

Abstract
The E3 ligase membrane-associated ring-CH-type finger 6 (MARCH6) is a polytopic enzyme bound to the membranes of the endoplasmic reticulum. It controls levels of several known protein substrates, including a key enzyme in cholesterol synthesis, squalene monooxygenase. However, beyond its own autodegradation, little is known about how MARCH6 itself is regulated. Using CRISPR/Cas9 gene-editing, MARCH6 overexpression, and immunoblotting, we found here that cholesterol stabilizes MARCH6 protein endogenously and in HEK293 cells that stably express MARCH6. Conversely, MARCH6-deficient HEK293 and HeLa cells lost their ability to degrade squalene monooxygenase in a cholesterol-dependent manner. The ability of cholesterol to boost MARCH6 did not seem to involve a putative sterol-sensing domain in this E3 ligase, but was abolished when either membrane extraction by valosin-containing protein (VCP/p97) or proteasomal degradation was inhibited. Furthermore, cholesterol-mediated stabilization was absent in two MARCH6 mutants that are unable to degrade themselves, indicating that cholesterol stabilizes MARCH6 protein by preventing its autodegradation. Experiments with chemical chaperones suggested that this likely occurs through a conformational change in MARCH6 upon cholesterol addition. Moreover, cholesterol reduced the levels of at least three known MARCH6 substrates, indicating that cholesterol-mediated MARCH6 stabilization increases its activity. Our findings highlight an important new role for cholesterol in controlling levels of proteins, extending the known repertoire of cholesterol homeostasis players.
Keywords: protein stability, E3 ubiquitin ligase, protein degradation, post-transcriptional regulation, cholesterol, proteasome, INSIG-2, RGS2, squalene monooxygenase, TEB4, Type 2 iodothyronine deiodinase
Introduction
Cholesterol is essential for mammals, with too little associated with developmental problems (1). However, too much is toxic, leading to diseases such as atherosclerosis and certain cancers. Hence, levels of this lipid are under tight control (2). To regulate cholesterol levels, cells must balance its efflux, uptake, and synthesis. Statins target an early step in cholesterol synthesis (catalyzed by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR)3), and their success in treating cardiovascular disease clearly demonstrates the clinical significance of this pathway. We previously showed that a critical rate-limiting enzyme in cholesterol synthesis, squalene monooxygenase (SM), is degraded by the proteasome in the presence of excess cholesterol (3). We found that this is mediated through ubiquitination in the first 100 amino acids of SM (SM N100) by the E3 ubiquitin ligase MARCH6 (membrane-associated really interesting new gene (RING) finger (C3HC4) 6) (4). MARCH6 is also one of several E3 ligases implicated in the degradation of HMGCR, another rate-limiting cholesterol synthesis enzyme (4–6). A mutant form of the cholesterol-trafficking protein, NPC1, is targeted to the proteasome by MARCH6-catalyzed ubiquitination (7), and a protein involved in lipid-droplet formation, PLIN2, is also targeted by MARCH6 (8). MARCH6 additionally affects transcriptional regulation in cholesterol homeostasis, ultimately reducing expression of the inducible-degrader of LDL receptor (IDOL) (9), another E3 ligase, which degrades lipoprotein receptors. MARCH6 therefore affects cholesterol homeostasis at several different points.
MARCH6, also known as TEB4 or RNF176, is a 103-kDa, 14-transmembrane domain (TMD) protein (10) localized to the endoplasmic reticulum (ER) (11). The yeast homolog, Doa10, is one of two key E3 ligases that mediate ER-associated degradation. E3 ligases attach ubiquitin moieties to their target substrates to promote their degradation, typically through the proteasome. Substrates of MARCH6, in addition to SM, HMGCR, and mutant NPC1, include a key regulator of thyroid hormone production (type 2 iodothyronine deiodinase, D2) (12), a regulator of G protein signaling involved in diseases such as cancer (RGS2) (13), and a mutant form of a bile salt exporter pump (Bsep*) (14). MARCH6 also autoubiquitinates itself, leading to its own degradation (15); accordingly, a mutation in its RING domain that renders it inactive (C9A) increases its levels (4). MARCH6 is regulated post-translationally by the deubiquitinase USP19 (16), but little else is known about its control. Therefore, we investigated possible mechanisms controlling MARCH6 levels.
Considering that MARCH6 helps control levels of the two rate-limiting enzymes in cholesterol synthesis, HMGCR and SM (4), we reasoned MARCH6 levels may be affected by sterol status through a feedback mechanism, i.e. high sterol levels increase MARCH6 levels, thus decreasing SM and HMGCR and consequently cholesterol synthesis. As precedents for sterol-mediated regulation of E3 ligases, both IDOL and RNF145 are transcriptionally up-regulated by sterols through liver X receptor (LXR) (17, 18), and TRC8 is post-translationally down-regulated by sterols (19, 20). Interestingly, TRC8 and MARCH6 were recently found to act together to facilitate degradation of select substrates (21).
One way that sterols are sensed by cells is through a five-TMD region known as a sterol-sensing domain (SSD) (2, 22). HMGCR (23), Scap (24, 25), NPC1 (26, 27), NPC1L1 (28), Patched (29), and Dispatched (30, 31) all have reported SSDs, as do the E3 ligases TRC8 (32) and RNF145 (6, 17). Because MARCH6 also has a large number of TMDs and is involved in cholesterol homeostasis, we hypothesized that it may also contain an SSD.
Here, we report that MARCH6 is stabilized by cholesterol, with subsequent decreases in levels of its substrates. This stabilization appears to be independent of a putative SSD but mediated through inhibition of MARCH6 autodegradation, probably due to a change in its conformation. This is the first time an E3 ligase has been shown to be post-translationally stabilized by cholesterol, introducing a novel mode of controlled protein demolition.
Results
MARCH6 gene expression levels are not affected by sterol status
We first considered whether MARCH6 gene expression levels are regulated by changing sterol status, as is the case with IDOL (18) and RNF145 (17). To test this, we employed previously generated cDNA sets (33) derived from cells in which the sterol status had been manipulated and levels of SREBP-2 and LXR target genes had been compared with another gene of interest. Here, we measured MARCH6 mRNA levels and found that these did not change under varying sterol conditions or in response to a synthetic LXR agonist (Fig. 1). To provide context, we have presented the data demonstrating the changing expression levels of SREBP-2 and LXR target genes as observed previously (33). These genes are highly responsive to changing sterol levels, whereas MARCH6 was unchanged (not statistically significantly different) across all conditions and cell lines, indicating that it is not transcriptionally regulated by sterols.
Figure 1.
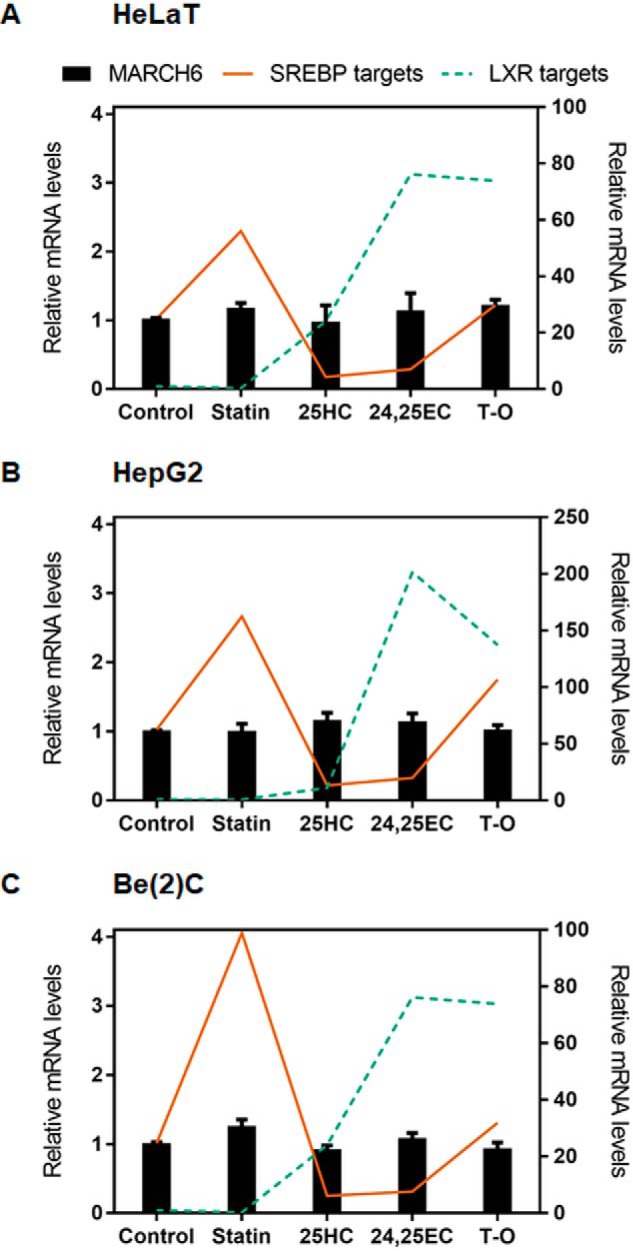
MARCH6 gene expression is not affected by changing sterol status. cDNA samples were from HeLaT (A), HepG2 (B), and Be(2)C (where Be(2)C are brain neuroblastoma cells) (C) cells treated for 24 h with statin, 25HC, 24(S),25-epoxycholesterol (24,25EC), or the synthetic LXR agonist TO-01317 (T-O) as described previously (33). MARCH6 mRNA levels were measured using qRT-PCR and normalized to PBGD housekeeping levels. mRNA levels are relative to the control condition, which was set to 1 in each cell line. Data are presented as mean ± S.E. from three separate experiments for HepG2 and Be(2)C, and two separate experiments for HeLaT, where each experiment was performed with triplicate cultures. SREBP-2 targets (solid lines) represents the average values of HMGCR and LDLR as published previously, and LXR targets (dashed lines) represent the average values of ABCA1 and ABCG1 (33). MARCH6 and SREBP-2 targets are plotted on the left y axis, and LXR targets are plotted on the right y axis.
