

Abstract
Improper protein folding and trafficking are common pathological events in neurodegenerative diseases that result in the toxic accumulation of misfolded proteins within the lumen of the endoplasmic reticulum (ER). While low-level stimulation of the unfolded protein response (UPR) is protective, sustained UPR activation resulting from prolonged ER stress can promote neurotoxicity. The cell-autonomous mechanisms of the UPR have been extensively characterized. However, the cell-extrinsic role of the UPR under physiological and pathological states in the central nervous system (CNS) remains to be elucidated. To begin to address this, we evaluated if transferring conditioned media between ER stressed astrocytes and neurons could modulate their functional characteristics. Our results indicate that ER stressed astrocytes and neurons secrete a molecule(s) with lipid characteristics which regulates both inflammatory and ER stress responses in other astrocytes, neurons, and microglia in vitro. Initial exposure to this stress factor(s) confers resistance against subsequent ER stress to neurons. However, persistent exposure to this unidentified mediator(s) suppresses the initial protective effect and becomes cytotoxic. Overall, these findings provide insight into the cell-nonautonomous influence of ER stress on cells of the central nervous system.
Keywords: Astrocytes, neurons, ER stress, inflammation, unfolded protein response
Summary Figure:
Previous studies have shown that endoplasmic reticulum (ER) stress can have cell-nonautonomous effects on neighboring and distal tissues. ER stress is also a common feature of neurological diseases. Therefore, we hypothesize that ER stress is transmissible between neural and glial cells. Our data suggest that neurons and glia under ER stressed conditions can secrete factor(s) that promote an ER stress response in unstressed cells. Transmission of ER stress can stimulate an inflammatory reaction, adaptation or cell death depending on cell type and duration. These finding may point to mechanism by which ER stress contributes to neuronal demise in neurodegenerative diseases.
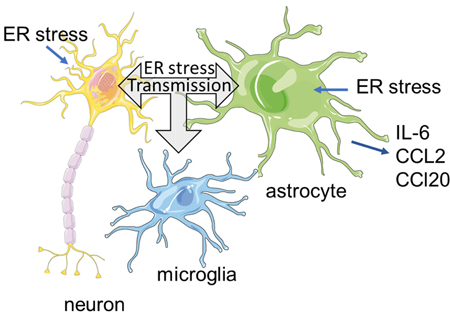
Introduction
Improper protein folding is a common pathological feature observed in various cell types in neurodegenerative diseases, such as Alzheimer’s disease, Parki2nson’s disease and Amyotrophic Lateral Sclerosis (Ciechanover & Kwon 2015, Sprenkle et al. 2017). The endoplasmic reticulum (ER) serves as a specialized compartment that coordinates multiple events to effectively facilitate the folding of secretory and membrane proteins, and to remove terminally misfolded proteins within the lumen of the ER (Araki & Nagata 2011). However, the protein quality control mechanisms of the ER are impaired during the course of neurodegeneration, resulting in the accumulation of misfolded proteins within the ER lumen. Mammalian cells respond to disturbances in ER homeostasis, or ER stress, by initiating a highly conserved cellular stress response called the unfolded protein response (UPR) to restore ER homeostasis (Ron & Walter 2007). The UPR is mediated by the ER transmembrane proteins protein kinase R-like ER kinase (PERK), inositol requiring enzyme 1 (IRE-1α) and activating transcription factor (ATF)6, each of which transduces distinct signaling cascades to coordinate transcriptional and translational alterations that enhance protein folding and promote adaptation to ER stress (Walter & Ron 2011).
Astrocytes, microglia and oligodendrocytes constitute a large proportion of the glial cell population within the central nervous system (CNS), and by definition are integral components which mediate neuronal function and integrity (Araque & Navarrete 2010). Biological challenges which overwhelm the compensatory mechanisms of the UPR can initiate astrocyte-derived inflammatory stress signaling pathways to mediate tissue-wide stress responses. This mode of intercellular communication is highly beneficial within the CNS, as it promotes debris clearance and aids in tissue repair after acute insult (Glass et al. 2010). However, prolonged proteome dysfunction resulting from the cell’s inability to resolve protein misfolding in the ER lumen engenders sustained UPR-dependent inflammation, which may contribute to the neurotoxic milieu commonly associated with neuropathology (Meares et al. 2014). We recently reported that ER stressed astrocytes could elicit a feed-forward inflammatory loop by polarizing microglia to an inflammatory phenotype that produce cytokines which can synergize with ER stress in astrocytes (Meares et al. 2014). Recently, neuronal ER stress has been linked to microglia activation in a model of traumatic brain injury (Harvey et al. 2015). Moreover, the highly secretory nature of oligodendrocytes predisposes them to ER stress-induced apoptosis (Lin & Popko 2009). In all, perturbations in glial cell function can aggravate neuropathology by orchestrating neuronal death through cell-nonautonomous means.
It is known that ER stress can be ‘transmitted’ between distinct cell types and may contribute to chronic disease pathology. For instance, ER stressed tumor cells can upregulate canonical ER stress responses in myeloid cells to promote tumor growth and cancer progression through the secretion of an unknown soluble molecule (Mahadevan et al. 2011). Interestingly, low-level activation of the UPR plays an essential role in maintaining vital biological processes against upcoming stress by heightening the resolution potential of the ER stress response (Inagi et al. 2008, Tsang et al. 2010). Additionally, expression of the UPR-mediator sXBP1 in pro-opiomelanocortin neurons engages distal metabolic pathways to regulate glucose homeostasis (Williams et al. 2014). The fact that transmissible ER stress has been observed across different cell populations suggests that it may be important for conferring cellular protection to surrounding cells in response to various genres of organismal stress (Zanetti et al. 2016, Zhang et al. 2017, Mahadevan et al. 2011, Taylor et al. 2014). Under diseased states, however, this homeostatic mechanism maybe compromised to promote disease progression.
