

Abstract
Type IA topoisomerases cleave single-stranded DNA and relieve negative supercoils in discrete steps corresponding to the passage of the intact DNA strand through the cleaved strand. Although it is assumed type IA topoisomerases accomplish this strand passage via a protein-mediated DNA gate, opening of this gate has never been observed. We developed a single-molecule assay to directly measure gate opening of the E. coli type IA topoisomerases I and III. We found that following cleavage of single-stranded DNA, the protein gate opens by as much as 6.6 nm and can close against forces in excess of 16 pN. Key differences in the cleavage, ligation and gate dynamics of these two enzymes provide insights into their different cellular functions. The single-molecule results are broadly consistent with conformational changes obtained from molecular dynamics simulations. These results allow us to develop a mechanistic model of type IA topoisomerase-ssDNA interactions.
Introduction
DNA topology in the cell is controlled by DNA-remodeling enzymes known as topoisomerases1,2. Topoisomerases are divided into two classes: type II topoisomerases cleave both strands of duplex DNA (dsDNA) and pass a second duplex through the transient break3, whereas type I topoisomerases cleave single-stranded DNA (ssDNA) and either permit rotation of the intact strand (Type IB)4,5 or pass the intact strand through the transient nick (Type IA)6,7. Type IA topoisomerases are ubiquitous and essential enzymes that include the bacterial topoisomerases I8 and III9, the archaeal reverse gyrase10, and the eukaryotic topoisomerases IIIɑ11,12 and IIIβ13. In prokaryotes type IA topoisomerases are involved in relaxation of excess negative supercoils14 and resolution of replication and recombination intermediates15,16. In eukaryotes, these enzymes are involved in accurate chromosome segregation during mitosis12,17,18, non-crossover resolution of double Holliday junctions19,20 and promoting expression from long mRNAs21,22.
Type IA topoisomerases bind and cleave ssDNA then pass a second single strand or duplex through the transient break7. This strand passage mechanism has long been assumed to involve a protein-mediated DNA gate23–25. In the gate model, ssDNA is bound across domains I and IV of the topoisomerase and is cleaved by the active site tyrosine in domain III (Fig. 1a–b)26. Domain III then moves away from domain I, creating an opening through which a second strand of DNA can pass. The enzyme must then return to the closed state, religate the DNA backbone, and open again to release the trapped DNA (Fig. 1b). Although structural,25,26 biochemical,6,27 computational28, and single-molecule experiments7 support this model, direct evidence for the gate mechanism has proved elusive. No structures or other definitive evidence of an open state exist, leaving open the possibility that the strand passage relies on an alternative conformational change29.
Figure 1.
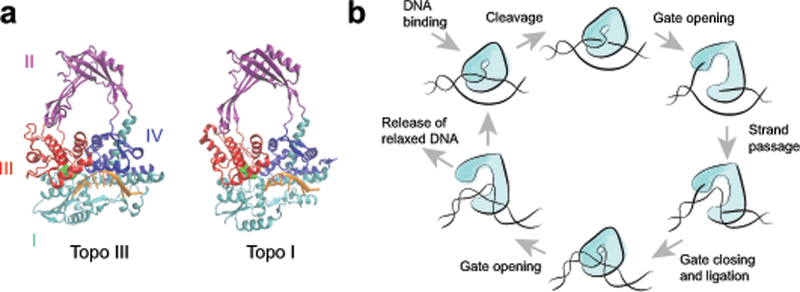
Proposed mechanism of type IA topoisomerase activity. (a) Crystal structures of the E. coli type IA topoisomerases, topo III (pdbid: I17D)48 and topo I (pdbid: 1MW8)32. DNA bound to the active site is shown in orange and the catalytic tyrosines are highlighted in green. For simplicity, the C-terminal zinc finger domains of topo I are not shown. (b) Proposed model of type IA topoisomerase protein-mediated DNA gate mechanism for strand passage.
Here we describe the direct observation of type IA topoisomerase gate opening dynamics of two E. coli type IA topoisomerases, topo I and topo III, interacting with ssDNA (Fig. 2a and 3a). By analyzing the force- and magnesium- dependent kinetics of the gate opening and closing, we delineate the catalytic cycle of type IA topoisomerases. These results reveal differences in the gate dynamics of topo I and topo III that may underlie their distinct biochemical activities. Molecular dynamics simulations reveal the potential structure of the open conformation and confirm that the experimentally observed extension changes are consistent with opening of the gate between domains I and III. Our results provide a detailed description of the kinetics of type IA topoisomerase-ssDNA interactions and establish an experimental paradigm to study the conformational dynamics of type IA topoisomerases.
Figure 2.
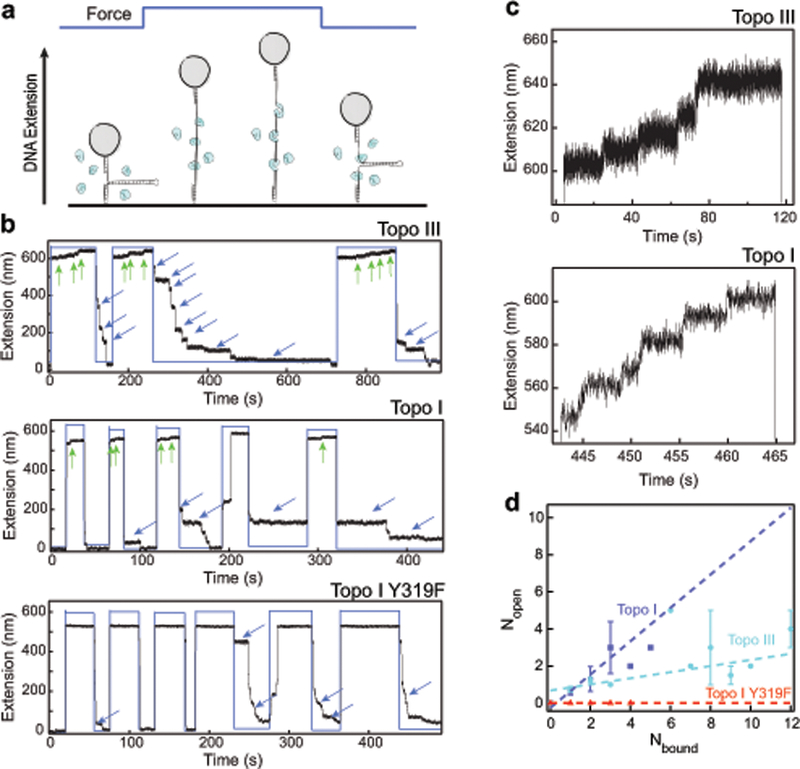
Magnetic tweezers measurements of topoisomerase binding and gate opening. (a) Experimental design (not to scale). Hairpin DNA was unfolded at high force in the presence of topoisomerase. Gate opening was detected as an increase in DNA extension beyond the fully extended state. Binding was detected as pauses in refolding upon lowering the force. (b) Example experimental traces for topo III, topo I, and topo I Y319F. Black lines are DNA extension. Blue lines represent the force, alternating between ~9 pN and ~22–24 pN. Green arrows indicate gate opening events. Blue arrows indicate binding. Differences in the baseline extension of the unfolded state are due to variation in the applied force. (c) Expanded examples of gate opening events for topo III and topo I. Extension increases were approximately 6.6 ± 0.7 nm for topo III and 6.6 ± 1.0 nm for topo I, F ~ 22–24 pN (Supplementary Fig. 1). Note the difference in time scales. (d) Correlation between gate opening and topoisomerase binding events. The average number of opening events (Nopen) for a given number of bound proteins (Nbound) is plotted. Topo III is shown in cyan (ntethers = 4, nunfolding-refolding cycles = 58), topo I in dark blue (ntethers = 5, ncycles= 25), and topo I catalytic mutant Y319F in red (ntethers = 5, ncycles = 18). Error bars are standard deviations. Dashed lines indicate linear fits to the data with slope 0.16 ± 0.05 for topo III, 0.9 ± 0.5 for topo I, and 0.0 for topo I Y319F.