Cholesterol increases MARCH6 post-translationally
We have previously found that transiently and massively overexpressed proteins do not recapitulate normal regulation (3), and we observed no sterol-mediated change in MARCH6 protein levels when it was transiently overexpressed (4). Here, we instead employed stable expression to examine potential post-translational regulation of MARCH6. We created HEK293 stable cell lines expressing MARCH6 tagged with a Myc or V5 epitope. Under sterol-depleted conditions we typically employ for SM, MARCH6 levels were very low and difficult to visualize, but we noted that addition of cholesterol increased MARCH6 protein (Fig. 2A). To maximize MARCH6 protein levels, we next treated cells with or without statins, which decrease sterol status and also up-regulate SM transcriptionally. We included the proteasomal inhibitor MG132 for 2 h prior to treatment with cholesterol (in the absence of MG132). This result clearly demonstrated that the MARCH6 protein was increased following cholesterol addition (Fig. 2B), a first with respect to E3 ligases. The increase in MARCH6 levels was associated with decreased levels of SM, a typical cholesterol-regulated substrate (Fig. 2, A and B). Similar MARCH6 increases by cholesterol were observed in nine out of 10 clones tested for these MARCH6-expressing stable cell lines (data not shown) with either a Myc or V5 epitope tag.
Figure 2.

Cholesterol stabilizes MARCH6 post-translationally. A, HEK293–MARCH6–Myc cells were pretreated overnight with statin, then treated with or without Chol/CD for 8 h before harvesting. Protein levels were analyzed by Western blotting with Myc (MARCH6), SM, and vinculin as a loading control. B, HEK293–MARCH6–Myc cells were pretreated with or without statin overnight, then treated with 10 μm MG132 for 2 h to increase basal levels of MARCH6, then treated for 6 h with or without Chol/CD before harvesting. Protein levels were analyzed by Western blotting with Myc (MARCH6), SM, and vinculin as a loading control. C, HEK293 cells were treated for 4 h with or without 20 μg/ml Chol/CD before harvesting. Protein levels were analyzed by Western blotting with endogenous MARCH6 antibody, SM, and vinculin as a loading control. D, RNA was harvested from HEK–CRISPR, HEK293, or HEK–MARCH6–V5 cells and cDNA analyzed for gene expression levels of MARCH6 normalized to the housekeeping gene PBGD. HEK293 control levels have been set to 1. Data are presented as mean ± S.E. from three separate experiments, where each experiment was performed with triplicate cultures. E, HEK–CRISPR, HEK293, or HEK–MARCH6–V5 cells transfected for 24 h with 1 μg of pTK-SM N100-GFP-V5 plasmid, then treated for 4 h with or without 20 μg/ml Chol/CD before harvesting. Protein levels were analyzed by Western blotting with V5 (MARCH6 and SM N100) and GAPDH as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the −Chol/CD condition in HEK–MARCH6–V5 cells, which has been set to 1 for each protein. Data are presented as mean ± S.E. from at least three separate experiments. F, HeLa–CRISPR or HeLa cells were transfected for 24 h with 2 μg of pTK-SM N100-GFP-V5 plasmid, pretreated overnight, then treated for 8 h with or without 20 μg/ml Chol/CD before harvesting. Protein levels were analyzed by Western blotting with V5 (SM N100) and β-actin as a loading control. Blots are representative of three independent experiments. Relative protein levels were measured using GeneTools and normalized to the −Chol/CD condition in HeLa cells, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments.
We next employed a commercially available antibody to endogenous MARCH6. This antibody detected a number of bands by Western blotting (Fig. S1), including a cholesterol-responsive band (Fig. 2C) running at approximately the same size as for epitope-tagged MARCH6 (Fig. S1). However, this was the only antibody of three commercially available antibodies and our own in-house–produced antibodies that yielded anything remotely promising. The poor quality of endogenous MARCH6 antibodies has also been observed by others (21), who instead used SM levels as a proxy. Therefore, we instead relied upon our overexpressing cell line, which has only ∼20% more MARCH6 mRNA than its parent cell line (Fig. 2D). This means our MARCH6 levels are close to endogenous levels and likely reflect endogenous MARCH6 regulation, as indicated in the endogenous MARCH6 Western blotting, where MARCH6 levels are comparable between the overexpressing and parent cell lines, and both are cholesterol-responsive (Fig. S1).
For additional evidence that the effects we observed were not a result of overexpressed MARCH6, we compared a MARCH6-deficient (CRISPR) cell line and a MARCH6-overexpressing cell line (MARCH6–V5) with the parental HEK293 cell line. We examined cholesterol-mediated degradation of the MARCH6 substrate SM N100 (4). This showed comparable reductions of SM N100 in response to cholesterol in both the HEK293- and MARCH6-expressing cells (Fig. 2E), which was lost in MARCH6-deficient cells. The dependence on MARCH6 of the reduction of SM N100 levels in response to cholesterol was repeated independently in HeLa cells CRISPR modified at exon 1 of the MARCH6 locus (Fig. 2F). Together, these data are consistent with endogenous levels of MARCH6 being regulated by cholesterol. The relative levels of SM N100 were higher in our modified HEK293 cells than our modified HeLa cells, which could be due to differences in the parental cell lines, as we have observed previously (4), and/or the different media used for treatments.
Sterol intermediates and oxysterols stabilize MARCH6
We next examined the rate of accumulation of MARCH6 over time following addition of cholesterol to cells. We found a significant increase in MARCH6 protein already by 2 h in these cells relative to those without cholesterol added, with relative accumulation increasing up to 8 h (Fig. 3A).
Figure 3.

A variety of sterol intermediates and oxysterols stabilize MARCH6. A, HEK–MARCH6–V5 cells were treated with or without 20 μg/ml Chol/CD for the indicated time (0–8 h) before harvesting. Protein levels were analyzed by Western blotting with V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative MARCH6 protein levels were measured using ImageStudio Lite and normalized to the 8-h −Chol/CD condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments. *, p < 0.05; **, p < 0.01, by Student's paired t test. B, HEK–MARCH6–V5 cells were treated with or without 20 μg/ml Chol/CD or the indicated sterol complexed to cyclodextrin or 1 μg/ml oxysterol for 8 h before harvesting. Protein levels were analyzed by Western blotting with V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the control condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments. *, p < 0.05; **, p < 0.01, by Student's paired t test. For full sterol names please see Table 1.
We have previously noted clear differences in the sterol signals for degradation of SM compared with HMGCR (3); therefore, we sought to establish which sterols increase MARCH6, a degrader of both of these enzymes. We found that several sterol intermediates and oxysterols tended to increase MARCH6 levels, with lanosterol, desmosterol, β-sitosterol, and 25-hydroxycholesterol reaching statistical significance (Fig. 3B).
Cholesterol inhibits degradation of MARCH6
To examine turnover of MARCH6, we used the protein synthesis inhibitor cycloheximide and followed the rate of MARCH6 disappearance. This showed that MARCH6 is turned over fairly rapidly (t½ ∼2–3 h) (Fig. 4A), but this can be at least partially rescued by addition of cholesterol (Fig. 4B). This result indicates that stabilization of MARCH6 occurs independently of protein synthesis.