In this study, we hypothesize that ER stressed astrocytes and neurons can transmit ER stress and enhance stress resistance in receiver cells. Here we show that ER stressed astrocyte conditioned media (ACM) upregulates the expression of proteins associated with all three branches of the UPR. Further, interleukin-6 (IL-6), C-C motif chemokine ligand (CCL)2 and CCL20 mRNA expression was significantly enhanced in resting astrocytes incubated with ER stressed ACM in a PERK-dependent fashion. Mild exposure to ER stressed ACM conferred stress resistance to neurons against acute thapsigargin (Thaps)-induced ER stress, as neurons expressed less pro-apoptotic factors and exhibited significantly less caspase-3 activity. Nevertheless, this protective effect was partially lost after prolonged exposure to Thaps. Collectively, these findings present novel insights into the cell-extrinsic regulatory functions of the UPR.
Materials and Methods
Reagents
Ac-DEVD-AMC (N-Acetyl-Asp-Glu-Val-Asp-7-amido-4-Methylcoumarin; 556449) was purchased from BD Biociences. Antibody (Ab) for XBP1 (RRID:AB_778939) was from Abcam. Ab for GAPDH (MAB374, RRID:AB_2107445) was from EMD Millipore Corp. Abs for JNK (9252, RRID:AB_2250373), P-JNK (4668, RRID:AB_823588), GFAP (12389, RRID:AB_2631098), P-eIF2α (3389) and eIF2α (5324) were from Cell Signaling Technology. Abs for ATF4 (sc-200, RRID:AB_2058752), CHOP (sc-7351, RRID:AB_627411) and GRP78 (sc-13968, RRID:AB_2119991) were from Santa Cruz Biotechnology, Inc, Iba1 was from Novus Biologicals (NB100–1028SS, RRID:AB_521594). Antibodies were validated by immunoblotting. Thapsigargin (Thaps; 586005–1MG) and tunicamycin (65–438-010MG) were from Calbiochem. Dulbecco’s modified Eagle’s medium (DMEM; MT10013CV), fetal bovine serum (FBS; MT25010CV), HEPES (MT25060CI), nonessential amino acids (MT25025CI), L-glutamine (MT2500CI), and penicillin-streptomycin (MT30002CI) were from Cellgro. Gentamicin (G1522–10ML) was purchased from Lonza. Immobilized proteinase K (82452–1G) was purchased from Sigma-Aldridge. The Pierce Lactate Dehydrogenase (LDH) Cytotoxicity Assay Kit (88953) was purchased from ThermoFisher Scientific. The probe-based qPCR primers that were purchased from Integrated DNA Technologies are as follows: CHOP (151620129), GRP78 (151620137), IL-6 (193420201), CCL2 (196214166), CCL20 (196214174), ATF4 (195069594), HPRT (193420197)
Mice and Primary Cell Preparations
C57BL/6J mice (Jackson Laboratories, RRID:IMSR_JAX:000664) were bred and housed in the animal facility at West Virginia University under the care of the animal resources program. Mice were housed at 21°c on a 12 h light-dark cycle with free access to food and water. One-day old pups were euthanized by decapitation. All procedures were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study was approved by the WVU animal care and use committee (#15–0307). Primary murine astrocyte cultures were prepared as previously described (Meares et al. 2014). Briefly, cerebra were removed from one day old pups and placed in cold media. Meninges were removed and tissue triturated to generate a cell suspension. The cell suspension was passed through a 100 μm strainer and washed 2× with media, centrifuging at 300 × g between washes. Astrocytes were then cultured in DMEM with 10% FBS, 16 mM HEPES, 1 × nonessential amino acids, 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamicin. Astrocytes were separated from microglia by shaking at 200 rpm for 2 h. The HT-22 mouse hippocampal neuronal cell line (RRID CVCL_0321) was provided by The Salk Institute for Biological Research. HT-22 cells were cultured in DMEM with 5% FBS, 16 mM HEPES, 1 × nonessential amino acids, 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin.
Conditioned Media
Primary murine astrocyte cultures were treated with 1.0 μM Thaps for 2 h. Cells were washed 3X with Dulbecco’s phosphate-buffered saline (PBS), and then incubated in fresh DMEM media for 24 h. Conditioned media was centrifuged at 1,600 rpm for 10 min to pellet cellular debris. Media was collected and used for conditioned media experiments. Untreated cells concomitantly underwent the procedure. The vehicle (control) was treated with DMSO.
Immunoblotting Analysis
Cells were washed twice with PBS and lysed with immunoprecipitation (IP) lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, and 1 × phosphatase/protease inhibitor cocktail (Pierce). Protein concentrations were determined using the bicinchonicic acid assay (BCA) assay (Pierce). Equal amounts of protein from each sample were solubilized in Laemmli sample buffer (2% SDS) and heated for 5 min at 95°C. Proteins were separated by 8% or 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose, and the membranes were blocked in 5% milk in wash buffer (20 mM Tris base, 137 mM NaCl and 0.05% Tween-20 (TBST)) followed by an overnight incubation at 4°C with primary Ab diluted in 5% bovine serum albumin (BSA) or milk, according to the manufacturer’s recommendation. Primary antibodies were diluted as follows; GRP78 1:250, XBP1 1:1000, CHOP 1:250, GAPDH 1:25000, ATF4 1:1000, Iba1 1:1000, GFAP 1:3000, PERK 1:2000, P-eIF2α 1:2000, eIF2α 1:3000, P-JNK 1:2000 and JNK 1:5000. Membranes were washed for 1 h with frequent changes to TBST. Horseradish peroxidase-conjugated donkey anti-rabbit or donkey anti-mouse (1:2000 dilution) secondary Ab was incubated for 1 h at room temperature. Membranes were washed for 1 h with frequent changes to TBST, followed by detection with enhanced chemiluminescence.
Reverse Transcription-quantitative Polymerase Chain Reaction (qRT-PCR)
RNA was isolated using TRIzol (Fisher). RNA was quantified using a NanoDrop system (Fisher). One microgram of RNA was used for cDNA synthesis using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega). The cDNA was analyzed by quantitative PCR performed using probe-based gene expression assays using Agilent Stratagene Mx3005P or Quant studio 5. Reactions were carried out in 20 μl and analyzed using the threshold cycle (ΔΔCT) method.
