

Abstract
The membrane-associated RING-CH (MARCH) family of membrane-bound E3 ubiquitin ligases regulates the levels of cell-surface membrane proteins, many of which are involved in immune responses. Although their role in ubiquitin-dependent endocytosis and degradation of cell-surface proteins is extensively documented, the features of MARCH proteins and their substrates that drive the molecular recognition events leading to ubiquitin transfer remain poorly defined. In this study, we sought to determine the features of human MARCH9 that are required for regulating the surface levels of its substrate proteins. Consistent with previous studies of other MARCH proteins, we found that susceptibility to MARCH9 activity is encoded in the transmembrane (TM) domains of its substrates. Accordingly, substitutions at specific residues and motifs within MARCH9's TM domains resulted in varying degrees of functional impairment. Most notably, a single serine-to-alanine substitution in the first of its two TM domains rendered MARCH9 completely unable to alter the surface levels of two different substrates: the major histocompatibility class I molecule HLA-A2 and the T-cell co-receptor CD4. Solution NMR analysis of a MARCH9 fragment encompassing the two TM domains and extracellular connecting loop revealed that the residues contributing most to MARCH9 activity are located in the α-helical portions of TM1 and TM2 that are closest to the extracellular face of the lipid bilayer. This observation defines a key region required for substrate regulation. In summary, our biochemical and structural findings demonstrate that specific sequences in the α-helical MARCH9 TM domains make crucial contributions to its ability to down-regulate its protein substrates.
Keywords: immunology, receptor endocytosis, ubiquitin ligase, membrane protein, nuclear magnetic resonance (NMR), major histocompatibility complex (MHC), T-cell, E3 ligase, membrane-associated RING-CH (MARCH), transmembrane domain
Introduction
The membrane-associated RING-CH (MARCH)5 proteins are a family of E3 ubiquitin ligases that regulate the cell-surface expression of proteins with important roles in immunity (1–3). Six mammalian MARCH proteins (MARCH1–4, MARCH8, and MARCH9) share a common structural organization featuring an N-terminal RING-CH domain that facilitates ubiquitin transfer, two predicted transmembrane (TM) domains connected by a short extracellular loop, and a C-terminal cytosolic tail of variable length (1). Biologically, the best characterized member of this family is MARCH1, whose biological roles include targeting major histocompatibility class II (MHC-II) complexes and the T-cell co-stimulatory molecule CD86 (B7.2) to endocytotic vesicles in antigen-presenting cells via ubiquitin-dependent pathways (4–9). In dendritic cells, when maturation is triggered by the uptake of antigenic cargo and/or exposure to Toll-like receptor ligands, MARCH1 activity is opposed by CD83 expression (10) and MARCH1 transcription stops (9, 11), resulting in MHC-II and CD86 up-regulation at the cell surface and increased CD4+ T-cell stimulatory capacity. A similar role for MARCH8 has recently been identified in thymic epithelial cells (12, 13), where it plays a key role in the development of CD4+ T-cells. The dynamic control of MARCH proteins thus has profound consequences for adaptive immune responses.
The mammalian MARCH proteins were originally identified by their homology to viral immune evasion proteins expressed by herpesviruses (1, 2, 14–19). The Kaposi's sarcoma herpesvirus (KSHV) genome encodes the modulators of immune recognition (MIR)1 and MIR2 proteins, both of which down-regulate MHC-I from the surface of infected cells to avoid detection by the host's immune system (14, 18, 19). Additionally, MIR2 down-regulates CD86 and intercellular adhesion molecule (ICAM)-1 (15, 16, 18), facilitating viral evasion of cytotoxic T lymphocyte responses. These and other viral MIR proteins share a similar domain organization with their mammalian counterparts, which strongly suggests that these proteins have been acquired by the virus from host genomes and modified to avoid endogenous regulation.
The proteins that can be regulated by mammalian MARCH family members also have key roles in innate and adaptive immunity as well as other important biological processes. MARCH1 and MARCH8 proteins are highly homologous and share an overlapping pool of substrates that include MHC-II molecules (1, 6, 7, 9, 11–13, 20–22), CD86 (2, 4, 23, 24), Fas (1), and transferrin receptor (1, 25). MARCH1 has also recently been shown to regulate insulin signaling through control of basal-state insulin receptor levels in adipocytes (26). MARCH2 and MARCH3 are less well-studied, but have been shown to mediate ubiquitin-dependent down-regulation of multispanning membrane proteins such as the β2-adrenergic receptor (27) and the cystic fibrosis transmembrane conductance regulator (MARCH2) (28), as well as the inhibitory antibody Fc receptor FcγRIIb (MARCH3) (29). The MARCH4 and MARCH9 proteins are closely related to one another and down-regulate MHC-I and the T-cell co-receptor CD4 (1) as well as the natural killer cell activating ligand Mult1 (30). MARCH9 has additionally been shown to regulate ICAM-I (31) and a large number of B-cell surface proteins identified in a proteomic study of MARCH9-overexpressing cells (32). How this unusual family of membrane-bound E3 ligases recognizes this wide variety of substrate proteins and what, specifically, identifies a membrane protein for MIR/MARCH-mediated regulation remain open questions.
Several lines of evidence indicate that substrates are recognized through interactions encoded in the membrane-associated domains of substrates and MIR/MARCH proteins. Multiple groups have reported that susceptibility to MIR- or MARCH-mediated ubiquitination can be transferred to nonsubstrate proteins through replacement of their TM and cytoplasmic domains with those of known substrates (2, 15, 16, 18, 23). A study using CD8 fusion proteins carrying the human MHC-II subunit HLA-DRβ TM and cytoplasmic regions identified sequences in the extracellular juxtamembrane (JM) region and the cytosolic TM–JM junction of DRβ that were required for maximal MARCH8-mediated down-regulation (20). However, the requirement for specific complementary sequences in MARCH8 was not investigated. Domain-swap experiments in the KSHV MIR proteins showed that the expanded substrate repertoire of MIR2 could be transferred to MIR1 by replacing only its TM–loop–TM sequence with that of MIR2 (33). MIR2 recognition of CD86 involves both the substrate TM and extracellular JM regions (33, 34), and specific residues in the MIR2 loop region (Phe-119/Ser-120) appear to make particularly important contributions (34). Taken together, these observations indicate that the substrate preferences of MIR/MARCH proteins are dependent on the specific substrate TM and flanking sequences, and that the membrane-associated domains of MIR/MARCH proteins themselves are implicated in determining this specificity.
Consistent with a possible role in direct interactions with substrate proteins, the TM domains of MIR and MARCH proteins contain many sequences that are commonly associated with specific TM–TM interfaces in membrane proteins (35). These include glycophorin A-like small amino acid motifs (glycine-XXX-glycine and derivatives containing alanine, serine, or threonine), strongly polar (glutamine and asparagine), polar/aromatic (tyrosine and tryptophan), and ionizable (lysine, aspartic acid, glutamic acid, and histidine) residues that frequently drive TM interactions through close helix–helix packing, hydrogen-bonds, and strong ionic interactions (36–44). However, there are very few experimental data available on whether any of these potential interaction motifs contribute to MIR/MARCH-mediated substrate recognition. To the best of our knowledge, the only reported point mutation in a MIR or MARCH TM domain that has been shown to affect function is in MARCH9, where a D231N substitution in the second predicted TM domain caused a partial reduction in MHC-I down-regulation but did not affect activity toward ICAM-I (31). Here, we examined the roles of specific sequences in the human MARCH9 TM domains by monitoring down-regulation of two different substrates, the human MHC-I molecule HLA-A2 and the T-cell co-receptor CD4, expressed in 293T cells. We show that a single serine residue in the first TM domain of MARCH9 was absolutely required for activity against both substrate proteins, whereas two other positions in the second TM domain (a glycine and a tyrosine) caused partial defects. An initial structural analysis using solution nuclear magnetic resonance (NMR) showed that these key residues are located in the α-helical portions of TM1 and TM2 that are closest to the extracellular face of the lipid bilayer, strongly implicating this particular region in MARCH9-mediated membrane protein regulation.