Figure 4.

MARCH6 is stabilized by cholesterol by inhibiting its degradation. A, HEK–MARCH6–V5 cells were treated with or without 10 μg/ml cycloheximide (CHX) for the indicated time (0–4 h) before harvesting. B, HEK–MARCH6–V5 cells were treated with or without 10 μg/ml cycloheximide and 20 μg/ml Chol/CD for 4 h before harvesting. C, left, HEK–MARCH6–V5 cells were treated with or without 20 μg/ml Chol/CD and inhibitors against the proteasome (MG132;10 μm), VCP inhibitor (CB5083; 5 μm), DUBs (PR619; 10 μm), or lysosomes (NH4Cl, 20 μm) for 4 h before harvesting. Right, HEK–MARCH6–V5 cells were transfected with 25 nm indicated siRNA for 24 h and then treated with or without 20 μg/ml Chol/CD for 4 h before harvesting. D, HEK–MARCH6–V5 cells were transfected with 25 nm indicated siRNA for 24 h, then RNA harvested and cDNA analyzed for gene expression levels of GP78, RNF145, TRC8, and VCP normalized to the housekeeping gene PBGD and to the control siRNA condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments, where each experiment was performed with triplicate cultures. E, HEK–MARCH6–V5 cells were transfected with 25 nm indicated siRNA for 24 h, then treated with or without 20 μg/ml Chol/CD for 4 h before harvesting. For all blots, protein levels were analyzed by Western blotting with V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the control condition in each blot, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments. A, *, p < 0.05; **, p < 0.01, by Student's paired t test. E, *, p < 0.01 by Student's paired t test compared with control −Chol/CD condition. All +Chol/CD conditions were significantly different from −Chol/CD conditions, except TRC8, where p = 0.1.
To determine whether cholesterol-mediated stabilization of MARCH6 is due to reduced degradation by the ubiquitin–proteasome system, we employed inhibitors targeting the proteasome (MG132), deubiquitinases (PR619), or valosin-containing protein (VCP/p97), which helps extract ubiquitinated proteins from the membrane (VCP inhibitor, CB5083) (Fig. 4C). Inhibition of the proteasome with MG132, or inhibition of VCP with CB5083, raised MARCH6 levels in basal conditions, and cholesterol addition did not further increase these levels, supporting the hypothesis that cholesterol raises MARCH6 levels by preventing its degradation through the proteasome. A different result was found using the general deubiquitinase (DUB) inhibitor PR619; PR619 prevented the increase in MARCH6 protein over its low basal levels in response to cholesterol addition (Fig. 4C). Presumably, autoubiquitination of MARCH6 is no longer reversed by cellular DUB activity, allowing its rapid degradation by the proteasome even in the presence of cholesterol. The relevant DUB might be USP19 (16).
As part of ER-associated degradation, the AAA ATPase valosin-containing protein (VCP, also known as p97) recognizes polyubiquitinated substrates and extracts them from the ER into the cytosol (34). Interestingly, the VCP inhibitor increased MARCH6 protein levels even further than proteasomal inhibition (Fig. 4C). To confirm this result, we employed VCP siRNA and found similar effects (Fig. 4, C and D). This could suggest that an additional mode of regulation is involved in controlling MARCH6 levels. Because VCP may also be involved in the degradation of proteins by the lysosome (35), we tested a lysosomal inhibitor (ammonium chloride), but this did not increase MARCH6 levels (Fig. 4C).
We next determined whether ER-associated degradation E3 ligases other than MARCH6 itself are involved in the cholesterol-dependent stabilization of MARCH6. We knocked down GP78 (also known as AMFR), TRC8, or RNF145, which together with INSIG play a role in sterol-mediated HMGCR degradation (5, 6, 36). Furthermore, TRC8 is degraded in response to sterols (19, 20), and RNF145 is transcriptionally up-regulated by sterols as an LXR target (17, 37). The siRNAs used knocked down their gene targets by at least 63% (Fig. 4D), but there was little effect on MARCH6 protein levels (Fig. 4E). GP78 may play a minor role in degradation of MARCH6, as knocking down this E3 ligase increased MARCH6 above control values by ∼56% (p = 0.039 by Student's paired t test for the −Chol/CD condition). But this increase occurred both with and without cholesterol, and so GP78 is unlikely to be a major contributor to cholesterol-mediated regulation of MARCH6.
MARCH6 may contain a sterol-sensing domain
SSDs are regions of five TMDs that play a role in sterol sensing (2, 22). By comparing sequence homology of the putative SSDs of a number of proteins with the classic SSDs in HMGCR, NPC1, and Scap, we found that our own predicted SSD in MARCH6 (amino acids 330–543; TMD4–8) may be bona fide (Fig. 5A), also having similar TMD spacing and orientation with those in other SSD-containing proteins. Furthermore, the SSD motif YIYF found in HMGCR and Scap revealed a potentially critical tyrosine residue in MARCH6, Tyr-340, which is similarly located within the first TMD of MARCH6's predicted SSD (Fig. 5, B and C). This sequence (YILL in MARCH6) was similar to the YISL in NPC1 and NPC1L1. There is also a leucine residue (Leu-366) that bears similarities to the critical Leu-315 in the second TMD of Scap (25) (Fig. 5, B and C).
Figure 5.

MARCH6 may contain a sterol-sensing domain. A, % similarity of SSDs from the indicated proteins with HMGCR, NPC1, or SCAP. B, alignment of MARCH6's Tyr-340 and Leu-366 with the indicated proteins. C, predicted topology of MARCH6 indicating the putative SSD. D, HEK–MARCH6–V5 or indicated mutant cell lines were treated with or without 20 μg/ml Chol/CD for 4 h and protein lysates subjected to Western blotting for V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the WT −Chol/CD condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments.
To determine whether MARCH6 contains a functional SSD, we tested two constructs containing either just the SSD and adjacent loops or also including the RING domain and C-terminal element. However, these constructs were not expressed. This was probably due to misfolding and degradation by the quality control system, as low levels of these proteins could just barely be discerned but only after MG132 treatment (data not shown). Instead, we tested stable cell lines expressing MARCH6 with either a Y340A mutation or an L366F mutation, which we predicted would perturb cholesterol regulation by disrupting the putative SSD. However, both of these mutants were still stabilized by cholesterol (Fig. 5D).
INSIGs are unlikely to be involved in the cholesterol-mediated stabilization of MARCH6
SSDs for ER resident proteins have been proposed as INSIG-interacting domains (2), and because 25HC, which binds INSIGs, stabilized MARCH6 (Fig. 3B), we investigated the requirement for INSIGs in cholesterol-dependent stabilization of MARCH6. Using siRNA to knock down INSIG-1, INSIG-2, or both (Fig. 6A), we found that MARCH6 was still stabilized by cholesterol (Fig. 6B). However, INSIG-2 plays a role in basal degradation of MARCH6 (p < 0.02 for the −Chol/CD condition by Student's paired t test).
Figure 6.
