

Abstract
Aspergillus flavus is an important zoonotic pathogen and a well-known aflatoxin producer. Aspergillus flavus strains that are prevalent in Japanese environments are reported to be non-aflatoxigenic, although their aflatoxin productivity, especially among clinical isolates, has not been thoroughly investigated to date. In this study, we sequenced the genomes of ten strains of A. flavus isolated in Japan and compared their sequences with each other as well as with those of Aspergillus oryzae RIB40 and A. flavus NRRL3357. The phylogenetic analysis based on identified SNPs indicated that five strains were closer to A. oryzae RIB40 than to A. flavus NRRL3357. In contrast, of those isolates that were closer to A. flavus NRRL3357 than to A. oryzae RIB40, three were found to possess either the entire or partial aflatoxin biosynthesis gene cluster of NRRL3357-type. Furthermore, two of the three actually produced either aflatoxin B1 or an intermediate of the reaction leading to aflatoxin formation. Three of the ten strains we isolated were identified to possess part of the aflatoxin gene cluster, while five others retained the A. oryzae RIB40-type cluster. The genome data thus obtained may be further explored and utilized for comparative analysis of aflatoxin production in environmental and clinical isolates of A. flavus.
Keywords: Aspergillus flavus, gene cluster for secondary metabolites, aflatoxin production
1. Introduction
Aspergillus flavus is a major plant pathogen that is widely distributed throughout the world. This organism is the causative agent of aspergillosis, an important zoonotic mycosis. Worldwide, A. flavus is the second leading cause of invasive and non-invasive aspergillosis.1,2 Tashiro et al. reported that about ∼5% of respiratory positive Aspergillus spp. cultures in Japan are identified as A. flavus.3 However, clinical A. flavus isolates have not been sufficiently investigated, and its virulence factors remain largely unknown.
Aspergillus flavus is also a well-known aflatoxin producer. Aflatoxins have serious hepatotoxic, hepatocarcinogenic, and immunosuppressive effects on humans and animals.4,5 Aflatoxigenic A. flavus is not prevalent in the environment in Japan,6 and as such, aflatoxin production in Japanese clinical A. flavus isolates has not been investigated and the genome characteristics and pathogenicity of these strains are unknown. The present study aims to determine the genome sequences and phenotypes, including pathogenicity, of A. flavus strains clinically isolated in Japan.
2. Materials and methods
2.1. Strains and preparation of conidia
The ten strains of A. flavus used in this study (Table 1) were obtained from the Medical Mycology Research Center (MMRC), Chiba University, through the National BioResource Project (http://www.nbrp.jp (15 November 2018, date last accessed)). rDNA internal transcribed spacer sequence (ITS) or partial β-tubulin (benA) sequence of A. flavus IFM57535, IFM59975, IFM60519, IFM60655, IFM61224, and IFM61226 were analyzsed to identify as A. flavus in MMRC. Other strains might be identified as A. flavus based on the morphology. Genome sequence data for A. flavus strains NRRL33577 and Aspergillus oryzae RIB408 were used in this study. These strains were routinely cultured at 25°C for 2 weeks on potato dextrose agar, followed by conidia collection as described previously.9 We observed very little A. flavus IFM59975, IFM61224, or IFM61226 conidia formation. Recovered conidia were used in further experiments.
Table 1.
Source of strains investigated in this study
| Strain | Isolation source |
|---|---|
| A. flavus IFM54693 | Clinical; gingiva |
| A. flavus IFM55053 | Clinical; paranasal sinus |
| A. flavus IFM57535 | Clinical; sputum |
| A. flavus IFM58503 | Clinical; paranasal sinus |
| A. flavus IFM59894 | Clinical; abdominal drain |
| A. flavus IFM59975 | Clinical; aural discharge |
| A. flavus IFM60519 | Clinical; sputum |
| A. flavus IFM60655 | Clinical; sputum |
| A. flavus IFM61224 | Clinical; aural discharge |
| A. flavus IFM61226 | Clinical; sputum |
| A. flavus NRRL3357 | Environment; peanut cotyledons in U.S. |
| A. oryzae RIB40 | Environment; fava bean in soy sauce factory in Japan |
2.2. Whole-genome sequencing
A ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research, Irvine, CA, USA) was used to prepare genomic DNA from A. flavus and A. oryzae strains. Five micrograms of genomic DNA were broken into fragments of, on average, 500 bp using a Covaris S2 sonication system (Covaris, Inc., Woburn, MA, USA). Sequencing libraries were prepared using a NEBNext DNA library prep kit for Illumina (New England Biolabs, Ipswich, MA, USA). A total of 300 cycles of paired-end sequencing were carried out using a MiSeq system (Illumina, San Diego, CA, USA) according to the manufacturer’s specifications. The sequencing reads were trimmed using CLC Genomics Workbench ver. 6.5 (Qiagen, Hilden, Germany) with the following parameters: Phred quality score > 30; remove 15 terminal nucleotides from the 5’ end and 2 terminal nucleotides from the 3’ end; remove truncated reads less than <100 nucleotides in length. Trimmed reads were assembled using CLC Genomics Workbench ver. 6.5 (Qiagen). Accession numbers in the DDBJ Sequence Read Archive and the scaffold accession number are shown in Supplementary Table S1. Assembled scaffolds were mapped to the genome data of A. flavus NRRL3357 and A. oryzae RIB40 using MUMmer. Trimmed read data sets by ea-utils10 were mapped to the assembled scaffolds or reference genomes using Bowtie2 v.2.3.211 with ‘–very-sensitive’ option. The number of reads is shown in Supplementary Table S2. Scaffold data were analysed using b-MiGAP12; assembled scaffold numbers, total read lengths, and the number of CDS regions are summarized in Supplementary Table S2. For genome-wide phylogenetic analysis, trimmed reads including SRR1835311 as the short read of A. oryzae RIB40 were mapped to the reference genome of A. flavus NRRL3357 using Bowtie2. Single nucleotide polymorphisms (SNPs) were identified using samtools v. 1.813, and filtered with 10-fold coverage, 30 mapping quality, and 75% consensus using in-house scripts.14,15. Repetitive regions of A. flavus NRRL3357 identified by RepeatMasker v. open-4.0.7 (http://repeatmasker.org (15 November 2018, date last accessed)) were excluded from the phylogenetic analysis. A maximum likelihood tree of 12 strains including A. flavus NRRL3357 and A. oryzae RIB40 was constructed based on 268, 510 SNPs using RAxML v. 8.2.1116 with 1, 000 bootstrap, and visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/ (11 July 2017, date last accessed)).
