

Abstract
The human cytosolic sulfotransferases (SULTs) comprise a 13-member enzyme family that regulates the activities of hundreds, perhaps thousands of signaling small molecules via regiospecific transfer of the sulfuryl-moiety (-SO3) from PAPS (3’-phosphoadenosine 5’-phosphosulfate) to the hydroxyls and amines of acceptors. Signaling molecules regulated by sulfonation include numerous steroid and thyroid hormones, epinephrine, serotonin, and dopamine. SULT1A1, a major phase II metabolism SULT isoform, is found at high concentration in liver and has recently been show to harbor two allosteric-binding sites, each of which binds a separate and complex class of compounds - the catechins (naturally occurring polyphenols) and NSAIDS. Among catechins, epigallocatechin gallate (EGCG) displays high affinity and specificity toward SULT1A1. The allosteric network associated with either site has yet to be defined. Here, using equilibrium binding and presteady state studies, the network is shown to involve fourteen distinct complexes. ECGG binds both the allosteric site and, relatively weakly, the active site of SULT1A1. It is not a SULT1A1 substrate, but is sulfonated by SULT2A1. EGCG binds 17-fold more tightly when the active-site cap of the enzyme is closed by the binding of nucleotide. When nucleotide is saturating, EGCG binds in two phases. In the first, it binds to the cap-open conformer; in the second, it traps the cap in the closed configuration. Cap-closure encapsulates the nucleotide, preventing its release; hence, the EGCG-induced cap stabilization slows nucleotide release, inhibiting turnover. Finally, a comprehensive quantitative model of the network is presented.
Keywords: allostery, inhibition, epigallocatechin gallate, EGCG, sulfotransferase, SULT, mechanism, ligand, binding, presteady, fluorescence, structure
Graphical Abstract
Transfer of the sulfuryl-moiety to specific sites on small-molecule scaffolds modulates, often profoundly, the interactions between signaling small molecules and their targets (1–3). These reactions are catalyzed by the human cytosolic sulfotransferases (SULTs) — a thirteen member family of enzymes that transfer the sulfuryl-moiety (-SO3) from PAPS (3’-phosphoadenosine 5’-phosphosulfate) to the hydroxyls and amines of thousands of small-molecule metabolites (4). SULTs are expressed in highly specific tissue and developmental patterns (5–7) and play critical roles in processes as diverse as adipogenesis (8, 9), andrenarchy (10), bone development (11, 12), and neuronal signaling (3, 13, 14).
SULT substrate specificities are typically broad, somewhat overlapping, and focused on different areas of metabolism. SULT1A1 is among the broadest specificity SULTs and is responsible for regulating and detoxifying endogenous metabolites and xenobiotics. It is found at its highest levels in liver (7), where it plays a key role in first-pass phase II metabolism (15, 16), and in the ileum of the small intestine (7, 17), where it is located mainly in brush-border enterocytes (17). These mature enterocytes form a single-cell layer that lines the lumen and acts as a biological sieve that selects and modifies nutrients as they pass into plasma.
Recent studies reveal that SULT1A1, a dimer, is allosterically regulated by two distinctly different mechanisms. Nucleotide binding is highly anti-cooperative, and different nucleotide occupancies are linked to different conformations of the active-site caps of the enzyme, which have widely different substrate selectivities (16). In addition to these homotropic interactions, SULT1A1 harbors two allosteric-inhibitor binding pockets, each of which binds a separate and complex class of compounds — the catechins and nonsteroidal anti-inflammatory drugs (NSAIDS) (18–20).
Catechins, a large natural-product family of compounds, are abundant in coffee (21), tea (22) and cocoa (23). Epigallocatechin gallate (EGCG) comprises ~12% of the weight of dry tea leaves (24) and is often associated with the health benefits of tea (25). Among the catechins tested, EGCG is the most potent SULT inhibitor and is highly specific for the SULT1A1 isoform, Ki = 34 nM (19). Each day, hundreds of millions of individuals drink tea (26). In a typical cup of green tea, the concentration of EGCG is ~ 1.4 mM (41,000 • Ki) (24); in human plasma, it is ~0.4 μ, or 12 • Ki (27, 28). These levels suggest in vivo roles for EGCG in determining the efficacy of drugs that are highly sulfonated by SULT1A1 (15), and in inhibiting SULT1A1-catalyzed activation of procarcinogens (29, 30).
The allosteric interactions between catechins and substrates, and the ways in which the SULT1A1 scaffold responds to ligands and mediates their interactions are not well understood, yet these interactions are likely involved in drug metabolism, drug-drug interactions and procarcinogen activation (18). Here, EGCG is used to explore this allosteric network and the results are used to construct a comprehensive model for the isoform specific allosteric regulation of STJLT1A1.
Materials and Methods
The materials and sources used in this study are as follows: dithiothreitol (DTT), 17-β-estradiol (E2), ethylenediaminetetraacetic acid (EDTA), L-glutathione (reduced), 1-hydroxypyrene (1-HP), 4-hydroxytamoxifen (TAM), imidazole, isopropyl-thio-β-D-galactopyranoside (IPTG), Lysogeny broth (LB), lysozyme, pepstatin A, raloxifene (Ral), and sodium phosphate were the highest grade available from Sigma. Ampicillin, HEPES, KCl, KOH, MgCl2, NaCl and phenylmethylsulfonyl fluoride (PMSF) were purchased from Fisher Scientific. Epigallocatechin gallate (EGCG) and epigallocatechin (EGC) were obtained from Santa Cruz Biotechnology, Inc. Anion exchange HPLC was performed using an Eprogen, AX100 (5μm) column. Glutathione- and nickel-chelating resins were obtained from GE Healthcare. Competent E. coli (BL21(DE3)) was purchased from Novagen. PAPS and PAP were synthesized in house as previously described (16, 31) and were > 98% pure as assessed by anion-exchange high- performance liquid chromatography.
Protein Purification.
A codon optimized SULT1A1 coding region was inserted into a pGEX-6P expression vector containing a PreScission-protease-cleavable N-terminal His/GST/MBP-tag. SULT expression and purification were performed as described previously (32). Briefly, cells were grown in LB with ampicillin (100 μg/ml) at 37 °C to an OD600 of 0.6, and induced with IPTG (0.30 mM) overnight at 18 °C. The cells were then pelleted, resuspended in NaPO4 (25 mM), KCl (0.40 M), PMSF (0.29 mM), pepstatin A (1.5 μM), and lysozyme (0.10 mg/ml), pH 7.5 sonicated, and centrifuged at 10,000g for 1.0 hr to remove debris. The supernatant was loaded onto a Chelating Sepharose Fast Flow column charged with Ni2+. The enzyme was then eluted with imidazole (10 mM) onto a Glutathione Sepharose column followed by elution with glutathione (10 mM). The fusion protein was digested with PreScission protease and dialyzed against HEPES/K+ (25 mM), DTT (1.5 mM), and KCl (75 mM), pH 7.5. The protein mixture was then passed through a second GST column to remove His/GST/MBP tag and PreScission Protease. All purification procedures were performed at 4°C. The enzyme was concentrated using 10K cutoff centrifugation filters. Protein purity was > 95 %, as determined by Coomassie staining of SDS-PAGE gels. The enzyme was aliquoted, flash frozen, and stored at −80 °C. The catalytic integrity of the enzyme was assessed by determining its initial rate parameters using para-nitrophenol (PNP). The parameters, which agree well with literature values (18, 33, 34), are as follows: kcat = 60 (± 1.8), Km (PNP) =1.4 (± 0.1), Ki = 6.6 (± 0.3). The experiments were performed under the following conditions: PAPS (100 μM), NaPO4 (50 mM), pH 7.2, 25 ± 2 °C.