Determining Cell Viability
Caspase-3 Activity Assay
Cleaved caspase-3 assay was carried out using a modified method previously described (Meares et al. 2010, Carrasco et al. 2003). Briefly, 200 μL of assay buffer (150 mM HEPES, 450 mM NaCl, 150 mM KCl, 30 mM MgCl2, 1.5% NP-40 alternative, 1.2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), 0.03% 3-[(3-cholamidopropyl)dimethylammonio-1-propanessulonate (CHAPS), and 30% sucrose, 1.5 mM of the fluorogenic substrate Ac-DEVD-AMC, 1 M Dithiothreitol (DTT) and 100 mM Phenylmethysulfonyl (PMSF)) was added to cells plus 400 μL media following treatment. After incubation at 37°C for 1 h, the release of AMC was measured with excitation at 360 nm and emission at 460 nm using fluorescence spectrophotometry (BioTek).
LDH Assay
The LDH assay was carried out according to the manufacturer’s instructions and as describe previously (Korzeniewski & Callewaert 1983). In brief, 50 μL supernatant from cells treated with the indicated compounds/conditioned media and 50 uL of reaction mixture (Substrate mix and Assay Buffer) was transferred to one well in a 96-well plate. Following incubation at room temperature for 30 min, 50 uL stop solution was added to each well. Extracellular LDH in the media was quantified by measuring the absorbance of released formazan dye at 490 nm with a subtraction wavelength of 680 nm. Relative LDH activity was calculated using the following equation:
Treatment LDH activity (treated – media alone) / maximal LDH activity (lysis buffer – media alone)
Annexin V Assay
The realtime Glo assay was used to assess phosphatidylserine externalization (Promega JA1011). Twenty thousand cells were plated in a 96-well clear bottom plate and incubated with astrocyte conditioned media for 18 h. Assay reagents were then diluted in the respective medium and added to the cells for an additional 6 h (24 h total exposure to conditioned media). Luminescence was quantified using a Synergy HTX plate reader from BioTek.
Microparticle Isolation by Ultracentrifugation
Conditioned media was collected from cells and centrifuged at 1,600 × g at 4°C for 5 minutes to remove cellular debris. The centrifuged conditioned media was collected separately from the pellet and used for ultracentrifugation. Samples were spun at 160,000 × g at 4°C for 1 h using Nalgene Oak Ridge conical tubes (Thermo Fisher Scientific). Conditioned media was collected and the pellet was reconstituted in Dulbecco’s PBS. Both the ultracentrifuged media and microparticale-containing solution was used for the conditioned media experiments. Enrichment of microparticles was quantified using NanoSight analysis.
Treatment of Conditioned Media with Proteinase K or Cleanascite
Proteinase K (Sigma), which contained proteinase K enzyme covalently attached to agarose beads, was used to digest protein molecules within the conditioned media. In brief, a 1.5 mL tube containing 1 mL B-27 supplemented media serum-free conditioned media and 100 μL proteinase K slurry (proteinase K + PBS) incubated in a 37°C thermomixer at 500 rpm for 1 h. Samples were then centrifuged at 2,000 × g for 2 min to pellet the agarose, and thus the proteinase K enzyme. The proteinase K-treated conditioned media was incubated with the indicated receiver cells to determine if the ER stress-inducing stress factor was a protein. Similarly, 100 μl of Cleanascite slurry was added to 1 ml of conditioned medium and incubated at RT with end-over-end mixing for 1 h followed by centrifugation.
Statistical Analysis and Study Design
The study was not pre-registered. Researchers were not blinded to the experimental conditions. Animals were only used for isolation of astrocytes and thus were not randomized. Statistical differences between experimental groups were determined using GraphPad Prism. Shapiro-Wilk test was used to determine if data were normally distributed, no test for outliers was conducted. One-way ANOVA was used to determine statistically significant differences between the means of three or more independent biological groups (N) in which the data were normally distributed. Post-hoc Tukey’s test was used for multiple comparisons. Two-way ANOVA with Bonferroni’s multiple comparisons test was used to determine statistically significant differences between groups with more than one independent variable in which the data were normally distributed. Mann-Whitney U test was used when data were non-parametric. Data are means of 3 or more independent experiments ± standard deviation. p < 0.05 was considered statistically significant.
Data Availability
Data sets are available from the corresponding author on request.
Results
ER stress is Transmissible among Cells of the CNS in vitro.
We recently demonstrated that ER stressed astrocytes can upregulate inflammatory gene expression in microglia (Meares et al. 2014). Because it has been shown that transmissible ER stress maybe a relevant phenomenon in cancer (Mahadevan et al. 2011), we investigated whether ER stress generated in astrocytes could trigger the secretion of molecules which, in turn, regulate ER stress responses in unstressed CNS cells. To do this, we produced, then transferred ER stressed astrocyte conditioned medium (ACM) to unstressed astrocytes and neurons. As described in Figure 1A, astrocytes were stimulated with the ER stress-inducing agent thapsigargin (Thaps), a non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (Lytton et al. 1991), for 2 h to induce transient ER stress. The ability of Thaps to induce ER stress in astrocytes has been shown in previous studies (Meares et al. 2014, Guthrie et al. 2016). We followed the treatment with a 24 h wash out period with fresh media. This step is critical as any mediator secreted from ER stressed astrocytes would be collected in the media without the presence of Thaps. Therefore, we could determine whether ER stressed astrocytes could modulate ER stress responses in unstressed cells through a paracrine mechanism by incubating different receiver cells with the conditioned media. No difference in lactate dehydrogenase (LDH) activity was detected in the conditioned media, indicating that the secretory profile of the astrocytes during the washout period was not a product of cell death (Figure 1A). This finding is consistent with our previous observations that astrocyte viability was not compromised when stimulated with various concentrations of Thaps for 24 h (Meares et al. 2014).
Figure 1. ER stressed astrocytes secrete a molecule(s) which upregulate ER stress responses in astrocytes and neurons.