2.3. Partial sequencing of α-amylase gene
A partial α-amylase gene, including mismatches between the original copy and extra copies in A. oryzae, was amplified with a forward primer (5′-CTAACCATCACATAGTTGGACTATATCCAG-3′), a reverse primer (5′-GTCGGTCGGGTAGCCCGAGAGCCAGGTTGC-3′), and EmeraldAmp PCR Master Mix (Takara Bio Inc., Shiga, Japan). Amplified fragments were purified with FastGene Gel and a PCR Extraction Kit (NIPPON Genetics Co., Ltd., Tokyo, Japan) and sequenced using a BigDye terminator v3.1 Cycle Sequencing Kit and an Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Prediction of gene clusters for secondary metabolite biosynthesis
Secondary metabolite biosynthesis gene cluster sequences were predicted using antiSMASH (https://antismash.secondarymetabolites.org/ (21 April 2017, date last accessed)) from annotated data for non-ribosomal peptide synthases (NRPS), PKS, dimethylallyl tryptophan synthase(DMTS), and terpene synthase. A total of 87 clusters were found (Supplementary Table S3). In 34 of the 87 clusters in A. oryzae RIB40, almost the entirety of each region was covered with reads obtained from each strain.
2.5. Detection of aflatoxin
To prepare A. flavus and A. oryzae extracts, conidia were inoculated on sterilized maize grain and cultured for 4–5 days at 25°C. After cultivation, each grain covered with fungal cells was dipped into 1 ml 70% wt/vol, methanol, and secondary metabolites, including aflatoxins, were extracted by mixing vigorously. The supernatants obtained from each strain were used in the following analyses. Extract aflatoxin content was estimated using a MycoJudge Total Aflatoxin ELISA kit (NH Foods Ltd., Osaka, Japan) according to the manufacturer’s instructions and detected by liquid chromatography quadrupole-time-of-flight mass spectrometry (LC-QTOF) as described below. Samples were separated by LC using the Agilent 1260 infinity system (Agilent Technologies, Santa Clara, CA, USA) equipped with a ZORBAX Eclipse Plus C18 column (Agilent Technologies) maintained at 40°C. Ammonium acetate (5 mM) (A) and methanol (B) were used as mobile phases. Each 3 μLμl sample was separated with LC, using a gradient of 10% A/90% B to 100% B over 30 min with a flow rate of 0.2 mLml/min. The Agilent 6550 iFunnel QTOF LC/MS system (Agilent Technologies) was used for MS analysis.
2.6. Detection of ustiloxin B by LC-MS/MS
The procedures used to prepare extracts and detect ustiloxin B produced by each strain are the same as those used for aflatoxin detection, as described above. Because we could not obtain a standard sample of ustiloxin B, we used an estimation based on the measured accurate mass of the protonated molecular ion [M + +H]+ that indicated relative mass errors within ± 2 ppm and high isotope scores (> 85%).
2.7. Visualization of sequence data
Sequence alignments and phylogenetic analyses were performed using MEGA7.17 The Integrative Genomic Viewer18, 19 (Broad Institute) was used to search the mapped reads and scaffolds of reference genomes. J-Circos (Australian Prostate Cancer Research Centre)20 was used to visualize the mapped reads for each strain. The Artemis Comparison Tool (Sanger Institute) was used for visualization by synteny plot.21
3. Results and discussion
3.1. Mapping of A. flavus data on the reference genomes of A. flavus NRRL3357 and A. oryzae RIB40
The read data from each clinical isolate of A. flavus were mapped to A. oryzae RIB40 and A. flavus NRRL3357 reference genome data and scaffolds of ten A. flavus strains. Mapping rates are shown in Table 2. Mapped reads on A. oryzae RIB40 chromosomes were visualized with J-Circos,20 as shown in Fig. 1. Some large gaps were found compared with A. oryzae RIB40. Additional notes about large gaps are included in the Supplementary Text.
Table 2.