Results
Sensitivity to MARCH9 down-regulation can be conferred by a substrate TM domain
To quantify MARCH9-mediated down-regulation of target molecules from the cell surface, we designed a cell-based assay in which 293T cells endogenously expressing HLA-A2 were transduced with a lentiviral vector containing a doxycycline (dox)-inducible human MARCH9 expression cassette and mCherry expressed from a constitutive promoter. Addition of dox to cells transduced with WT MARCH9 caused a near-complete removal of HLA-A2 from the cell surface, which was measured by flow cytometry after staining with a fluorescent anti-HLA-A2 antibody (Fig. 1A). As a negative control, we used a RING-CH mutant of MARCH9 (W143A) (32), which is analogous to the W41A mutation of MIR1 that destroys the E2-binding site to prevent ubiquitin transfer to substrate (45). Cells expressing the MARCH9 W143A mutant were unable to down-regulate HLA-A2 and were indistinguishable from cells harboring the empty vector encoding only the fluorescent mCherry marker, demonstrating that substrate down-regulation depends on the presence of a functioning RING-CH domain, as shown previously for other substrates (32). To simplify the quantitative representation of MARCH9 activity, the mean fluorescence intensity (MFI) of HLA-A2 staining before and after dox addition for each transduced cell line was used to calculate the percent substrate remaining after MARCH9 induction (Fig. 1, B and C). WT and W143A values (gray dashed lines) are used in Fig. 1, B and C, to define full activity and no activity.
Figure 1.

Susceptibility to MARCH9-mediated down-regulation can be transferred by the CD4 TM domain sequence. A, representative histograms showing 293T cells transduced with lentivirus-encoding dox-inducible WT (left), W143A RING mutant (middle), or no MARCH9 (right) with a constitutively expressed mCherry marker. Transduced cells were incubated with or without dox for 48 h, and surface levels of endogenous HLA-A2 on mCherry+ cells were measured by flow cytometry. B and C, 293T cells stably expressing human CD4 were transduced with lentiviral constructs shown in A, and surface levels of HLA-A2 in B and CD4 in C were measured on mCherry+ cells after 48 h of culture with or without dox. Data are presented as percentage of substrate remaining on the surface of dox-treated cells relative to untreated cells from the same transduction. Dashed lines indicate mean substrate level remaining on dox-treated cells expressing control WT (lower) and RING mutant W143A (upper) MARCH9. D and E, 293T cells stably expressing human CD4, human CD19, or a chimeric CD19 protein containing the TM domain of CD4 (CD19–CD4TM) were transduced with WT MARCH9 and cultured with and without doxycycline for 48 h. Surface levels of CD4/CD19 (D) and HLA-A2 (E) were measured on mCherry+ cells by flow cytometry and presented as in B and C. In all graphs, lines represent the mean and S.E. of three independent experiments performed on different days (n = 3), and each point represents data from one experiment. Unpaired t test: ns, not significant; **, p < 0.02. F, human CD4 and CD19 partial amino acid sequences showing the residues that were swapped in the CD19–CD4TM chimera. Predicted TM domains underlined. Lysine residues in cytoplasmic tails highlighted in bold represent possible ubiquitination sites.
MARCH9 has previously been reported to down-regulate the T-cell co-receptor molecule CD4 (35), but not the B-cell antigen CD19 (32). To confirm these observations, we generated 293T cell lines stably expressing either CD4 or CD19 and transduced them with dox-inducible MARCH9. The addition of dox caused robust CD4 down-regulation from the cell surface, while CD19 levels were not significantly altered (Fig. 1D). Simultaneous measurement of endogenous HLA-A2 levels confirmed that MARCH9 was active in each cell line (Fig. 1E). We then used this system to test whether MARCH9 recognition of CD4 depended on its TM sequence by generating a chimeric CD19 molecule that had its 23-amino acid TM domain replaced with the corresponding sequence from CD4 (Fig. 1F). Down-regulation of the CD19–CD4TM chimera was indistinguishable from that observed for CD4 after MARCH9 induction (Fig. 1D). This result shows that the CD4 TM domain is sufficient to allow MARCH9 recognition of CD19 and that the CD19 cytoplasmic domain supports MARCH9-mediated down-regulation, but its native TM domain does not.
Replacing MARCH9 TM domains with generic hydrophobic sequences abrogates function
We hypothesized that the MARCH9 TM–loop–TM domain is responsible for substrate recognition, as has been shown for viral MIRs (33) and for MARCH1 (23). To test this hypothesis, we used the Transmembrane Hidden Markov Model (TMHMM) prediction tool (46) to estimate the boundaries of the TM domains of MARCH9 (Fig. 2A) and used this prediction as a guide for designing MARCH9 TM domain mutants. We replaced the native TM domains of MARCH9 with generic hydrophobic stretches in the form of polyleucine (polyL) sequences (Fig. 2B). Two cysteines in the second predicted TM domain were retained as these represent potential S-acylation sites that might influence MARCH9 trafficking or membrane sub-localization. Replacing MARCH9 TM domains either together (polyL–TMs) or separately (polyL–TM1 and polyL–TM2) eliminated MARCH9 activity against both HLA-A2 (Fig. 2C) and CD4 (Fig. 2D). All mutants were statistically indistinguishable from W143A, except in the case of the polyL–TM mutant, which was even less active than W143A against CD4 (Fig. 2D).
Figure 2.

MARCH9 TM sequences contain features that are absolutely required for substrate down-regulation. A, TMHMM (46) prediction of the locations of two TM domains in human MARCH9. Shaded bars are 50% confidence or better, and boundary residues for TM1 (Val-183–Ser-205) and TM2 (Leu-218–His-240) are marked above the graph. B, sequences of mutants analyzed in this figure. Red bold font indicates substituted amino acids. Two cysteines in predicted TM2 were not substituted. MARCH9_ScramLoop was generated by randomizing the 12-amino acid loop sequence such that no position contained the native amino acid but the overall amino acid content was unchanged. C and D, 293T cells stably expressing human CD4 were transduced with the indicated lentiviral constructs, and surface levels of HLA-A2 (C) and CD4 (D) were measured after 48 h culture with or without dox as in Fig. 1. Dashed lines indicate mean substrate level remaining on dox-treated cells expressing control WT (lower) and RING mutant W143A (upper) MARCH9. Unpaired t test: ns, not significant; *, p < 0.05. E, Western blot analysis of MARCH9 expression. WT and mutant MARCH9 sequences with C-terminal HA tag were transiently expressed in 293T cells under the strong EF1α promoter by calcium phosphate transfection and harvested 24–48 h later. Whole-cell lysates were separated by reducing SDS-PAGE, transferred to PVDF, and sequentially immunoblotted using α-GAPDH and α-HA antibodies. + marks the positions of a cellular product detected by SA-HRP; arrow marks the position of the HA-tagged MARCH9 protein. Table shows expression normalized to GAPDH loading control and relative to MARCH9-WT, quantitated by densitometry.
The extracellular loop of KSHV MIR2 has previously been shown to contribute to CD86 down-regulation (34). To test the role of the MARCH9 loop region, we scrambled the 12-residue sequence such that the amino acid composition was preserved but no single residue was in its native location (Fig. 2B, ScramLoop mutant). This significantly reduced MARCH9 activity (Fig. 2, C and D), but substantial down-regulation was still observed, indicating that the EC loop may play a role in MARCH9 function but its specific sequence may not be crucial. To ensure that the MARCH9 mutants with reduced or abrogated activity were expressed in 293T cells, we appended a C-terminal HA tag to the WT and mutant MARCH9 sequences and performed Western blot analysis on whole-cell lysates. WT MARCH9 expression was not detectable by Western blotting in dox-treated lentiviral transductants (data not shown) despite clear activity, so these experiments were performed in transiently transfected cells (Fig. 2E). The expression of the RING-CH mutant W143A was similar to WT and the polyL–TM mutants similar or higher, whereas the ScramLoop mutant was expressed at lower levels compared with WT. These results show that the TM domains of MARCH9 contain specific sequences that make a crucial contribution to MARCH9-mediated substrate down-regulation beyond targeting of the E3 ligase activity to the membrane, for which the polyL–TMs would be sufficient. Although scrambling the EC loop also resulted in reduced substrate down-regulation, this may be related to reduced expression levels rather than a sequence-specific functional defect (see below).