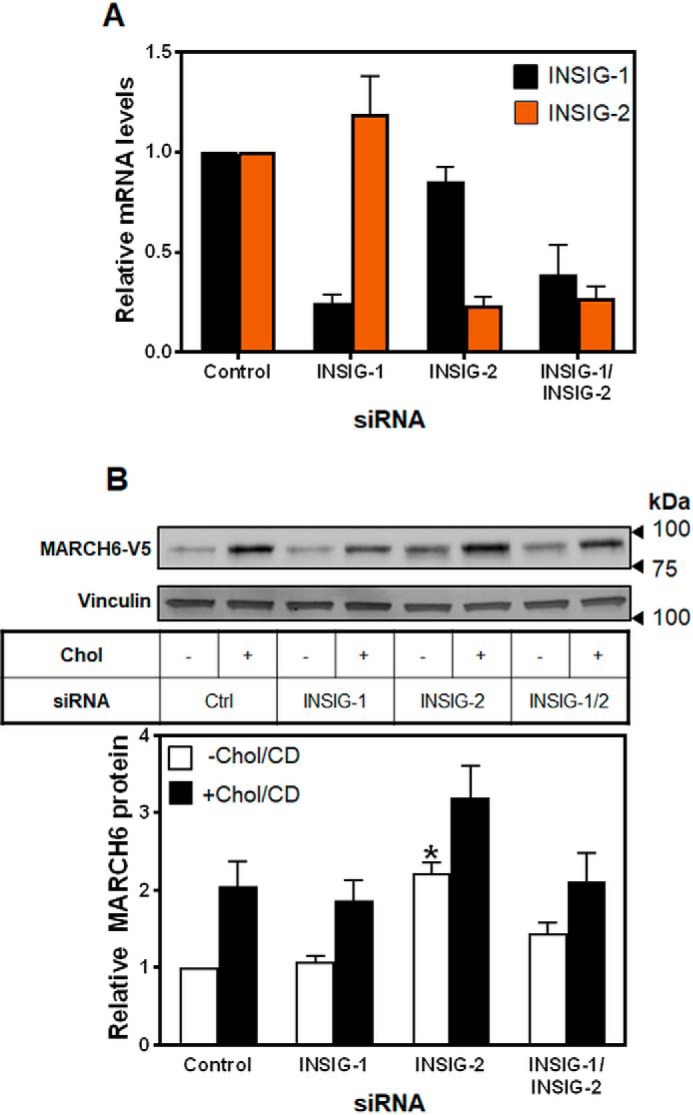
INSIGs are unlikely to be involved in the sterol-mediated stabilization of MARCH6. A, HEK–MARCH6–V5 cells were transfected with 25 nm indicated siRNA for 24 h, then RNA harvested, and cDNA analyzed for gene expression levels of INSIG-1, and INSIG-2, normalized to the housekeeping gene PBGD, and to the control siRNA condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments, where each experiment was performed with triplicate cultures. B, HEK–MARCH6–V5 cells were transfected with 25 nm indicated siRNA for 24 h, then treated with or without 20 μg/ml Chol/CD for 4 h before harvesting. Protein levels were analyzed by Western blotting with V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the control −Chol/CD condition, which has been set to 1. Data are presented as mean ± S.E. from nine separate experiments. *, p < 0.01 by Student's paired t test. All +Chol/CD conditions were significantly different from −Chol/CD conditions.
Stabilization of MARCH6 by cholesterol occurs via inhibition of self-destruction
MARCH6 catalyzes its own ubiquitination and degradation. To investigate whether the stabilization of MARCH6 by cholesterol occurs via inhibition of this ability to self-destruct, we next tested whether mutations at sites critical for MARCH6 autoubiquitination were similarly stabilized by cholesterol. We tested both a RING mutation, C9A, and a C-terminal element mutation, N890A, which also prevents its own degradation (11). We again employed stable overexpression of these mutants, and we found that both were stabilized in the absence of cholesterol, due to the prevention of self-degradation, but neither was further stabilized in the presence of cholesterol (Fig. 7A), indicating that cholesterol likely prevents degradation of MARCH6 mediated by its ubiquitin ligase activity.
Figure 7.

Cholesterol prevents self-destruction of MARCH6, likely through a conformational change. A, HEK–MARCH6–V5 or mutant cell lines were treated with or without 20 μg/ml Chol/CD for 4 h before harvesting. Protein lysates were subjected to Western blotting for V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the control −Chol/CD condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments. B–D, HEK–MARCH6–V5 cells were pretreated overnight with the indicated concentration of chemical chaperone (glycerol, betaine, or proline), then treated with or without 20 μg/ml Chol/CD and the indicated concentration of chemical chaperone for 4 h before harvesting. Protein lysates were subjected to Western blotting for V5 (MARCH6) and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the control −Chol/CD condition, which has been set to 1. Data are presented as mean ± S.E. from at least three separate experiments.
Cholesterol may cause a conformational change in MARCH6
To determine whether cholesterol changes the conformation of MARCH6 to prevent its self-destruction, we employed three chemical chaperones: glycerol, proline, and betaine. These chaperones cause proteins to favor their native conformation (38). Treatment with these chaperones resulted in lower levels of MARCH6 protein, which could be partially overcome by addition of cholesterol (Fig. 7, B–D). This suggests that MARCH6 preferentially adopts a conformation that makes it prone to rapid degradation, presumably through enhanced auto-ubiquitination, but addition of cholesterol changes this conformation to one that limits auto-degradation. We cannot fully rule out indirect effects of these chaperones, but a direct effect on MARCH6 is the simplest explanation.
Cholesterol-dependent stabilization of MARCH6 is accompanied by a decrease in levels of its substrates
As well as the cholesterol synthesis enzymes SM and HMGCR, MARCH6 has a number of known other substrates, including D2 (12) and RGS2 (13). Considering that increasing cholesterol status stabilizes MARCH6 protein levels, we hypothesized that this would also result in a decrease in levels of its substrates, as we have shown for one substrate, SM (Fig. 2). To investigate this, we obtained plasmids for RGS2 and D2 expression, and we tested them for cholesterol responsiveness under our previously optimized conditions for SM. D2 and RGS2 levels decreased in response to increased sterol levels (Fig. 8). These changes were small but significant, even in our transient expression system. This is consistent with another group's finding that D2 has increased activity following statin treatment (with sterol deprivation decreasing MARCH6) (39). Endogenous SM has a more pronounced decrease because it is also subjected to transcriptional control by sterols (40). Taken together, these findings identify a new role for cholesterol in the post-translational regulation of D2 and RGS2. It is likely that other substrates of MARCH6 are similarly regulated by cholesterol through its effects on MARCH6 turnover.
Figure 8.
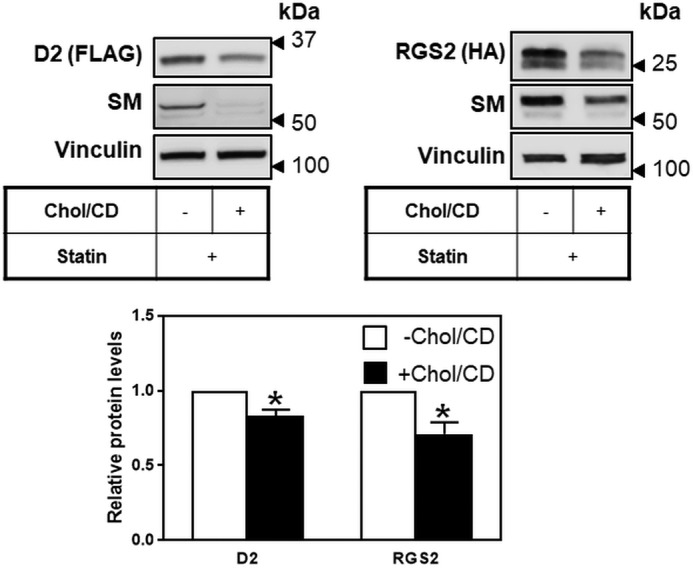
Cholesterol regulates two additional MARCH6 substrates. HEK293 cells were transfected for 24 h with 0.5 μg of D2-FLAG or 0.5 μg of RGS2-HA plasmid, followed by statin pretreatment overnight, then treated with 10 μm MG132 for 2 h to increase basal levels, and then treated for 6 h with or without 20 μg/ml Chol/CD before harvesting. Protein levels were analyzed by Western blotting with FLAG (D2), HA (RGS2), SM and vinculin as a loading control. Blots are representative of at least three independent experiments. Relative protein levels were measured using ImageStudio Lite and normalized to the −Chol/CD condition, which has been set to 1 in each experiment. Data are presented as mean ± S.E. from at least three separate experiments. *, p < 0.05, by Student's t test.
Discussion
Here, we have demonstrated that cholesterol increases protein levels of the E3 ligase MARCH6 by preventing its autodegradation, possibly via a sterol-induced conformational change. This in turn increases MARCH6 activity as shown by reduced levels of at least three of its substrates, revealing a new mode of regulation by cholesterol.
Considering that MARCH6 affects cholesterol homeostasis at several points, it seemed plausible that its levels may be affected by sterol status, in a classic feedback mechanism. As precedents, two other RING-containing E3 ligases, IDOL and RNF145, are both LXR gene targets and are up-regulated in response to increasing sterol status (17, 18). Although sterol status does not alter mRNA levels of MARCH6 (Fig. 1), protein levels are markedly increased (Figs. 2–7). We had previously shown that protein levels were unaffected (4). However, this was likely due to the nature of transient and massive overexpression masking any regulation by cholesterol, as we have previously observed for SM (3).
Here, we instead used stable MARCH6 expression systems (either Myc- or V5-tagged enzyme) expressing MARCH6 at levels comparable with the parent cell line and observed robust cholesterol-mediated stabilization. We have observed this effect in more than 50 experiments, measuring a consistent 2-fold effect. A doubling of the level of an E3 ligase like MARCH6 is biologically significant, as indicated by the reduced levels of two substrates ostensibly unrelated to cholesterol metabolism (Fig. 8).