Mapping of reads to scaffold and reference genomea
| Scaffold | Read mapping rate (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IFM54693 | IFM55053 | IFM57535 | IFM58503 | IFM59894 | IFM59975 | IFM60519 | IFM60655 | IFM61224 | IFM61226 | |
| IFM54693 | 74.90 | 89.18 | 89.22 | 89.11 | 87.68 | 90.86 | 89.09 | 89.74 | 89.98 | 89.08 |
| IFM55053 | 76.22 | 98.69 | 95.38 | 95.94 | 91.78 | 95.94 | 95.99 | 93.74 | 94.60 | 96.27 |
| IFM57535 | 76.93 | 96.16 | 98.50 | 96.20 | 92.51 | 96.59 | 95.80 | 94.62 | 95.92 | 95.71 |
| IFM58503 | 76.40 | 96.22 | 95.64 | 98.28 | 91.74 | 96.09 | 95.58 | 93.97 | 94.87 | 95.98 |
| IFM59894 | 77.13 | 95.05 | 94.93 | 94.69 | 98.41 | 96.00 | 94.85 | 95.36 | 95.71 | 94.65 |
| IFM59975 | 77.67 | 95.81 | 95.68 | 95.68 | 92.72 | 98.90 | 95.37 | 95.30 | 96.01 | 95.66 |
| IFM60519 | 76.88 | 96.84 | 95.84 | 96.18 | 92.46 | 96.37 | 98.69 | 94.60 | 95.70 | 96.09 |
| IFM60655 | 78.32 | 95.46 | 95.52 | 95.43 | 94.08 | 97.15 | 95.44 | 98.55 | 96.35 | 95.41 |
| IFM61224 | 76.38 | 93.74 | 94.32 | 93.81 | 91.79 | 95.31 | 94.03 | 93.75 | 96.93 | 93.54 |
| IFM61226 | 75.89 | 95.87 | 94.50 | 95.33 | 91.08 | 95.43 | 94.86 | 93.32 | 94.16 | 97.66 |
| NRRL3357a | 72.53 | 92.65 | 91.60 | 93.00 | 88.63 | 92.96 | 92.50 | 90.65 | 90.72 | 92.84 |
| RIB40 | 78.12 | 96.16 | 96.38 | 96.01 | 93.43 | 97.26 | 96.07 | 96.23 | 96.64 | 95.78 |
The sequence reads of the isolates were mapped onto their scaffolds as well as onto the genome sequence of RIB40, and the percentages of successful mapping are shown. The highest mapping (black cells) and the second highest mapping (grey cells) are indicated.
Figure 1.

Whole-genome comparison of ten clinical isolates of A. flavus analysed as described in the text. Solid circles indicate putative centromeres and centripetal solid lines on the outermost grey circle indicate chromosome boundaries.
Phylogenic analysis was performed based on identified 268, 510 SNPs, which indicated ten strains used in this study were roughly divided into three groups (Fig. 2). Aspergillus flavus NRRL3357 was grouped in a same group with A. flavus IFM55053, IFM58503, and IFM61226 (Fig. 2, shaded with green). On the other hand, A. oryzae RIB40 was grouped in a same group with A. flavus IFM54693, IFM59894, IFM59975, IFM60655, and IFM61224 (Fig. 2, shaded with blue). Aspergillus flavus IFM57535 and IFM60519 were away from both groups.
Figure 2.

An unrooted phylogenetic tree of ten clinical isolates of A. flavus, along with A. flavus NRRL3357 and A. oryzae RIB40, calculated on the basis of SNPs identified. The light gray-shaded strains include A. flavus NRRL3357, while the gray-shaded ones include A. oryzae RIB40.
More unmapped reads were observed in A. flavus IFM54693 than in the other strains (Table 2). One possible reason for this is that the total scaffold length of A. flavus IFM54693 was shortest of all ten strains and ∼1.5 Mb shorter than the average of the other nine strains. A possible cause of short total scaffold length is that scaffolds smaller than 1 kb were not included in this analysis. The impaired region, therefore, might be a cause of reads unmapped to the strain’s own scaffold and to those of other strains. Additionally, many unmapped reads were across two different scaffolds (data not shown). Consistent with this finding, most unmapped reads were mapped on strains’ own scaffolds using the ‘local’ option in Bowtie2 (which improved the mapping rate from 74.90% to 96.16%), suggesting that A. flavus IFM54693 has some rearrangement regions, which could be a cause of the low mapping rate. Further investigation using long-read sequence technologies is warranted to determine the precise reason for many unmapped A. flavus IFM54693 reads.
3.2. Comparison of strains by molecular phylogenetic analysis of cmdA
Partial gene sequences of β-tubulin (benA), the second largest subunit of RNA polymerase II (rpb2), and a calmodulin gene (cmdA) similar to the rDNA internal transcribed spacer (ITS1–5.8S-ITS2 region) have been used as molecular identification markers to distinguish species, and the calmodulin gene has been proposed as a secondary identification marker.22 The partial benA and rpb2 genes of the ten A. flavus strains used in this study were identical or had single base substitutions compared with the sequences of A. flavus NRRL3357 and A. oryzae RIB40. The partial cmdA sequences were more diverse than those of benA or rpb2 among the ten strains. Molecular phylogenetic analysis showed that the ten strains could be divided into three groups (Fig. 3), which is consistent with the findings of Okoth et al.23
Figure 3.