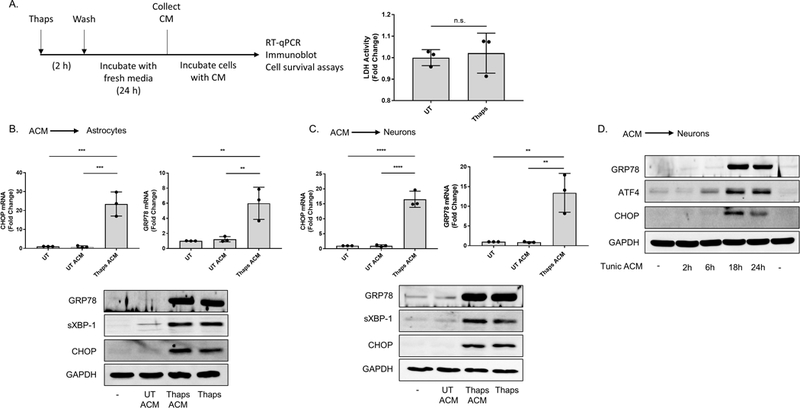
(A) Outline for the astrocyte conditioned media (ACM) experiments. Following treatment with the vehicle (DMSO) or 1.0 μM thapsigargin (Thaps) for 2 h, primary murine astrocyte cultures were washed with PBS three times, then incubated in fresh media for 24 h. After this period, the media (ACM) was collected. Lactate dehydrogenase (LDH) activity in the collected ACM was measured using the LDH assay. N = 3, p = 0.7 determined by Mann Whitney test. Not significant (n.s.) (B-C) Primary murine astrocytes (B) and murine HT-22 hippocampal neurons (C) incubated with control (UT ACM) or ER stressed ACM (Thaps ACM) for 6 h. CHOP and GRP78 mRNA expression was quantified using RT-qPCR. Immunoblot analysis was used to examine GRP78, spliced XBP-1 (sXBP-1) and CHOP protein expression. 1.0 μM (B) or 0.1 μM (C) Thaps was used as the positive control for B and C, respectively. GAPDH was used as the loading control. N = 3 independent cell culture preparations, **P ≤ 0.01, ***P ≤ 0.001, **** P ≤ 0.0001 determined by one-way ANOVA with Tukey’s multiple comparisons test. (D) HT-22 neurons incubated with control or ER stressed ACM from tunicamycin (Tunic)-treated primary murine astrocytes for the indicated time points. Immunoblot analysis was used to examine GRP78, ATF4 and CHOP protein expression. GAPDH was used as the loading control.
To determine if ER stress could initiate the secretion of ER stress-inducing molecules from astrocytes, we incubated primary murine astrocytes and the murine HT-22 neuronal cell line (Davis & Maher 1994) with ER stressed ACM for 6 h. The contents within the ER stressed ACM significantly upregulated the expression of signaling components associated with all three arms of the UPR relative to cells incubated with control ACM (Figure 1B-C). We performed a similar experiment using ER stressed ACM derived from astrocytes stimulated with tunicamycin (Tunic), an N-glycosylation inhibitor. Similar to our findings using Thaps, ER stressed ACM from Tunic-treated astrocytes increased ATF4, C/EBP homologous protein (CHOP) and glucose regulated protein (GRP)78 protein expression in HT-22 neurons, between 6 – 24 h (Figure 1D). We next tested if neuronal cells could also transfer ER stress. As shown in Figures 2A and 2B, conditioned media from Tunic treated HT-22 cells stimulated an ER stress response in HT-22 cells and primary astrocytes. We also tested if NCM could stimulate an ER stress response in primary microglia. As shown in Figure 2C, conditioned media from HT-22 cells stimulated robust CHOP expression in microglia. To verify efficient isolation of microglia from astrocytes, Iba1 was immunoblotted and observed in only the microglial cells. In contrast, astrocyte marker GFAP was present in astrocytes and not microglia. Together, these data indicate that ER stress can be transferred between astrocytes, neurons and microglia.
Figure 2. ER stressed neurons secrete a molecule(s) that upregulate ER stress responses in neurons, astrocytes, and microglia.
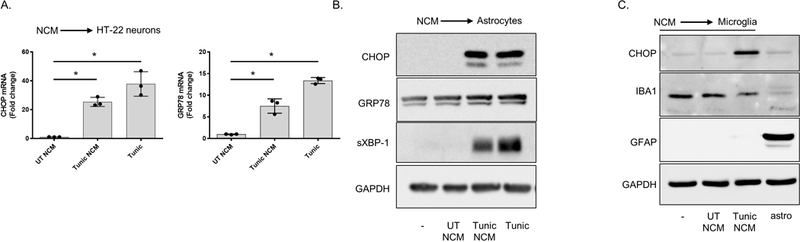
HT-22 cells were treated with 5 μM tunic for 2 h followed by washing in PBS then incubated in fresh media for 24 h. After this period, the neuron conditioned media (NCM) was collected. (A) HT-22 cells were cultured in NCM from UT or tunic treated HT-22 cells for 6 h or treated directly with tunic (5 μM) for 6 h followed by measurement of CHOP and GRP78 by qPCR. N = 3 independent cell culture preparations, *P ≤ 0.05, **P ≤ 0.01, determined by one-way ANOVA with Tukey’s multiple comparisons test. (B) Astrocytes were cultured in NCM from UT or tunic treated HT-22 cell for 6 h followed by immunoblotting. (C) Primary microglia were treated with NCM as in B, followed by immunoblotting. Astrocyte lysate (astro, lane 4) was included to confirm efficient separation of microglia and astrocytes.
Previously, we have shown that ER stress promotes PERK-dependent inflammatory gene expression in astrocytes (Meares et al. 2014). Therefore we hypothesized that conditioned medium from ER stressed neurons would similarly drive an inflammatory reaction in astrocytes. To test this, astrocytes were transfected with control or PERK siRNA for 48 h, which substantially reduced PERK expression and prevented thaps-induced phosphorylation of eIF2α (Figure 3A). As shown in Figure 3B, conditioned media from ER stressed HT-22 cells stimulates IL-6, CCL2 and CCL20 expression in astrocytes and knockdown of PERK significantly reduces the expression of these inflammatory genes. These data show that ER stressed neurons stimulate a PERK-dependent inflammatory reaction in astrocytes.
Figure 3. Neuronal ER stress promotes PERK-dependent inflammatory signaling in astrocytes.