This cholesterol-mediated reduction in D2 and RGS2 is consistent with cholesterol stabilizing endogenous MARCH6. Indeed, we have presented several lines of evidence that, like ectopic MARCH6, endogenous MARCH6 is very likely to be regulated by cholesterol. Although the utility of current endogenous MARCH6 antibodies is questionable (Fig. S1), we observed cholesterol-mediated stabilization of the probable MARCH6 band. In the absence of a reliable endogenous antibody, we relied on ectopic expression in most experiments, but importantly, MARCH6 mRNA levels were similar between the parental HEK293 cells and our MARCH6–V5–expressing cells (Fig. 2C). In addition, we found that cholesterol no longer induced degradation of SM N100, a normally cholesterol-responsive derivative of SM, in two MARCH6-deficient cell lines independently generated in two different laboratories by CRISPR–Cas9 technology. We have previously used this technology to generate a HeLa cell line with an epitope-tagged endogenous MARCH6 (11), but less than 1% of the cells had detectable tagged MARCH6 based on immunofluorescence. This cell line is therefore unsuitable for biochemical work.
Although a range of the sterols tested tended to increase MARCH6 levels, about half of them had no significant effect. Notably, lathosterol neither stabilizes MARCH6 nor causes degradation of HMGCR or SM (Fig. 3B and Table 1). It could be argued that sterol stabilization of MARCH6 shows less specificity than sterol-mediated degradation of HMGCR and SM (Table 1). However, considering that these experiments on MARCH6, HMGCR, and SM have been performed across a number of cell types in different laboratories, using variable concentrations and incubation times, it is possible that the differences in sterol specificity are partially explained by these and other confounding factors. For example, the degradation of SM in response to cholesterol may require both a conformational change in SM (41) as well as stabilization of MARCH6 for maximal regulation. Head to head comparisons may be needed to better determine the relative sterol specificities of MARCH6 stabilization and HMGCR and SM degradation.
Table 1.
Sterol specificity
A comparison of the effects of the indicated sterols on degradation of HMGCR and SM and the stabilization of MARCH6 is shown. − indicates no effect; + indicates effect (degradation for HMGCR and SM, stabilization for MARCH6); −/+ indicates nonsignificant effect; ND indicates not determined.
| HMGCR degradation | SM degradation | M6 stabilization | |
|---|---|---|---|
| Cholesterol (Chol) | −a | +b | + |
| Lanosterol (Lano) | +c | −b | + |
| 24,25-Dihydrolanosterol (24,25 DHL) | +c | −b | −/+ |
| Lathosterol (Lath) | −c | −b | − |
| 7-Dehydrocholesterol (7DHC) | −c, +d | −b | − |
| Desmosterol (Desmo) | −c | −b | + |
| β-Sitosterol (β-sito) | −c | ND | + |
| 25-Hydroxycholesterol (25HC) | +a | −b | + |
| 27-Hydroxycholesterol (27HC) | +a | −b | − |
| 7α-Hydroxycholesterol (7αHC) | −c | +b | − |
| 7-Ketocholesterol (7KC) | −c | +b | − |
To further investigate the mechanism by which cholesterol regulates MARCH6, we explored the possibility that MARCH6 contains an SSD. A number of cholesterol-related proteins contain SSDs, and comparisons of the TMD patterns in these proteins indicated that MARCH6 may similarly possess an SSD comprising 5 of its 14 TMDs (Fig. 5). Importantly, two critical residues from other SSDs were identified in MARCH6, but neither of these residues appears critical for stabilization of MARCH6 by cholesterol, as mutants of either remained sterol-responsive (Fig. 5D). However, it is possible that these tagged mutant proteins oligomerize with endogenous untagged MARCH6 bearing the normal SSD, masking any effect of the mutations.
SSDs have been proposed as INSIG-interacting domains, at least for Scap, HMGCR, and the E3 ligase RNF145 (6). However, our results indicated that INSIGs are unlikely to be responsible for the cholesterol-dependent stabilization of MARCH6 (Fig. 6). Interestingly, knockdown of INSIG-2 resulted in a significant increase in MARCH6 protein levels, but knockdown of both INSIG-1 and INSIG-2 blunted this increase. It is possible that a complex interplay between the INSIGs and other degradation machinery are exquisitely balanced. For example, GP78 also plays a minor role in basal degradation of MARCH6 (Fig. 4E) and interacts with INSIGs, particularly INSIG-1 (42). In the absence of both INSIGs, GP78 could be more available to degrade MARCH6, blunting the increase in MARCH6 levels observed with INSIG-2 knockdown alone. Further work is required to determine whether there is a bona fide SSD in MARCH6 and whether INSIG-2 directly interacts with this domain.
MARCH6 is rapidly turned over in cells (Figs. 2 and 4), and this appears to be largely due to autodegradation, as mutants that prevent this (C9A and N890A) have much higher expression levels (Fig. 7A) (4, 11). We found that neither of these mutants was further stabilized by cholesterol (Fig. 7A), indicating that cholesterol likely prevents autodegradation of MARCH6. Because MARCH6's substrate protein levels are decreased following cholesterol treatment (Fig. 8), it is likely that cholesterol alters the specificity of MARCH6 for its substrates, similar to the N890A mutant, which can no longer cause autodegradation but still targets some of its substrates (11). Future work should explore ubiquitination levels on MARCH6 following treatment with cholesterol.
Employing three different small molecule chaperones, we found that cholesterol prevents MARCH6's autodegradation through a conformational change (Fig. 7, B–D), as has been found for other cholesterol-responsive proteins (41, 43). The structure of MARCH6 remains to be solved, and so how precisely cholesterol modulates the shape of this 14-TMD protein in and out of the ER membrane is unknown, although it is worth noting that cholesterol can directly bind to MARCH6 (44).
Two of MARCH6's substrates, D2 and RGS2, were decreased by cholesterol under conditions previously used to assess post-translational degradation of SM (Fig. 8). Endogenous SM is subject to transcriptional regulation by sterols as well as post-translational regulation (3), explaining the larger effect size on this substrate. This finding that two substrates of MARCH6 not directly involved in cholesterol metabolism are degraded in response to cholesterol highlights a new role for cholesterol in regulating other aspects of cell homeostasis beyond the known repertoire. D2 is a deiodinase that activates thyroid hormone by converting the pro-hormone T4 to T3, the biologically active thyroid hormone (12), and RGS2 is a regulator of G proteins that lowers blood pressure by decreasing signaling through Gαq (13). The other established substrates of MARCH6 are itself, a mutant bile salt exporter pump (14), mutant NPC1 (7), the perilipin PLIN2 (8), and the cholesterol synthesis enzymes SM and HMGCR (4). Cholesterol synthesis is especially energy-intensive (45), and interestingly, bile acids induce energy expenditure by promoting intracellular thyroid hormone activation via induction of D2 (46). Also of note, human RGS2 polymorphisms are linked to weight gain and increased susceptibility to metabolic syndrome (47, 48). PLIN2 is a constitutive and ubiquitously expressed lipid droplet protein that is involved in adipocyte differentiation and lipid droplet dynamics. Loss of PLIN2 protects against the development of obesity in both genetic and pharmacological intervention models (49). Therefore, there are likely to be overlapping metabolic networks mediated by MARCH6, and cholesterol may in turn influence these through stabilizing MARCH6.
In the presence of cholesterol, another E3 ligase, TRC8, is degraded, while MARCH6 is stabilized. Considering that these two ER membrane-bound E3 ligases have recently been shown to act together in the degradation of select substrates (21), our new finding of MARCH6 cholesterol regulation needs to be considered. It is possible that TRC8 activity is favored in low cholesterol conditions, while MARCH6 dominates when cholesterol levels are high. Thus, a spectrum of substrate regulation could exist between the two extremes of cholesterol status within cells.
In conclusion, we have identified a new role for cholesterol in controlling protein demolition through stabilization of an E3 ubiquitin ligase, MARCH6. This is likely to affect levels of MARCH6's other substrates, opening up unexplored territory for regulation of cell homeostasis by cholesterol levels. Furthermore, we have shown that MARCH6 is highly entwined with cholesterol and possibly energy metabolism and should be considered a possible candidate E3 ligase for other cholesterol-related and metabolically related proteins that are degraded proteasomally.