Molecular phylogenic analysis of partial cmdA sequences by the maximum likelihood method based on the Kimura 2-parameter model.37 The tree with the highest log likelihood (−817.8602) is shown. The number at each of the branch points indicates percentage of association of the clustered sequences listed. Initial tree(s) for the heuristic search were obtained automatically by applying neighbour-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood approach and then selecting the topology with superior log likelihood value. A discrete γ distribution was used to model evolutionary rate differences among sites [5 categories (+G, parameter = 0.0500)]. The tree is drawn to scale, with branch lengths based on the number of substitutions per site. The analysis involved 20 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 516 positions in the final data set. Evolutionary analysis was conducted as in MEGA7.17
3.3. Two clinical isolates possess one or two extra copies of the α-amylase gene
Aspergillus oryzae RIB40 possesses one original α-amylase gene and two extra identical α-amylase gene copies.8 On the other hand, A. flavus NRRL3357 has only an original α-amylase gene. Three nucleotide mismatches have been identified between the original α-amylase gene and the two extra α-amylase copies.24 To detect the extra α-amylase copies, we performed sequencing a partial region of α-amylase genes. As shown in Fig. 4, the 791st nucleotide residue of A. flavus NRRL3357, indicating that the strain did not contain extra α-amylase copies (Fig. 4, top). No clinical isolates, except A. flavus IFM54693 and IFM60655, included these mismatches, suggesting that they do not possess the extra α-amylase copies found in the A. oryzae RIB40 strain. On the other hand, A. flavus IFM54693 and IFM60655 showed thymine and cytosine peaks at the 791st nucleotide residue in the α-amylase gene (Fig. 4). As consistent with that, we found reads possessing thymine or cytosine at the residue of both strains. The peak heights of the thymine and cytosine nucleotides of A. flavus IFM60655 were similar each other, suggesting that an extra α-amylase gene was presence in the genome of A. flavus IFM60655 similarly to A. oryzae NBRC30105.24 On the other hand, the thymine peak was higher than the cytosine peak at the same site in A. flavus IFM54693 similar to A. oryzae RIB40 (Fig. 4), suggesting that A. flavus IFM54693 has two extra α-amylase copies. As shown in Fig. 2, A. flavus IFM54693 and IFM60655 are phylogenetically close to A. oryzae RIB40, suggesting that these strains might be classified as A. oryzae. Although A. flavus IFM59975 is also included in a group with A. flavus IFM54693 and IFM60655, the strain did not possess extra α-amylase gene, suggesting that A. flavus IFM59975 is closer to native strain in Japan than the other extra-α-amylase acquired strains or lose extra α-amylase genes spontaneously.
Figure 4.
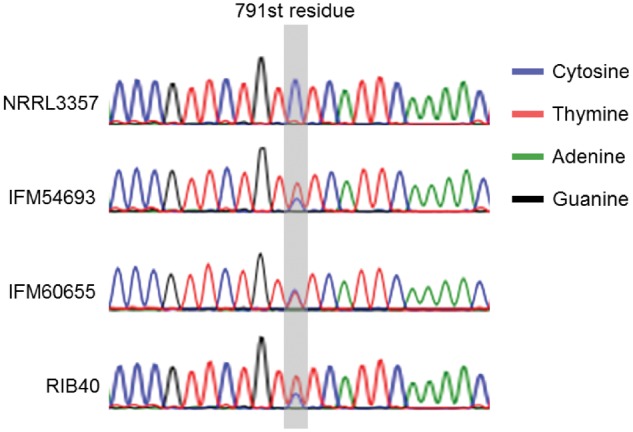
Sanger sequencing revealed a difference between the original and a secondary α-amylase gene.
3.4. Most clinical isolates possess the RIB40-type aflatoxin biosynthesis gene cluster
The clusters homologous to the aflatoxin biosynthesis gene cluster in ten clinical isolates were compared with those of A. oryzae RIB40 and A. flavus NRRL3357. As shown in Fig. 5, the region from fas-1 to norB could clearly be divided into three groups: A. flavus NRRL3357-type, A. oryzae RIB40-type, or another group including A. flavus IFM59894 and IFM60655. The clusters from hypA to norB in A. flavus IFM57535 and IFM60519 are nearly identical to those in A. oryzae RIB40. Those strains were separated from each other in the phylogenetic (Fig. 2). The clusters in A. flavus IFM54693, IFM59975, and IFM61224 are highly similar to those in A. oryzae RIB40 in the region from avnA to norB, and these strains were included in a group with A. oryzae RIB40 (Fig. 2). On the other hand, the A. flavus IFM61226 cluster from hypA to norB is highly similar to that of A. flavus NRRL3357. The A. flavus IFM58503 cluster and the A. flavus IFM55053 show high identity from ver-1 to norB and hypA to aflR in the A. flavus NRRL3357 aflatoxin biosynthesis cluster, respectively. As shown in phylogenetic analysis based on SNPs, those A. flavus strains, IFM55053, IFM58503, and IFM61226 were included in a group with A. flavus NRRL3357 (Fig. 2). The clusters in IFM55053, IFM59894, and IFM60655 are partially deleted (Fig. 5).
Figure 5.

Comparison of aflatoxin biosynthesis gene clusters. White arrows indicate genes showing higher similarity to A. flavus NRRL3357, while black arrows indicate those more similar to A. oryzae RIB40. Grey arrows indicate genes that share <99% identity with those in A. flavus NRRL3357 and A. oryzae RIB40. Striped arrows indicate ≥99% identity with A. flavus NRRL3357 and A. oryzae RIB40. Dotted line arrows corresponding to omtB possess a 50-base insertion. Dotted lines corresponding to the region containing cypA and norB indicate a 612-base deletion including the start codons for cypA and norB.