Mutation of a single serine residue in TM1 of MARCH9 abrogates substrate down-regulation
To determine whether MARCH9 activity could be attributed to specific sequence motifs within the MARCH9 TM domains, we took a candidate-based mutagenesis approach. We first mutated a block of three serine residues predicted to be in the core membrane-spanning region of TM1 (Ser-192/Ser-198/Ser-200) to alanine (Fig. 3A). This triple-serine mutant (3SA) was completely unable to down-regulate HLA-A2 (Fig. 3B) or CD4 (Fig. 3C) and was statistically indistinguishable from the RING mutant W143A. Conversely, mutation of a second block of four serines located at the extracellular end of TM1 and in the loop sequence (Ser-205/Ser-206/Ser-208/Ser-210; mutant 4SA) had little effect on MARCH9 activity (Fig. 3, B and C) and was statistically indistinguishable from WT MARCH9. To determine whether a single position was responsible for the Ser-192/Ser-198/Ser-200 triple mutant phenotype, we generated the serine to alanine mutations separately. The activities of the S192A and S200A mutants were unperturbed, whereas the S198A mutant, like the triple mutant, was completely unable to down-regulate HLA-A2 (Fig. 3D) or CD4 (Fig. 3E). Substitution of Ser-198 with threonine (S198T) resulted in WT levels of activity against both substrates, whereas its nonpolar near-isostere valine (S198V) yielded no activity. These data indicate that the hydroxyl group is critical at this position, but a small increase in steric bulk is well tolerated. Western blot analysis (Fig. 3F) showed that S198A was expressed slightly lower than WT MARCH9 in whole-cell lysates, whereas the triple-serine and S198V mutants were expressed at higher levels. Together, these results show that a small hydroxyl-bearing residue at position 198 in TM1 is absolutely required for MARCH9 activity against two different substrate proteins, and the defect associated with the S198A mutation is not related to poor expression.
Figure 3.

Serine 198 in the first predicted TM domain of MARCH9 is absolutely required for substrate down-regulation. A, sequences of mutants analyzed in this figure. Red bold font indicates substituted amino acids. Vertical box highlights the position of Ser-198. B–E, 293T cells stably expressing human CD4 were transduced with the indicated lentiviral constructs, and surface levels of HLA-A2 (B and D) and CD4 (C and E) were measured after 48 h of culture with or without dox as in Fig. 1. Serine-to-alanine substitutions were examined in blocks of three or four (3S-A and 4S-A mutants; B and C) and separately for the single-serine mutants as indicated (D and E). Dashed lines indicate mean substrate level remaining on dox-treated cells expressing control WT (lower) and RING mutant W143A (upper) MARCH9. Unpaired t test: ns, not significant. F, Western blot analysis of MARCH9 WT and inactive mutant expression, performed as in Fig. 2E. + marks the position of a cellular product detected by SA-HRP; arrow marks the position of MARCH9 protein. Table shows expression normalized to GAPDH loading control and relative to MARCH9-WT, quantitated by densitometry.
Subcellular localization of MARCH9 S198 mutants
We next considered whether the lack of activity in MARCH9 S198A and S198V mutants was the result of altered subcellular localization. Lehner and co-workers (31) previously reported that a MARCH9–GFP fusion localized to punctate vesicular structures containing the lysosomal protease cathepsin D, indicating that MARCH9 resides primarily in lysosomes. We observed a similar punctate vesicular distribution using MARCH9–ZsGreen fusions expressed in 293T cells (Fig. 4), and we found no activity-correlated differences among WT and mutant MARCH9 distribution. Active WT and S198T proteins as well as inactive mutants S198A, S198V, and W143A (RING mutant) were visible in punctate vesicular structures in transfected cells, and none displayed concentrated perinuclear staining that would indicate failure to exit the endoplasmic reticulum. Together with the Western blotting data, the apparently normal subcellular localization of inactive mutants indicates that their functional defects do not arise from gross expression or trafficking defects.
Figure 4.

Serine 198 mutations do not alter MARCH9 subcellular localization. Representative images are shown for 293T cells expressing free cytosolic ZsGreen protein (empty vector, for reference) or MARCH9–ZsGreen fusion proteins with the indicated mutations. Cell preparations were stained and imaged by confocal microscopy 48 h after transient transfection. Color channels are ZsGreen (yellow), F-actin stain (Alexa 555-phalloidin, magenta), and nuclear stain (DAPI, cyan). All images contain both transfected and untransfected cells.
Mutation of tryptophan and small amino acid motifs in TM1 of MARCH9 yields milder phenotypes
To test whether other sequences in TM1 contribute to MARCH9 function, we also mutated two tryptophan residues (Trp-201/Trp-204) and a glycophorin A–like small amino acid motif (Ala-187/Gly-191) (Fig. 5A). W201A and W204A mutations each caused a partial defect in HLA-A2 and CD4 down-regulation compared with WT MARCH9, and combination of the two mutations (2WA) caused a stronger defect (Fig. 5, B and C). In contrast, simultaneous mutation of the two small amino acids in the glycophorin A-like motif to leucine (AG-LL mutant) had no effect on activity against HLA-A2 (Fig. 5D), and the small effect on CD4 down-regulation did not reach statistical significance (Fig. 5E). Thus, the tryptophan residues located near Ser-198 at the extracellular end of TM1 may also contribute to MARCH9 down-regulation of HLA-A2 and CD4, whereas the small amino acid motif near the intracellular end of TM1 does not.
Figure 5.

Mutation of tryptophans in TM1 of MARCH9 causes moderate defects, but mutation of a glycophorin A-like motif does not. A, sequences of mutants analyzed in this figure. Red bold font indicates substituted amino acids. B–E, 293T cells stably expressing human CD4 were transduced with the indicated lentiviral constructs, and surface levels of HLA-A2 (B and D) and CD4 (C and E) were measured after 48 h culture with or without dox as in Fig. 1. Dashed lines indicate mean substrate level remaining on dox-treated cells expressing control WT (lower) and RING mutant W143A (upper) MARCH9. Unpaired t test: ns, not significant; *, p < 0.05; **, p < 0.02; ***, p < 0.001; ****, p < 0.0001.
Polar and small amino acids in TM2 also contribute to MARCH9 function
The results above indicate that the end of TM1 closest to the extracellular membrane face contains functionally important sequences. We therefore tested the same region of TM2 for positions that could form part of a composite molecular surface that is required for MARCH9 activity. Based on the TMHMM prediction, we generated mutations in the extracellular half of TM2, including Q221M, C223A, Y224F, G225V, and Y227F (Fig. 6A). Although C223A was indistinguishable from WT MARCH9, Q221M, Y224F, and G225V all exhibited small but statistically significant reductions in activity against HLA-A2 (Fig. 6B) and CD4 (Fig. 6C). The Y227F defect was more severe, and combination with the Y224F mutation (mutant 2YF) moderately exacerbated the defect against HLA-A2 but not CD4. Finally, we tested alanine substitution of an aspartic acid residue (Asp-231) in TM2 (Fig. 6A) that was previously reported to play a role in MHC-I but not ICAM-1 down-regulation (31). MARCH9 D231A exhibited a mild but reproducible defect in HLA-A2 down-regulation (Fig. 6D), whereas alterations in CD4 down-regulation did not reach statistical significance (Fig. 6E). These data indicate that, like TM1, the most severe functional defects are seen in mutations at positions in the end of TM2 closest to the extracellular membrane face.
Figure 6.

Mutation of small, polar, and aromatic residues in TM2 of MARCH9 affects substrate down-regulation. A, sequences of mutants analyzed in this figure. Red bold font indicates substituted amino acids. B–E, 293T cells stably expressing human CD4 were transduced with the indicated lentiviral constructs, and surface levels of HLA-A2 (B and D) and CD4 (C and E) were measured after 48 h of culture with or without dox as in Fig. 1. Dashed lines indicate mean substrate level remaining on dox-treated cells expressing control WT (lower) and inactive mutant S198A (upper) MARCH9. Unpaired t test: ns, not significant; *, p < 0.05; **, p < 0.02; ***, p < 0.001; ****, p < 0.0001.