Equilibrium Binding Studies.
The binding of ligands to SULT1A1 was monitored via changes in the intrinsic fluorescence of the enzyme (Xgx = 290 nm, Xgm = 370 nm). Conditions were as follows: SULT1A1 (0.010 – 5.0 μM, dimer), PAP (0 – 0.50 mM), EGCG (0 – 12 μM), TAM (0 – 60 μM), E2 (0 – 20 μM), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2 °C. EGCG inner filter effects were corrected using a standard curve (described below). Ligand stock solutions were prepared in ethanol or DMSO, controls confirmed that the addition of ethanol or DMSO did not cause detectible fluorescence change. Titrations were performed in duplicate or triplicate. Data were averaged and least-squares fit to a model that assumed a single binding site per monomer. In all cases, ligand concentrations were varied from approximately 0.10 to 20 ▪ Kd.
EGCG Inner Filter Effects.
The absorbance spectrum of EGCG overlaps both the excitation and emission spectra of SULT1A1 (see, Fig S1), and thus can cause inner filter effects by absorbing excitation and/or emitted light. To minimize these effects, the excitation wavelength used in the EGCG titrations (290 nm) was selected to maximize the SULT1A1/EGCG absorbance ratio, and the emission wavelength was set at 370 nm, where EGCG absorbance is negligibly small (ε370 =0.16 mM−1 cm−1). To distinguish inner filter from binding effects, an inner-filter standard curve was constructed using ATP, which absorbs at 290 nM (ε290 ATP = 0.054 mM−1 cm−1), but does not bind SULT1A1. A standard curve of SULT1A1 fluorescence intensity vs OD290 (λex = 290 nm, λex =370 nm) was constrUcted using ATp. The EGCG extinction coefficient (ε290 EGCG 10 mM−1 cm−1) was used to recast the ATP standard curve in terms of EGCG concentration (see, Fig S2). The resulting curve was least-squares fit using a single-exponential equation, which was used to correct the raw titration data to reveal the effects due solely to binding. The corrected titrations were then fit to obtain the EGCG affinity constants. At the highest EGCG concentrations used the titrations (20 μM) the inner filter effect ~ 25% that of the binding effects.
EGCG Binding to the SULT1A1 Active Site.
EGCG binding was measured by competitive displacement of 1-HP, which was monitored via changes in 1-HP fluorescence anisotropy (λex = 385 nm, λex = 430 nm). Titrations were performed by successive addition of EGCG to a solution containing SULT1A1 (8.0 μM, monomer, 1.2 ▪ Kd (1-HP)), 1-HP (10 μM, 1.5 ▪ Kd (1-HP)), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, T = 25 ± 2 °C. Dilution at titration endpoints was < 2.0 %. The affinity and maximum fluorescence anisotropy of 1-HP were determined by titrating SULT1A1 into a solution containing 1-HP (see, Fig S3). The fraction of bound 1-HP was calculated from the ratio of the anisotropy at a given condition to the anisotropy of the fully bound ligand (anisotropy ~ 0 at [SULT1A1] = 0). The absorbance of EGCG is low enough (ε385 =0.034 mM−1 cm−1) that EGCG inner-filter effects were negligible. Titrations were performed in triplicate. The data were averaged and least-squares fit according to competitive binding model (35).
EGCG is not a Substrate.
[35S]PAPS was used to test whether EGCG is a SULT1A1 substrate. Reactions conditions were: SULT1A1 or 2A1 (30 μM monomer, 84 ▪ Km PAPS), EGCG (250 μM, 37- Kd active sites), 35S-PAPS (0.30 μM, SA 0.69 Ci/mmol), MgCl2 (5.0 mM), NPO4 (50 mM), pH 7.5, and 25 ± 2 °C. Reactions were initiated by the addition of EGCG. Reactions were run for 2.0, 5.0, 10, and 30 min, quenched by NaOH (0.10 mM, final) and neutralized with HCl. Reaction mixtures were heated in a boiling water bath for 1.0 min to denature the enzyme, centrifuged for 5 min at 12,000 ▪ g, and the supernatants were spotted onto anion exchange TLC plate. Radiolabeled reactants were separated using a 0.90 M LiCl mobile phase and quantitated by STORM imaging (36). Controls were identical except that they lacked EGCG. Anion exchange was used to determine the purity of the EGCG. The chromatographic protocol was as follows: column (Eprogen, AX100, 5μm), Buffer A (KPO4 (50mM), pH7.5), Buffer B (KPO4 (50mM), NaCl (1.0M), pH7.5), flow rate (1.0 ml/min). EGCG eluted at 72% Buffer B; its elution was monitored at 290 nM. The EGCG was > 98% pure and was baseline separated from EGC (epigallocatechin), which eluted at 46% Buffer B.
EGCG Presteady State Binding Studies.
Presteady state binding experiments were performed using an Applied Photophysics SX20 stopped-flow spectrofluorimeter. SULT1A1 fluorescence was excited at 290 nm and detected above 325 nm using a cutoff filter. Reactions were initiated by rapidly mixing [1:1 (v:v)] a solution containing SULT1A1 (0 – 5.0 μM, dimer), EGCG (0 – 12 μM), PAP (0 – 0.50mM), MgCl2 (5.0 mM), NaPO4 (50 mM) at pH 7.5, 25 ± 2 °C with a solution that was identical except that it lacked enzyme and contained EGCG. kobs values were obtained by fitting progress curves that were the average of ~ 6 binding reactions. Each value was determined in triplicate, and the average is presented in the figures. Data were fit using Pro-K analysis software (76). It should be noted that any inner filter effects of EGCG will remain fixed throughout the binding reaction and thus will not alter the time dependent change in signal that is used to obtain kobs.
PAP Presteady State Binding Studies.