As shown by Tominaga et al.25 and Kiyota et al.,26 some of the genes that differ between A. oryzae RIB40 and A. flavus NRRL3357 play roles in aflatoxin production. In A. oryzae RIB40, AflT has a large deletion of the C-terminal region due to a TC nucleotide insertion. All of the A. flavus strains, except IFM58503 and IFM61226, also have the AflT C-terminal deletion, as does A. oryzae RIB40. In norA of A. oryzae RIB40, a frameshift mutation was reported to be the cause of truncation.25 Among A. flavus clinical strains, this frameshift mutation was not found. The intergenic region and 5’-coding sequences of cypA and norB, which are deleted in A. oryzae strains, including RIB40 and several A. flavus strains,25,27 are retained in A. flavus IFM58503, IFM61226, and NRRL3357. AflJ is an aflatoxin pathway regulator that binds to another regulator, AflR,28 to cooperatively regulate genes in the aflatoxin biosynthesis cluster.26,29 Kiyota et al. reported that AflJ in A. oryzae had four unique amino acid substitutions as compared with A. flavus strains, indicating that these substitutions induce inactivation of AflJ.26 As shown in Supplementary Fig. S1, A. flavus IFM54693, IFM57535, IFM59975, IFM60519, and IFM61224 share four amino acids with A. oryzae RIB40 at the 8th, 22nd, 268th, and 354th nucleotide residues of AflJ, suggesting that AflJ is inactivated in those strains.
Production of aflatoxin or its intermediates were analysed by enzyme-linked immunosorbent assay (ELISA) and mass spectrometry. Consistent with grouping with the region from fas-1 to norB, aflatoxins were not detected in the extracts from the strains with an RIB40-type cluster (Fig. 5). In A. flavus IFM58503 extracts, two aflatoxin intermediates, sterigmatocystin and O-methylsterigmatocystin (OMST), were detected by mass spectrometry (data not shown). OMST is converted into aflatoxin B1 and G1 by OrdA. OrdA of A. flavus IFM58503 has an A110E substitution, which is conserved among A. flavus NRRL3357, A. oryzae RIB40, and nine other isolates. Because the amino acid is also conserved among OrdA homologues in Aspergillus ochraceoroseus, Aspergillus parasiticus, and Aspergillus nomius, the mutation might contribute to the non-aflatoxigenicity of the strain. Aflatoxin B1 was detected in A. flavus IFM61226 extracts by mass spectrometry but not by ELISA (data not shown). The aflatoxin production of A. flavus IFM61226 was significantly lower than that of A. flavus NRRL3357, suggesting that some factors inside or outside of the A. flavus IFM61226 cluster affect production.
Aflatoxin production was not detected in the studied strains, except in A. flavus IFM61226. This result reflects the prevalence of non-aflatoxigenic A. flavus in the environment in Japan. Aspergillus flavus IFM61226 possesses an aflatoxin biosynthetic cluster sequence highly similar to that of A. flavus NRRL3357. Aspergillus flavus IFM55053, IFM59894, and IFM60655 have lost a part of this cluster, as shown in Fig. 5. Although A. flavus IFM54693, IFM57535, IFM59975, IFM60519, and IFM61224 possess the entire cluster region, aflatoxin production was impaired in these strains. The clusters in these strains show high identity with A. oryzae RIB40 suggesting that the substitutions identical to A. oryzae RIB40 are the cause of the loss of aflatoxin production in these strains.
3.5. RIB40-type sesquiterpene cluster is prevalent among A. flavus clinical isolates
Next, we focussed on the sesquiterpene cluster described by Gibbons et al.30 This cluster differed markedly between A. oryzae RIB40 and A. flavus NRRL3357.30 All A. oryzae strains and three of eight A. flavus strains are reported to carry the ‘9-gene cluster’ allele.30 In contrast, other A. flavus strains, including NRRL3357, contain the ‘6-gene cluster’ allele. The clusters of the ten clinical isolates investigated in this study were divided into 3 groups. Seven of the ten strains maintained a 9-gene cluster similar to that of A. oryzae RIB40 (Fig. 6). No 6-gene clusters were found among the ten A. flavus strains. Aspergillus flavus IFM59975 retained the longest cluster among the studied clinical isolates. In comparison with A. flavus IFM59975, the A. flavus IFM55053 and IFM58503 clusters lacked the region containing AFL2G_05186, AFL2G_05187, and AFL2G_05186 genes (Fig. 7). These three strains retained a unique region in the cluster. Analysis using Augustus (http://bioinf.uni-greifswald.de/augustus/(9 March 2018, date last accessed)) predicted a large gene in the unique region (Supplementary Fig. S2). Preliminary transcriptomic analysis of A. flavus IFM58503 showed that the region was transcribed to mRNA (Supplementary Fig. S3). Three protein sequences of those predicted genes are highly conserved (Supplementary Fig. S4). BLAST homology analysis (Supplementary Fig. S4) identified an ankyrin repeat protein in A. parasiticus (accession number: KJK61165) and a hypothetical protein in Aspergillus bombycis (accession number: OHM48268).
Figure 6.

