga Prasad S. V. (2007) Dynamic regulation of phosphoinositide 3-kinase-γ activity and β-adrenergic receptor trafficking in end-stage human heart failure. Circulation 116, 2571–2579 10.1161/CIRCULATIONAHA.107.706515 [DOI] [PubMed] [Google Scholar]
- 18. Hershko A., and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 19. Jean-Charles P. Y., Snyder J. C., and Shenoy S. K. (2016) Chapter one–ubiquitination and deubiquitination of G protein-coupled receptors. Prog. Mol. Biol. Transl. Sci. 141, 1–55 10.1016/bs.pmbts.2016.05.001 [DOI] [PubMed] [Google Scholar]
- 20. Shenoy S. K., McDonald P. H., Kohout T. A., and Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 10.1126/science.1063866 [DOI] [PubMed] [Google Scholar]
- 21. Shenoy S. K., Xiao K., Venkataramanan V., Snyder P. M., Freedman N. J., and Weissman A. M. (2008) Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the β2-adrenergic receptor. J. Biol. Chem. 283, 22166–22176 10.1074/jbc.M709668200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao K., and Shenoy S. K. (2011) β2-Adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. J. Biol. Chem. 286, 12785–12795 10.1074/jbc.M110.203091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berthouze M., Venkataramanan V., Li Y., and Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 28, 1684–1696 10.1038/emboj.2009.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kommaddi R. P., Jean-Charles P. Y., and Shenoy S. K. (2015) Phosphorylation of the deubiquitinase USP20 by protein kinase A regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 290, 8888–8903 10.1074/jbc.M114.630541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dores M. R., and Trejo J. (2012) Ubiquitination of G protein-coupled receptors: functional implications and drug discovery. Mol. Pharmacol. 82, 563–570 10.1124/mol.112.079418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borodovsky A., Ovaa H., Meester W. J., Venanzi E. S., Bogyo M. S., Hekking B. G., Ploegh H. L., Kessler B. M., and Overkleeft H. S. (2005) Small-molecule inhibitors and probes for ubiquitin- and ubiquitin-like-specific proteases. Chembiochem 6, 287–291 10.1002/cbic.200400236 [DOI] [PubMed] [Google Scholar]
- 27. Hewings D. S., Flygare J. A., Bogyo M., and Wertz I. E. (2017) Activity-based probes for the ubiquitin conjugation-deconjugation machinery: new chemistries, new tools, and new insights. FEBS J. 284, 1555–1576 10.1111/febs.14039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naik E., and Dixit V. M. (2016) Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCβ kinase activity. J. Immunol. 196, 3438–3451 10.4049/jimmunol.1403165 [DOI] [PubMed] [Google Scholar]
- 29. Jean-Charles P. Y., Zhang L., Wu J. H., Han S. O., Brian L., Freedman N. J., and Shenoy S. K. (2016) Ubiquitin-specific protease 20 regulates the reciprocal functions of β-arrestin2 in toll-like receptor 4-promoted nuclear factor κB (NFκB) activation. J. Biol. Chem. 291, 7450–7464 10.1074/jbc.M115.687129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou J., Lal H., Chen X., Shang X., Song J., Li Y., Kerkela R., Doble B. W., MacAulay K., DeCaul M., Koch W. J., Farber J., Woodgett J., Gao E., and Force T. (2010) GSK-3α directly regulates β-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J. Clin. Invest. 120, 2280–2291 10.1172/JCI41407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mangmool S., Shukla A. K., and Rockman H. A. (2010) β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β(1)-adrenergic receptor stimulation. J. Cell Biol. 189, 573–587 10.1083/jcb.200911047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morisco C., Zebrowski D., Condorelli G., Tsichlis P., Vatner S. F., and Sadoshima J. (2000) The Akt-glycogen synthase kinase 3β pathway regulates transcription of atrial natriuretic factor induced by β-adrenergic receptor stimulation in cardiac myocytes. J. Biol. Chem. 275, 14466–14475 10.1074/jbc.275.19.14466 [DOI] [PubMed] [Google Scholar]