Relationship between MARCH9 mutant protein levels and substrate down-regulation
As we had noted that the scrambled loop MARCH9 mutant had lower total cellular protein levels that could account for its reduced activity compared with WT, we took a more systematic approach to determine the relationship between protein and activity levels for MARCH9 mutants that showed substantially reduced activity toward one or both substrates. Quantitation of MARCH9 levels by Western blotting, as shown in Figs. 2 and 3, was performed for three independent transfections per construct, and relative protein levels (normalized to GAPDH loading control) were plotted against relative activity levels (for HLA-A2) in Fig. 7. This analysis shows that the lower activity levels of ScramLoop mutant and most TM mutants were concomitant with lower protein levels, suggesting that reduced substrate down-regulation may be due to expression defects or protein instability rather than specific functional defects. However, like the RING-CH mutant W143A, the MARCH9 TM mutants S198A, G225V, and Y227F (Fig. 7, red dots) produced protein levels that were similar to (or greater than) WT yet displayed reduced or abrogated activity. We therefore conclude that these three positions make specific contributions to MARCH9 function that are likely to be independent from effects on protein expression or stability in 293T cells.
Figure 7.
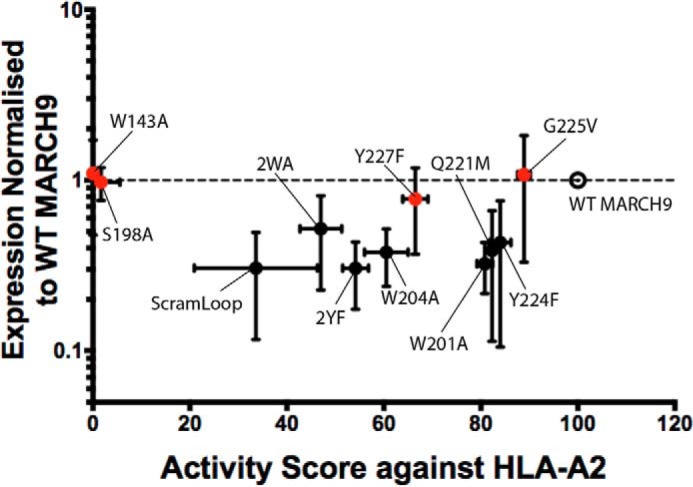
Relationship between MARCH9 mutant activity and protein levels. Expression levels for the indicated mutants (y axis) were analyzed by transient transfection in 293T cells as shown in Figs. 2 and 3, and MARCH9-specific bands were quantitated by densitometry. MARCH9 expression was normalized to GAPDH signal and plotted relative to WT (open circle). Activity score (x axis) was determined by the reduction in surface levels of HLA-A2 after dox treatment (data shown in Figs. 2–5) and expressed as a percentage of WT activity. Dashed line represents expression of WT MARCH9, which was set to 1 for comparison. Vertical error bars represent S.D. of at least three independent transfections (n ≥ 3). Horizontal error bars represent S.E. of three independent experiments (n = 3). Red symbols highlight mutants for which functional defects are likely to be independent of total cellular protein levels.
Ser-198, Gly-225, and Tyr-227 are located in the α-helical regions of TM1 and TM2
To determine more precisely the locations of these key residues within the structural organization of MARCH9, we produced a 65-amino acid fragment encompassing the TM–loop–TM region (Ile-180–Ser-244) for solution NMR studies (MARCH9–TM). MARCH9–TM rapidly aggregated or precipitated out of samples prepared in pure alkyl phosphocholine detergents (dodecyl phosphocholine and tetradecyl phosphocholine (TDPC)), the short-chain phospholipid dihexanoyl phosphatidylcholine (DHPC), and bicelles composed of DHPC and dimyristoyl phosphatidylcholine (DMPC), but produced stable samples with well-resolved 1H–15N HSQC spectra in a 5:1 TDPC/SDS mixture (Fig. S1) and in pure lyso-myristoyl phosphatidylglycerol (LMPG) (Fig. 8A). Complete backbone assignments were obtained under both conditions using a standard suite of transverse relaxation (TROSY)-optimized (47) triple-resonance experiments (48) recorded on a uniformly 15N,13C,2H (85%)-labeled peptide. Analysis of secondary chemical shifts in LMPG (Fig. 8B) using TALOS+ (49) identified a region of continuous α-helical structure (Lys-182–Ser-206) that was in very good agreement with the TMHMM prediction (Val-183–Ser-205) for the location of TM1, whereas most of the loop region and N- and C-terminal (intracellular) ends were predicted to be unstructured. A second α-helical region (Arg-215–Val-239) was also in good general agreement with the TMHMM prediction for the location of TM2 (Leu-218–His-240). However, backbone dihedral angle predictions (Fig. 8C) were at low-confidence around the locations of two glycine residues (Gly-228 and Gly-236), and there was an increasing loss of helicity toward the C-terminal end (Ile-235–Ile-238). Results in TDPC/SDS samples were very similar (Fig. S1). Backbone amide relaxation (T1/T2) measurements in LMPG (Fig. 8D) also yielded significantly different average T1 values for TM1 and TM2 regions (1318 and 934 ms, respectively). Together, these data indicate that the C-terminal half of TM2 is unstable and that TM1 and TM2 undergo independent motions within the micelle and are therefore unlikely to form a well-defined interhelical interface. These features notwithstanding, our solution NMR data show that Ser-198, Gly-225, and Tyr-227 (highlighted with vertical shaded boxes in Fig. 8, C and D) are located within the stable α-helical regions of the MARCH9 TM–loop–TM segment. This defines two functionally relevant sites in TM1 and TM2 that are likely to be located at similar depths in the membrane and may therefore represent a composite surface that governs molecular interactions involved directly or indirectly in substrate binding.
Figure 8.

Structural analysis of a MARCH9 TM–loop–TM fragment by solution NMR. A, assigned 1H–15N HSQC spectrum of a uniformly 15N,13C,2H 80%-labeled, 65-amino acid MARCH9 TM–loop–TM peptide (1 mm) in 250 mm LMPG, 20 mm phosphate buffer, pH 6.8, 5% D2O at 600 MHz and 40 °C. The TM–loop–TM peptide contained mutations C223S, M226V, M230V, and C234S to aid in production and an I239V substitution that is present in the mouse sequence (see “Experimental procedures”). B, secondary structure prediction from assigned backbone chemical shifts using TALOS+ (49). Confidence scores for residues predicted to be part of a helix (red bars) or random coil (blue bars) are superimposed with TMHMM predictions (black lines/dots) for comparison. C, backbone torsion angle ϕ (dots) and ψ (triangles) predictions from TALOS+. Values are in degrees, and error bars represent the estimated S.D. of the prediction error. D, backbone 15N T1 (top) and T2 (bottom) relaxation times (milliseconds) determined for a uniformly 15N-labeled MARCH9–TM sample prepared as in A and analyzed at 600 MHz and 40 °C. Helix 1 and Helix 2 are highlighted in large shaded boxes. The positions of key residues Ser-198, Gly-225, and Tyr-227 are highlighted in vertical shaded boxes.
