

Abstract
Summary
CABS-flex standalone is a Python package for fast simulations of protein structure flexibility. The package combines simulations of protein dynamics using CABS coarse-grained protein model with the reconstruction of selected models to all-atom representation and analysis of modeling results. CABS-flex standalone is designed to allow for command-line access to the CABS computations and complete control over simulation process. CABS-flex standalone is equipped with features such as: modeling of multimeric and large-size protein systems, contact map visualizations, analysis of similarities to the reference structure and configurable modeling protocol. For instance, the user may modify the simulation parameters, distance restraints, structural clustering scheme or all-atom reconstruction parameters. With these features CABS-flex standalone can be easily incorporated into other methodologies of structural biology.
Availability and implementation
CABS-flex standalone is distributed under the MIT license, which is free for academic and non-profit users. It is implemented in Python. CABS-flex source code, wiki with examples of use and installation instructions for Linux, macOS and Windows are available from the CABS-flex standalone repository at https://bitbucket.org/lcbio/cabsflex.
1 Introduction
Structural flexibility of a protein may play an important role in its biological function. Experimental investigation of protein flexibility is often difficult or impossible, hence the important role of computer simulations. Unfortunately, capabilities of contemporary computers limit the applicability of classical simulations (all-atom Molecular Dynamics, MD) to small system sizes and short timescales (Kmiecik et al., 2016). In practice, for the majority of biologically relevant protein systems, classical simulations of structure flexibility require huge computational resources (i.e. using high performance computer clusters). The CABS-flex standalone provides a tool for fast simulations of protein structure dynamics that overcomes system-size limitations of classical all-atom MD: the CABS-flex has been estimated to be 3 to 4 orders of magnitude faster than all-atom MD (Jamroz et al., 2013).
CABS-flex method is based on a well-established CABS coarse-grained protein model. The CABS design was originally described in detail by Kolinski (Kolinski, 2004) and its applications have been recently reviewed by Kmiecik et al. (2016). The picture of CABS Monte Carlo dynamics proved to be consistent with the dynamics seen in (nanosecond time scale) MD simulations of folded globular proteins (Jamroz et al., 2013), fluctuations observed in NMR ensembles (Jamroz et al., 2014) and various experimental data on protein folding (Kmiecik et al., 2016). The CABS-flex method has been implemented as publicly available web server (Jamroz et al., 2013) and recently updated to version 2.0 (Kuriata et al., 2018) available at http://biocomp.chem.uw.edu.pl/CABSflex2. The CABS-flex is also used as a dynamics modeling component of Aggrescan3D method (Zambrano et al., 2015) for structure-based predictions of protein aggregation propensities. Finally, CABS-flex methodology was successfully adapted for modeling flexibility of protein-peptide complexes during docking simulations (Kurcinski et al., 2015), including large-scale conformational changes of a peptide and a protein receptor (Ciemny et al., 2016).
2 Features
The new CABS-flex standalone is implemented as a Python 2.7 object-oriented package, combined with a Fortran-based CABS algorithm implementation. The package provides an easy-to-use command-line script, which offers full control over the CABS-flex program.
The CABS-flex standalone pipeline in the default mode is presented in Figure 1. The only required input for CABS-flex is the protein structure data in the PDB file format. The default output consists of: (i) a trajectory and an ensemble of representative protein structures that reflect the protein flexibility in PDB format file, (ii) RMSF-profile of the protein (both a SVG file with the plot and a CSV with the data), (iii) residue-residue contact maps for the simulation trajectory, clusters of similar models found in the trajectory and the representative structures (a SVG file with the plot and a CSV with the data for each map).
Fig. 1.