Experimental procedures
Plasmids
Our previously described MARCH6–Myc plasmid (4) was subcloned into a pcDNA5/FRT construct, and the Myc tag was replaced with a V5 tag to yield pcDNA5/MARCH6–V5/FRT. Mutations of this construct (C9A, Y340A, L366F, and N890A) were created using site-directed mutagenesis, and primer sequences are available on request. pcDNA3.1 encoding human RGS2-HA was a generous gift from Professor Yaping Tu (50). D10 encoding full-length human D2-FLAG with a double selenocysteine to cysteine mutation and the D15 expression vector were generous gifts from Dr. Ann Marie Zavacki (12). A plasmid expressing the first 100 amino acids of SM (SM N100) fused to GFP, pTK-SM N100-GFP-V5, was generated previously (3). The px459-gRNA1-MARCH6-KO was generated by inserting annealed oligonucleotides JMB414 (CACCGCGCCGACTTACCTTCCTCCG) and JMB415 (AAACCGGAGGAAGGTAAGTCGGCGC) into px459 as described previously (11, 51). The px458-gRNA2-MARCH6-KO plasmids containing AGACAAGATGGACACCGCGG and CGGCGCGGGCGGGAAACGGC were similarly generated.
Cell lines
HEK293 cells stably overexpressing MARCH6 with a Myc or V5 epitope tag (HEK–MARCH6–Myc or HEK–MARCH6–V5) or MARCH6–V5 mutants were created using the FlpIn system as we have done previously (4). Cells were grown in 10% (v/v) fetal calf serum (FCS)/DMEM high glucose with 20 or 50 μg/ml hygromycin B. HeLa cells (ATCC) were cultured in 10% (v/v) FCS/DMEM.
CRISPR/Cas9 modification of chromosomal MARCH6
HEK293 cells
HEK293 cells were seeded at 1.35 × 106 cells/well in 10-cm dishes. The next day, cells were transfected with 10 μg of px458-gRNA2-MARCH6-KO plasmids for 24 h using Lipofectamine/LTX reagent in a 1:4 ratio. Cells were washed and refreshed for a further 24 h before being sorted by FACS into 96-well plates at a density of one cell/well using GFP selection. Single colonies were expanded, and MARCH6 genomic disruption was verified in the selected CRISPR clone by PCR using primers CCTCTCTCGCACCTGAGCGT and GCGGGGACAAGGGAGGCCAC (Fig. S2).
HeLa cells
HeLa cells were plated at 30–40% confluency in a 6-well plate, transfected after 24 h with px459-gRNA1-MARCH6-KO, and diluted after 48 h at low density in 15-cm2 plates to generate single colonies. Isolated single colonies were expanded and screened for MARCH6 gene modification by PCR using primers JMB439 (CGCGGGAGCCTCGTG) and JMB440 (GGGTACGAACCCGGGCG) to amplify a 120-nucleotide fragment disrupted by the isolated CRISPR clone (Fig. S2).
Plasmid transfections
For RGS2 transfections (in a 6-well plate), cells were transfected with 0.5 μg of pcDNA3.1 and 0.5 μg of RGS2-HA using 2 μl of Lipofectamine LTX (ThermoFisher Scientific). For D2 transfections (in a 6-well plate), cells were transfected with 0.5 μg of D2 plasmid and 0.5 μg of D15 plasmid using 2 μl of Lipofectamine LTX. For SM N100 transfections, HEK–MARCH6–CRISPR cells, HEK293 cells, or HEK–MARCH6–V5 cells were seeded in 6-well plates and transfected with 1 μg of pTK-SM N100-GFP-V5, 1.5 μl of Lipofectamine-3000 reagent, and 2 μl of P3000 reagent. Transfections were for 24 h, then treatments as described. HeLa cells and HeLa–MARCH6–CRISPR cells seeded in 6-well plates were transfected with 2 μg of pTK-SM N100-GFP-V5 and 6 μl of X-tremeGENE HP (Roche Applied Science).
Treatments
For treatment with 20 μg/ml sterols, these were complexed to methyl-β-cyclodextrin. Oxysterols were added in ethanol. Inhibitors were added in DMSO. Solvents were kept constant between conditions and did not exceed 0.2% (v/v). Cells were treated in 10% (v/v) FCS/DMEM high glucose, except for statin pretreatments where cells were switched to 10% (v/v) lipoprotein-deficient FCS/DMEM high glucose containing 50 μm mevalonate and 5 μm mevastatin or 10 μm lovastatin. For treatments with chemical chaperones (glycerol, betaine, or proline), cells were pretreated overnight with the indicated concentration of chemical chaperone. Cells were then treated with the indicated concentration of chemical chaperone with or without Chol/CD (20 μg/ml).
siRNA transfection
HEK–MARCH6–V5 cells were transfected for 24 h using 25 nm siRNA targeted to human INSIG-1 (52) and/or INSIG-2 (53), GP78 and TRC8 (4), RNF145 (Sigma), and VCP (target sequence, GGUAAUCUCUUCGAGGUAU) using Lipofectamine RNAiMAX (ThermoFisher Scientific). Following this, cells were either harvested to measure mRNA levels or treated as indicated and protein harvested for Western blotting.
qRT-PCR
mRNA levels were measured using primers specific to Insig-1 and Insig-2 (53), gp78, TRC8, and MARCH6 (4), RNF145 (forward, GGTATATGGCCTTCTCGCCTTGGG, and reverse, GTAAGGAGTGCTGCAGCATTCCGC), VCP (forward, GGAAGCGTATCGACCCATCC, and reverse, CTCGTTTGATAGGCTCCCCTT), and porphobilinogen deaminase (PBGD) (54) using SYBR Green, as we have described previously (33).
Western blotting
After treatment, protein was harvested from HEK293 cells as we have described previously (4). Briefly, cells were lysed in modified RIPA (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% (w/v) SDS, 1.5% (w/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, and 2 mm MgCl2) supplemented with 2% (v/v) protease inhibitor mixture (Sigma), passed through a 22-gauge needle 20 times, rotated at 4 °C for 30 min, and then centrifuged at 17,000 × g at 4 °C for 15 min. HeLa cells were harvested by scraping in ice-cold PBS, transferred to microcentrifuge tubes, pelleted at 500 × g for 4 min at 4 °C, flash-frozen in liquid nitrogen, and stored at −80 °C. Cell pellets were thawed on ice and then resuspended in lysis buffer (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1% (v/v) Triton X-100, 2 mm EDTA, cOmplete EDTA-free protease inhibitor tablet (Roche Applied Science)). Lysates were rotated for 10 min followed by centrifugation at 11,200 × g for 10 min at 4 °C. The resulting supernatants for both HEK293 and HeLa cells were normalized for protein content and subjected to 10% (w/v) SDS-PAGE and Western blotting with the following: mouse anti-V5 (1:5000, ThermoFisher Scientific, R96025); mouse anti-β-actin (1:10,000, Sigma, A5441); rabbit anti-MARCH6 (1:5000, Bethyl Laboratories, BETHA304-171A); rabbit anti-vinculin (1:2000, Abcam, ab129002); rabbit anti-GAPDH (1:2000, Cell Signaling Technology, 2118L); goat anti-mouse IgG-HRP (1:10,000, Jackson ImmunoResearch, 115-035-003); donkey anti-rabbit IgG-HRP (1:10000, Jackson ImmunoResearch, 711-035-152); or IRDye 680RD donkey anti-rabbit IgG (1:20,000, Millennium Science) as indicated in figure legends. MARCH6 runs at an apparent molecular mass of ∼80 kDa during SDS-PAGE, which is lower than its expected mass of ∼100 kDa. This is likely due to its 14 TMDs, which confer a greater capacity to bind SDS (55). Blots were imaged using Clarity ECL substrate (Bio-Rad) on a LAS500 imager (GE Healthcare) or, for GAPDH and vinculin, using an Odyssey-CLx imager (LI-COR Bioscience). Densitometry was performed using ImageStudio Lite version 5.2. For HeLa and HeLa–MARCH6–CRISPR experiments, primary antibodies were probed with peroxidase-coupled anti-IgG antibodies (1:10,000, GE Healthcare, NA931V and NA934V), which were activated by enhanced chemiluminescence, imaged on a G:Box system (Syngene), and quantified using Gene Tools (Syngene).
Data presentation
All quantitative data are presented as mean ± S.E. from at least three independent experiments. Statistical significance (p < 0.05) was determined using Student's paired t test.