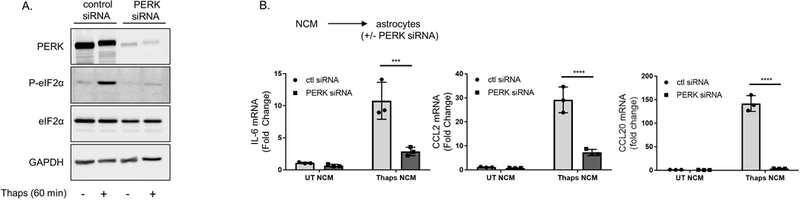
(A) Astrocytes were transfected with non-targeting (control) or PERK siRNA for 48 h followed by treatment with thaps (1 μM) for 60 min. Cell lysates were then immunoblotted for the indicated protiens. (B) Astrocytes were transfected as in A for 48 h prior to incubating with control or ER stressed HT-22 conditioned media (UT or Thaps NCM) for 6 h. IL-6, CCL2 and CCL20 mRNA expression was quantified using RT-qPCR. N = 3 independent cell culture preparations, **P ≤ 0.01, *** P ≤ 0.001 determined by two-way ANOVA with Bonferroni’s multiple comparisons test.
The Secreted Stress Factor(s) is not a Microparticle, is Resistant to Proteinase K and has Lipid-like Properties
We next sought to characterize the molecular determinant(s) secreted from ER stressed astrocytes that evoked ER stress responses in receiver cells. To exclude the possibility that inflammatory cytokines secreted by activated astrocytes were responsible for inducing ER stress in neurons, we stimulated HT-22 neurons with IL-1β, IL-6 or TNF-α for 6 h (Figure 4A). Contrary to previous findings with hepatocytes (Zhang et al. 2006), IL-6, IL-1β or tumor necrosis factor α (TNFα) did not induce ER stress in neurons after 6 h in vitro, even when using a supraphysiological concentration (10 ng/mL) (Figure 4A). This finding supports a previous observation where transmissible ER stress between tumor cells and macrophages did not involve the IL-6 receptor (Mahadevan et al. 2011).
Figure 4. The stress factor(s) secreted by ER stressed astrocytes is not a microparticle or proteinase K-sensitive protein.
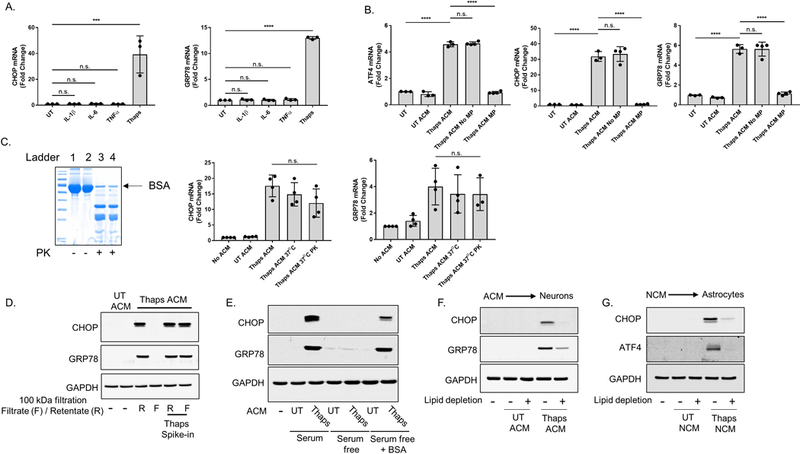
(A) Murine HT-22 hippocampal neurons were stimulated with 10 ng/mL of IL-1β, IL-6 or TNFα for 6 h. CHOP and GRP78 mRNA expression was quantified using RT-qPCR. Thaps (1μM) was used as a positive control. (B) Microparticles (MP) were isolated from the ER stressed astrocyte conditioned media (ACM) via ultracentrifugation (160,000 × g for 1 h). The supernatant (sup) was collected, and the pellet at the bottom of the tube (contains MPs) was resuspended in PBS. The centrifuged ACM (thapsigargin (Thaps) ACM Sup) and the solution containing the MPs (Thaps ACM MP pellet) were incubated with astrocytes for 6 h. ATF4, CHOP and GRP78 mRNA was quantified using RT-qPCR. (C) ER stressed ACM was treated with immobilized proteinase K for 1 h at 37°C. After which, the ACM was centrifuged at 1000 × g for 2 min to separate the beads and the media. The proteinase K-free ACM was collected, then incubated with astrocytes for 4 h. CHOP and GRP78 mRNA was quantified using RT-qPCR. To confirm that proteinase K was functional, 1 mg/mL of BSA was treated with proteinase K for 1 h at 37°C. Proteins from the BSA (lanes 1 and 2) or proteinase K-treated BSA (lanes 3 and 4) solutions were stained using Coomassie Blue staining following SDS-PAGE. A & B (N = 3), C (N = 4) independent cell culture preparations, ***P ≤ 0.001, **** P ≤ 0.0001 determined by one-way ANOVA with Tukey’s multiple comparisons test. (D) ACM from control or Thaps treated astrocytes was filtered through a 100 kDa molecular weight cut off filter. In a separate sample, Thaps was added directly to the ACM prior to filtration. The filtrate (F) or retentate (R) were then added to HT-22 cells (final concentration of 50% ACM) for 18 h followed by immunoblotting. (E) Astrocytes were cultured in media with serum, without serum or without serum + 100 ug/ml BSA to produce ACM from UT or Thaps treated astrocytes. The media was incubated with HT-22 cells (final concentration of 50% ACM) for 18h followed by immunoblotting. (F) ACM was incubated at RT for 1 h without or with Cleanascite, then centrifuged at 2000 × g and added to HT-22 cells (final concentration of 50% ACM) for 18h followed by immunoblotting. (G) NCM was incubated at RT for 1 h without or with Cleanascite, then centrifuged at 2000 × g and added to astrocytes for 18h followed by immunoblotting.
Extracellular vesicle secretion provides a means of intercellular communication in the CNS and is essential for the interactive exchanges of cargo that maintain neuronal competency (Budnik et al. 2016). Under conditions of neurodegeneration, however, toxic substrates can potentially be incorporated into extracellular vesicles and perturb CNS homeostasis (Thompson et al. 2016). To determine whether microparticles (MP) were responsible for inducing ER stress in receiver cells, we enriched MPs from the ER stressed ACM using ultracentrifugation. Unstressed astrocytes incubated with the centrifuged ER stressed ACM showed a similar magnitude of induction in the expression of ATF4, CHOP and GRP78 mRNA compared to astrocytes incubated with ER stressed ACM (Figure 4B). In contrast, astrocytes stimulated with the enriched MP-containing solution did not show any signs of UPR activation (Figure 4B). The MP-enriched sample contained approximately 5-fold more MP compared to the respective supernatant following ultracentrifugation (1.1 × 109 vs. 5.1 × 109 particles/ml) These findings indicate that the extracellular vesicles in the conditioned media were not the stress factor(s) responsible for inducing ER stress in astrocytes or neurons.