Discussion
The viral MIR and mammalian MARCH proteins regulate the cell-surface levels of immunologically important membrane proteins. Although their RING-CH domains facilitate the transfer of ubiquitin to lysine residues in the substrate cytoplasmic tails, the TM–loop–TM region is believed to be the primary determinant of substrate recognition. Whether this occurs through direct interactions between MIR/MARCH and substrate TM domains or by other means is unknown. Previous studies have shown that substrate specificity can be transferred between MIR1 and MIR2 by exchange of the TM–loop–TM region (33) and that the TM and cytoplasmic domains of at least some substrates are sufficient for down-regulation by MIR1/2 (15, 18) and MARCH1/8 (2, 23). Our data show that susceptibility to MARCH9-mediated down-regulation can be transferred to the nonsubstrate CD19 by strict exchange of only its 23-amino acid TM domain with that of the substrate protein CD4. Thus, although the extracellular and intracellular sequences of CD19 appear to be fully compatible with MARCH9-mediated down-regulation, any contributions they make to MARCH9 susceptibility are insufficient for detectable down-regulation in the absence of a compatible TM domain. We also showed that specific sequences in the TM domains of MARCH9 are required for down-regulation of HLA-A2 and CD4 because replacement of native MARCH9 TM domains with polyleucine sequences, which are highly favorable in the membrane (50–52) but support only weak, nonspecific TM–TM interactions (53), eliminated all detectable activity despite higher than WT expression levels.
All mammalian MARCH proteins contain a high density of TM sequences that may contribute to specific protein interactions within the lipid bilayer, including strongly polar (Lys, Asp, Glu, and Gln), weakly polar (Ser and Thr), polar/aromatic (Tyr, Trp, and His), and small amino acid (Gly and Ala) motifs (35). The TM domains of MARCH9 contain examples of all of these motifs and display near-absolute conservation among mammals; the 72 mammalian sequences available in Ensembl are identical in the region corresponding to human Ile-180–Ser-244 except for position 239 at the intracellular end of TM2, which can be Ile or Val. Very little mutagenesis has been reported examining the functional importance of specific MARCH TM sequences, but strongly polar and glycophorin A-like small amino acid motifs might be expected to make the largest contributions to intramembrane interactions based on energetic considerations (38, 39, 44, 50, 53). Surprisingly, we found that mutation of the most strongly polar MARCH9–TM residue, Asp-231 in the middle of TM2 (mutated to alanine), had little effect on its activity. A more conservative D231N mutation was previously reported to cause a modest defect in down-regulation of MHC-I but not ICAM-1 (31), consistent with the small but statistically significant defect we observed against HLA-A2 but not CD4. Similarly, a glycophorin A-like small amino acid motif in the intracellular half of TM1 (187AXXXG191) did not appear to contribute significantly to HLA-A2 or CD4 down-regulation. Mutation of other polar and polar/aromatic residues resulted in significant defects, but for most of these we could not discount their lower expression levels as the primary cause of their reduced activity. Amino acid substitutions at three different positions produced WT or near-WT protein levels yet still displayed significant functional defects against both substrates analyzed: S198A/S198V, G225V, and Y227F. Mutations at Ser-198 in TM1 of MARCH9 had the strongest effect of any residue that we tested, causing a complete loss of function against both HLA-A2 and CD4 despite similar (S198A) or higher (S198V) expression levels compared with WT. The nature of these mutations (to nonpolar alanine or valine) coupled with the full activity observed for the threonine substitution shows that the polar hydroxyl group at this position is the key determinant of activity, and the defect was entirely position-specific because mutation of two other adjacent serines (Ser-192 and Ser-200) in TM1 and a block of four serines in the TM1–to–loop region had no effect. To the best of our knowledge, these are the only TM point mutations reported to completely abrogate function of a MIR or MARCH family E3 ubiquitin ligase, and the severity of their defects underscores the crucial role of specific MARCH9 TM sequences in its function.
How might Ser-198 contribute to MARCH9 function? Our fluorescence microscopy data rule out gross defects in subcellular localization for Ser-198 mutants, but they do not rule out a role for this position in membrane sub-domain localization. It could further play a role in internal structural stabilization, formation of oligomeric MARCH9 structures (31), or interactions with adaptor proteins that may be required to bring MARCH9 and substrates together in the membrane, the latter being a role recently ascribed to the Salmonella protein SteD in MARCH8-mediated MHC-II regulation in infected cells (54). Perhaps most interestingly, Ser-198 may represent the site of a specific interaction with a complementary site that is available in both HLA-A2 and CD4 TM domains. Although there are no polar residues in the extracellular half of these substrate TM sequences, a notable concentration of small amino acids (Gly and Ala) in this region of both HLA-A2 and CD4 (but not CD19) could expose polar main-chain groups as potential hydrogen-bonding partners. The failure to identify a point mutation in TM2 that recapitulates the complete functional defect observed for S198A/S198V mutants argues against a critical TM1–TM2 interaction that is as simple as a hydrogen bond with a single partner side chain. We therefore sought to structurally characterize the MARCH9 TM–loop–TM region to gain further insight into the locations of functionally relevant amino acids.
There are no experimentally determined structures available for any MIR or MARCH membrane-associated domains. Our NMR analysis of a MARCH9 TM–loop–TM fragment in mixed detergent (TDPC/SDS) or lysolipid (LMPG) micelles shows two α-helical regions that are consistent with the predicted locations of TM1 and TM2 and are connected by an unstructured loop region of approximately nine residues. This indicates that our sample conditions support the expected secondary structural organization of a helical hairpin. The functionally critical residue Ser-198 is located approximately two helical turns into the membrane from the extracellular end of TM1. The only other point mutations we identified as having functional defects that are likely to be independent from reduced expression (G225V and Y227F) are located at a similar position in TM2, approximately three helical turns into the membrane from the extracellular end. Although the data presented here do not provide information regarding whether these represent directly interacting sites in a native cell membrane, their location at similar depths in the membrane suggests that they could form a composite surface that engages in molecular interactions required for MARCH9 function. The structural deterioration and independent motion we noted in TM2 indicate that it is unlikely to form a stable interface with TM1 under the NMR sample conditions used here. Although these observations may be strictly related to the suboptimal membrane-mimicking properties of the micellar environment (55), it is also possible that no stable interhelical structure exists in the absence of a substrate TM helix, a MARCH9 oligomeric assembly, or other partner proteins, and this could reflect a physiologically relevant feature that supports interaction with many different substrates. More detailed structural studies of MARCH9 and MARCH9–substrate complexes in a lipid bilayer environment will be required to provide further insights.
Together, our biochemical results and structural analysis provide clear evidence that specific sequences in the α-helical MARCH9 TM domains contribute to function. These lay the foundations for future functional and structural studies to further define the nature of their role in down-regulation of substrate proteins.
Experimental procedures
Cells and antibodies
Human embryonic kidney (HEK) 293T cells were mycoplasma-free and maintained in Dulbecco's modified Eagle's media containing 4.5 g/liter d-glucose (Gibco), supplemented with 10% bovine fetal calf serum, 2 mm l-glutamine, and incubated in 10% CO2 at 37 °C. Cells were routinely passaged every 3 days. Phycoerythrin-conjugated anti-HLA-A2 (clone BB7.2), allophycocyanin (APC)-conjugated anti-CD19 (clone HIB19), and APC-conjugated anti-CD4 (clone OTK4) used in flow cytometry analysis were from BioLegend (San Diego). Western blotting reagents anti-GAPDH–peroxidase (GAPDH-71.1) and streptavidin-peroxidase were from Sigma. Biotin-conjugated anti-HA (clone 3F10) was from Roche Applied Science.
Plasmids and constructs
The human MARCH9-coding sequence (accession no. NM_138396.4) was supplied in the pCMV-XL5 vector (SC120445; Origene). A GC-rich region in the native sequence interfered with downstream cloning steps and was codon-modified to reduce GC content without altering protein sequence. Site-directed mutagenesis was performed by PCR using overlapping primers (IDT) that encoded mutations at specific sites. Synthetic G-blocks (IDT) encoding polyleucine sequences were used to replace native MARCH9 TM domains using SexAI and SphI restriction sites. For functional assays with doxycycline control, MARCH9 variants were cloned into pTRE-tight (Clontech) and shuttled into the lentiviral backbone pFUGW-mCherry (56, 57) using PacI sites. For expression analysis by Western blotting, MARCH9 was modified with a C-terminal HA tag and cloned into pHAGE-EF1α-IRES-ZsGreen (Harvard Gene Therapy Initiative), which enabled constitutive expression of MARCH9 under the control of the EF1α promoter. For fluorescence microscopy experiments, the IRES was deleted from this construct, and the ZsGreen sequence was fused directly to the native MARCH9 C terminus (without HA tag) before the stop codon.