Author contributions
L. J. S. and A. J. B. conceptualization; L. J. S., V. H., N. A. S., W. L., L. P., and J. M. B. data curation; L. J. S., V. H., N. A. S., L. P., and J. M. B. formal analysis; L. J. S., M. H., and A. J. B. supervision; L. J. S., V. H., N. A. S., and A. J. B. validation; L. J. S., V. H., N. A. S., W. L., L. P., J. M. B., and A. J. B. investigation; L. J. S. visualization; L. J. S., V. H., and J. M. B. methodology; L. J. S. and A. J. B. writing-original draft; L. J. S. and A. J. B. project administration; L. J. S., V. H., N. A. S., W. L., L. P., J. M. B., M. H., and A. J. B. writing-review and editing; M. H., J. M. B., and A. J. B. funding acquisition; A. J. B. resources.
Supplementary Material
Acknowledgment
We thank Dr. Anika Prabhu for help with CRISPR.
This work was supported by National Health and Medical Research Council Grant 1060515 (to A. J. B.), Australian Research Council Grant DP170101178 (to A. J. B.), a Gold Star award from UNSW Sydney (to A. J. B.), and in part by National Institutes of Health Grants R01 GM046904 (to M. H.) and F32 GM113456 (to J. M. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S2.
- HMGCR
- 3-hydroxy-3-methylglutaryl coenzyme A reductase
- D2
- type 2 iodothyronine deiodinase
- MARCH6
- membrane-associated RING finger (C3HC4) 6
- RGS2
- regulator of G protein signaling-2
- SM
- squalene monooxygenase
- SSD
- sterol-sensing domain
- TMD
- transmembrane domain
- VCP
- valosin-containing protein
- LXR
- liver X receptor
- 25HC
- 25-hydroxycholesterol
- ER
- endoplasmic reticulum
- qRT-PCR
- quantitative RT-PCR
- Chol/CD
- cholesterol complexed to cyclodextrin
- PBGD
- porphobilinogen deaminase
- FCS
- fetal calf serum
- DMEM
- Dulbecco's modified Eagle's medium
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HRP
- horseradish peroxidase
- DUB
- deubiquitinase.
References
- 1. Prabhu A. V., Luu W., Li D., Sharpe L. J., and Brown A. J. (2016) DHCR7: a vital enzyme switch between cholesterol and vitamin D production. Prog. Lipid Res. 64, 138–151 10.1016/j.plipres.2016.09.003 [DOI] [PubMed] [Google Scholar]
- 2. Brown M. S., Radhakrishnan A., and Goldstein J. L. (2018) Retrospective on cholesterol homeostasis: the central role of scap. Annu. Rev. Biochem. 87, 783–807 10.1146/annurev-biochem-062917-011852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gill S., Stevenson J., Kristiana I., and Brown A. J. (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 13, 260–273 10.1016/j.cmet.2011.01.015 [DOI] [PubMed] [Google Scholar]
- 4. Zelcer N., Sharpe L. J., Loregger A., Kristiana I., Cook E. C., Phan L., Stevenson J., and Brown A. J. (2014) The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell. Biol. 34, 1262–1270 10.1128/MCB.01140-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jo Y., Lee P. C., Sguigna P. V., and DeBose-Boyd R. A. (2011) Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. U.S.A. 108, 20503–20508 10.1073/pnas.1112831108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang L. Y., Jiang W., Tian N., Xiong Y. N., Liu J., Wei J., Wu K. Y., Luo J., Shi X. J., and Song B. L. (2018) Ring finger protein 145 (RNF145) is a ubiquitin ligase for sterol-induced degradation of HMG-CoA reductase. J. Biol. Chem. 293, 4047–4055 10.1074/jbc.RA117.001260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schultz M. L., Krus K. L., Kaushik S., Dang D., Chopra R., Qi L., Shakkottai V. G., Cuervo A. M., and Lieberman A. P. (2018) Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 9, 3671 10.1038/s41467-018-06115-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nguyen K. T., Lee C. S., Mun S. H., Nhung T. T., Park S. K., and Hwang C. S. (2018) N-terminal acetylation and the N-end rule pathway control degradation of the lipid droplet protein PLIN2. J. Biol. Chem. 293, 10.1074/jbc.RA118.005556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loregger A., Cook E. C., Nelson J. K., Moeton M., Sharpe L. J., Engberg S., Karimova M., Lambert G., Brown A. J., and Zelcer N. (2016) A MARCH6 and IDOL E3 ubiquitin ligase circuit uncouples cholesterol synthesis from lipoprotein uptake in hepatocytes. Mol. Cell. Biol. 36, 285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kreft S. G., Wang L., and Hochstrasser M. (2006) Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI). J. Biol. Chem. 281, 4646–4653 10.1074/jbc.M512215200 [DOI] [PubMed] [Google Scholar]
- 11. Zattas D., Berk J. M., Kreft S. G., and Hochstrasser M. (2016) A conserved C-terminal element in the yeast Doa10 and human MARCH6 ubiquitin ligases required for selective substrate degradation. J. Biol. Chem. 291, 12105–12118 10.1074/jbc.M116.726877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zavacki A. M., Arrojo E Drigo R., Freitas B. C., Chung M., Harney J. W., Egri P., Wittmann G., Fekete C., Gereben B., and Bianco A. C. (2009) The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol. Cell. Biol. 29, 5339–5347 10.1128/MCB.01498-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park S. E., Kim J. M., Seok O. H., Cho H., Wadas B., Kim S. Y., Varshavsky A., and Hwang C. S. (2015) Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 347, 1249–1252 10.1126/science.aaa3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang L., Dong H., Soroka C. J., Wei N., Boyer J. L., and Hochstrasser M. (2008) Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 48, 1558–1569 10.1002/hep.22499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hassink G., Kikkert M., van Voorden S., Lee S. J., Spaapen R., van Laar T., Coleman C. S., Bartee E., Früh K., Chau V., and Wiertz E. (2005) TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 388, 647–655 10.1042/BJ20041241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakamura N., Harada K., Kato M., and Hirose S. (2014) Ubiquitin-specific protease 19 regulates the stability of the E3 ubiquitin ligase MARCH6. Exp. Cell Res. 328, 207–216 10.1016/j.yexcr.2014.07.025 [DOI] [PubMed] [Google Scholar]
- 17. Cook E. C., Nelson J. K., Sorrentino V., Koenis D., Moeton M., Scheij S., Ottenhoff R., Bleijlevens B., Loregger A., and Zelcer N. (2017) Identification of the ER-resident E3 ubiquitin ligase RNF145 as a novel LXR-regulated gene. PLoS One 12, e0172721 10.1371/journal.pone.0172721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zelcer N., Hong C., Boyadjian R., and Tontonoz P. (2009) LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 10.1126/science.1168974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Irisawa M., Inoue J., Ozawa N., Mori K., and Sato R. (2009) The sterol-sensing endoplasmic reticulum (ER) membrane protein TRC8 hampers ER to Golgi transport of sterol regulatory element-binding protein-2 (SREBP-2)/SREBP cleavage-activated protein and reduces SREBP-2 cleavage. J. Biol. Chem. 284, 28995–29004 10.1074/jbc.M109.041376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee J. P., Brauweiler A., Rudolph M., Hooper J. E., Drabkin H. A., and Gemmill R. M. (2010) The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol. Cancer Res. 8, 93–106 10.1158/1541-7786.MCR-08-0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stefanovic-Barrett S., Dickson A. S., Burr S. P., Williamson J. C., Lobb I. T., van den Boomen D. J., Lehner P. J., and Nathan J. A. (2018) MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep. 19, e45603 10.15252/embr.201745603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuwabara P. E., and Labouesse M. (2002) The sterol-sensing domain: multiple families, a unique role? Trends Genet. 18, 193–201 10.1016/S0168-9525(02)02640-9 [DOI] [PubMed] [Google Scholar]
- 23. Sever N., Yang T., Brown M. S., Goldstein J. L., and DeBose-Boyd R. A. (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 11, 25–33 10.1016/S1097-2765(02)00822-5 [DOI] [PubMed] [Google Scholar]
- 24. Nohturfft A., Brown M. S., and Goldstein J. L. (1998) Sterols regulate processing of carbohydrate chains of wild-type SREBP cleavage-activating protein (SCAP), but not sterol-resistant mutants Y298C or D443N. Proc. Natl. Acad. Sci. U.S.A. 95, 12848–12853 10.1073/pnas.95.22.12848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yabe D., Xia Z. P., Adams C. M., and Rawson R. B. (2002) Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc. Natl. Acad. Sci. U.S.A. 99, 16672–16677 10.1073/pnas.262669399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies J. P., and Ioannou Y. A. (2000) Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J. Biol. Chem. 275, 24367–24374 10.1074/jbc.M002184200 [DOI] [PubMed] [Google Scholar]