Figure 3.
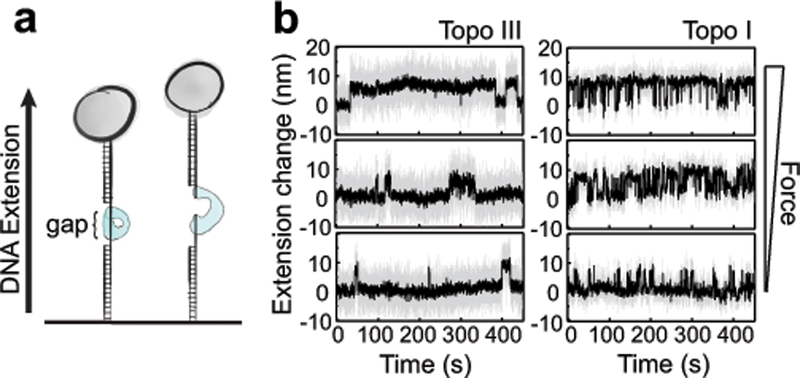
Force dependence of gate opening dynamics. (a) Experimental design (not to scale). Duplex DNA with a 37 nt gap was held at constant force and gate opening/closing detected as transient increases in extension of ~ 6 nm. (b) Example traces showing extension change of single DNA molecules as a function of time for topo III and topo I at 16, 14, and 12 pN. Gray lines are raw data. Black lines are smoothed data using a 4th order Savitzky-Golay algorithm over 51 points.
Results
Cleavage by type IA topoisomerases increases ssDNA extension.
We first measured ssDNA binding by type IA topoisomerases using a magnetic tweezers30 hairpin rezipping assay (Fig. 2a)31. A 537 bp hairpin was mechanically unfolded by applying high force (~22–24 pN) in the presence of 500 pM E. coli topo III or topo I (Fig. 2b). The DNA was held at high force for 30–120 s after which the force was reduced to ~ 9pN, allowing the hairpin to refold. Binding of topoisomerase to the ssDNA was detected as pauses in hairpin refolding (Fig. 2b, blue arrows). Dissociation of the topoisomerase resulted in further refolding of the hairpin. Pauses in refolding of the hairpin occurred at multiple distinct locations, indicating stable binding of multiple proteins.
At high force, we observed topoisomerase-dependent increases in the bead height beyond the extension of the fully unfolded hairpin (Fig. 2b and Supplementary Fig. 1a–b). These extension increases were on average 6.6 ± 0.7 nm for topo III and 6.6 ± 1.0 nm for topo I (Fig. 2c, Supplementary Fig. 1c–f). We interpreted these increases in DNA extension as cleavage of the DNA by the topoisomerase followed by a conformational change that separated the cleaved ends of the DNA. To verify that the extension increases were cleavage-dependent, we repeated the hairpin experiments with a cleavage deficient mutant of topo I, Y319F (Fig. 2b)32. We observed refolding pauses characteristic of protein binding (blue arrows), but no discrete increases in extension at high force. Protein-mediated cleavage associated with increases in extension was confirmed by rapid loss of DNA tethers that had lengthened in the presence of topoisomerase following denaturation of the protein by the addition of SDS33 (Supplementary Note 1, Supplementary Fig. 1g).
Comparing the number of ~6.6 nm extension increases (Nopen) with the number of binding events (Nbound) for each unfolding-refolding cycle indicates that they are linearly related (Fig. 2d). Interestingly, nearly all the bound topo I molecules cleaved and opened the DNA, whereas only a small fraction of topo III molecules did. We observed no opening events in the absence of bound protein. Based on these results, we concluded that the 6.6 nm extension increases correspond to the opening of a topoisomerase-mediated DNA gate.
Force dependence of gate dynamics
To accurately characterize the conformational changes and kinetics of the gate dynamics, we developed an assay that allowed us to measure gate dynamics of a single topoisomerase bound to ssDNA as function of force. Force selectively affects the kinetic transitions involving motion by altering the underlying free energy profile34. For these experiments, a 2.5 kb dsDNA with a 37 nt gap was attached to a coverslip on one end and a magnetic bead on the other (Fig. 3a).
Opening and closing of the topoisomerase gate was observed as transient increases in the DNA extension (Fig. 3b) (ntethers = 8 for topo III, ntethers = 7 for topo I). The average size of the extension change was 5.5 ± 0.4 nm for topo III, independent of the applied force (8 to 16 pN) (Supplementary Fig. 2, Supplementary Table 1). For topo I, the extension change increased slightly with applied force (12 to 18 pN) (Supplementary Fig. 2, Supplementary Table 1), with an average value of 5.9 ± 0.6 nm. Surprisingly, the topoisomerases were able to close the gate and religate DNA against forces of 18 pN.
To obtain kinetic states from the extension data, we used vbFRET35 (Fig. 4), which applies an unbounded hidden Markov model to determine the number of states in single-molecule time traces. The program reliably found two extension states for both topo III and topo I. However, analysis of the lifetime distributions revealed a third state; in addition to the open state and a long-lived closed state, there is a short-lived closed state (Fig. 4a–b).
Figure 4.
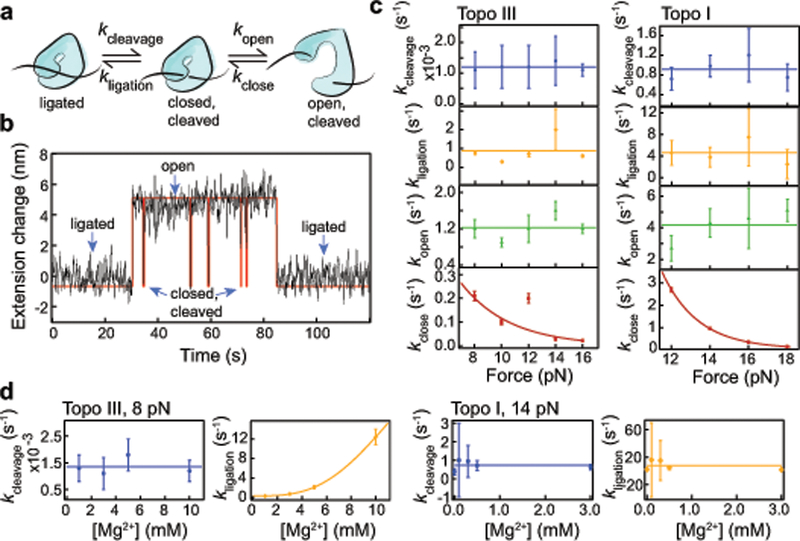
Three-state kinetics of topoisomerase IA gate dynamics. (a) Three-state kinetic model. (b) Example of a single kinetic cycle of topo III from cleavage to transient opening and closing of the gate, followed by religation. Black line is smoothed data, red line is the HMM fit from vbFRET. (c) Force dependent kinetic analysis of kcleavage (blue), kligation (orange), kopen (green), and kclose (red), ntethers = 8 for topo III, ntethers = 7 for topo I. Values were calculated from exponential fits of the lifetimes of each state (Supplementary Fig. 3–4). Error bars represent standard deviations of the fit coefficients. For kcleavage, kligation, and kopen, lines represent average values. kclose values were fit with k = k0exp(−FΔx/kBT). The fit returned Δx and k0 values of 1.10 ± 0.04 nm and 1.8 ± 0.3 s−1 respectively, for topo III and 2.2 ± 0.1 nm and 1642 ± 549 s−1 for topo I. (d) Magnesium concentration dependence of ligation and cleavage rates for topo III at 8 pN force (ntethers = 3, 8, 3, and 2 for 1, 3, 5, and 10 mM Mg2+, respectively) and topo I at 12 pN force (ntethers = 6, 4, 3, 5, and 2 for and 0, 0.1, 0.3, 0.5 and 3 mM Mg2+). The topo III magnesium-dependent ligation rate was fit with a sigmoid with a Km of 8.3 ± 1.2 mM Mg2+ and a Vmax of 17 ± 5 s−1.