- 33. Noma T., Lemaire A., Naga Prasad S. V., Barki-Harrington L., Tilley D. G., Chen J., Le Corvoisier P., Violin J. D., Wei H., Lefkowitz R. J., and Rockman H. A. (2007) β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Invest. 117, 2445–2458 10.1172/JCI31901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rockman H. A., Ross R. S., Harris A. N., Knowlton K. U., Steinhelper M. E., Field L. J., Ross J. Jr., and Chien K. R. (1991) Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 88, 8277–8281 10.1073/pnas.88.18.8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao M., Fajardo G., Urashima T., Spin J. M., Poorfarahani S., Rajagopalan V., Huynh D., Connolly A., Quertermous T., and Bernstein D. (2011) Cardiac pressure overload hypertrophy is differentially regulated by β-adrenergic receptor subtypes. Am. J. Physiol. Heart Circ. Physiol. 301, H1461–H1470 10.1152/ajpheart.00453.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Colomer J. M., Mao L., Rockman H. A., and Means A. R. (2003) Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol. Endocrinol. 17, 183–192 10.1210/me.2002-0350 [DOI] [PubMed] [Google Scholar]
- 37. Skavdahl M., Steenbergen C., Clark J., Myers P., Demianenko T., Mao L., Rockman H. A., Korach K. S., and Murphy E. (2005) Estrogen receptor-β mediates male-female differences in the development of pressure overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 288, H469–H476 10.1152/ajpheart.00723.2004 [DOI] [PubMed] [Google Scholar]
- 38. Tachibana H., Naga Prasad S. V., Lefkowitz R. J., Koch W. J., and Rockman H. A. (2005) Level of β-adrenergic receptor kinase 1 inhibition determines degree of cardiac dysfunction after chronic pressure overload-induced heart failure. Circulation 111, 591–597 10.1161/01.CIR.0000142291.70954.DF [DOI] [PubMed] [Google Scholar]
- 39. Tachibana H., Perrino C., Takaoka H., Davis R. J., Naga Prasad S. V., and Rockman H. A. (2006) JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem. Biophys. Res. Commun. 343, 1060–1066 10.1016/j.bbrc.2006.03.065 [DOI] [PubMed] [Google Scholar]
- 40. Shcherbakova O. G., Hurt C. M., Xiang Y., Dell'Acqua M. L., Zhang Q., Tsien R. W., and Kobilka B. K. (2007) Organization of β-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J. Cell Biol. 176, 521–533 10.1083/jcb.200604167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Asano K., Zisman L. S., Yoshikawa T., Headley V., Bristow M. R., and Port J. D. (2001) Bucindolol, a nonselective β1- and β2-adrenergic receptor antagonist, decreases β-adrenergic receptor density in cultured embryonic chick cardiac myocyte membranes. J. Cardiovasc. Pharmacol. 37, 678–691 10.1097/00005344-200106000-00005 [DOI] [PubMed] [Google Scholar]
- 42. Bristow M. R., Abraham W. T., Yoshikawa T., White M., Hattler B. G., Crisman T. S., Lowes B. D., Robertson A. D., Larrabee P., and Gilbert E. M. (1997) Second- and third-generation β-blocking drugs in chronic heart failure. Cardiovasc. Drugs Ther. 11, Suppl. 1, 291–296 [DOI] [PubMed] [Google Scholar]