The equipment and procedures are described in the preceding paragraph. Reactions were initiated by rapidly mixing [1:1 (v:v)] a solution containing SULT1A1 (0.15 μM, dimer), EGCG (12 μM), MgCh2 (5.0 mM), NaPO4 (50 mM) at pH 7.5, 25 ± 2 °C with a solution that was identical except that it contained PAP and was without enzyme.
Results and Discussion
The SULT1A1 Mechanism - A Brief Review.
Like most SULTs, SULT1A1 harbors a dynamic, ~30-residue active site cap whose structure responds to ligands and mediates their interactions. Nucleotide binding fosters cap closure. When it closes, nucleotide is encapsulated and cannot escape, and a “pore” forms at the acceptor-binding site. The pore acts as a molecular sieve to sterically screen acceptors, selecting them based on their size and geometry. The affinities of acceptors small enough to pass through the pore are not affected by cap closure. Those that are too large must “wait” for the cap to open to enter; consequently, their affinities decrease by a factor that is essentially equal to the change in the cap-closure equilibrium constant that occurs when nucleotide binds (32, 37–39). This isomerization equilibrium constant (Kiso) has been determined for 1A1 in the absence of nucleotide, in which case the cap favors the open state (Kiso = [E]closed/[E]open ≤ 0.05 (32), and at saturation, where Kiso = 18 (32). These values predict that the affinity of large substrates will decrease 18-fold, relative to unliganded enzyme, at saturating nucleotide - which is what is observed. It should be noted that the effects of PAP and PAPS on Kiso and the binding of small and large acceptors are experimentally indistinguishable (16).
Human SULT1A1 is a dimer of identical subunits that are highly interactive with respect to the binding of nucleotide (16, 31). Binding of the first molecule of PAPS causes the cap to close over the PAPS-bound subunit; the cap of the unoccupied subunit remains open. The second molecule binds 88-fold more weakly than the first and causes both caps to open. As the second site becomes occupied, the catalytic efficiency (kcat/Km) of the enzyme increases 8-fold toward small substrates, whose Km values are independent of cap closure, and 144-fold toward large substrates, whose Km values decrease 18-fold when the caps are opened by the binding of the second nucleotide. Nucleotide affinities for the first and second sites are 0.35 and 31 μM, respectively (16).
PAPS concentrations in the cytosols of cells in tissues exposed to low xenobiotic loads are sufficient to saturate only the high-affinity site, while those in tissues that experience high loads are likely to saturate both (40, 41). Hence, the specificity of SULT1A1 toward small and large substrates is expected to be highly tissue dependent, and to be broadest in those tissues challenged with xenobiotics - an enormous, structurally diverse family of compounds.
EGCG Binding and Stoichiometry at the Allosteric Site.
EGCG binding can be monitored via changes in the intrinsic fluorescence of SULT1A1. Binding to the ligand-free enzyme is monophasic (Fig 1A), suggesting that both binding sites of the dimer behave identically toward EGCG, and Kd for this interaction is 0.68 ± 0.05 μM (Table 1). The EGCG-binding stoichiometry, 1.0 per subunit, was determined by titrating EGCG at [SULT 1A1] = 15 ▪ Kd (Fig 1B). The trailing-downward slope in the titration (open circles) seen at stoichiometric excess of EGCG is caused by the relatively weak binding of EGCG (6.8 μM) at the active site (see following section), and was corrected by performing the titration (blue dots) in the presence of saturating estradiol (E2), a SULT1A1 acceptor, at 15 μM (26 ▪ Kd) above the active-site concentration. Controls ensured that E2 did not affect the affinity of EGCG.
Figure 1. EGCG Binding Studies.

A.) Binding at the SULT1A1 Allosteric Site. Binding was monitored via changes in the intrinsic fluorescence of the enzyme (λex = 290 nm, λem = 370 nm). Fluorescence intensity is given relative to the intensity in the absence of ligand (I/I0). Conditions: SULT1A1 (0.25 μM, dimer), NaPO4 (50 mM), pH 7.5, 25 ± 2 °C. B.) Allosteric Site Binding Stoichiometry. Fluorescence measurements were as described in Panel A. Conditions: SULT1A1 (10 μM, monomer), E2 (0 μM (white dots), or 15 μM (26 ▪ Kd, blue dots)), NaPO4 (50 mM), pH 25 ± 2 °C. C.) Binding at the SULT1A1 Active Site. Binding was monitored via changes in the fluorescence anisotropy (λex = 385 nm, λem = 430 nm) of 1-HP — a competitive ligand. Conditions: SULT1A1 (6.0 μM, active site, 1.0 ▪ Kd 1-HP), 1-HP (10 μM, 1.5 ▪ Kd 1-HP), NaPO4 (50 mM), pH 7.5, 25 ± 2 °C. The fraction of enzyme-bound 1-HP was calculated from anisotropy measurements. For each titration, each point is the average of three independent determinations. The curves passing through the data in panels A and B are predicted by best-fit single-site and competitive-binding models, respectively. The stoichiometry was obtained from the point- of-intersection of the sub- and super-stoichiometric regions of the binding curve. All data were corrected for EGCG inner filter effects (see Materials and Methods).
Table 1.
Ligand Binding to SULT1A1
| Enzyme Form | Kd (μM) | |
|---|---|---|
| EGCG | PAP | |
| E | 0.68 (0.05)a | 0.35 (0.04)b |
| E·PAP | 0.044 (0.004) | 31 (2)b |
| E·(PAP)2 | 0.039 (0.003) | |
| E·EGCGPAP | 0.81 (0.07) | 31 (2) |
| E·(EGCG)2 | 0.024 (0.002) | |
| E·(EGCG)2PAP | 0.78 (0.06) | |
Values in parentheses indicate error.
Determined previously (16).
EGCG Binding and Turnover at the Active Site.
To assess its binding at the SULT1A1 active site, EGCG was used in a competitive binding experiment to displace 1-hydroxypyrene (1-HP) - a fluorescent 1A1 substrate whose binding and initial-rate parameters are known (42). Fluorescence anisotropy, which distinguishes free from bound ligand on the basis of the ligand’s rotational correlation time (43), was used to measure the fraction of bound 1-HP. Anisotropy measurements were chosen to avoid EGCG inner filter effects (see, Materials and Methods). The EGCG-dependent displacement of 1-HP is shown in Fig 1C. The data were least-squares fit using the root of the third-order polynomial that describes the competition at a fixed 1-HP. The line through the data is the behavior predicted by the best-fit EGCG dissocation constant, 6.8 μM. This experiment monitors binding of the third and fourth molecules of EGCG to the 1A1 dimer, the first two molecules bind at the allosteric sites and are saturated at concentrations well below those used in this experiment.