Phylogenetic analysis of sesquiterpene biosynthesis gene clusters in 13 nucleotide sequences containing a total of 14,081 positions. Evolutionary relationship was inferred using the neighbour-joining method.38 An optimal tree with the sum of branch length = 0.10317173 is shown. The tree is drawn to scale with bootstrap values per 1,000 replicates shown at the branches.39 The evolutionary distances were computed using the Tamura–Nei method.40 The rate variation among sites was modelled with a γ distribution (shape parameter = 1). Evolutionary analysis was conducted as in MEGA7.17
Figure 7.

Comparison of sesquiterpene biosynthesis gene clusters of A. oryzae RIB40, A. flavus IFM55053, IFM58503, IFM59975, and NRRL3357. The unique regions shared by IFM55053, IFM58503, and IFM59975 are hatched.
Seven of the ten isolates were classified as carrying a 9-gene cluster allele comparable to that of A. oryzae RIB40. Interestingly, three A. flavus strains possessing the 9-gene cluster allele, SRRC1357, SRRC2112, and SRRC2524 described by Gibbons et al.30 were of Eurasian origin. Together with their data, our findings suggest that the 9-gene cluster, but not the 6-gene cluster, are prevalent in Eurasia and Japan. Three strains share a unique region that encodes a hypothetical protein homologous to an Ankyrin repeat protein. The products of these clusters remain unknown. The cluster that includes a unique gene is expected to produce a novel sesquiterpene. Further investigation is warranted to identify the product.
3.6. Intergenic region between ustT and ustR is conserved in all studied clinical strains
The ustiloxin biosynthesis gene cluster was found in all clinical A. flavus isolates. Ustiloxin, originally found as a secondary metabolite of Ustilaginoidea virens, inhibits microtubule assembly.31,32 Umemura et al. have identified a gene cluster responsible for the synthesis of ustiloxin B in A. flavus.33,34 As shown previously,33 the intergenic region between ustT and ustR is absent in A. oryzae RIB40, which leads faults in ustiloxin B production. Our sequence data indicate that the intergenic region is retained in all strains. Transcriptomic analysis of A. flavus IFM58503, a strain confirmed by mass spectrometry to be involved in ustiloxin B production, indicated that ustR transcription starts about ∼200 bp upstream of the start codon (data not shown). This region is missing in the A. oryzae RIB40 genome, suggesting that the absence of ustiloxin B production in this strain is induced during domestication and causes the fault in ustR transcription.
3.7. Other clusters and regions showing important differences between A. flavus NRRL3357 and A. oryzae RIB40
Nine regions with clear differences between A. oryzae RIB40 and A. flavus NRRL3357 are listed in Table 3. AFL2G_11806 and AFL2G_04006 are NRPS in cluster #34 and PKS in cluster #52, respectively. These genes are found in neither the ten clinical isolates nor A. oryzae RIB40. A ∼23 kb region containing cluster #34 of A. flavus NRRL3357 was not found in the ten clinical isolates or in A. oryzae RIB40. Instead, A. oryzae RIB40 and the ten clinical isolates have a shared region. In cluster #52, a ∼13 kb region was found in neither the ten clinical isolates nor in A. oryzae RIB40. Cluster #49 is known as a homologue of the 10, 11-dehydrocurvularin biosynthetic cluster in A. terreus35 and was not found in A. oryzae RIB40. The cluster is located in the telomeric region of the left arm of chromosome 3 (Supplementary Fig. S5a). Two PKS genes, afcurS2 and afcurS1, are encoded by the cluster.35 All strains except for A. flavus IFM60655 retained these genetic regions (Supplementary Fig. S5b). Among these, the A. flavus IFM57535, IFM58503, and IFM61226 clusters were highly conserved compared with that of A. flavus NRRL3357 (Supplementary Fig. S5b). Half or more strains did not retain the A. flavus NRRL3357-unique region in four other clusters, except in cluster #75, as shown in Table 3. Additionally, ∼63 and ∼19 kb regions were retained in only one and two clinical strains, respectively. On the other hand, some A. oryzae RIB40-specific regions were found on the genome. A ∼25 kb region, including an NRPS AO090026000585, (cluster #61) was only found in A. flavus IFM54693. On chromosome 8 of A. oryzae RIB40, reads from each strain were mapped infrequently to a ∼300-kb region from AO090010000234 to AO090010000317, which was also not found in A. flavus NRRL3357. Those differences might become a factor of strain individuality.
Table 3.
Retention of each region in isolates
| NRPS AFL2G_11806 in cluster #34 ∼ 23 kb | PKS AFL2G_04006 in cluster #52 ∼ 13 kb | Cluster #49 > 30 kb | AFL2G_08888- AFL2G_08903 in cluster #62 ∼ 32 kb | AFL2G_09850 in cluster #72 ∼ 18 kb | AFL2G_09859 in cluster #72 ∼ 24 kb | NRPS AFL2G_12207 in cluster #75 ∼ 31 kb | AFL2G_05014- AFL2G_05031 ∼ 63 kb | AFL2G_06401- AFL2G_06406 ∼ 19 kb | |
|---|---|---|---|---|---|---|---|---|---|
| IFM54693 | |||||||||
| IFM55053 | |||||||||
| IFM57535 | |||||||||
| IFM58503 | |||||||||
| IFM59894 | |||||||||
| IFM59975 | |||||||||
| IFM60519 | |||||||||
| IFM60655 | |||||||||
| IFM61224 | |||||||||
| IFM61226 | |||||||||
| NRRL3357 | + | + | + | + | + | + | + | + | + |
| RIB40 | − | − | − | − | − | − | − | − | − |
| Retention of the region | 0/10 | 0/10 | 9/10 | 3/10 | 5/10 | 5/10 | 6/10 | 1/10 | 2/10 |
Grey and white cells indicate the retention and the absence of each region, respectively.