- 43. Yoshikawa T., Port J. D., Asano K., Chidiak P., Bouvier M., Dutcher D., Roden R. L., Minobe W., Tremmel K. D., and Bristow M. R. (1996) Cardiac adrenergic receptor effects of carvedilol. Eur. Heart J. 17, Suppl. B, 8–16 [DOI] [PubMed] [Google Scholar]
- 44. Aarons R. D., and Molinoff P. B. (1982) Changes in the density of β adrenergic receptors in rat lymphocytes, heart and lung after chronic treatment with propranolol. J. Pharmacol. Exp. Ther. 221, 439–443 [PubMed] [Google Scholar]
- 45. Sigmund M., Jakob H., Becker H., Hanrath P., Schumacher C., Eschenhagen T., Schmitz W., Scholz H., and Steinfath M. (1996) Effects of metoprolol on myocardial β-adrenoceptors and Giα-proteins in patients with congestive heart failure. Eur. J. Clin. Pharmacol. 51, 127–132 10.1007/s002280050172 [DOI] [PubMed] [Google Scholar]
- 46. Yue T. L., McKenna P. J., Ruffolo R. R. Jr, and Feuerstein G. (1992) Carvedilol, a new β-adrenoceptor antagonist and vasodilator antihypertensive drug, inhibits superoxide release from human neutrophils. Eur. J. Pharmacol. 214, 277–280 10.1016/0014-2999(92)90130-V [DOI] [PubMed] [Google Scholar]
- 47. Feuerstein G., Liu G. L., Yue T. L., Cheng H. Y., Hieble J. P., Arch J. R., Ruffolo R. R. Jr., and Ma X. L. (1998) Comparison of metoprolol and carvedilol pharmacology and cardioprotection in rabbit ischemia and reperfusion model. Eur. J. Pharmacol. 351, 341–350 10.1016/S0014-2999(98)00326-4 [DOI] [PubMed] [Google Scholar]
- 48. Cleland J. G., Charlesworth A., Lubsen J., Swedberg K., Remme W. J., Erhardt L., Di Lenarda A., Komajda M., Metra M., Torp-Pedersen C., Poole-Wilson P. A., and COMET Investigators. (2006) A comparison of the effects of carvedilol and metoprolol on well-being, morbidity, and mortality (the “patient journey”) in patients with heart failure: a report from the Carvedilol Or Metoprolol European Trial (COMET). J. Am. Coll. Cardiol. 47, 1603–1611 10.1016/j.jacc.2005.11.069 [DOI] [PubMed] [Google Scholar]
- 49. Dargie H. J. (2001) Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 357, 1385–1390 10.1016/S0140-6736(00)04560-8 [DOI] [PubMed] [Google Scholar]
- 50. Flesch M., Ettelbrück S., Rosenkranz S., Maack C., Cremers B., Schlüter K. D., Zolk O., and Böhm M. (2001) Differential effects of carvedilol and metoprolol on isoprenaline-induced changes in β-adrenoceptor density and systolic function in rat cardiac myocytes. Cardiovasc. Res. 49, 371–380 10.1016/S0008-6363(00)00277-7 [DOI] [PubMed] [Google Scholar]
- 51. Böhm M., Ettelbrück S., Flesch M., van Gilst W. H., Knorr A., Maack C., Pinto Y. M., Paul M., Teisman A. C., and Zolk O. (1998) β-Adrenergic signal transduction following carvedilol treatment in hypertensive cardiac hypertrophy. Cardiovasc. Res. 40, 146–155 10.1016/S0008-6363(98)00099-6 [DOI] [PubMed] [Google Scholar]
- 52. Han S. O., Xiao K., Kim J., Wu J. H., Wisler J. W., Nakamura N., Freedman N. J., and Shenoy S. K. (2012) MARCH2 promotes endocytosis and lysosomal sorting of carvedilol-bound β(2)-adrenergic receptors. J. Cell Biol. 199, 817–830 10.1083/jcb.201208192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Komander D., Clague M. J., and Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 10.1038/nrm2731 [DOI] [PubMed] [Google Scholar]