This transient closed state is consistent with a three-state model in which the protein-ssDNA complex is in a ligated state (L), a closed cleaved state (C), or an open cleaved state (O) (Fig. 4b), resulting in the following kinetic scheme;
In this model, the short-lived closed states correspond to state C, which the protein may visit multiple times before religating the DNA and returning to the longer-lived closed state L. Although we have assigned three states to our data, it is likely that there are additional states that cannot be directly observed.
The gate kinetics as a function of force for both topo III and topo I are plotted in Fig. 4c. For topo III, the lifetimes of the long- and short-lived closed states are well-separated and the individual lifetime distributions were fit to single exponentials to determine kopen and kcleavage (Supplementary Fig. 3a–b). For topo I, the short and long closed-state lifetimes were comparable, and the closed state lifetime distribution was best fit with a double exponential (Supplementary Fig. 3c). The distribution of open state lifetimes was fit to an exponential to determine kclose (Supplementary Fig. 4a, 4b).
To estimate the ligation rate, we consider the kinetic competition between ligation and opening from the closed state C. The probability of opening from state C depends on the rates of these two competing pathways:
We determine Popen from the number of opening events (short-lived closed states) relative to the total number of closed state events.
For both topoisomerases, kcleavage and kligation were insensitive to the applied force (Fig. 4c, Supplementary Table 2). Surprisingly, the opening rates were also unaffected by the applied force (Fig. 4c, Supplementary Table 2). This lack of force dependence indicates that the opening rate reflects a rate-limiting step that precedes the mechanical opening of the gate. The closing rates exhibited an exponential force dependence (Fig. 4c), k(F) ≈ k0exp(−F∆x/kBT), where k0 is the zero-load closing rate, ∆x is the distance from the open state to the transition state, and kBT is the thermal energy. The distances to the transition state, ∆xclose, obtained from exponential fits are 1.10 ± 0.01 nm for topo III and 2.2 ± 0.1 nm for topo I. The zero-load closing rates are 1.8 ± 0.3 s−1 for topo III and 1642 ± 549 s−1 for topo I (Table 1).
Table 1.
Equilibrium rates and barriers associated with Figure 6. Zero-load values for kcleavage, kligation, and the rate limiting conformation changes associated with gate opening kopen, and closing, kclose. Δxclose values calculated from experiments and simulations are also shown. In the case of Topo III, this corresponds to Δxclose**, the distance between the fully open state and the barrier associated with formation of an open-state stabilizing salt bridge.
| kcleavage (s−1) | kligation (s−1) | kopen (s−1) | kclose (s−1) | Δxexp (nm) | Δxsim (nm) | |
|---|---|---|---|---|---|---|
| Topo III | 0.0012 ± 0.0004 | 1.5 ± 0.7 | 1.07 ± 0.17 | 1.8 ± 0.3 | 1.10 ± 0.04 | 1.3 ± 0.4 |
| Topo I | 0.9 ± 0.2 | 4.6 ± 1.0 | 4.1 ± 1.0 | 1642 ± 549 | 2.2 ± 0.1 | 2.9 ± 0.4 |
The lifetime of the open state is several orders of magnitude longer for topo III than topo I (Fig. 4c, Table 1). Biochemical and single-molecule studies demonstrate that topo I is generally more effective at relaxing negatively supercoiled DNA than topo III36, whereas topo III is more effective at unlinking DNA catenanes37,38. Relaxation of negative supercoils requires passage of a single DNA strand through the protein-mediated DNA gate. In contrast, decatenation requires passage of dsDNA. The more stable open state of topo III could facilitate capture of duplex DNA required for decatenation, whereas the faster dynamics of topo I are compatible with capturing a local ssDNA segment. The fast closing rate also explains the difficulty in capturing the open conformation of topo I; only by slowing gate closing through the application of significant opposing forces could the open state configuration be observed.
The estimated rate of a full catalytic cycle based on extrapolated zero force kinetics of the individual steps for topo I is 0.64 ± 0.38 s−1, which is comparable to previous relaxation rate measurements (Table 1, Supplementary Table 3). Conversely the estimated rate of the topo III catalytic cycle is 0.0011 ± 0.0004 s−1. This unexpectedly slow rate, which is dominated by the cleavage rate, is much slower than measured relaxation rates. The slow cleavage rate is consistent with previously observed long lag times (up to ~129 s for negatively supercoiled DNA) between relaxation events for topo III36,38. However, these experiments also demonstrated much faster processive relaxation by topo III (between ~19 s−1 and 129 s−1, Table 1, Supplementary Table 3). One possible explanation for the discrepancy is the absence of a second DNA strand in our experiments. If topo III cleavage and ligation rates are stimulated by the presence of a second ss- or ds-DNA strand, then passage of an initial strand could lead to the fast bursts of processive activity previously observed36. The requirement of a second strand of DNA would align with kinetic studies of S. solfatarius topo III in which ligation of cleaved ssDNA was facilitated by annealing of a complementary strand39.
Magnesium dependent kinetics
Although magnesium is necessary for the activity of type IA topoisomerases, the distinct roles that magnesium plays in cleavage and religation remain under debate. Previous studies have shown that increasing magnesium concentration increases the cleavage product, but magnesium is not required for cleavage40. Religation requires magnesium, but the magnesium dependence of the religation rate is unknown 41,42. We determined the effect of magnesium on the cleavage and religation rates by measuring the gate kinetics as a function of magnesium concentration (Fig 4d, Supplementary Table 4).
The cleavage rate of both proteins was independent of the magnesium concentration (Fig 4d, Supplementary Table 4). The ligation rate of topo III depended strongly on the magnesium concentration (Fig. 4d, Supplementary Table 4), exhibiting a sigmoidal relationship, with a Km of 8.3 ± 1.2 mM Mg2+ and a Vmax of 17 ± 4 s−1. The fact that the relationship is sigmoidal rather than hyperbolic is consistent with biochemical evidence suggesting that the catalytic cycle depends on two coordinating magnesium ions43,44. In contrast, the ligation rate of topo I showed no magnesium dependence (Fig. 4d, Supplementary Table 4). The differences in magnesium dependence of religation may reflect differences in the active sites of the two topoisomerases. Topo III has a strictly conserved Lysine residue (K8) that interacts with the putative scissile phosphate in the active site44, and as a result may not bind magnesium as tightly as topo I. Based on a recently determined structure of M. tuberculosis topo I with bound ssDNA and Mg2+, it was proposed that the observed Mg2+ may play a role in positioning the 3′-hydroxyl group relative to the phosphotyrosine linkage for DNA religation45. The potential role of a second Mg2+ remains to be determined.
For topo III we observed a decrease in binding affinity and reduced religation below 1 mM Mg2+. Topo I showed decreased binding below 0.3 mM, but could cleave and religate DNA, even without magnesium in the buffer. Only when EDTA was added to the reaction to chelate trace amounts of metal was ligation by topo I suppressed (Supplementary Fig. 5). These results suggest that magnesium-dependent DNA binding may contribute to observed effects of magnesium on the ratio of cleavage to religation in ensemble measurements.