4. Conclusive remarks
This study provides the genome sequences of A. flavus isolated from clinical specimens in Japan. The phylogenetic analysis based on identified SNPs showed that five of the ten isolates were grouped with A. oryzae RIB40, while other 3 isolates with A. flavus NRRL3357. Two isolates, A. flavus IFM57535 and IFM60519, were placed between the two groups. To distinguish between A. flavus and A. oryzae is difficult by partial sequencing of regions such as ITS. As shown in the review by Frisvad36, A. oryzae is a domesticated strain of A. flavus. The fact that A. flavus IFM59975, IFM59894, and IFM61224 were grouped with A. oryzae RIB40 in phylogenetic analysis suggests that they are likely to be wild forms of A. oryzae in Japan and perhaps the causative agent of aspergillosis.
Specimens isolated through the oral cavity, such as gingival and sputum samples, may possess the risk of contamination of oral fungi, because ‘koji’-based foods are commonly consumed in Japan and might result in contamination in oral specimens. As discussed previously, the presence of multiple copies of α-amylase is a common characteristic of A. oryzae24.oryzae.24 Although A. flavus IFM54693 and IFM60655 have previously been identified as A. flavus based on the ITS sequences, those strains might be reclassified as domesticated A. oryzae because of their possession of multiple α-amylase genes. The difference in sequence between the original and additional copies of α-amylase genes would be useful to discriminate between the wild and domesticated strains.
In summary, the genome data obtained in this study will be useful for further analysis of the genomes of clinical isolates of A. flavus and A. oryzae obtained from oversea and Japanese environments. The phylogenetic analysis based on SNPs might enable us to differentiate between A. flavus and A. oryzae.
Supplementary Material
Acknowledgements
We would like to thank the editor for his valuable suggestions to improve expressions in our manuscript. We would like to thank Daisuke Hagiwara, Myco Umemura, Akira Watanabe, and Takashi Yaguchi, for fruitful discussion.
Accession numbers
DRA006279, DRA006280, DRA006281, DRA006282, DRA006283, DRA006284, DRA006285, DRA006286, DRA006287, and DRA006288.
Funding
This study was supported by a Cooperative Research Grant of the Genome Research for BioResource, NODAI Genome Research Center, Tokyo University of Agriculture and Joint Usage/Research Program of Medical Mycology Research Center, Chiba University (16–16).
Conflict of interest
None declared.
References
- 1. Krishnan S., Manavathu E.K., Chandrasekar P.H.. 2009, Aspergillus flavus: an emerging non-fumigatus Aspergillus species of significance, Mycoses, 52, 206–22. [DOI] [PubMed] [Google Scholar]
- 2. Yu J. 2012, Current understanding on aflatoxin biosynthesis and future perspective in reducing aflatoxin contamination, Toxins (Basel), 4, 1024–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tashiro T., Izumikawa K., Tashiro M., et al. 2011, Diagnostic significance of Aspergillus species isolated from respiratory samples in an adult pneumology ward, Med . Mycol., 49, 581–7. [DOI] [PubMed] [Google Scholar]
- 4. Corrier D.E. 1991, Mycotoxicosis: mechanisms of immunosuppression, Vet . Immunol. Immunopathol., 30, 73–87. [DOI] [PubMed] [Google Scholar]
- 5. Jakab G.J., Hmieleski R.R., Zarba A., Hemenway D.R., Groopman J.D.. 1994, Respiratory aflatoxicosis: suppression of pulmonary and systemic host defenses in rats and mice, Toxicol . Appl. Pharmacol., 125, 198–205. [DOI] [PubMed] [Google Scholar]
- 6. Manabe M., Tsuruta O.. 1978, Geographical distribution of aflatoxin-producing fungi inhabiting in Southeast Asia, JARQ Japan Agric . Res. Q., 12, 224–7. [Google Scholar]
- 7. Nierman W.C., Yu J., Fedorova-Abrams N.D., et al. 2015, Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed, Genome Announc., 3, e00168–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Machida M., Asai K., Sano M., et al. 2005, Genome sequencing and analysis of Aspergillus oryzae, Nature, 438, 1157–61. [DOI] [PubMed] [Google Scholar]
- 9. Toyotome T., Adachi Y., Watanabe A., Ochiai E., Ohno N., Kamei K.. 2008, Activator protein 1 is triggered by Aspergillus fumigatus beta-glucans surface-exposed during specific growth stages, Microb . Pathog., 44, 141–50. [DOI] [PubMed] [Google Scholar]
- 10. Aronesty E. 2013, Comparison of sequencing utility programs, Open Bioinformatics J., 7, 1–8. [Google Scholar]
- 11. Langmead B., Trapnell C., Pop M., Salzberg S.L.. 2009, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome, Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sugawara H., Ohyama A., Mori H., Kurokawa K.. 2009, Microbial Genome Annotation Pipeline (MiGAP) for diverse users. In: 20th International Conference Genome Informatics, S001–1–2. [Google Scholar]