Next, we pretreated ER stressed ACM with immobilized proteinase K to digest the protein molecules within the sample. The ACM in these experiments contained 1% B27 supplement in place of 10% FBS. As shown in Figure 4C, this proteinase K readily digests BSA in solution. However, proteinase K-treated ER stressed ACM still induces CHOP and GRP78 gene expression, suggesting that the stress factor(s) responsible for inducing cell-nonautonomous UPR activation is not a protein or is resistant to proteinase K digestion (Figure 4C). We next used size-exclusion filtration with a 100 kDa filter to obtain information on the general molecular weight of the molecule(s) responsible for transmission. ACM was filtered and the filtrate and retentate were incubated with astrocytes. As shown in Figure 4D, the filtrate did not induce ER stress suggesting the molecule(s) are greater than 100 kDa. To confirm that the filter was not binding small molecules non-specifically, Thaps was added directly to the ACM (thaps spike-in) prior to filtration. As expected, Thaps was not removed by the filter and induced ER stress in receiver astrocytes (Figure 4D). Additionally, this demonstrates that Thaps carry-over in the conditioned media is not responsible for ER stress transmission. While working to characterize the mechanism of ER stress transmission, we made the unexpected observation that removal of serum from the culture medium prevented ER stress transmission (Figure 4E). However, ER stress was readily transmissible if the medium contained B27 supplement. This suggested that growth factors and/or a carrier molecule is needed to transmit ER stress. Consistent with this, the addition of BSA, the main protein component of B27, to serum free medium restored the ability to transmit ER stress (Figure 4E). Lipids often require carrier molecules and BSA regularly accommodates this role. Therefore, we tested if lipids were involved in the transmission of ER stress by lipid removal. As shown in Figure 4F, depletion of lipids from ACM using Cleanascite abrogated transmission of ER stress. To test if transmission from ER stressed neurons uses a similar mechanism, NCM was depleted of lipids which abrogated the induction of CHOP and ATF4 in astrocytes (Figure 4G). Collectively these data suggest that transmission of ER stress is mediated by a proteinase K-resistant lipid-like molecule.
ER Stressed Astrocytes Initially Protect Neurons against Subsequent Stress
Recent work has shown that cancer cells exposed to ER stressed conditioned media are resistant to subsequent stress (Rodvold et al. 2017). Therefore, we tested if ER stressed astrocytes were able to confer stress resistance to neurons. HT-22 neurons exposed to ER stressed ACM for 24 h activate caspse-3, indicating they are undergoing apoptosis (Figure 5A). No differences in caspase-3 activity were observed between the groups incubated with ER stressed ACM relative to the control after 6 h, indicating that the factor(s) within the ACM became cytotoxic to neurons after 6 h (Figure 5A). The apoptotic effects of the ER stressed ACM were confirmed by measurements of phosphatidylserine externalization based on annexin V binding, which was significantly increased after 24 h of exposure to ER stressed ACM (Figure 5B). Next, we sought to identify the conditions necessary for ER stressed ACM to induce a hormetic response in neurons. We chose to follow the accepted model proposing that the low-level activation of the ER stress sensors promotes an adaptive state by selectively upregulating the expression of pro-survival proteins, without the concomitant expression of UPR-associated apoptotic factors (Rutkowski et al. 2006a). To this point, we set our preconditioning standards where neurons highly expressed chaperone proteins (GRP78, pro-survival (Racek et al. 2008)) and do not express CHOP, which is considered apoptotic (Li et al. 2014, Marciniak et al. 2004, Song et al. 2008).
Figure 5. Mild exposure to the stress factor(s) promotes an adaptive state in neurons.
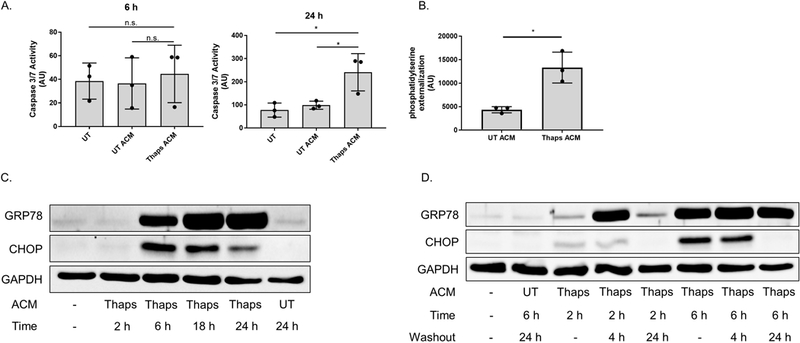
(A) Murine HT-22 hippocampal neurons were stimulated with control or ER stressed astrocyte conditioned media (UT or Thaps ACM) for 6 or 24 h. Caspase-3 activity was assessed by measuring the fluorescence intensity of the cleaved capase-3 substrate Ac-DEVD-AMC. Caspase-3 activity was expressed as arbitrary fluorescence units (AU). N = 3, *P ≤ 0.05 determined by one-way ANOVA with Tukey’s multiple comparisons test. (B) HT-22 neurons were treated with astrocyte conditioned media (UT or Thaps ACM) for 24 h followed by measuring phosphatidylserine externalization using a split luciferase coupled to Annexin V (Real-time Glo), data are expressed as arbitrary luminescent units (AU). N = 3 independent cell culture preparations, *P ≤ 0.05 determined by Student’s T test. (C) HT-22 neurons incubated with 25% control or ER stressed ACM for the indicated time points. GRP78 and CHOP protein expression was examined using immunoblot analysis. (D) HT-22 neurons incubated with 25% control or ER stressed ACM for 2 or 6 h, followed with a 4 or 24 h washout period. GRP78 and CHOP protein expression was examined using immunoblot analysis. GAPDH was used as the loading control. UT, untreated; UTA, control ACM; TA, Thaps ACM.