Transient transfections and lentiviral transductions
For production of lentivirus, HEK-293T cells were transiently transfected with a 1:1.2:2:4 ratio of pVSVg, pRSV, pMdi, and pFUGW-mCherry MARCH9 using standard calcium phosphate precipitation. After 48 h, viral particles were harvested from the supernatant and used to transduce 293T cells by spin infection in the presence of Polybrene transfection reagent (4 μg/ml; Millipore). Transient transfection of 293T cells was performed by complexing DNA with calcium phosphate, and transfected cells were assayed 24–48 h post-transfection for MARCH9 expression analysis by Western blotting and fluorescence microscopy.
Down-regulation assay and flow cytometry analysis
At 7 days post-transduction, 293T cells harboring transduced pFUGW-mCherry MARCH9 were seeded at 60% confluence, and dox was added 24 h later (1 μg/ml final concentration) to induce expression of MARCH9. 48 h after doxycycline addition, cells were harvested and stained with fluorophore-conjugated primary antibodies against HLA-A2, CD4, or CD19 on ice for 30 min in PBS containing 0.5% BSA (w/v), 2 mm EDTA and washed twice before analysis. Data acquisition was performed on a Fortessa X20 or Fortessa1 flow cytometer (BD Biosciences) and analyzed in FlowJo version 10.3. Substrate % remaining after MARCH9 induction was calculated for mCherryHi cells as (x− N/y − N)·100, where x = MFI of substrate on dox-treated cells; y = MFI of substrate on a matched sample that did not receive dox; and n = MFI of unstained control cells. Activity score against HLA-A2 (Fig. 6) was calculated by normalizing the % remaining after MARCH9 induction of each mutant against that of WT MARCH9. Graphs and statistical analyses were performed in Prism 7 (Graphpad). Statistical differences in activity of MARCH9 variants were calculated in an unpaired Student's t test performed using data acquired from three independent experiments performed on separate days. p ≤ 0.05 were considered significant for all statistical calculations.
Western blotting
Transiently transfected 293T cells were harvested 24–48 h post-transfection and washed with cold PBS. Cells were lysed on ice in RIPA buffer (50 mm Tris, pH 8, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) containing 1% protease inhibitor mixture (Sigma P8340). Cleared lysates were boiled in LDS sample buffer (Invitrogen) containing dithiothreitol (DTT, 100 mm). Proteins were separated by SDS-PAGE using NuPAGE 12% BisTris acrylamide gels (Invitrogen) in MES buffer (200 V, 40 min) and transferred to polyvinylidene fluoride (PVDF) membranes. PVDF membranes were blocked for at least 1 h in Tris-buffed saline (TBS) containing 5% skim milk powder while rocking. Membranes were washed three times in TBS containing 0.1% Tween 20 (TBST) prior to application of antibodies. Loading controls were established by blotting the membrane with 1:20,000 dilution of anti-GAPDH–peroxidase (GAPDH-71.1; Sigma) and developing with Western Lightning Plus ECL (PerkinElmer Life Science). Peroxidase activity was subsequently quenched by soaking membranes in 0.5% sodium azide in TBS for 30 min at room temperature and washing with TBST overnight at 4 °C before re-blotting with 0.1 μg/ml biotin-conjugated anti-HA (3F10; Roche Applied Science) in TBST for 1 h while rocking. Three TBST washes were performed before incubating with 1:5000 dilution of streptavidin-peroxidase (Sigma) for at least 30 min while rocking. Membranes were again visualized by adding Western Lightning Plus ECL (PerkinElmer Life Science) directly onto the membrane and detecting peroxidase signals in a ChemiDoc XRS+ Molecular Imager (Bio-Rad).
Fluorescence microscopy
293T cells were cultured on sterile coverslips prior to transient transfection with MARCH9–ZsGreen fusion constructs or empty vector expressing cytosolic ZsGreen. 48 h after transfections, coverslips were fixed in 4% paraformaldehyde in PBS for 1 h at room temperature and washed twice with PBS/Tween 20 (PBST) solution. F-actin was stained with Alexa Fluor 555/phalloidin (ThermoFisher Scientific) in a 1:500 dilution for 1 h on ice. Cells were then sequentially washed twice with PBST and twice more with PBS. Coverslips were mounted onto slides with Fluoroshield-DAPI (Sigma) and sealed with nail varnish. Samples were imaged on a Zeiss LSM780 confocal microscope and processed in FIJI (ImageJ).
NMR
The 65-amino acid TM–loop–TM peptide produced for NMR was based on the mouse MARCH9 sequence, which differs from human MARCH9 at only one position in this region (Ile-239 in human MARCH9 is Val in mouse MARCH9). The sequence IEKVQIAAIVLGSLFLVASISWLIWSSLSPSAKWQRQDLLFQICYGMYGFMDVVCIGLIVHEGSS corresponding to mouse MARCH9 Ile-180–Ser-244 (predicted TM domains underlined) was adapted for production by changing two internal methionines and two internal cysteines (bold) to valine and serine, respectively. The resulting sequence IEKVQIAAIVLGSLFLVASISWLIWSSLSPSAKWQRQDLLFQISYGVYGFVDVVSIGLIVHEGSS was produced in Escherichia coli BL21(DE3) as a His9-tagged trpLE fusion, as described previously (58), dissolved in 6 m guanidine HCl, pH 8, 1% Triton X-100, and purified from inclusion bodies on nickel-nitrilotriacetic acid affinity resin (HIS-Select, Sigma). The MARCH9 TM peptide was released from the histidine-tagged trpLE sequence by cyanogen bromide digest (0.5 m cyanogen bromide in 80% trifluoroacetic acid (TFA)) overnight at room temperature, dialyzed extensively to water, and lyophilized. Digest products were separated by reversed-phase HPLC on an Agilent Zorbax SB-300 C3 column using a mobile-phase gradient of 20% acetonitrile, 10% trifluoroethanol (TFE), 0.3% TFA in water (solvent A) to 90% 2-propanol, 10% TFE, 0.3% TFA (solvent B). Peptide identity and purity were confirmed by SDS-PAGE and matrix-assisted laser desorption ionization (MALDI) MS. NMR samples were prepared by dissolving lyophilized, stable isotope-labeled peptide to 1 mm with the indicated lipid/detergent in 20 mm phosphate buffer, pH 6.8, and 5% D2O. All data were collected on a Bruker Avance III 600 MHz spectrometer equipped with triple-resonance cryoprobe at 40 °C and processed in TopSpin 3.2 (Bruker). Backbone assignments were obtained from transverse relaxation-optimized (TROSY) versions (47) of the HNCA, HNCO, HN(CO)CA, and HNCACB experiments (48) using the CARA (Computer-Aided Resonance Assignment) software package. Backbone chemical shift-based secondary structure analysis was performed using the TALOS+ (49) web server. Backbone 15N T1 and T2 relaxation measurements were derived from pseudo–three-dimensional HSQC experiments with variable delay times.
Author contributions
C. T. formal analysis; C. T., E. F. B., C. A.-C., M. J. C., and M. E. C. investigation; C. T., M. J. C., and M. E. C. methodology; C. T., E. F. B., C. A.-C., M. J. C., and M. E. C. writing-review and editing; M. J. C. and M. E. C. conceptualization; M. J. C. and M. E. C. supervision; M. J. C. and M. E. C. funding acquisition; M. J. C. and M. E. C. writing-original draft; M. J. C. and M. E. C. project administration.
Supplementary Material
Acknowledgment
We acknowledge the use of the Bio21 Magnetic Resonance Facility at the University of Melbourne, Parkville, Victoria, Australia.
This work was supported in part by Australian National Health and Medical Research Council Grant GNT1030902 and IRIISS Infrastructure Support and Human Frontier Science Program Grant RGP0064/2011. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
The NMR chemical shift data of this paper are available from the Biological Magnetic Resonance Data Bank under BMRB accession numbers 27543 and 27709.