EGCG is sulfonated at low levels in humans (44) and the SULT isoform(s) responsible for its sulfonation has not yet been identified. To assess whether SULT1A1 sulfonates EGCG, the following high-sensitivity assay was performed. Briefly, the enzyme active-site concentration was set high enough (30 μM, 84 ▪ Km PAPS) to adsorb virtually all of the 35S-PAPS (0.30 μM) in solution, and the EGCG concentration was saturating (250 μM, 37 ▪ Kd active site). The reactions were quenched and radiolabeled reactants were separated chromatographically. Parallel experiments were performed with SULT1A1 and 2A1. Thirty-four percent of PAPS was converted to sulfonated EGCG by SULT2A1 in 1.0 min; sulfonation was not detected in the SULT1A1 reaction over 30 min (see, Materials and Methods). The lower detection limit is ~5 % of PAPS conversion; hence, SULT1A1 turns over EGCG at less than 0.16% min−1. EGCG does not appear to be a substrate for SULT1A1.
Due to the high sensitivity of the assay, it was necessary to confirm that EGCG, rather than a low-level contaminant, is in fact a substrate for SULT2A1. To do so, a reaction was run under conditions designed to convert to product a fraction that is well above contaminant levels. Anion exchange HPLC analysis of starting material indicated that EGCG was > 98% pure (see, Materials and Methods). At EGCG (30 μM), 35S-PAPS (30 μM), SULT2A1 (0.50 μM, dimer), 12% of EGCG was converted to product in 2 min. Thus, EGCG is clearly a substrate for SULT2A1, which likely contributes to its sulfonation in vivo.
EGCG Interactions with ENucleotide Complexes.
As discussed above, the allosteric interactions of nucleotides (PAP or PAPS) determine both the nucleotide affinity and the open/closed-status of the SULT1A1 caps. The first nucleotide to add (K1 = 0.35 μM) closes its cap, and stabilizes the adjacent cap in the open position. The affinity of the second nucleotide (K2 = 31 μM) is 88-fold weaker than the first, thus allowing the single- (E·PAP) and double-nucleotide (E-(PAP)2) species to be studied largely independently.
The E·PAP complex.
The binding of EGCG to E·PAP at equilibrium is shown in Fig 2A. The affinity of EGCG for the complex (Kd = 0.044 μM) is 15-fold greater than its affinity for E (Table 1). In the Fig 2B titration, the [SULT1A1]dimer is 1.0 μM, or 23-times Kd EgcG for binding to E·PAP, and the PAP concentration (4.0 μM) is such that the enzyme is predominantly in the E·PAP form - the resulting distribution of species at zero EGCG is as follows: E (10%), E·PAP (82%), E-(PAP)2 (8%). As expected under these conditions, EGCG adsorption is quantitative at sub-stoichiometric EGCG concentrations. The titration shows a distinct break at a stoichiometry of 1.0 per dimer (see inset) and further addition of EGCG reveals a second, low-affinity site with an EGCG affinity that is indistinguishable from that of the unliganded enzyme (Table 1).
Figure 2. EGCG Binding to SULT1A1PAP Complex.

A.) EGCG Binding to the Nucleotide- bound Subunit. Binding was monitored via changes in the intrinsic fluorescence of SULT1A1 (λex = 290 nm, λgm = 370 nm). Fluorescence intensity is given relative to that in the absence of EGCG (I/I0). Conditions: SULT1A1 (75 nM, dimer), PAP (4.0 μM, 11 ▪ Kd PAP high affinity site and 0.10 ▪ Kd PAP low affinity site), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C. B.) Binding is Biphasic. Fluorescence was measured and plotted as describe in Panel A. Conditions: SULT1A1 (1.0 μM, dimer), PAP (4.0 μM, 11 ▪ Kd PAP high affinity site, 0.10 ▪ Kd PAP low affinity site), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C. The Panel B inset reveals the stoichiometry of EGCG binding at the PAP-containing subunit. The titrations were corrected for EGCG inner filter effects (see, Materials and Methods). Each data point is the average of three independent measurements, and the curved lines are the behaviors predicted by a best-fit, singlesite binding model.
As mentioned previously, one of the caps of E·PAP complex is open, the other is closed (16), and both caps of the unliganded enzyme (E) are open. The coincident affinities of EGCG for E, and one of the E·PAP subunits suggest that the cap of that subunit is open, and, by default, that the high-affinity site resides on the closed-cap subunit.
The E (PAP)2 complex.
EGCG binding to the double-nucleotide complex (E-(PAP)2) was also studied via ligand induced changes in SULT1A1 intrinsic fluorescence. Binding at equilibrium is monophasic (Fig S4) with a stoichiometry of 1.0 EGCG/subunit (Fig S4). The affinity of EGCG (39 ± 3 nM) is indistinguishable from that for the cap-closed subunit of the single-nucleotide species (44 ± 4 nM). This finding seems at odds with our previous work showing that the caps of the E-(PAP)2 complex are open (16). A plausible explanation for this discrepancy is that the caps are open when EGCG binds, and close subsequently. If so, a second phase might be observed in the ligand-binding reaction. Stopped-flow fluorescence studies of the reaction reveal that EGCG binding is indeed bi-phasic (Fig 3A).
Figure 3. EGCG binding to the E·(PAP)2 Complex.

A.) The Binding-Reaction Progress Curve. Binding-induced fluorescence changes were monitored using a stopped-flow fluorimeter (λex = 290 nm, λem ≥ 325 nm (cutoff filter). Reactions were initiated by rapidly mixing (1:1 v/v) a solution containing SULT1A1 (0.25μM, active sites), PAP (0.50 mM), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C with a solution that was identical except that it lacked SULT1A1 and contained EGCG. The curve through the data (red line) is the behavior predicted by the best fit to a double-exponential model. The insert shows the residuals from a single- (black) and doubleexponential (red) fit. The single-exponential model deviates substantially from the data, while the bi-phasic model provides an excellent fit. B.) kobs vs [EGCG] for the First (Fast) Phase. kobs values were obtained at the four indicated EGCG concentrations under the conditions given in Panel A. Each value is the average of three independent determinations. The reactions were pseudo-first-order in EGCG in all cases. C.) EGCG Dissociation from E·(PAP)2·(EGCG)2 Complex. A pulse solution containing SULT1A1 (20 μM, monomer), EGCG (24 μM (4.0 μM free, 100 × Kd)), and PAP (500 μM, 16 ▪ Kd low affinity site), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C, was diluted 200:1 into a chase solution containing 75 μM 4-hydroxytamoxifen (TAM, 100 ▪ Kd cap open form), PAP (0.50 mM, 16 ▪ Kd low affinity site), NaPO4 (50 mM), MgCl2 (5.0 mM), pH 7.5, 25 ± 2°C. The final EGCG concentration is 0.12 μM (0.20 ▪ Kd cap open form). Dissociation was monitored by a change in SULT1A1 fluorescence (λex = 290 nm, λem = 370 nm). Three progress curves (black dots) are superposed. The curve through the data is the behavior predicted by a best-fit, single-exponential model.