Simulations of gate dynamics
Our experiments indicate a separation between the cleaved DNA ends of 5.5 – 6.6 nm, which is unexpectedly large. Single-stranded DNA needs a gap of only ~1 nm to pass through the break, whereas duplex DNA needs ~2 nm. Given the relatively high forces used in our experiments, it is possible that the enzyme normally undergoes a much smaller conformational change in the absence of force on the DNA. To test this possibility, we conducted umbrella sampling46,47 molecular dynamics simulations of topo III bound to ssDNA (Fig. 5, Supplementary Video 1, Supplementary Fig. 6a–d)48. In these simulations, the distance between the center of mass of domain III (shown in red in Fig. 5a) and the center of mass of domain IV (shown in blue) was restrained using a harmonic potential, , where r is the restrained distance, and r0 is the center of the potential well for a given window. To open the gate, r0 was gradually increased over a series of windows. Domains III and IV must move away from each other for the gate to open, but the direction of this motion may not be aligned with our experimentally imposed force and displacement axis. Restraining the distance between the domains rather than pulling along a vector allows the system to follow an energetically unbiased pathway.
Figure 5.
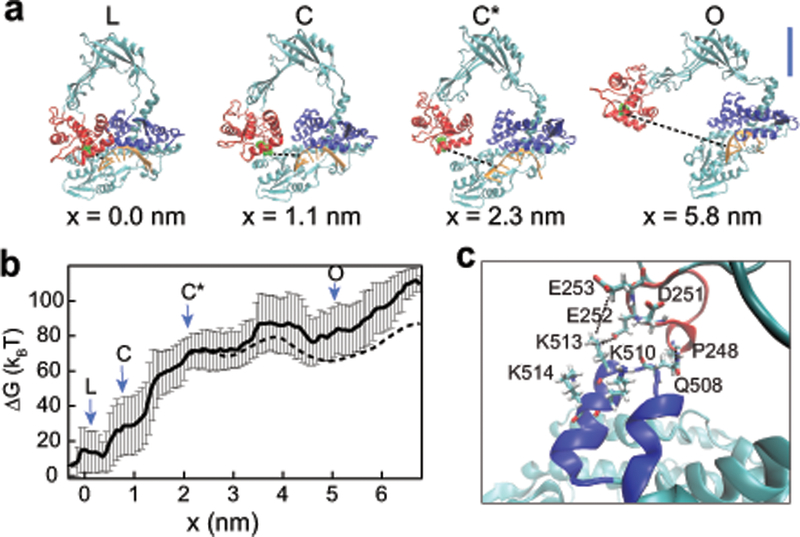
Molecular dynamics simulations of topo III gate opening. (a) Topo III structures from molecular dynamics umbrella sampling simulations. Representative structures are shown for the closed state (L), the state in which contacts between domains III and IV are broken (C), the state in which contacts between domains III and I are broken (C*), and the fully open structure (O). x (dashed black line) is the change in distance between the oxygen of the catalytic tyrosine, Y328, and the cleavage site of the DNA backbone. Blue scale bar represents 2 nm. (b) Relative free energy change as a function of gate opening (x). Arrows indicate the location of the states represented in (a). The distance between the closed state and C*, ∆xopen is 2.2 ± 0.3 nm. Δxclose is 2.9 ± 0.4 nm. The dashed line is the estimated free energy for an applied force of 22 pN under the assumption that the transition from L to C* is independent of force (Supplementary Fig. 6). (c) Structure of the decatenation latch. The decatenation loop is shown in blue and the acidic loop in red. Key residues in forming the latch are highlighted. Dashed lines indicate specific contacts. K513 forms contacts with both E253 and E252, while Q508 interacts with the backbone of P248. Two other basic residues in the decatenation loop, K510 and K514, and a third acidic residue in the acidic loop, D251, are also involved in loop contacts at other points in the simulations (Supplementary Video 2).
The simulations reveal that domain III initially moves along domain I rather than outward, first breaking contacts with domain IV (Fig. 5a, structure C) before breaking contacts with domain I (structure C*). As a result of this sliding motion, the protein gate is not large enough to accommodate ssDNA until the distance between the cleavage site and the catalytic tyrosine exceeds 3.8 nm. For dsDNA to pass through the DNA break, a separation of ~5.8 nm was required (Fig. 5a, structure O), in close agreement with our experiments.
The umbrella sampling method can be used to calculate equilibrium free energy profiles along any reaction coordinate, provided the conformational space of the coordinate is fully sampled during the simulations49. In this way, we could estimate the free energy profile along the experimental coordinate defined by the applied force (Fig. 5b, Supplementary Note 1, Supplementary Figure 6a–c). The initial barrier to opening is defined by breaking of contacts between domain III and domains IV and I. Once these contacts are broken, the free energy profile is relatively flat except for a small barrier between ~3.5 and 4.5 nm that corresponds to interactions between the decatenation loop of topo III and a second smaller loop on domain II. Both loops are absent in topo I (Fig. 5c, Supplementary Video 2). The decatenation loop has a cluster of conserved basic residues50, which form contacts with acidic residues in the second loop. The interaction presumably stabilizes the open state and suggests a structural role for the decatenation loop, which is required for the unlinking activity of topo III50. Stabilization of the open state explains why the closing rate is slower for topo III in our experiments (Fig. 4c, Supplementary Table 2). The distance of this barrier from the open state well is 1.3 ± 0.4 nm, which agrees with the experimental topo III Δxclose value of 1.10 ± 0.01 nm. The experimental Δxclose for topo I, 2.2 ± 0.1 nm, agrees with the distance between the open state well and state C*, 2.9 ± 0.4 nm (Fig. 5b).
Mechanistic Model of Type IA topoisomerase-ssDNA interactions
Whereas the three-state model is consistent with the number of observed states, analysis of the force-dependent rates, combined with simulation results, suggest that there is at least one additional kinetic state in the gate dynamics pathway. The lack of force dependence of the experimental opening rate (Fig. 4c, Supplementary Table 2) indicates that the opening rate is dominated by a rate limiting step that precedes the mechanical opening of the gate. This additional rate limiting kinetic transition is supported by the fact that the initial barrier associated with breaking contacts between domains III and I in the simulations is too high to be readily thermally accessible. This high energy barrier suggests that opening is limited by a conformational change that lowers the transition energy barrier but that is too slow to be sampled in our simulations.
To accommodate these results, we extended the model of topoisomerase-ssDNA interactions (Fig. 6, Table 1) to include an additional state, C* that is inferred but not directly observed.
Cleavage and ligation rates in this scenario correspond to the chemical reactions, but the opening and closing rates each represent a convolution of two rates from C to O via C* and vice versa. Accordingly, the measured kopen is dominated by the rate of a conformational change (kc-c*) that allows domain III to escape contacts with domain I. The actual mechanical opening rate (kopen*) is force dependent but too fast to observe. Similarly, kc*-c is assumed faster than kclose* and kopen*, such that the measured closing rate is dominated by kclose*, and the equilibrium between C* and C is biased towards state C from which religation can occur. In this scenario, the gate can undergo multiple transitions between states C and C* before opening, in which case kopen would represent the escape rate, rather than kc-c*. The measured force dependence of the closing rate supports the hypothesis that the transition from state C* to C is fast (much faster than the fastest closing rate of 2.7 ± 0.1 s−1). Furthermore, the lack of force dependence of the religation and opening rates is consistent with the hypothesis that the rate kc*-c exceeds all other rates.
Figure 6.