- 54. Sahtoe D. D., and Sixma T. K. (2015) Layers of DUB regulation. Trends Biochem. Sci. 40, 456–467 10.1016/j.tibs.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 55. Perrino C., Naga Prasad S. V., Mao L., Noma T., Yan Z., Kim H. S., Smithies O., and Rockman H. A. (2006) Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Invest. 116, 1547–1560 10.1172/JCI25397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bristow M. R., Ginsburg R., Minobe W., Cubicciotti R. S., Sageman W. S., Lurie K., Billingham M. E., Harrison D. C., and Stinson E. B. (1982) Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 307, 205–211 10.1056/NEJM198207223070401 [DOI] [PubMed] [Google Scholar]
- 57. Francis G. S., Goldsmith S. R., Ziesche S. M., and Cohn J. N. (1982) Response of plasma norepinephrine and epinephrine to dynamic exercise in patients with congestive heart failure. Am. J. Cardiol. 49, 1152–1156 10.1016/0002-9149(82)90039-X [DOI] [PubMed] [Google Scholar]
- 58. Thomas J. A., and Marks B. H. (1978) Plasma norepinephrine in congestive heart failure. Am. J. Cardiol. 41, 233–243 10.1016/0002-9149(78)90162-5 [DOI] [PubMed] [Google Scholar]
- 59. Chidsey C. A., Braunwald E., and Morrow A. G. (1965) Catecholamine excretion and cardiac stores of norepinephrine in congestive heart failure. Am. J. Med. 39, 442–451 10.1016/0002-9343(65)90211-1 [DOI] [PubMed] [Google Scholar]
- 60. Jean-Charles P. Y., Yu S. M., Abraham D., Kommaddi R. P., Mao L., Strachan R. T., Zhang Z. S., Bowles D. E., Brian L., Stiber J. A., Jones S. N., Koch W. J., Rockman H. A., and Shenoy S. K. (2017) Mdm2 regulates cardiac contractility by inhibiting GRK2-mediated desensitization of β-adrenergic receptor signaling. JCI Insight 2, 95998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morisco C., Marrone C., Galeotti J., Shao D., Vatner D. E., Vatner S. F., and Sadoshima J. (2008) Endocytosis machinery is required for β1-adrenergic receptor-induced hypertrophy in neonatal rat cardiac myocytes. Cardiovasc. Res. 78, 36–44 10.1093/cvr/cvn008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Salazar N. C., Vallejos X., Siryk A., Rengo G., Cannavo A., Liccardo D., De Lucia C., Gao E., Leosco D., Koch W. J., and Lymperopoulos A. (2013) GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun. Signal. 11, 64 10.1186/1478-811X-11-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nienaber J. J., Tachibana H., Naga Prasad S. V., Esposito G., Wu D., Mao L., and Rockman H. A. (2003) Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J. Clin. Invest. 112, 1067–1079 10.1172/JCI18213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Oudit G. Y., Crackower M. A., Eriksson U., Sarao R., Kozieradzki I., Sasaki T., Irie-Sasaki J., Gidrewicz D., Rybin V. O., Wada T., Steinberg S. F., Backx P. H., and Penninger J. M. (2003) Phosphoinositide 3-kinase γ-deficient mice are protected from isoproterenol-induced heart failure. Circulation 108, 2147–2152 10.1161/01.CIR.0000091403.62293.2B [DOI] [PubMed] [Google Scholar]
- 65. Harding V. B., Jones L. R., Lefkowitz R. J., Koch W. J., and Rockman H. A. (2001) Cardiac β ARK1 inhibition prolongs survival and augments β blocker therapy in a mouse model of severe heart failure. Proc. Natl. Acad. Sci. U.S.A. 98, 5809–5814 10.1073/pnas.091102398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Naga Prasad S. V., Barak L. S., Rapacciuolo A., Caron M. G., and Rockman H. A. (2001) Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by β-adrenergic receptor kinase 1. A role in receptor sequestration. J. Biol. Chem. 276, 18953–18959 10.1074/jbc.M102376200 [DOI] [PubMed] [Google Scholar]