The simplest descriptions of biphasic ligand-binding reactions involve a two-step mechanism in which isomerization occurs prior to or following ligand addition. In either case, the on-rate constant for the addition of ligand (ki) is given by the slope of the kobs vs [EGCG] plot for the fast phase of the reaction (Fig 3B), and the y-axis intercept is equal to the sum of the remaining three rate constants (k-1 + k2 + k.−1). Given three independent equations containing these unknowns, each can be calculated by substitution. A second such equation is given by the expression for EGCG binding to the E(PAP)2 complex: Kd = (k-1 · k-2) / (k1 · k2). The third is given by the algebraic description of the EGCG dissociation experiment shown in Fig 3C. Here, a solution in which EGCG is > 99% bound to the E(PAP)2 complex is diluted 200-fold into a solution containing a competitive inhibitor of EGCG (4-hydroxytamoxifen, TAM) at 100-times its Kd. (TAM / EGCG interactions are described in Supplemental). The result is an essentially irreversible dissociation of EGCG for which an observed rate constant (kobs) can be obtained by fitting using a single-exponential equation.
If isomerization occurs subsequent to binding, kobs = k-2 · k-1 / (k-1 + k-2). The four rate constants associated with this mechanism are compiled in Table 2 and shown in Fig 4. These values (i.e., k-1/k1) predict that Kd for EGCG binding to the non-isomerized E(PAP)2 complex, 0.67 μM, is equal, within error, to Kd for binding to the unliganded enzyme (0.68 μM). Here again, the coincidence of values argues strongly in favor of EGCG binding to a cap-open form with subsequent isomerization. If, instead, isomerization occurred prior to binding, the rate constants for binding would be given by the slope and kobs values associated with the data shown in Figs 3A and C, respectively. These constants predicted a Kd of 38 nM, which is within error equal to the Kd measured by equilibrium titration, 39 nM. These values are equivalent only if the enzyme resides entirely in the isomerized form prior to binding, in which case, the second phase is not observed.
Table 2.
Rate Constants for EGCG Binding to E·(PAP)2
| Stepa | Binding | Isomerization |
|---|---|---|
| kfor | 9.9 (0.6) × 106M−1s−1 | 172 s−1 |
| krev | 6.5 s−1 | 10 s−1 |
| Kd | 0.66 μM | |
| Kiso | 17 |
The mechanism is depicted in Fig 4.
Figure 4. The Two-Step Binding Model for EGCG Binding to the E(PAP)>2 Complex.

L and E represent EGCG and E(PAP)2, respectively. The constants were obtained as described in Results and Discussion. The second step appears to be the opening and closure of the active-site cap.
The rate constants associated with the isomerization predict an isomerization equilibrium constant of 17, which is precisely the value associated with the EGCG-induced stabilization of the cap-closed complex. Thus, it is quite likely that the observed isomerization corresponds to cap closure. If so, the second phase is monitoring cap dynamics, and its associated rate constants report on the kinetics of cap opening and closure. The closure rate constant, 172 s−1, is a measure of the rate at which the cap is trapped by EGCG, not the rate at which an open cap “flips” into the closed conformation, which may be far greater. It notable that the cap-closure rate constant is nearly 10-fold greater than chemistry, as determined in presteady state burst studies (31, 45). Hence, closure does not rate-limit chemistry.
Nucleotide Interactions with EEGCG Complexes.
To complete the description of the interactions in this system, PAP binding to EEGCG forms of the enzyme was studied. As is seen in Fig 5A, the equilibrium binding of PAP to E-(EGCG)2 is bi-phasic, and the inset reveals that PAP binds stoichiometrically (1:1) at the high-affinity site. Kd at the low-affinity site, 0.78 μM, was obtained by fitting the titration data at PAP concentrations under which the first site is saturated. Kd at the high-affinity site, 24 nM, was obtained by fitting the Fig 5B data.
Figure 5. PAP Binding to SULT1A1(EGCG)2.

A.) Binding is Biphasic. Binding was monitored via ligand-induced changes in the intrinsic fluorescence of the enzyme (λex = 290 nm, λex = 370 nm). Fluorescence is plotted relative the intensity in the absence of PAP (i.e., I/Io). Conditions: SULT1A1 (0.60 μM, dimer), EGCG (12 μM, 15 ▪ Kd), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2 °C. PAP binds each subunit of the dimer with different affinities. The insert reveals the stoichiometry of PAP binding at the high affinity site; the super-stoichiometric PAP concentrations were used to obtain the affinity at the low-affinity site. B.) PAP Binding to the High-Affinity Site. Binding was monitored as described in Panel A. Conditions: SULT1A1 (50 nM, dimer), EGCG (12 μM, 15 ▪ Kd), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2 °C. Each titration data point is the average of three determinations. Lines through the data are predicted by a best-fit, single-site binding model.
Binding of the first nucleotide to the E-(EGCG)2 complex causes a 33-fold decrease in the affinity of the second. While this value is smaller than the 88-fold decrease associated with binding to unliganded enzyme (16), the allosteric interactions between the nucleotides clearly remain intact, and PAP affinities increase markedly at saturating EGCG — 15- and 40-fold at the high- and low-affinity sites, respectively.
Cap closure encapsulates the nucleotide and prevents its escape (16, 38, 45). Thus, the enhanced nucleotide affinity may be due to EGCG-induced cap closure. This was tested by determining the on- and off-rate constants for PAP binding to the high- and low-affinity sites of the E-(EGCG)2 complex. These rate constants report on the open/closed status of the cap. If, for example, EGCG binding to a cap-open form of the enzyme does not affect the PAP on-rate constant, the disposition of the cap has not been changed - it remains open. In this scenario, any differences in PAP affinities are due to changes in off-rate constants, which provide a quantitative measure of changes in Kiso caused by the binding of EGCG.
PAP on- and off-rate constants for binding to the E-(EGCG)2 complex were obtained from the kobs vs [PAP] data shown in Fig 6. Each kobs value is the average of three independent determinations each obtained from single-exponential fitting of 6–9 averaged, ligand-binding progress curves monitored by stopped-flow fluorescence. As predicted by the 33-fold difference in PAP affinity for the two sites in this complex, the kobs vs [PAP] plot is biphasic, Fig 6A. Experimental conditions were tailored to obtain the rate constants from the two sites, and the results are presented in the Panels B and C. The rate constants, obtained from the slopes and intercepts of the data, are compiled in Table 3. The accuracy of the constants was assessed by using them to predict the kobs values associated with Fig 6A. The predicted values, indicated by red dots, match the experimental data well.
Figure 6. Presteady State binding of PAP to SULT1A1(EGCG)2.