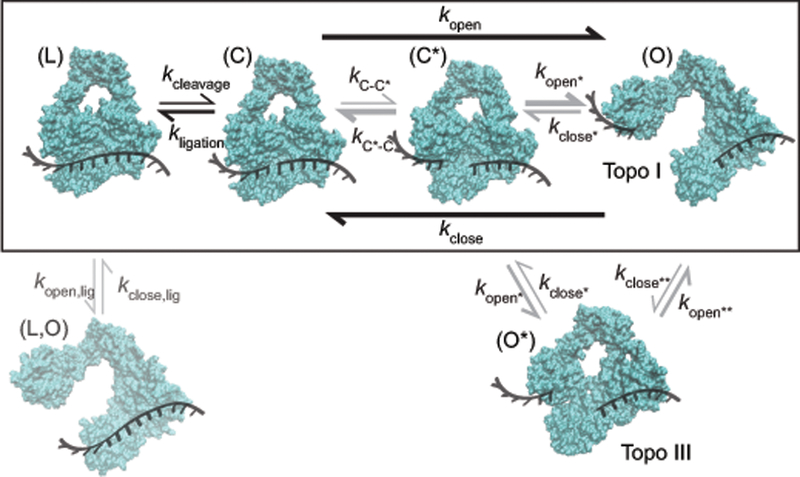
Kinetic scheme of type IA topoisomerase gate dynamics. Structures of topo III from MD simulations representing protein-ssDNA states: (L,O) open gate with intact DNA (invisible in our experiments); (L) closed gate intact (ligated) DNA; (C) closed gate with cleaved DNA; (C*) closed gate with cleaved DNA after a rate-limiting conformational change in which domain I moves relative to domain III; and (O) open gate with cleaved and separated DNA. The black box designates states common to topo I and topo III that are directly observed (L,C,O) or inferred (C*). (O*) is a topo III specific intermediate, identified from MD simulations, in which the decatenation loop forms contacts with a second loop in domain II (Fig. 5). Experimentally measured rates (black) are shown in Table 1. Rates that are inferred but not directly measured are shown in gray. The weight of the arrows represents relative kinetic rates.
Topo III has an additional state, O*, between C* and O, in which the decatenation loop forms a bridge that creates a barrier to closing. This additional barrier observed in the simulated free energy profile is supported by the significantly slower gate closing rate, and by the shorter distance between the open state and the transition state obtained from the force-dependent closing rate of topo IIII in comparison to topo I. The long lifetimes of the topo III ligated state also suggest the possibility that the protein could open and close while bound to ligated DNA (Fig. 6), contributing to the slower apparent cleavage rate.
It is important to note that this model is based exclusively on interactions with a single strand of DNA and lack the second strand that is presumably passed through the gate during relaxation. It is probable that this strand affects the gate kinetics. This is especially true for topo III, which has a faster relaxation rate than our gate dynamics measurements would predict.
Discussion
Since the first structure of a type IA topoisomerase was solved, it has been predicted that these enzymes utilize a protein-mediated DNA gate mechanism for strand passage, similar to type II topoisomerases23–25. In the intervening 24 years, no structures of the protein in an open state have been solved, though crystal structures of a fragment suggest that domain II could bend, separating domains I and III25 (Supplementary Fig. 6e–f). Biochemical studies have shown that domains I and III move relative to each other in order for strand passage to occur6. The same studies demonstrated that circular DNA can be trapped in a complex with the protein by crosslinking domains I and III, suggesting that the passed strand resides within the protein cavity6. Ensemble fluorescence quenching assays have similarly linked strand passage to a conformational change between domains I and III27. More recent combined magnetic tweezers – fluorescence experiments also indicate a conformational change prior to strand passage29, though the results were interpreted as domain closure rather than domain opening. Attempts to estimate gate opening of topo I from force-dependent relaxation kinetics resulted in a ∆x value of ~ 10 nm, too large to be reflective of gate opening7. These studies are consistent with, but did not conclusively demonstrate, the existence of the putative protein-mediated DNA gate or the gate dynamics.
By measuring the extension of individual DNA molecules containing ssDNA regions we observed conformational changes corresponding to the separation of cleaved DNA by 5.5–6.6 nm in two bacterial type IA topoisomerases. These experiments provide direct evidence for the protein-gate model of topoisomerase IA activity. Although the magnitude of gate opening was unexpectedly large, molecular dynamics simulations indicate that a 5.8 nm separation is required to pass duplex DNA through the gate. The fact that the gate opening and ssDNA cleavage are reversible and occur with comparable kinetics as the strand passage reaction suggest that the observed open state is biochemically relevant. This conclusion is bolstered by the close agreement between the experiments and simulations, and the fact that topo III and topo I remain catalytically active under applied loads as high as 12 pN (Supplementary Note 1, Supplementary Fig. 7).
These results offer new insight into type IA topoisomerase activity. The differences in gate dynamics between topo III and topo I suggest a possible mechanism for their different biochemical activities and roles in the cell37,51. Whereas topo I removes negative supercoils, topo III functions primarily as a decatenase37, an activity which requires passing duplex DNA through the break in a single strand. The fast dynamics of topo I could favor efficient relaxation of negatively supercoiled DNA, whereas the slower closing rate of topo III could facilitate capture of double stranded DNA. This is supported by the gate stabilizing interaction between the decatenation loop50 of topo III and an acidic loop in domain II observed in our simulations. A similar role in promoting the open state has been proposed for the eukaryotic protein RMI1, which is required for the decatenation activity of topo IIIα19,52. Once DNA has entered the cavity, it may disrupt this interaction, leading to fast closing of the gate and potentially facilitating the processive bursts of relaxation activity observed by Terekhova et al36.
Because of their importance in genome maintenance and replication, topoisomerases are a potential target for both anti-bacterial and anti-cancer drugs53–55. Topo I is the only type IA topoisomerase present in mycobacteria. Mycobacterium tuberculosis topo I has been validated genetically and chemically as a TB drug target 56–58. Poison inhibitors targeting type IA topoisomerases in other bacterial pathogens are also expected to be bactericidal since trapping of a small number of the covalent cleavage complexes on the chromosomal DNA would be sufficient for bacterial cell killing53,59,60. It is therefore highly desirable to identify poisons that can inhibit DNA ligation following the DNA cleavage step. Currently no drugs are in use that target type IA topoisomerases. Design of such drugs has been hampered by the inability to experimentally probe the details of the full strand passage reaction. Key residues that drive the conformational changes for the opening and closing of the DNA “gate” have not yet been identified. The magnetic tweezers assays described here have the potential to be powerful tools in testing the effects of mutations and small molecule interactions on this class of proteins.
Methods
Protein purification
Recombinant topoisomerase III was purified as previously described in Seol et al. (2013)61. E. coli topo III containing a histidine tag and tobacco etch virus (TEV) cleavage site was expressed in Rosetta (DE3) Competent Cells (EMD4Bioscience). Cells were grown at 37 °C in LB Medium containing 50 μg/mL carbenicillin and 34 μg/mL chloramphenicol and induced with 1 mM IPTG for 20 h. Cells were harvested by centrifugation and resuspended in 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, and 10 % v/v glycerol. Cells were emulsified using a high-pressure cell homogenizer (Avestin). Lysate was centrifuged at 40 krpm at 4 °C for 1 h in a Ti45 rotor (Beckman). The supernatant was loaded onto a 12 ml Ni-Nitrilotriacetic acid (NTA) column (Qiagen) and eluted with an imidazole gradient (Buffer A: 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1mM β-mercaptoethanol, and 10 % v/v glycerol, Buffer B: 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1mM β-mercaptoethanol, 500 mM imidazole and 10 % v/v glycerol). Eluted protein was concentrated and buffer exchanged into Buffer A. The protein was incubated with 0.5 U/μg TEV protease for 6 hours to remove the his-tag. Digested protein was loaded onto a His-Trap FF 5ml Ni-NTA column (GE) and collected in the flowthrough. The cleaved protein was concentrated and loaded onto a Hiload 16/60 Superdex 200 column (GE) and eluted with Buffer A. Size and purity was confirmed by SDS-PAGE gel.