- MARCH
- membrane-associated RING-CH
- TM
- transmembrane
- JM
- juxtamembrane
- EC
- extracellular
- HLA
- human leukocyte antigen
- MHC
- major histocompatibility complex
- KSHV
- Kaposi's sarcoma herpes virus
- MIR
- modulator of immune recognition
- ICAM-1
- intercellular adhesion molecule-1
- dox
- doxycycline
- MFI
- mean fluorescence intensity
- TMHMM
- Transmembrane Hidden Markov Model
- polyL
- polyleucine mutant
- ScramLoop
- scrambled loop sequence mutant
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- TDPC
- tetradecyl phosphocholine
- DHPC
- dihexanoyl phosphatidylcholine
- DMPC
- dimyristoyl phosphatidylcholine
- LMPG
- lyso-myristoyl phosphatidylglycerol
- TROSY
- transverse relaxation-optimized spectroscopy
- TALOS
- Torsion Angle Likeliness Obtained from Shifts and Sequence Similarity
- HEK
- human embryonic kidney
- APC
- allophycocyanin
- HA
- influenza hemagglutinin epitope tag (YPYDVPDYA)
- IRES
- internal ribosome entry site
- TFE
- trifluoroethanol
- PVDF
- polyvinylidene fluoride
- HRP
- horseradish peroxidase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HSQC
- heteronuclear single-quantum correlation
- DAPI
- 4,6-diamidino-2-phenylindole.
References
- 1. Bartee E., Mansouri M., Hovey Nerenberg B. T., Gouveia K., and Früh K. (2004) Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 78, 1109–1120 10.1128/JVI.78.3.1109-1120.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goto E., Ishido S., Sato Y., Ohgimoto S., Ohgimoto K., Nagano-Fujii M., and Hotta H. (2003) c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J. Biol. Chem. 278, 14657–14668 10.1074/jbc.M211285200 [DOI] [PubMed] [Google Scholar]
- 3. Ohmura-Hoshino M., Goto E., Matsuki Y., Aoki M., Mito M., Uematsu M., Hotta H, and Ishido S. (2006) A novel family of membrane-bound E3 ubiquitin ligases. J. Biochem. 140, 147–154 10.1093/jb/mvj160 [DOI] [PubMed] [Google Scholar]
- 4. Baravalle G., Park H., McSweeney M., Ohmura-Hoshino M., Matsuki Y., Ishido S., and Shin J.-S. (2011) Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J. Immunol. 187, 2966–2973 10.4049/jimmunol.1101643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Furuta K., Walseng E., and Roche P. A. (2013) Internalizing MHC class II–peptide complexes are ubiquitinated in early endosomes and targeted for lysosomal degradation. Proc. Natl. Acad. Sci. U.S.A. 110, 20188–20193 10.1073/pnas.1312994110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsuki Y., Ohmura-Hoshino M., Goto E., Aoki M., Mito-Yoshida M., Uematsu M., Hasegawa T., Koseki H., Ohara O., Nakayama M., Toyooka K., Matsuoka K., Hotta H., Yamamoto A., and Ishido S. (2007) Novel regulation of MHC class II function in B cells. EMBO J. 26, 846–854 10.1038/sj.emboj.7601556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oh J., Wu N., Baravalle G., Cohn B., Ma J., Lo B., Mellman I., Ishido S., Anderson M., and Shin J.-S. (2013) MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J. Exp. Med. 210, 1069–1077 10.1084/jem.20122695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walseng E., Furuta K., Goldszmid R. S., Weih K. A., Sher A., and Roche P. A. (2010) Dendritic cell activation prevents MHC class II ubiquitination and promotes MHC class II survival regardless of the activation stimulus. J. Biol. Chem. 285, 41749–41754 10.1074/jbc.M110.157586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young L. J., Wilson N. S., Schnorrer P., Proietto A., ten Broeke T., Matsuki Y., Mount A. M., Belz G. T., O'Keeffe M., Ohmura-Hoshino M., Ishido S., Stoorvogel W., Heath W. R., Shortman K., and Villadangos J. A. (2008) Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat. Immunol. 9, 1244–1252 10.1038/ni.1665 [DOI] [PubMed] [Google Scholar]
- 10. Tze L. E., Horikawa K., Domaschenz H., Howard D. R., Roots C. M., Rigby R. J., Way D. A., Ohmura-Hoshino M., Ishido S., Andoniou C. E., Degli-Esposti M. A., and Goodnow C. C. (2011) CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 208, 149–165 10.1084/jem.20092203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Gassart A., Camosseto V., Thibodeau J., Ceppi M., Catalan N., Pierre P., and Gatti E. (2008) MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH1 down-regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 3491–3496 10.1073/pnas.0708874105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H., Jain R., Vuong V., Ishido S., La Gruta N. L., Gray D. H., Villadangos J. A., and Mintern J. D. (2016) Ubiquitin ligase MARCH8 cooperates with CD83 to control surface MHCII expession in thymic epithelium and CD4 T cell selection. J. Exp. Med. 213, 1695–1703 10.1084/jem.20160312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. von Rohrscheidt J., Petrozziello E., Nedjic J., Federle C., Krzyzak L., Ploegh H. L., Ishido S., Steinkasserer A., and Klein L. (2016) Thymic CD4 T cell selection requires attenuation of MARCH8-mediated MHCII turnover in cortical epithelial cells through CD83. J. Exp. Med. 213, 1685–1694 10.1084/jem.20160316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coscoy L., and Ganem D. (2000) Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains enhancing their endocytosis. Proc. Natl. Acad. Sci. U.S.A. 97, 8051–8056 10.1073/pnas.140129797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coscoy L., and Ganem D. (2001) A viral protein that selectively downregulates ICAM-1 and B7–2 and modulates T cell costimulation. J. Clin. Invest. 107, 1599–1606 10.1172/JCI12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coscoy L., Sanchez D. J., and Ganem D. (2001) A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273 10.1083/jcb.200111010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishido S., Choi J.-K., Lee B.-S., Wang C., DeMaria M., Johnson R. P., Cohen G. B., and Jung J. U. (2000) Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374 10.1016/S1074-7613(00)00036-4 [DOI] [PubMed] [Google Scholar]
- 18. Ishido S., Wang C., Lee B.-S., Cohen G. B., and Jung J. U. (2000) Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309 10.1128/JVI.74.11.5300-5309.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stevenson P. G., Efstathiou S., Doherty P. C., and Lehner P. J. (2000) Inhibition of MHC class I-restricted antigen presentation by γ2-herpesvirus. Proc. Natl. Acad. Sci. U.S.A. 97, 8455–8460 10.1073/pnas.150240097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jahnke M., Trowsdale J., and Kelly A. P. (2012) Structural requirements for recognition of major histocompatibility complex class II by membrane-associated RING-CH (MARCH) protein E3 ligases. J. Biol. Chem. 287, 28779–28789 10.1074/jbc.M112.381541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lapaque N., Jahnke M., Trowsdale J., and Kelly A. P. (2009) The HLA-DRα chain is modified by polyubiquitination. J. Biol. Chem. 284, 7007–7016 10.1074/jbc.M805736200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohmura-Hoshino M., Matsuki Y., Aoki M., Goto E., Mito M., Uematsu M., Kakiuchi T., Hotta H., and Ishido S. (2006) Inhibition of MHC class II expression and immune responses by c-MIR. J. Immunol. 177, 341–354 10.4049/jimmunol.177.1.341 [DOI] [PubMed] [Google Scholar]
- 23. Corcoran K., Jabbour M., Bhagwandin C., Deymier M. J., Theisen D. L., and Lybarger L. (2011) Ubiquitin-mediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1). J. Biol. Chem. 286, 37168–37180 10.1074/jbc.M110.204040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jabbour M., Campbell E. M., Fares H., and Lybarger L. (2009) Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J. Immunol. 183, 6500–6512 10.4049/jimmunol.0901521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita H., Iwabu Y., Tokunaga K., and Tanaka Y. (2013) Membrane-associated RING-CH (MARCH) 8 mediates the ubiquitination and lysosomal degradation of the transferrin receptor. J. Cell Sci. 126, 2798–2809 10.1242/jcs.119909 [DOI] [PubMed] [Google Scholar]