A.) kobs vs [PAP]. The binding- reaction progress curves were monitored using a stopped-flow fluorimeter (λex = 290 nm, λem ≥ 325 nm (cutoff filter). kobs values (black dots) were obtained by fitting 6–9 averaged progress curves using a single-exponential equation. Each kobs value is the average of three independent determinations. Red dots indicate kobs values predicted using the kon and koff values obtained from the experiments associated with Panels B and C. B.) Binding at the High-Affinity Site (kobs vs [PAP]). Reactions were initiated by mixing (1:1) a solution containing SULT1A1 (75 nM, dimer), EGCG (12 μM, 18 ▪ Kd cap open form), MgCh2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C, with a solution that was identical except that it lacked SULT1A1 and contained PAP at the indicated concentrations. C.) Binding at the Low-Affinity Site (kobs vs [PAP]). Reactions were initiated by rapidly mixing (1:1) a solution containing SULT1A1 (1.0 μM, dimer), 1.0 μM PAP (42 ▪ Kd high affinity and 1.3 ▪ Kd low affmity), EGCG (12 μm, 18 ▪ Kd cap open form), MgCl2 (5.0 mM), NaPO4 (50 mM), pH 7.5, 25 ± 2°C, with a solution that was identical except that it lacked SULT1A1 and contained PAP at the indicated concentrations. Pre-equilibration at [PAP] = [SULT1A1]dimer saturates the high-affinity nucleotide-binding site and thus prevents it from contributing to the low-affinity binding measurements.
Table 3.
Ligand-Binding Rate Constants
| Ligand | Enzyme Species | kon(μM−1s−1) | koff (s−1) | Kd(μM) (koff/Kon) |
|---|---|---|---|---|
| EGCG | E | 9.8 (0.3)a | 6.9 (0.2) | 0.71 (0.05) |
| EGCG | E·PAP | 13.5 (0.7) | 0.62 (0.03) | 0.046 (0.005) |
| EGCG | E·PAP·EGCG | 17.1 (0.3) | 13.9 (0.2) | 0.81 (0.02) |
| PAPb | E | 2.0 (0.1) | 0.70 (0.05) | 0.37 (0.04) |
| PAPb | E·PAP | 0.96 (0.01) | 29 (1) | 30 (3) |
| PAP | E·(EGCG)2 | 1.7 (0.1) | 0.038 (0.002) | 0.022 (0.003) |
| PAP | E·(EGCG)2·PAP | 0.88 (0.06) | 0.77 (0.04) | 0.88 (0.1) |
Values in parentheses indicate standard error.
Determined previously (16).
In interpreting the rate-constants, it is important to realize that if the PAP binding sites are identical toward addition of the first nucleotide, the on-rate constant for the first step will appear to be twice that of the second because the second site is not present at detectible levels until the first nucleotide has added, at which point, the concentration of binding sites has decreased by a factor of two. This is precisely what is observed. kon for binding to the first site (1.7 μM−1 s−1) is twice that of the second (0.88 μM−1 s−1). This also true for PAP binding to E (2.0 μM−1 s−1, and 0.70 μM−1 s−1) - a species in which both caps are open (16). Here again, coincident values of kon for PAP binding to E and E-(EGCG)2 strongly suggests that the caps of the E-(EGCG)2 complex are open.
The differences in PAP affinity for the high- and low-affinity sites of E-(EGCG)2 are due solely to differences in off-rate constants. A similar pattern is seen when comparing the binding of PAP to enzyme forms in which the cap eithers closes normally or is “held open” by saturation with a large substrate (16). The on-rate constants are identical for both species; however, when the cap is allowed to close normally, the off-rate constant decreases relative to that of the held- open complex by a factor of 21, which is equal to the equilibrium constant for cap closure (Kiso). Based on these data and structures indicating that the closed cap prevents nucleotide release, it was concluded that PAP departs exclusively from cap-open forms (16). Extending this model to the E・(EGCG)2 complex: koff for PAP departure from the high affinity site (0.038 s−1) of the complex is 18-fold less than that from E; hence, its cap closes 18-fold more “tightly,” and Kiso for the cap of the high-affinity subunit in the E・(EGCG)2 complex is 378 (i.e., 21 ▪ 18).
SULT1A1 turnover varies considerably with acceptor (46). “Good” substrates turnover at ~ 80 s−1, “poor” substrates are much slower, ~ 1s−1. The rate constant for PAP release at saturating EGCG, 0.038 s−1, is far slower that either class of acceptor. Thus, it is likely that the trapping of PAP by EGCG and its subsequent slow release is the primary mechanism of EGCG inhibition.
EGCG Affects Acceptor Binding and Cap Closure.
To independently assess the effects of EGCG on cap closure at acceptor1binding pockets, the affinities of large and small substrates for the E and E·(PAP)2 complexes were determined in equilibrium1binding studies in the presence and absence of saturating EGCG. The affinities of TAM and E2 (large and small acceptors, respectively) for E were quite similar, neither was affected by addition of EGCG (Table 4), indicating the cap is open in these complexes. On the contrary, the affinity of the large acceptor decreases substantially (23-fold) when EGCG binds to E·(PAP)2, and the affinity of the small acceptor was not affected. EGCG clearly closes the cap of the E·(PAP)2 complex, but does not do so detectibly in the nucleotide1free complex. Thus, although EGCG clearly stabilizes the cap1 closed form of the enzyme, this stabilization is not sufficient to shift a detectible fraction of the enzyme into the cap1closed conformation.
Table 4.
EGCG Effects on Cap Closure
| Enzyme Form | E | E·(EGCG)2 | E·(PAP)2 | E·(PAP)2·(EGCG)2 |
|---|---|---|---|---|
| Acceptor | Kd(μM) | |||
| TAMa | 0.65 (0.05)c | 0.67 (0.01) | 0.70 (0.05) | 16 (1.4) |
| E2b | 1.0 (0.06) | 1.0 (0.04) | 1.0 (0.2) | 1.0 (0.1) |
TAM is a large substrate.
E2 is a small substrate.
Values in parentheses indicate error.
The Complete Binding Scheme.
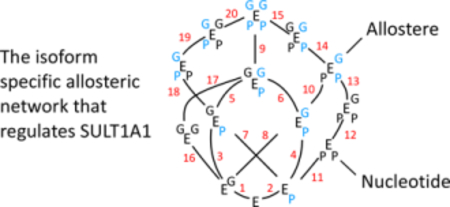
The foregoing experiments reveal that at least fourteen enzyme forms are involved in the binding and interactions of EGCG and PAP. These species, depicted in Fig 7, are interconnected by twenty steps, which are labelled, and the associated equilibrium constants and ΔG values are compiled in Table S1. The right- and left-hand sides of each E symbol represent different subunits of the dimer, and the upper and lower corners represent EGCG (G) and PAP (P) binding pockets. If a ligand is blue, the cap on the subunit to which it is bound is closed; if it’s black, it’s open. The caps of the unliganded enzyme are open. The system is fully determined hence the energetic differences between any of the 193 possible pairs of species can be calculated. Equilibria 3, 5 and 7 involve the G E P complex, in which G and P are bound at apposing subunits. This species cannot be detected due to the fact that G binds 17-fold more tightly to the P-bound subunit because of its closed cap. These constants can, however, be calculate by conservation of energy. Similarly, the energetics of reaction 8 were calculated because E G is not readily isolated from G E G due to the fact that the first and second G add with identical affinities.