Recombinant E. coli topoisomerase I and the Y319F mutant were purified as previously described62. Topo I was overexpressed in E. coli strain MV1190 (BioRad). Cells were grown at 37 °C in LB medium containing 100 μg/mL ampicillin and induced with 400 μM IPTG for 3 h. Cells were harvested by centrifugation and resuspended in 10 mM Tris-HCl pH 8.0, 400 mM KCl, and 1 mg/mL lysozyme. Cells were lysed by freezing and thawing 3 times. Lysate was centrifuged at 5080g in a Ti45 rotor (Beckman) at 4 °C for 3 h. Extract was dialyzed overnight against 20 mM potassium phosphate, pH 7.4, 200 mM KCl,1 mM dithiothreitol (DTT), 1 mM EDTA, and 10 % v/v glycerol at 4 °C. Extract was mixed with equal volume DEAE-51 (Whatman) and stirred for 1 h at 4 °C. Extract was centrifuged at 5080g in a GSA rotor (Sorvall) for 10 min at 4 °C. Supernatant was loaded onto a phosphocellulose column (Whatman) and eluted with a 0.2–1 M KCl gradient. Fractions containing protein were pooled and diluted in 20 mM potassium phosphate, pH 7.4, 1 mM DTT, 1 mM EDTA, and 10 % v/v glycerol to achieve a final KCl concentration of 100 mM. Pooled fractions were loaded onto a 10 ml ssDNA cellulose column (Sigma) and eluted with a 0.1–1M KCl gradient. Size and purity were confirmed by SDS-PAGE gel.
DNA constructs for magnetic tweezers experiments
Hairpin construct
For the hairpin assay, we generated a 537-base hairpin with dsDNA handles on both ends as described in Seol et at. (2016)63. DNA handles were by generated by first separately annealing the top and bottom handles. Complementary strands were incubated at 94°C for 5 minutes and then gradually cooled to room temperature (−1°C/30 s) in 100 mM Potassium acetate and 30 mM HEPES, pH 7.5. The top handle was modified with biotin on the 3′ end for attachment to a streptavidin coated magnetic bead and the bottom handle was modified with three digoxigenins at the 5´ end for attachment to the coverslip. The handles contained complementary overhangs and were subsequently annealed to each other. To create the hairpin, a 500 bp segment was generated by PCR of λ DNA using primers containing BsaI recognition sequences The DNA was digested with BsaI-HF (New England Biolabs) to create 3′ overhangs. The digested product was purified using a PCR purification kit (Roche) and ligated to a 12 bp hairpin with a 5′ overhang complementary to the 500 bp duplex using T4 DNA ligase (New England Biolabs). The 500 bp hairpin was then ligated to the annealed handles. Oligonucleotides for hairpin generation were purchased from Eurofins MWG Operon.
Gapped DNA construct
Linear 2.5-kb dsDNA with a 37-nt gap with a single biotin or digoxigenin at each 5′-end was prepared as described in Seol et al. (2013)61. The 2.5-kb PCR product was digested with Nt.BbvCI (New England Biolabs) to create two nicks at positions 2258 and 2294, near the middle of the DNA template. The ssDNA was engineered to have a CTTC sequence, which is expected to be preferentially cleaved by topoisomerases.
Magnetic tweezers experiments
Magnetic tweezers sample cell preparation
Sample cell preparation is described in detail in Seol et al. (2016)63. Briefly, 5–10 × 10−15 mol DNA substrate was mixed with ~100 ng anti-digoxygenin antibody (Sigma-Aldrich) in PBS and incubated in a sample cell overnight at 4° C. Unbound DNA was washed out with 200 μL of wash buffer (PBS supplemented with 0.1% v/v Tween-20 and 0.3% w/v BSA). 1-μm streptavidin-coated magnetic beads (MyOne, Invitrogen) were introduced into the sample cell and allowed to bind DNA for 5 minutes. The sample cell was then washed with 1 mL wash buffer, followed by 400–1000 μL reaction buffer (50 mM Tris-HCL (pH 8.0), 30 mM monopotassium L-glutamic acid, 0.1 % Tween, 0.03% BSA, + 0–10 mM Mg L-glutamic acid). Measurements were performed at room temperature on a commercial Picotwist instrument. An average force calibration versus magnet position for 1 μm MyOne magnetic beads was obtained using Brownian motion analysis of 11 kb DNA tethers. Force calibration measurements over many beads indicate that relying on the average force calibration introduces an error in force of ~ 10%. Bead position in three dimensions was acquired at 60 Hz.
Hairpin assay
Topo III or topo I was diluted to 1 μM in reaction buffer lacking Mg (50 mM Tris-HCL (pH 8.0), 30 mM monopotassium L-glutamic acid, 0.1 % Tween-20, 0.03% BSA) and incubated at 37 °C for 30–60 minutes. Samples were then diluted to 500 pM in 1 mM Mg reaction buffer. Hairpin DNA tethers were characterized before the addition of protein by applying a range of forces to determine the critical melting force of each individual tether, as slight differences in bead magnetization could affect this value. Following hairpin characterization, 200 μL of topoisomerase sample was flowed into to the experimental sample cell. A force of 8–10 pN was applied to determine a baseline for the folded state of the hairpin, and then a force or 22–24 pN was applied to mechanically melt the DNA. The DNA was held at high force for 30s – 5 min and then the force was returned to the baseline level. Upon full refolding of the hairpin, the force was again raised to 22–24 pN and the measurement repeated. Hairpin unfolding experiments were also conducted in buffer containing 0 Mg L-glutamic acid to determine the Mg-dependence of binding.
Gapped DNA assay
Topoisomerase was diluted to 1 μM in 0 Mg reaction buffer and incubated at 37 °C for 30–60 minutes, then diluted to 250 pM in reaction buffer containing 0–10 mM Mg L-glutamic acid and 200 μL of sample was introduced to the experimental sample cell. Data was taken at 3 mM Mg2+ for topo III and 0.5 mM (ntethers = 5) and 3 mM (ntethers = 2) for topo I. Forces of 8, 10, 12, 14, 16, and 18 pN were applied. Data from multiple tethers (ntethers = 8 for topo III, ntethers = 7 for topo I) was combined to reduce force errors. Data for each tether was taken at multiple forces ranging from 8 to 18 pN, although we do not have data for the full range of forces for every tether due to the fact that tethers are lost over time. Below 12 pN, closing rates for topo I were too fast to be sampled accurately with the time resolution of our experiments. Closing rates were too slow to provide adequate statistics at forces higher than 16 pN for topo III and 18 pN for topo I. For magnesium titration experiments, measurements were conducted at 8 pN on 250 pM topo III in reaction buffer containing 1 mM, 3 mM, 5 mM, or 10 mM Mg L-glutamic acid and at 14 pN for topo I at 0, 0.1 mM, 0.3 mM, 0.5 mM, and 3 mM Mg L-glutamic acid. Additional experiments were conducted for topo I in reaction buffer containing no Mg L-glutamic acid and 1 mM EDTA.
Molecular dynamics simulations
Simulations were conducted in NAMD64 v. 2.12 using the Charmm 36 force field65,66. Topo III bound to ssDNA (pdbid: I17D)48 was solvated with 148,705 TIP3P67 water molecules. Na and Cl ions were added to a concentration of 50 mM. The fully solvated system was minimized for 2500 steps with the protein atoms restrained with a force constant of 5 kcal/mol/Å2 and an additional 5000 steps without restraints. After minimization, the system was equilibrated at constant pressure and temperature for 500 ps. For the CPT equilibration step, pressure was held at 1.01325 Bar using Berendsen’s method and the protein atoms were harmonically restrained with a force constant of 5 kcal/mol/Å2. The full system was further equilibrated without restraints for 10 ns at constant volume and temperature. A Langevin thermostat was used to maintain a temperature of 300 K. From the closed structure, the protein gate was manually opened by applying a harmonic restraint of 4 – 10 kcal/mol/Å2 on the distance between the center of mass of residues 290 to 415 and 492 to 620. The distance was gradually increased over a series of windows from 2.5 nm to 6.5 nm in 0.1 nm increments. Umbrella sampling windows were run for 2 ns – 20 ns, with a 200 ps equilibration period for each new window. Coordinates were saved every 1 ps.