- 13. Li H. 2011, A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data, Bioinformatics, 27, 2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris S.R., Feil E.J., Holden M.T.G., et al. 2010, Evolution of MRSA during hospital transmission and intercontinental spread, Science ,327, 469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kusuya Y., Takahashi-Nakaguchi A., Takahashi H., Yaguchi T.. 2015, Draft genome sequence of the pathogenic filamentous fungus Aspergillus udagawae strain IFM 46973T, Genome Announc., 3, e00834–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar S., Stecher G., Tamura K.. 2016, MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets, Mol . Biol. Evol., 33, msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. 2013, Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration, Brief Bioinform., 14, 178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson J.T., Thorvaldsdóttir H., Winckler W., et al. 2011, Integrative genomics viewer, Nat . Biotechnol., 29, 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. An J., Lai J., Sajjanhar A., Batra J., Wang C., Nelson C.C.. 2015, J-Circos: an interactive Circos plotter, Bioinformatics, 31, 1463–5. [DOI] [PubMed] [Google Scholar]
- 21. Carver T.J., Rutherford K.M., Berriman M., Rajandream M.-A., Barrell B.G., Parkhill J.. 2005, ACT: the Artemis Comparison Tool, Bioinformatics ,21, 3422–3. [DOI] [PubMed] [Google Scholar]
- 22. Samson R.A., Visagie C.M., Houbraken J., et al. 2014, Phylogeny, identification and nomenclature of the genus Aspergillus, Stud . Mycol., 78, 141–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okoth S., Boevre De, Vidal M., A., et al. 2018, Genetic and toxigenic variability within Aspergillus flavus population isolated from maize in two diverse environments in Kenya, Front . Microbiol, 9, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hunter A.J., Jin B., Kelly J.M.. 2011, Independent duplications of α-amylase in different strains of Aspergillus oryzae, Fungal Genet . Biol. ,48, 438–44. [DOI] [PubMed] [Google Scholar]
- 25. Tominaga M., Lee Y., Hayashi R., et al. 2006, Molecular analysis of an inactive aflatoxin biosynthesis gene cluster in Aspergillus oryzae RIB strains molecular analysis of an inactive aflatoxin biosynthesis gene cluster in Aspergillus oryzae RIB strains, Appl . Environ. Microbiol., 72, 484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kiyota T., Hamada R., Sakamoto K., Iwashita K., Yamada O., Mikami S.. 2011, Aflatoxin non-productivity of Aspergillus oryzae caused by loss of function in the aflJ gene product, J . Biosci. Bioeng., 111, 512–7. [DOI] [PubMed] [Google Scholar]
- 27. Ehrlich K.C., Chang P.K., Yu J., Cotty P.J.. 2004, Aflatoxin biosynthesis cluster gene cypA is required for G aflatoxin formation, Appl . Environ. Microbiol., 70, 6518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chang P.-K. 2003, The Aspergillus parasiticus protein AFLJ interacts with the aflatoxin pathway-specific regulator AFLR, Mol . Genet. Genomics, 268, 711–9. [DOI] [PubMed] [Google Scholar]
- 29. Meyers D.M., Obrian G., Du W.L., Bhatnagar D., Payne G.A.. 1998, Characterization of aflJ, a gene required for conversion of pathway intermediates to aflatoxin, Appl . Environ. Microbiol., 64, 3713–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibbons J.G., Salichos L., Slot J.C., et al. 2012, The evolutionary imprint of domestication on genome variation and function of the filamentous fungus Aspergillus oryzae, Curr . Biol. ,22, 1403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koiso Y., Li Y., Iwasaki S., et al. 1994, Ustiloxins, antimitotic cyclic peptides from false smut balls on rice panicles caused by Ustilaginoidea virens, J . Antibiot., 47, 765–73. [DOI] [PubMed] [Google Scholar]
- 32. Koiso Y., Natori M., Iwasaki S., et al. 1992, Ustiloxin: a phytotoxin and a mycotoxin from false smuth balls on rice panicles, Tetrahedron Lett., 33, 4157–60. [Google Scholar]
- 33. Umemura M., Nagano N., Koike H., et al. 2014, Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus, Fungal Genet . Biol., 68, 23–30. [DOI] [PubMed] [Google Scholar]
- 34. Umemura M., Koike H., Nagano N., et al. 2013, MIDDAS-M: motif-independent de novo detection of secondary metabolite gene clusters through the integration of genome sequencing and transcriptome data, PLoS One, 8, e84028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu Y., Espinosa-Artiles P., Schubert V., et al. 2013, Characterization of the biosynthetic genes for 10, 11- dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus, Appl . Environ. Microbiol., 79, 2038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frisvad J.C., Hubka V., Ezekiel C.N., et al. 2019, Taxonomy of Aspergillus section Flavi and their production of aflatoxins, ochratoxins and other mycotoxins, Stud. Mycol., 93, 1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kimura M. 1980, A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences, J . Mol. Evol., 16, 111–20. [DOI] [PubMed] [Google Scholar]
- 38. Saitou N., Nei M.. 1987, The neighbor-joining method: a new method for reconstructing phylogenetic trees, Mol . Biol. Evol., 4, 406–25. [DOI] [PubMed] [Google Scholar]
- 39. Felsenstein J. 1985, Confidence limits on phylogenies: an approach using the bootstrap, Evolution, 39, 783–91. [DOI] [PubMed] [Google Scholar]
- 40. Tamura K., Nei M.. 1993, Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees, Mol . Biol. Evol., 10, 512–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.