Figure 7. The Complete Interaction Scheme.

The fourteen enzyme forms involved in the bindii and interactions of EGCG (G) and PAP (P) are shown. The enzyme symbol, E, is “sided” - the two right- and left-hand corners represent binding sites on separate subunits of the SULT1A1 dimer. Upper and lower corners represent the G- and P-binding sites, respectively. The 20 binding steps that interconvert the species are numbered, and the corresponding equilibrium constants can be found in Table S1. A blue letter indicates that that ligand is bound to a subunil whose cap is closed.
The allosteric web of interactions between EGCG and SULT1A1 is multifaceted, subunit specific and dictated by nucleotide occupancy and cap disposition. PAPS levels vary with tissue and cell type over a range of concentrations expected to produce both single and double nucleotide-bound species. Thus, EGCG inhibition will vary in a tissue-dependent fashion according to Fig 7 and Table S1.
Conclusions
EGCG and PAP affinities and stoichiometries were determined for a series of SULT1A1 complexes and these measurements culminated in a complete quantitative binding-and- interaction scheme for the two metabolites. EGCG was shown to bind, but not turnover, at the SULT1A1 active site, and to be a substrate for SULT2A1 - this is the first EGCG-cognate SULT isoform to be identified. EGCG binds 17-fold more tightly to cap-closed forms of the enzyme, and, in so doing, increases Kiso for cap closure by that same amount. Studies with the E·(PAP)2 complex, where both caps are open, reveal that EGCG binding is followed by an isomerization whose energetics exactly match the EGCG stabilization of the cap. Thus, EGCG appears to trap the cap in the closed position. It proved possible to directly monitor cap opening and closure and obtain the associated rate-constants, which reveal that closure does not rate limit chemistry. Finally, since cap closure encapsulates the nucleotide, EGCG stabilization of the cap reduces the rate at which nucleotide escapes from the enzyme to more than two orders of magnitude below the turnover of the uninhibited enzyme; thus, nucleotide encapsulation appears to be the mechanism of inhibition.
Among very closely sequence-related members of the human SULT1A isoforms, EGCG is highly selective for 1A1. The caps of these isoforms differ suggesting that specificity differences may correlate with changes in cap residues. If EGCG interacts directly with these residues, it may be possible to craft EGCG derivatives to control SULT caps in an isoform specific manner - powerful tools for controlling SULT activity and its associated biology. We are hopeful that our ongoing structural efforts will soon reveal the molecular basis of EGCG binding and specificity.
Supplementary Material
Acknowledgments
Supported by the National Institutes of Health Grant GM10158
Abbreviations:
- E2
estradiol
- HEPES
N-2-hydroxyethylpiperazine-N’−2-ethanesulfonic acid
- 1-HP
1-hydroxypyrene
- MBP
maltose binding protein
- PAP
3’, 5’-diphosphoadenosine
- TAM
4-hydroxytamoxifen
Footnotes
Supporting Information Available
- Study of the interaction between EGCG and TAM.
- Supplementary Figures
- Supplementary Table
References
- 1.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, and Gustafsson JA (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta., Endocrinology 138, 863–870. [DOI] [PubMed] [Google Scholar]
- 2.Visser TJ (1994) Role of sulfation in thyroid hormone metabolism, Chem Biol Interact 92, 293–303. [DOI] [PubMed] [Google Scholar]
- 3.Eisenhofer G, Coughtrie MW, and Goldstein DS (1999) Dopamine sulphate: an enigma resolved, Clin Exp Pharmacol Physiol Suppl 26, S41–53. [PubMed] [Google Scholar]
- 4.Blanchard RL, Freimuth RR, Buck J, Weinshilboum RM, and Coughtrie MW (2004) A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily, Pharmacogenetics 14, 199–211. [DOI] [PubMed] [Google Scholar]
- 5.Nowell S, and Falany CN (2006) Pharmacogenetics of human cytosolic sulfotransferases, Oncogene 25, 1673–1678. [DOI] [PubMed] [Google Scholar]
- 6.Pacifici GM, Franchi M, Colizzi C, Giuliani L, and Rane A (1988) Sulfotransferase in humans: development and tissue distribution, Pharmacology 36, 411–419. [DOI] [PubMed] [Google Scholar]
- 7.Riches Z, Stanley EL, Bloomer JC, and Coughtrie MW (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”, DrugMetab Dispos 37, 2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ihunnah CA, Wada T, Philips BJ, Ravuri SK, Gibbs RB, Kirisci L, Rubin JP, Marra KG, and Xie W (2014) Estrogen sulfotransferase/SULT1E1 promotes human adipogenesis, Mol Cell Biol 34, 1682–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wada T, Ihunnah CA, Gao J, Chai X, Zeng S, Philips BJ, Rubin JP, Marra KG, and Xie W (2011) Estrogen sulfotransferase inhibits adipocyte differentiation, Mol Endocrinol 25, 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura Y, Gang HX, Suzuki T, Sasano H, and Rainey WE (2009) Adrenal changes associated with adrenarche, Rev Endocr Metab Disord 10, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stelzer C, Brimmer A, Hermanns P, Zabel B, and Dietz UH (2007) Expression profile of Papss2 (3’-phosphoadenosine 5’-phosphosulfate synthase 2) during cartilage formation and skeletal development in the mouse embryo, Dev Dyn 236, 1313–1318. [DOI] [PubMed] [Google Scholar]
- 12.Lee SA, Choi JY, Shin CS, Hong YC, Chung H, and Kang D (2006) SULT1E1 genetic polymorphisms modified the association between phytoestrogen consumption and bone mineral density in healthy Korean women, Calcif Tissue Int 79, 152–159. [DOI] [PubMed] [Google Scholar]
- 13.Salman ED, Kadlubar SA, and Falany CN (2009) Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain, Drug Metab Dispos 37, 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dousa MK, and Tyce GM (1988) Free and conjugated plasma catecholamines, DOPA and 3-O-methyldopa in humans and in various animal species, Proc Soc Exp Biol Med 188, 427–434. [DOI] [PubMed] [Google Scholar]