Data analysis
Hairpin assay
For magnetic tweezers hairpin experiments, data analysis was performed using custom written routines in Igor Pro 6.3A. Binding events were defined as pauses in refolding lasting > 100 ms and opening events were defined as discrete extension increases of ~6 – 7 nm from the fully unfolded state. To determine the size of extension increases, the data was smoothed using a 2nd order Savitzky-Golay algorithm over 51 points then histogrammed and fit to a sum of Gaussian distributions using the multi-peak function in Igor. Extension increases were calculated by taking the difference between the centers of the individual Gaussian fits. Because of the fast nature of topo I opening, for some events the number of points was too few to appear in the distribution, leading to apparent increases of ~twice the average size. The data for each unfolding transition was combined and histogrammed with bin size 1 nm. The number of binding and opening events for each unfolding-refolding cycle was determined and the average number of opening events for a given number of bound topoisomerases was plotted.
Gapped DNA assay
To determine the size of gate opening, data was smoothed using a 2nd order Savitzky-Golay algorithm over 51 points then histogrammed using a bin size of 0.5 nm and fit to a double-Gaussian distribution using the multi-peak function in Igor Pro 6.3A. Extension increases were calculated by taking the difference between the centers of the individual Gaussian fits. KD values were determined from comparison of the area under the two Gaussians.
To determine kinetics, traces were analyzed using vbFRET35 to assign states and then further analyzed in Igor Pro. Default values of priors were used in vbFRET analysis (upi=1, mu=0.5, beta=0.25, W=50, v=5, ua=1, uad=0). Lifetime analysis of the idealized traces from vbFRET were calculated using a thresholding algorithm in Igor Pro 6.3A. The open and closed state lifetimes were histogrammed and fit to exponentials to determine cleavage, opening, and closing rates. For topo III, closed state lifetimes were treated as two separate data sets, with short time bins for short lifetime states (t ≤ 20 s) and longer time bins for the long lifetime states (t > 20 s). We assumed that the long-lifetime closed state corresponded to cleavage followed by an opening event (tlong = tcleavage + topen), whereas the short-lifetime closed states corresponded to opening events (tshort = topen). Because the short lifetimes were orders of magnitude less than the longer lifetimes for topo III, we assumed that kcleavage ~ klong. For topo I, the closed states were fit to a double exponential. We assigned the faster rate to kopen, the slower rate to klong, and calculated kcleavage as 1/(tlong – topen). Closing rates were calculated from single-exponential fits to histograms of the open state lifetimes. In some traces we observed two topo III molecules active simultaneously. Cleavage rates were adjusted accordingly. Closing rates for events where two molecules were open simultaneously were excluded from analysis. Ligation rates were calculated using the kinetic competition model described in the results section.
Molecular dynamics simulations
The reweighted free energy profile of the distance between the catalytic tyrosine and the cleavage site was calculated using a modification of the weighted histogram analysis method (WHAM)47 that allows for calculation of the free energy of a reaction coordinates orthogonal to the restrained coordinates49. Values for the experimental reaction coordinate, x, defined as the distance between the oxygen of the catalytic tyrosine (Y328) and the cleavage site on the DNA backbone (P7), were obtained from the coordinates saved from each simulation window using VMD v. 1.9.268. To determine the free energy along our experimental reaction coordinate x, we calculated two-dimensional histograms of the experimental and the restrained reaction coordinates for each simulation window. These histograms were then unbiased and the free energy profiles calculated using the program: WHAM: The weighted histogram analysis method, version 2.0.9.1, http://membrane.urmc.rochester.edu/content/wham69. The two-dimensional free energy profile was collapsed along the restrained coordinate to determine the free energy along x.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (HL001056-07 to K.C.N.) and a grant from National Institutes of Health (R01GM054226 to Y.T). This work utilized the computational resources of the NIH HPC Biowulf cluster. We thank Dr. Yeonee Seol for insightful discussions, experimental assistance, and for providing the DNA substrates. We thank Lynda Bradley for assistance with purifying topoisomerase III and Bokun Cheng for purified topoisomerase I.
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
Source data for Figure 4 is available with the paper online. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 70, 369–413 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Corbett KD & Berger JM Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. Biomol. Struct 33, 95–118 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Liu LF, Liu C-C & Alberts BM Type II DNA topoisomerases: Enzymes that can unknot a topologically knotted DNA molecule via a reversible double-strand break. Cell 19, 697–707 (1980). [DOI] [PubMed] [Google Scholar]
- 4.Seol Y, Zhang H, Pommier Y & Neuman KC A kinetic clutch governs religation by type IB topoisomerases and determines camptothecin sensitivity. Proc. Natl. Acad. Sci 109, 16125–16130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart L, Redinbo MR, Qiu X, Hol WG & Champoux JJ A model for the mechanism of human topoisomerase I. Science 279, 1534–1541 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Li Z, Mondragón A & DiGate RJ The Mechanism of Type IA Topoisomerase-Mediated DNA Topological Transformations. Mol. Cell 7, 301–307 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Dekker NH et al. The mechanism of type IA topoisomerases. Proc. Natl. Acad. Sci 99, 12126–12131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tse-Dinh YC Biochemistry of bacterial type I DNA topoisomerases. Adv. Pharmacol. San Diego Calif 29A, 21–37 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Srivenugopal KS, Lockshon D & Morris DR Escherichia coli DNA topoisomerase III: purification and characterization of a new type I enzyme. Biochemistry 23, 1899–1906 (1984). [DOI] [PubMed] [Google Scholar]
- 10.Confalonieri F et al. Reverse gyrase: a helicase-like domain and a type I topoisomerase in the same polypeptide. Proc. Natl. Acad. Sci 90, 4753–4757 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goulaouic H et al. Purification and characterization of human DNA topoisomerase IIIα. Nucleic Acids Res 27, 2443–50 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W & Wang JC Mammalian DNA topoisomerase IIIα is essential in early embryogenesis. Proc. Natl. Acad. Sci 95, 1010–1013 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seki T, Seki M, Onodera R, Katada T & Enomoto T Cloning of cDNA Encoding a Novel Mouse DNA Topoisomerase III (Topo IIIβ) Possessing Negatively Supercoiled DNA Relaxing Activity, Whose Message Is Highly Expressed in the Testis. J. Biol. Chem 273, 28553–28556 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Massé E & Drolet M Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J. Biol. Chem 274, 16654–16658 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Suski C & Marians KJ Resolution of Converging Replication Forks by RecQ and Topoisomerase III. Mol. Cell 30, 779–789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez‐Cheeks BA, Lee C, Hayama R & Marians KJ A role for topoisomerase III in Escherichia coli chromosome segregation. Mol. Microbiol 86, 1007–1022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortés F, Pastor N, Mateos S & Domínguez I. Roles of DNA topoisomerases in chromosome segregation and mitosis. Mutat. Res. Mutat. Res 543, 59–66 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Goodwin A, Wang S-W, Toda T, Norbury C & Hickson ID Topoisomerase III is essential for accurate nuclear division in Schizosaccharomyces pombe. Nucleic Acids Res 27, 4050–4058 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bocquet N et al. Structural and mechanistic insight into Holliday junction dissolution by Topoisomerase IIIα and RMI1. Nat. Struct. Mol. Biol 21, 261–268 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bussen W, Raynard S, Busygina V, Singh AK & Sung P Holliday junction processing activity of the BLM-Topo IIIalpha-BLAP75 complex. J. Biol. Chem 282, 31484–31492 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Ahmad M et al. Topoisomerase 3β is the major topoisomerase for mRNAs and linked to neurodevelopment and mental dysfunction. Nucleic Acids Res 45, 2704–2713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu D et al. Top3β is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci 16, 1238–1247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lima CD, Wang JC & Mondragón A Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature 367, 138–146 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Mondragón A & DiGate R The structure of Escherichia coli DNA topoisomerase III. Structure 7, 1373–1383 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Feinberg H, Lima CD & Mondragón A Conformational changes in E. coli DNA topoisomerase I. Nat. Struct. Biol 6, 918–922 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Baker NM, Rajan R & Mondragón A Structural studies of type I topoisomerases. Nucleic Acids Res 37, 693–701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leelaram MN et al. Type IA topoisomerase inhibition by clamp closure. FASEB J 27, 3030–3038 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Xiong B et al. The type IA topoisomerase catalytic cycle: A normal mode analysis and molecular dynamics simulation. Proteins Struct. Funct. Bioinforma 71, 1984–1994 [DOI] [PubMed] [Google Scholar]