As shown in Figure 5C, stimulating HT-22 neurons with ER stressed ACM for 6 h – 24 h increases the protein expression of GRP78 and CHOP. However, when stimulated with ACM for 6 h followed by a 24 h washout period, GRP78 protein expression was maintained, while CHOP protein expression dissipated, consistent with the short half-life of CHOP (Rutkowski et al. 2006b) (Figure 5D). Although stimulating neurons for 2 h with ER stressed ACM, followed with a 4 h washout period achieves a preconditioned state, the expression of GRP78 is significantly reduced after 24 h (Figure 5D). This would suggest that the adapted state induced under these conditions is not as established compared to when neurons are stimulated for 6 h, then allowed to recover for 24 h. Therefore, we used these conditions to determine whether preconditioning neurons promotes resistance against ER stress-induced cell death.
We stimulated preconditioned (6 h ACM followed by 24 h recovery) neurons with Thaps for the indicated time points and analyzed the protein expression of phosphorylated c-Jun N-terminal kinase (JNK) (P-JNK) and CHOP, pro-apoptotic factors induced during UPR-mediated apoptosis (Tabas & Ron 2011) (Figure 6A-B). Pretreating neurons with ER stressed ACM attenuates P-JNK and CHOP when neurons were stimulated with Thaps for 4 h (Figure 6A and 6B). The thaps-induced expression of CHOP was quantified and was significantly reduced in cells preconditioned with ER stressed ACM. However, the reduction in P-JNK, while consistent, did not reach statistical significance. Additionally, we assessed caspase-3 activity in preconditioned neurons after Thaps treatment and found that neurons preconditioned with ER stressed ACM exhibit less caspase-3 activity after 4 h or 18 h treatment with Thaps (Figure 6C and 6D). Taken together, these data indicate that ER stressed astrocytes initially confer stress resistance to neurons by engaging the adaptive mechanisms of the UPR to promote hormesis.
Figure 6. Conditioned media from ER stressed astrocytes confer stress resistance to neurons.
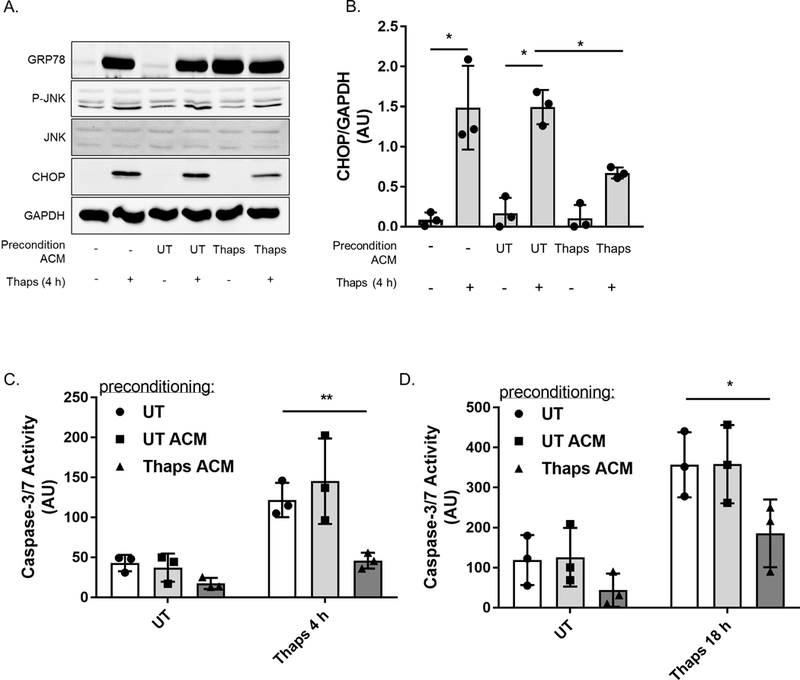
(A) Murine HT-22 hippocampal neurons were preconditioned with 25% control (UTA or UT ACM) or ER stressed ACM (TA or Thaps ACM) for 6 h, followed by a 24 h recovery period with fresh media. Cells were then treated with Thaps for 4 h. GRP78, phosphorylated JNK (P-JNK) and CHOP protein expression was assessed using immunoblot analysis. (B) CHOP proteins levels were quantified and normalized to GAPDH. N = 3 independent cell culture preparations, *P ≤ 0.05 determined by two-way ANOVA with Tukey’s multiple comparisons test. (C) Caspase-3 activity was analyzed by measuring the fluorescence intensity of the cleaved caspase-3 substrate Ac-DEVD-AMC after preconditioned HT-22 neurons were stimulated with either the vehicle (DMSO) or 0.1 μM thapsigargin (Thaps) for 4 h or (D) 18 h. N = 3, *P ≤ 0.05, **P ≤ 0.01 determined by two-way ANOVA with Tukey’s multiple comparisons test.
Discussion
Studies investigating how pathological UPR signaling contributes to neurodegenerative diseases have largely focused on neuronal cell-autonomous mechanisms. Nevertheless, there still remains a plethora of questions regarding the cell-nonautonomous role of the UPR under normal physiology, and how this mechanism is compromised in diseased states. To our knowledge, we provide the first evidence showing that ER stress is transmissible among mammalian CNS cells. Activation of the UPR has been shown to play an essential role in maintaining vital biological processes within the brain during cellular stress. In fact, moderate ER stress enhances cellular protection against subsequent ER stress-inducing stimuli by improving the adaptive capacity of the cell, a response called the hormetic response (Mollereau et al. 2014). These observations suggest that transmissible ER stress could be essential for alerting surrounding cells about upcoming distresses, and to prepare them for subsequent insult by initiating the adaptive signals of the UPR (Schinzel & Dillin 2015). In support of this concept, direct activation of sXBP-1, a downstream transcription factor of the IRE-1α pathway, in neurons has been shown to improve stress resistance in distal, non-neuronal cells by regulating UPR signaling components through the secretion of neurotransmitters in Caenorhabditis elegans (Taylor & Dillin 2013). In agreement with these findings, our data suggest that ER stressed astrocytes initially protect neurons by inducing mild, cell-nonautonomous ER stress, which in turn increases their resistance against subsequent ER stress (Figure 6).