- 15.Cook I, Wang T, Falany CN, and Leyh TS (2013) High accuracy in silico sulfotransferase models, J Biol Chem 288, 34494–34501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Cook I, and Leyh TS (2014) 3’-Phosphoadenosine 5’-phosphosulfate allosterically regulates sulfotransferase turnover, Biochemistry 53, 6893–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teubner W, Meinl W, Florian S, Kretzschmar M, and Glatt H (2007) Identification and localization of soluble sulfotransferases in the human gastrointestinal tract., Biochem J404, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook I, Wang T, Falany CN, and Leyh TS (2015) The allosteric binding sites of sulfotransferase 1A1, DrugMetab Dispos 43, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coughtrie MW, and Johnston LE (2001) Interactions between dietary chemicals and human sulfotransferases-molecular mechanisms and clinical significance, Drug Metab Dispos 29, 522–528. [PubMed] [Google Scholar]
- 20.Vietri M, De Santi C, Pietrabissa A, Mosca F, and Pacifici GM (2000) Inhibition of human liver phenol sulfotransferase by nonsteroidal anti-inflammatory drugs, Eur J Clin Pharmacol 56, 81–87. [DOI] [PubMed] [Google Scholar]
- 21.Renouf M, Guy P, Marmet C, Longet K, Fraering AL, Moulin J, Barron D, Dionisi F, Cavin C, Steiling H, and Williamson G (2010) Plasma appearance and correlation between coffee and green tea metabolites in human subjects, Br J Nutr 104, 1635–1640. [DOI] [PubMed] [Google Scholar]
- 22.Arts IC, van De Putte B, and Hollman PC (2000) Catechin contents of foods commonly consumed in The Netherlands. 2. Tea, wine, fruit juices, and chocolate milk, J Agric Food Chem 48, 1752–1757. [DOI] [PubMed] [Google Scholar]
- 23.Cordero-Herrera I, Martin M, Goya L, and Ramos S (2014) Cocoa flavonoids attenuate high glucose-induced insulin signalling blockade and modulate glucose uptake and production in human HepG2 cells, Food Chem Toxicol 64, 10–19. [DOI] [PubMed] [Google Scholar]
- 24.Sabhapondit S, Karak T, Bhuyan LP, Goswami BC, and Hazarika M (2012) Diversity of catechin in northeast Indian tea cultivars, ScientificWorldJournal 2012, 485193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfram S (2007) Effects of green tea and EGCG on cardiovascular and metabolic health, JAm CollNutr 26, 373S–388S. [DOI] [PubMed] [Google Scholar]
- 26.(2015) Tea Global Corporate Strategy: Diversity and Tea Experience, p 6, Euromonitor International Ltd, London, UK. [Google Scholar]
- 27.Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, and Alberts DS (2003) Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals, Clin Cancer Res 9, 3312–3319. [PubMed] [Google Scholar]
- 28.Lee MJ, Wang ZY, Li H, Chen L, Sun Y, Gobbo S, Balentine DA, and Yang CS (1995) Analysis of plasma and urinary tea polyphenols in human subjects, Cancer Epidemiol Biomarkers Prev 4, 393–399. [PubMed] [Google Scholar]
- 29.Glatt H (1997) Sulfation and sulfotransferases 4: bioactivation of mutagens via sulfation., FASEB J11, 314–321. [DOI] [PubMed] [Google Scholar]
- 30.Glatt H, Boeing H, Engelke CE, Ma L, Kuhlow A, Pabel U, Pomplun D, Teubner W, and Meinl W (2001) Human cytosolic sulphotransferases: genetics, characteristics, toxicological aspects., Mutat Res 482, 27–40. [DOI] [PubMed] [Google Scholar]
- 31.Sun M, and Leyh TS (2010) The human estrogen sulfotransferase: a half-site reactive enzyme, Biochemistry 49, 4779–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook I, Wang T, Almo SC, Kim J, Falany CN, and Leyh TS (2013) The gate that governs sulfotransferase selectivity, Biochemistry 52, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnett AC, Tsvetanov S, Gamage N, Martin JL, Duggleby RG, and McManus ME (2004) Active site mutations and substrate inhibition in human sulfotransferase 1A1 and 1A3, The Journal of biological chemistry 279, 18799–18805. [DOI] [PubMed] [Google Scholar]
- 34.Tyapochkin E, Cook PF, and Chen G (2009) para-Nitrophenyl sulfate activation of human sulfotransferase 1A1 is consistent with intercepting the E[middle dot]PAP complex and reformation of E[middle dot]PAPS, The Journal of biological chemistry 284, 29357–29364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang ZX (1995) An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule, FEBS Lett 360, 111–114. [DOI] [PubMed] [Google Scholar]
- 36.Wei J, Tang QX, Varlamova O, Roche C, Lee R, and Leyh TS (2002) Cysteine biosynthetic enzymes are the pieces of a metabolic energy pump, Biochemistry 41, 8493–8498. [DOI] [PubMed] [Google Scholar]
- 37.Cook I, Wang T, Almo SC, Kim J, Falany CN, and Leyh TS (2013) Testing the Sulfotransferase Molecular Pore Hypothesis, J Biol Chem 288, 8619–8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T, Cook I, Falany CN, and Leyh TS (2014) Paradigms of sulfotransferase catalysis: the mechanism of SULT2A1, J Biol Chem 289, 26474–26480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cook I, Wang T, Falany CN, and Leyh TS (2012) A Nucleotide-Gated Molecular Pore Selects Sulfotransferase Substrates, Biochemistry 51, 5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alnouti Y, and Klaassen CD (2006) Tissue distribution and ontogeny of sulfotransferase enzymes in mice., ToxicolSci 93, 242–255. [DOI] [PubMed] [Google Scholar]
- 41.Klaassen CD, and Boles JW (1997) Sulfation and sulfotransferases 5: the importance of 3’-phosphoadenosine 5’-phosphosulfate (PAPS) in the regulation of sulfation., Faseb J11, 404–418. [DOI] [PubMed] [Google Scholar]
- 42.Ma B, Shou M, and Schrag ML (2003) Solvent effect on cDNA-expressed human sulfotransferase (SULT) activities in vitro, Drug Metab Dispos 31, 1300–1305. [DOI] [PubMed] [Google Scholar]
- 43.Weber G (1953) Rotational Brownian motion and polarization of the fluorescence of solutions, Adv Protein Chem 8, 415–459. [DOI] [PubMed] [Google Scholar]
- 44.Sang S, Lee MJ, Yang I, Buckley B, and Yang CS (2008) Human urinary metabolite profile of tea polyphenols analyzed by liquid chromatography/electrospray ionization tandem mass spectrometry with data-dependent acquisition, Rapid Commun Mass Spectrom 22, 1567–1578. [DOI] [PubMed] [Google Scholar]
- 45.Wang T, Cook I, and Leyh T (2016) The Design and Interpretation of Human SULT1A1 Assays, Drug Metab Dispos 44, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook I, Wang T, and Leyh TS (2015) Sulfotransferase 1A1 Substrate Selectivity: A Molecular Clamp Mechanism, Biochemistry 54, 6114–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.