- 29.Gunn KH, Marko JF & Mondragón A An orthogonal single-molecule experiment reveals multiple-attempt dynamics of type IA topoisomerases. Nat. Struct. Mol. Biol 24, 484–490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seol Y & Neuman K Magnetic Tweezers for Single-Molecule Manipulation. in Single Molecule Analysis (eds. Peterman EJG & Wuite GJL) 265–293 (Humana Press, 2011). doi: 10.1007/978-1-61779-282-3_15 [DOI] [PubMed] [Google Scholar]
- 31.Mills M et al. RecQ helicase triggers a binding mode change in the SSB-DNA complex to efficiently initiate DNA unwinding. Nucleic Acids Res 45, 11878–11890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perry K & Mondragón A Structure of a Complex between E. coli DNA Topoisomerase I and Single-Stranded DNA. Structure 11, 1349–1358 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Dekker NH et al. Thermophilic Topoisomerase I on a Single DNA Molecule. J. Mol. Biol 329, 271–282 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Bell GI Models for the specific adhesion of cells to cells. Science 200, 618–627 (1978). [DOI] [PubMed] [Google Scholar]
- 35.Bronson JE, Fei J, Hofman JM, Gonzalez RL & Wiggins CH Learning Rates and States from Biophysical Time Series: A Bayesian Approach to Model Selection and Single-Molecule FRET Data. Biophys. J 97, 3196–3205 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terekhova K, Gunn KH, Marko JF & Mondragón A Bacterial topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res 40, 10432–10440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nurse P, Levine C, Hassing H & Marians KJ Topoisomerase III Can Serve as the Cellular Decatenase in Escherichia coli. J. Biol. Chem 278, 8653–8660 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Terekhova K, Marko JF & Mondragón A Single-molecule analysis uncovers the difference between the kinetics of DNA decatenation by bacterial topoisomerases I and III. Nucleic Acids Res gku785 (2014). doi: 10.1093/nar/gku785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Pan B, Li Z, Sheng Zhao X & Huang L Kinetic insights into the temperature dependence of DNA strand cleavage and religation by topoisomerase III from the hyperthermophile Sulfolobus solfataricus. Sci. Rep 7, 5494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domanico PL & Tse-Dinh YC Mechanistic studies on E. coli DNA topoisomerase I: Divalent ion effects. J. Inorg. Biochem 42, 87–96 (1991). [DOI] [PubMed] [Google Scholar]
- 41.Tse-Dinh YC Uncoupling of the DNA breaking and rejoining steps of Escherichia coli type I DNA topoisomerase. Demonstration of an active covalent protein-DNA complex. J. Biol. Chem 261, 10931–10935 (1986). [PubMed] [Google Scholar]
- 42.Sorokin EP et al. Inhibition of Mg2+ binding and DNA religation by bacterial topoisomerase I via introduction of an additional positive charge into the active site region. Nucleic Acids Res 36, 4788–4796 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu CX, Roche CJ & Tse-Dinh YC Effect of Mg(II) binding on the structure and activity of Escherichia coli DNA topoisomerase I. J. Biol. Chem 272, 16206–16210 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Bhat AG, Leelaram MN, Hegde SM & Nagaraja V Deciphering the distinct role for the metal coordination motif in the catalytic activity of Mycobacterium smegmatis topoisomerase I. J. Mol. Biol 393, 788–802 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Cao N, Tan K, Annamalai T, Joachimiak A & Tse-Dinh Y-C Investigating mycobacterial topoisomerase I mechanism from the analysis of metal and DNA substrate interactions at the active site. Nucleic Acids Res 46, 7296–7308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torrie GM & Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 23, 187–199 (1977). [Google Scholar]
- 47.Souaille M & Roux B Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun 135, 40–57 (2001). [Google Scholar]
- 48.Changela A, DiGate RJ & Mondragón A Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature 411, 1077–1081 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Minh DDL Multiple Potentials of Mean Force from Biased Experiments Along a Single Coordinate. J. Phys. Chem. B 111, 4137–4140 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Mondragón A, Hiasa H, Marians KJ & DiGate RJ Identification of a unique domain essential for Escherichia coli DNA topoisomerase III-catalysed decatenation of replication intermediates. Mol. Microbiol 35, 888–895 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Zechiedrich EL et al. Roles of Topoisomerases in Maintaining Steady-state DNA Supercoiling in Escherichia coli. J. Biol. Chem 275, 8103–8113 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Cejka P, Plank JL, Dombrowski CC & Kowalczykowski SC Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol. Cell 47, 886–896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tse-Dinh Y-C Targeting bacterial topoisomerase I to meet the challenge of finding new antibiotics. Future Med. Chem 7, 459–471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandhaus S et al. Small-Molecule Inhibitors Targeting Topoisomerase I as Novel Antituberculosis Agents. Antimicrob. Agents Chemother 60, 4028–4036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giles GI & Sharma RP Topoisomerase enzymes as therapeutic targets for cancer chemotherapy. Med. Chem. Shariqah United Arab Emir 1, 383–394 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Nagaraja V, Godbole AA, Henderson SR & Maxwell A DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov. Today 22, 510–518 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Ahmed W, Menon S, Godbole AA, Karthik PVDNB & Nagaraja V Conditional silencing of topoisomerase I gene of Mycobacterium tuberculosis validates its essentiality for cell survival. FEMS Microbiol. Lett 353, 116–123 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Ravishankar S et al. Genetic and chemical validation identifies Mycobacterium tuberculosis topoisomerase I as an attractive anti-tubercular target. Tuberc. Edinb. Scotl 95, 589–598 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Aedo S & Tse-Dinh Y-C Isolation and Quantitation of Topoisomerase Complexes Accumulated on Escherichia coli Chromosomal DNA. Antimicrob. Agents Chemother 56, 5458–5464 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pommier Y & Marchand C Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discov 11, 25–36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seol Y, Hardin AH, Strub M-P, Charvin G & Neuman KC Comparison of DNA decatenation by Escherichia coli topoisomerase IV and topoisomerase III: implications for non-equilibrium topology simplification. Nucleic Acids Res 41, 4640–4649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu C-X & Tse-Dinh Y-C Overexpression and Purification of Bacterial DNA Topoisomerase I. in DNA Topoisomerase Protocols 145–151 (Humana Press, 1999). doi: 10.1385/1-59259-259-7:145 [DOI] [PubMed] [Google Scholar]
- 63.Seol Y, Strub M-P & Neuman KC Single molecule measurements of DNA helicase activity with magnetic tweezers and t-test based step-finding analysis. Methods San Diego Calif 105, 119–127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalé L et al. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys 151, 283–312 (1999). [Google Scholar]
- 65.Brooks BR et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 30, 1545–1614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang Jing & MacKerell Alexander D. CHARMM36 all‐atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem 34, 2135–2145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neria E, Fischer S & Karplus M Simulation of activation free energies in molecular systems. J. Chem. Phys 105, 1902–1921 (1996). [Google Scholar]
- 68.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–28 (1996). [DOI] [PubMed] [Google Scholar]
- 69.Grossfield A WHAM: The weighted histogram analysis method, version 2.0.9.1, http://membrane.urmc.rochester.edu/content/wham.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.