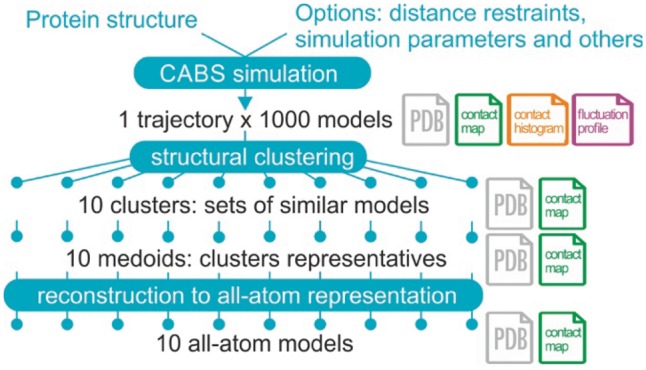
Pipeline of CABS-flex standalone in the default mode. CABS-flex combines CABS coarse-grained model with structural clustering of the simulation results and reconstruction of selected models to all-atom representation. Number of models generated at each modeling step can be set up by the user. In the default mode, CABS-flex trajectory counts 1000 models in coarse-grained representation that are clustered to 10 representative models, subsequently reconstructed to all-atom models. In addition to protein models in PDB format, CABS-flex standalone provides a number of analysis and visualization options, such as contact maps and histograms (marked in the figure, in the appropriate pipeline stages)
In comparison to the web server version (Kuriata et al., 2018), CABS-flex standalone introduces a number of new features that allow handling larger system-sizes and modifying the modeling protocol. The new features include command-line access to the full settings of: the CABS coarse-grained model, geometrical restrains used in the CABS simulations and the results analysis. The options for the results analysis include: (i) customization of the output parameters; (ii) optional MODELLER-based (Webb and Sali, 2016) all-atom reconstruction (including hydrogen atoms); (iii) customizable clustering and filtering; (iv) contact map calculation and visualization (contact maps provide residue-to-residue contact frequencies using a user-defined contact cut-off).
The user may additionally impose custom distance restraints (derived for example from experimental data) on the simulated models. Moreover, it is possible to manually modify the allowed flexibility of selected regions. The protein flexibility may be changed locally by tuning the flexibility factors of selected residues or globally by modifying the restraints generation scheme.
Descriptions of all CABS-flex options, examples of use and installation instructions are available in the CABS-flex standalone repository at https://bitbucket.org/lcbio/cabsflex.
3 Conclusions
CABS-flex standalone significantly extends the capabilities provided by the web server version (Jamroz et al., 2013; Kuriata et al., 2018) and provides an alternative to other coarse-grained based protein simulation packages like Rosetta (Chaudhury et al., 2010), UNRES (Liwo et al., 2014) or PRIMO (Kar and Feig, 2017). Since CABS-flex standalone is provided as a Python 2.7 package, it may be readily incorporated into other methodologies as an element of their pipelines. The large set of customizable options allows users to access the CABS simulation engine (Kmiecik et al., 2016). This makes CABS-flex standalone well suited for a number of applications in protein dynamics predictions, ranging from low-temperature structure refinement, near-native fluctuations and large-scale structural transitions, like protein folding mechanisms or dynamics of large disordered regions.
Funding
The authors acknowledge support from the National Science Centre (NCN), Poland, Grant [MAESTRO2014/14/A/ST6/00088]
Conflict of Interest: none declared.
References
- Chaudhury S. et al. (2010) PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics, 26, 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciemny M.P. et al. (2016) Protein-peptide molecular docking with large-scale conformational changes: the p53-MDM2 interaction. Sci. Rep., 6, 37532.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M. et al. (2013) CABS-flex: server for fast simulation of protein structure fluctuations. Nucleic Acids Res., 41, W427–W431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M. et al. (2014) CABS-flex predictions of protein flexibility compared with NMR ensembles. Bioinformatics, 30, 2150–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M. et al. (2013) Consistent view of protein fluctuations from all-atom molecular dynamics and coarse-grained dynamics with knowledge-based force-field. J. Chem. Theory Comput., 9, 119–125. [DOI] [PubMed] [Google Scholar]
- Kar P., Feig M. (2017) Hybrid all-atom/coarse-grained simulations of proteins by direct coupling of CHARMM and PRIMO force fields. J. Chem. Theory Comput., 13, 5753–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik S. et al. (2016) Coarse-grained protein models and their applications. Chem. Rev., 116, 7898–7936. [DOI] [PubMed] [Google Scholar]
- Kolinski A. (2004) Protein modeling and structure prediction with a reduced representation. Acta Biochim. Polonica, 51, 349–371. [PubMed] [Google Scholar]
- Kuriata A. et al. (2018) CABS-flex 2.0: a web server for fast simulations of flexibility of protein structures. Nucleic Acids Res, 46, W338–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurcinski M. et al. (2015) CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res., 43, W419–W424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liwo A. et al. (2014) A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model., 20, 2306.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B., Sali A. (2016) Comparative protein structure modeling using MODELLER. Current Protocols Prot. Sci., 86, 2 9 1-2 9 37. [DOI] [PubMed] [Google Scholar]
- Zambrano R. et al. (2015) AGGRESCAN3D (A3D): server for prediction of aggregation properties of protein structures. Nucleic Acids Res., 43, W306–W313. [DOI] [PMC free article] [PubMed] [Google Scholar]