- 26. Nagarajan A., Petersen M. C., Nasiri A. R., Butrico G., Fung A., Ruan H.-B., Kursawe R., Caprio S., Thibodeau J., Bourgeois-Daigneault M.-C., Sun L., Gao G., Bhanot S., Jurczak M. J., Green M. R., et al. (2016) MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat. Commun. 7, 12639 10.1038/ncomms12639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han S.-O., Xiao K., Kim J., Wu J.-H., Wisler J. W., Nakamura N., Freedman N. J., and Shenoy S. K. (2012) MARCH2 promotes endocytosis and lysosomal sorting of carvedilol-bound β2-adrenergic receptors. J. Cell Biol. 199, 817–830 10.1083/jcb.201208192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng J., and Guggino W. (2013) Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS ONE 8, e68001 10.1371/journal.pone.0068001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fatehchand K., Ren L., Elavazhagan S., Fang H., Mo X., Vasilakos J. P., Dietsch G. N., Hershberg R. M., Tridandapani S., and Butchar J. P. (2016) Toll-like receptor 4 ligands down-regulate Fcγ receptor IIb (FcγRIIb) via MARCH3 protein-mediated ubiquitination. J. Biol. Chem. 291, 3895–3904 10.1074/jbc.M115.701151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nice T. J., Deng W., Coscoy L., and Raulet D. H. (2010) Stress-regulated targeting of the NKG2D ligand Mut1 by a membrane-associated RING-CH family E3 ligase. J. Immunol. 185, 5369–5376 10.4049/jimmunol.1000247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoer S., Smith L., and Lehner P. J. (2007) MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett. 581, 45–51 10.1016/j.febslet.2006.11.075 [DOI] [PubMed] [Google Scholar]
- 32. Hör S., Ziv T., Admon A., and Lehner P. J. (2009) Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantification identify new substrates for the MARCH9 transmembrane E3 ligase. Mol. Cell. Proteomics 8, 1959–1971 10.1074/mcp.M900174-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanchez D. J., Coscoy L., and Ganem D. (2002) Functional organization of MIR2, a novel viral regulator of selective endocytosis. J. Biol. Chem. 277, 6124–6130 10.1074/jbc.M110265200 [DOI] [PubMed] [Google Scholar]
- 34. Kajikawa M., Li P.-C., Goto E., Miyashita N., Aoki-Kawasumi M., Mito-Yoshida M., Ikegaya M., Sugita Y., and Ishido S. (2012) The intertransmembrane region of Kaposi's sarcoma-associated herpesvirus modulator of immune recognition 2 contributes to B7-2 downregulation. J. Virol. 86, 5288–5296 10.1128/JVI.00219-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bauer J., Bakke O., and Morth J. P. (2017) Overview of the membrane-associated RING-CH (MARCH) E3 ligase family. N. Biotechnol. 38, 7–15 [DOI] [PubMed] [Google Scholar]
- 36. Call M. E., Schnell J. R., Xu C., Lutz R. A., Chou J. J., and Wucherpfennig K. W. (2006) The structure of the ζζ transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127, 355–368 10.1016/j.cell.2006.08.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Call M. E., Wucherpfennig K. W., and Chou J. J. (2010) The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol. 11, 1023–1029 10.1038/ni.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choma C., Gratkowski H., Lear J. D., and DeGrado W. F. (2000) Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7, 161–166 10.1038/72440 [DOI] [PubMed] [Google Scholar]
- 39. Gratkowski H., Lear J. D., and DeGrado W. F. (2001) Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. U.S.A. 98, 880–885 10.1073/pnas.98.3.880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Knoblich K., Park S., Lutfi M., van 't Hag L., Conn C. E., Seabrook S. A., Newman J., Czabotar P. E., Im W., Call M. E., and Call M. J. (2015) Transmembrane complexes of DAP12 crystallised in lipid membranes provide insights into control of oligomerization in immunoreceptor assembly. Cell Rep. 11, 1184–1192 10.1016/j.celrep.2015.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. MacKenzie K. R., Prestegard J. H., and Engelman D. M. (1997) A transmembrane helix dimer: structure and implications. Science 276, 131–133 10.1126/science.276.5309.131 [DOI] [PubMed] [Google Scholar]
- 42. Russ W. P., and Engelman D. M. (2000) The GxxxG motif: a framework for transmembrane helix–helix association. J. Mol. Biol. 296, 911–919 10.1006/jmbi.1999.3489 [DOI] [PubMed] [Google Scholar]
- 43. Trenker R., Call M. E., and Call M. J. (2015) Crystal structure of the glycophorin a transmembrane dimer in lipidic cubic phase. J. Am. Chem. Soc. 137, 15676–15679 10.1021/jacs.5b11354 [DOI] [PubMed] [Google Scholar]
- 44. Zhou F. X., Cocco M. J., Russ W. P., Brunger A. T., and Engelman D. M. (2000) Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat. Struct. Biol. 7, 154–160 10.1038/72430 [DOI] [PubMed] [Google Scholar]
- 45. Dodd R. B., Allen M. D., Brown S. E., Sanderson C. M., Duncan L. M., Lehner P. J., Bycroft M., and Read R. J. (2004) Solution structure of the Kaposi's sarcoma-associated herpesvirus K3 N-terminal domain reveals a novel E2-binding C4HC3-type RING domain. J. Biol. Chem. 279, 53840–53847 10.1074/jbc.M409662200 [DOI] [PubMed] [Google Scholar]
- 46. Krogh A., Larsson B., von Heijne G., and Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 47. Salzmann M., Wider G., Pervushin K., and Wüthrich K. (1999) Improved sensitivity and coherence selection for 15N,1H-TROSY elements in triple resonance experiments. J. Biomol. NMR 15, 181–184 10.1023/A:1008358030477 [DOI] [PubMed] [Google Scholar]
- 48. Kay L. E., Ikura M., Tschudin R., and Bax A. (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89, 496–514 10.1016/j.jmr.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 49. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elazar A., Weinstein J., Biran I., Fridman Y., Bibi E., and Fleishman S. J. (2016) Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. Elife 5, e12125 10.7554/eLife.12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feng J., Call M. E., and Wucherpfennig K. W. (2006) The assembly of diverse immune receptors is focused on a polar membrane-embedded interaction site. PLoS Biol. 4, e142 10.1371/journal.pbio.0040142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 10.1038/nature03216 [DOI] [PubMed] [Google Scholar]
- 53. Zhou F. X., Merianos H. J., Brunger A. T., and Engelman D. M. (2001) Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. U.S.A. 98, 2250–2255 10.1073/pnas.041593698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bayer-Santos E., Durkin C. H., Rigano L. A., Kupz A., Alix E., Cerny O., Jennings E., Liu M., Ryan A. S., Lapaque N., Kaufmann S. H. E., and Holden D. W. (2016) The Salmonella effector SteD mediates MARCH8-dependent ubiquitination of MHC II molecules and inhibits T cell activation. Cell Host Microbe 20, 584–595 10.1016/j.chom.2016.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chipot C., Dehez F., Schnell J. R., Zitzmann N., Pebay-Peyroula E., Catoire L. J., Miroux B., Kunji E. R. S., Veglia G., Cross T. A., and Schanda P. (2018) Perturbations of native membrane protein structure in alkyl phosphocholine detergents: a critical assessment of NMR and biophysical studies. Chem. Rev. 118, 3559–3607 10.1021/acs.chemrev.7b00570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aubrey B. J., Kelly G. L., Kueh A. J., Brennan M. S., O'Connor L., Milla L., Wilcox S., Tai L., Strasser A., and Herold M. J. (2015) An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 10, 1422–1432 10.1016/j.celrep.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 57. Lois C., Hong E. J., Pease S., Brown E. J., and Baltimore D. (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- 58. Sharma P., Kaywan-Lutfi M., Krshnan L., Byrne E. F., Call M. J., and Call M. E. (2013) Production of disulfide-stabilized transmembrane peptide complexes for structural studies. J. Vis. Exp. 2013, e50141 10.3791/50141 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.