- 67. Rockman H. A., Chien K. R., Choi D. J., Iaccarino G., Hunter J. J., Ross J. Jr, Lefkowitz R. J., and Koch W. J. (1998) Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7000–7005 10.1073/pnas.95.12.7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rockman H. A., Choi D. J., Akhter S. A., Jaber M., Giros B., Lefkowitz R. J., Caron M. G., and Koch W. J. (1998) Control of myocardial contractile function by the level of β-adrenergic receptor kinase 1 in gene-targeted mice. J. Biol. Chem. 273, 18180–18184 10.1074/jbc.273.29.18180 [DOI] [PubMed] [Google Scholar]
- 69. Koch W. J., Rockman H. A., Samama P., Hamilton R. A., Bond R. A., Milano C. A., and Lefkowitz R. J. (1995) Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science 268, 1350–1353 10.1126/science.7761854 [DOI] [PubMed] [Google Scholar]
- 70. Shenoy S. K., Modi A. S., Shukla A. K., Xiao K., Berthouze M., Ahn S., Wilkinson K. D., Miller W. E., and Lefkowitz R. J. (2009) β-Arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc. Natl. Acad. Sci. U.S.A. 106, 6650–6655 10.1073/pnas.0901083106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., and Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 10.1074/jbc.M506576200 [DOI] [PubMed] [Google Scholar]
- 72. Jean-Charles P. Y., Yu S. M., Abraham D., Kommaddi R. P., Mao L., Strachan R. T., Zhang Z. S., Bowles D. E., Brian L., Stiber J. A., Jones S. N., Koch W. J., Rockman H. A., and Shenoy S. K. (2017) Mdm2 regulates cardiac contractility by inhibiting GRK2-mediated desensitization of β-adrenergic receptor signaling. JCI Insight 2, e95998 10.1172/jci.insight.95998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hara M. R., Kovacs J. J., Whalen E. J., Rajagopal S., Strachan R. T., Grant W., Towers A. J., Williams B., Lam C. M., Xiao K., Shenoy S. K., Gregory S. G., Ahn S., Duckett D. R., and Lefkowitz R. J. (2011) A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature 477, 349–353 10.1038/nature10368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hegde A., Strachan R. T., and Walker J. K. (2015) Quantification of β adrenergic receptor subtypes in β-arrestin knockout mouse airways. PLoS ONE 10, e0116458 10.1371/journal.pone.0116458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Luo J., Deng Z. L., Luo X., Tang N., Song W. X., Chen J., Sharff K. A., Luu H. H., Haydon R. C., Kinzler K. W., Vogelstein B., and He T. C. (2007) A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2, 1236–1247 10.1038/nprot.2007.135 [DOI] [PubMed] [Google Scholar]
- 76. Jean-Charles P. Y., Wu J. H., Zhang L., Kaur S., Nepliouev I., Stiber J. A., Brian L., Qi R., Wertman V., Shenoy S. K., and Freedman N. J. (2018) USP20 (ubiquitin-specific protease 20) inhibits TNF (tumor necrosis factor)-triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 38, 2295–2305 10.1161/ATVBAHA.118.311071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Keys J. R., Greene E. A., Cooper C. J., Naga Prasad S. V., Rockman H. A., and Koch W. J. (2003) Cardiac hypertrophy and altered β-adrenergic signaling in transgenic mice that express the amino terminus of β-ARK1. Am. J. Physiol. Heart Circ. Physiol. 285, H2201–H2211 10.1152/ajpheart.00112.2003 [DOI] [PubMed] [Google Scholar]
- 78. Iaccarino G., Tomhave E. D., Lefkowitz R. J., and Koch W. J. (1998) Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by β-adrenergic receptor stimulation and blockade. Circulation 98, 1783–1789 10.1161/01.CIR.98.17.1783 [DOI] [PubMed] [Google Scholar]