- 27. Ohgami N., Ko D. C., Thomas M., Scott M. P., Chang C. C., and Chang T. Y. (2004) Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. U.S.A. 101, 12473–12478 10.1073/pnas.0405255101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J., Chu B. B., Ge L., Li B. L., Yan Y., and Song B. L. (2009) Membrane topology of human NPC1L1, a key protein in enterohepatic cholesterol absorption. J. Lipid Res. 50, 1653–1662 10.1194/jlr.M800669-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strutt H., Thomas C., Nakano Y., Stark D., Neave B., Taylor A. M., and Ingham P. W. (2001) Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr. Biol. 11, 608–613 10.1016/S0960-9822(01)00179-8 [DOI] [PubMed] [Google Scholar]
- 30. Burke R., Nellen D., Bellotto M., Hafen E., Senti K. A., Dickson B. J., and Basler K. (1999) Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 99, 803–815 10.1016/S0092-8674(00)81677-3 [DOI] [PubMed] [Google Scholar]
- 31. Kawakami T., Kawcak T., Li Y. J., Zhang W., Hu Y., and Chuang P. T. (2002) Mouse dispatched mutants fail to distribute hedgehog proteins and are defective in hedgehog signaling. Development 129, 5753–5765 10.1242/dev.00178 [DOI] [PubMed] [Google Scholar]
- 32. Gemmill R. M., West J. D., Boldog F., Tanaka N., Robinson L. J., Smith D. I., Li F., and Drabkin H. A. (1998) The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene, TRC8. Proc. Natl. Acad. Sci. U.S.A. 95, 9572–9577 10.1073/pnas.95.16.9572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zerenturk E. J., Sharpe L. J., and Brown A. J. (2012) Sterols regulate 3β-hydroxysterol Δ24-reductase (DHCR24) via dual sterol regulatory elements: Cooperative induction of key enzymes in lipid synthesis by sterol regulatory element binding proteins. Biochim. Biophys. Acta 1821, 1350–1360 10.1016/j.bbalip.2012.07.006 [DOI] [PubMed] [Google Scholar]
- 34. Huang E. Y., To M., Tran E., Dionisio L. T. A., Cho H. J., Baney K. L. M., Pataki C. I., and Olzmann J. A. (2018) A VCP inhibitor substrate trapping approach (VISTA) enables proteomic profiling of endogenous ERAD substrates. Mol. Biol. Cell 29, 1021–1030 10.1091/mbc.E17-08-0514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bug M., and Meyer H. (2012) Expanding into new markets–VCP/p97 in endocytosis and autophagy. J. Struct. Biol. 179, 78–82 10.1016/j.jsb.2012.03.003 [DOI] [PubMed] [Google Scholar]
- 36. Song B. L., Sever N., and DeBose-Boyd R. A. (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19, 829–840 10.1016/j.molcel.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 37. Zhang L., Rajbhandari P., Priest C., Sandhu J., Wu X., Temel R., Castrillo A., de Aguiar Vallim T. Q., Sallam T., and Tontonoz P. (2017) Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. eLife 6, e28766 10.7554/eLife.28766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wangeline M. A., and Hampton R. Y. (2018) “Mallostery”–ligand-dependent protein misfolding enables physiological regulation by ERAD. J. Biol. Chem. 293, 14937–14950 10.1074/jbc.RA118.001808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller B. T., Ueta C. B., Lau V., Jacomino K. G., Wasserman L. M., and Kim B. W. (2012) Statins and downstream inhibitors of the isoprenylation pathway increase type 2 iodothyronine deiodinase activity. Endocrinology 153, 4039–4048 10.1210/en.2012-1117 [DOI] [PubMed] [Google Scholar]
- 40. Howe V., Sharpe L. J., Prabhu A. V., and Brown A. J. (2017) New insights into cellular cholesterol acquisition: promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 647–657 10.1016/j.bbalip.2017.03.009 [DOI] [PubMed] [Google Scholar]
- 41. Howe V., Chua N. K., Stevenson J., and Brown A. J. (2015) The regulatory domain of squalene monooxygenase contains a re-entrant loop and senses cholesterol via a conformational change. J. Biol. Chem. 290, 27533–27544 10.1074/jbc.M115.675181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J. N., Song B., DeBose-Boyd R. A., and Ye J. (2006) Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J. Biol. Chem. 281, 39308–39315 10.1074/jbc.M608999200 [DOI] [PubMed] [Google Scholar]
- 43. Brown A. J., Sun L., Feramisco J. D., Brown M. S., and Goldstein J. L. (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 10.1016/S1097-2765(02)00591-9 [DOI] [PubMed] [Google Scholar]
- 44. Hulce J. J., Cognetta A. B., Niphakis M. J., Tully S. E., and Cravatt B. F. (2013) Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 10, 259–264 10.1038/nmeth.2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brown A. J., and Sharpe L. J. (2015) in Biochemistry of Lipids, Lipoproteins, and Membranes (Ridgway N. D., and McLeod R. S., eds) 6th Ed., pp. 327–358, Elsevier, Waltham, MA [Google Scholar]
- 46. Watanabe M., Houten S. M., Mataki C., Christoffolete M. A., Kim B. W., Sato H., Messaddeq N., Harney J. W., Ezaki O., Kodama T., Schoonjans K., Bianco A. C., and Auwerx J. (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489 10.1038/nature04330 [DOI] [PubMed] [Google Scholar]
- 47. Freson K., Stolarz K., Aerts R., Brand E., Brand-Herrmann S. M., Kawecka-Jaszcz K., Kuznetsova T., Tikhonoff V., Thijs L., Vermylen J., Staessen J. A., Van Geet C., and European Project on Genes in Hypertension Investigators (2007) −391 C to G substitution in the regulator of G-protein signalling-2 promoter increases susceptibility to the metabolic syndrome in white European men: consistency between molecular and epidemiological studies. J. Hypertens. 25, 117–125 10.1097/HJH.0b013e3280109c6c [DOI] [PubMed] [Google Scholar]
- 48. Sartori M., Ceolotto G., Dorigatti F., Mos L., Santonastaso M., Bratti P., Papparella I., Semplicini A., Palatini P., and HARVEST Group. (2008) RGS2 C1114G polymorphism and body weight gain in hypertensive patients. Metabolism 57, 421–427 10.1016/j.metabol.2007.10.021 [DOI] [PubMed] [Google Scholar]
- 49. Libby A. E., Bales E. S., Monks J., Orlicky D. J., and McManaman J. L. (2018) Perilipin-2 deletion promotes carbohydrate-mediated browning of white adipose tissue at ambient temperature. J. Lipid Res. 59, 1482–1500 10.1194/jlr.M086249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cao X., Qin J., Xie Y., Khan O., Dowd F., Scofield M., Lin M. F., and Tu Y. (2006) Regulator of G-protein signaling 2 (RGS2) inhibits androgen-independent activation of androgen receptor in prostate cancer cells. Oncogene 25, 3719–3734 10.1038/sj.onc.1209408 [DOI] [PubMed] [Google Scholar]
- 51. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sever N., Song B. L., Yabe D., Goldstein J. L., Brown M. S., and DeBose-Boyd R. A. (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278, 52479–52490 10.1074/jbc.M310053200 [DOI] [PubMed] [Google Scholar]
- 53. Prabhu A. V., Luu W., Sharpe L. J., and Brown A. J. (2016) Cholesterol-mediated degradation of 7-dehydrocholesterol reductase switches the balance from cholesterol to vitamin D synthesis. J. Biol. Chem. 291, 8363–8373 10.1074/jbc.M115.699546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kielar D., Dietmaier W., Langmann T., Aslanidis C., Probst M., Naruszewicz M., and Schmitz G. (2001) Rapid quantification of human ABCA1 mRNA in various cell types and tissues by real-time reverse transcription-PCR. Clin. Chem. 47, 2089–2097 [PubMed] [Google Scholar]
- 55. Rath A., Glibowicka M., Nadeau V. G., Chen G., and Deber C. M. (2009) Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 1760–1765 10.1073/pnas.0813167106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song B. L., and DeBose-Boyd R. A. (2004) Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J. Biol. Chem. 279, 28798–28806 10.1074/jbc.M402442200 [DOI] [PubMed] [Google Scholar]
- 57. Song B. L., Javitt N. B., and DeBose-Boyd R. A. (2005) Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 1, 179–189 10.1016/j.cmet.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 58. Fitzky B. U., Moebius F. F., Asaoka H., Waage-Baudet H., Xu L., Xu G., Maeda N., Kluckman K., Hiller S., Yu H., Batta A. K., Shefer S., Chen T., Salen G., Sulik K., et al. (2001) 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J. Clin. Invest. 108, 905–915 10.1172/JCI200112103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.