A number of CNS-resident cells, including astrocytes, microglia and neurons, secrete membrane-bound nanovesicles called exosomes under normal and pathological conditions (Gupta & Pulliam 2014). In a study evaluating prion disease, abnormally folded prion aggregates were found to associate with neuronal exosomes and infect both neuronal and non-neuronal cells without the need for direct cell-to-cell contact (Fevrier et al. 2004). It was tempting to speculate that the factor(s) secreted from ER stressed cells were extracellular vesicles containing a heterogeneous species of RNA, protein or lipid that could induce ER stress in unstressed cell types. However, our MP isolation experiments demonstrated that both exosomes and other microvesicles secreted by ER stressed astrocytes do not upregulate UPR markers in recipient astrocytes (Figure 4B). Similarly, MPs secreted from cancer cells are not responsible for triggering both ER stress and proinflammatory responses in receiver myeloid cells (Zanetti et al. 2016). Treating ER stressed ACM with proteinase K did not rescue receiver cells from ER stress, suggesting that the stress molecule(s) is not a protein. However, we cannot exclude the possibility that it may exist in a proteinase K-resistant form, as disease-associated prion proteins do (Saunders et al. 2007). To add to the characterization of this stress factor(s), it appears that it cannot be inactivated by heat (Mahadevan et al. 2011). The transmissible factor(s) is potentially a lipid as it was absorbed by the Cleanascite matrix which is selective for lipids. The factor was also removed by filtration through a 100 kDa cutoff filter. It is unlikely the molecule is over 100 kDa in size, but rather the lipids are forming micelles that are larger than the pore size and are therefore unable to pass through. However, we cannot rule out the possibility that the molecule is a large proteinase K-resistant protein that was lipid-modified and absorbed by the matrix. These characteristics are somewhat different from recent work describing neurotoxic astrocytes induced by IL-1α, TNF-α and the complement component C1q. Under these conditions, astrocytes secrete a neurotoxic factor that is large (> 30 kDa) but also heat and protease sensitive, strongly suggesting the toxic factor is a protein (Liddelow et al. 2017). This suggests that astrocytes are equipped with multiple mechanisms that are engaged in a stimulus-specific fashion to influence neuronal fate and survival.
While our data points toward a lipid-related molecule, the identity of the factor(s) responsible for ER stress transmission remains unknown. Identifying the soluble stress molecule(s) secreted by ER stressed cells will be essential in order to better understand the molecular mechanisms involved in the cell-extrinsic regulatory function of the UPR. Based on our findings, transmissible ER stress does not appear to require cell-to-cell contact, thus supporting the notion that ER stress transmission is mediated by a stress-induced soluble molecule(s) (Zhang et al. 2017). While ER stress in other cell types has been shown to trigger the secretion of ER stress-inducing molecules, the identity of this factor(s) still remains elusive (Mahadevan et al. 2011, Zanetti et al. 2016, Zhang et al. 2017). Our data using non-transformed primary astrocytes indicate that transmission of ER stress is not unique to cancer cells and suggest it may be a general feature of most cells. It is important to consider that this mediator(s) may not be a conventional DAMP, but rather intracellular constituents released from the cell in an effort to alleviate ER stress. The secretion of products from the cell could be the result of an autophagy-dependent unconventional secretion pathway that is induced during cellular stress. The UPR is a well-known trigger of autophagosome formation, and the secretion of alarmins through this non-canonical autophagy pathway has been shown to serve different extracellular functions (Ogata et al. 2006, Deretic et al. 2012). These secretory byproducts may, in turn, promote ER stress in neighboring cells. Further studies are needed to test this possibility.
Other studies investigating transmissible ER stress between mammalian cells discuss its potential role in disease progression. As represented in Figure 6, we propose that ER stressed astrocytes are able to confer resistance against upcoming ER stress-inducing insults to neurons by promoting the low-level activation of the UPR. In doing so, the pro-survival signals associated with the ER stress response are engaged (i.e. increased expression of molecular chaperones), thus enhancing the protein folding capacity of the cell. The perpetual presence of misfolded proteins in many neurodegenerative diseases may lead to ongoing ER stress. In such cases, chronic exposure to the uncharacterized stress factor(s) secreted by neurons and activated astrocytes may induce prolonged UPR signaling, which favors the initiation of apoptosis due to the prolong expression of apoptotic factors such as CHOP. In this way, a protective mechanism may be usurped to contribute to neuronal demise in neurodegenerative diseases. Additionally, the release of both inflammatory mediators (i.e. cytokines and ROS) and the ER stress-inducing factor(s) by dysfunctional astrocytes could contribute to the cell-nonautonomous mechanisms that drive the neurotoxic events observed in many neurodegenerative disorders.
These findings provide insight into the cell-extrinsic influence of the UPR on cells of the CNS. Considering that persistent ER stress likely contributes to disease pathology, understanding the molecular mechanisms underlying the cell-extrinsic functions of the UPR would present novel therapeutic opportunities to treat neurodegenerative diseases.
Acknowledgements
We would like to thank Mr. Akshay Kesari, Ms. Alicia Meyer, and Mr. John Nowery for technical assistance. This work was supported by a career transition award from the national multiple sclerosis society (TA 3050-A-1) to GPM and grants from the NIH (R01NS099304) (Stroke COBRE GM109098) (WVU CTSI U54GM104942).
Abbreviations:
- (PERK)
protein kinase R-like ER kinase
- (IRE-1α)
inositol requiring enzyme 1
- (ATF)
activating transcription factor
- (UPR)
unfolded protein response
- (CCL)
C-C motif chemokine ligand
- (tunic)
thapsigargin (thaps), tunicamycin
- (ACM)
astrocyte conditioned medium
- (NCM)
neuron conditioned medium
- (GRP)
glucose regulated protein
- (CHOP)
C/EBP homologous protein
- (JNK)
c-Jun N-terminal kinase
- (MP)
microparticles
- (RRID)
Research Resource Identifier
Footnotes
Conflict of interest
The authors declare they have no competing interests.
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. The complete Open Science Disclosure form for this article can be found at the end of the article. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/.
References:
- Araki K and Nagata K (2011) Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol, 3, a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A and Navarrete M (2010) Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci, 365, 2375–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Ruiz-Cañada C and Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci, 17, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco RA, Stamm NB and Patel BKR (2003) One-step cellular caspase-3/7 assay. Biotechniques, 34, 1064–1067. [DOI] [PubMed] [Google Scholar]
- Ciechanover A and Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med, 47, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JB and Maher P (1994) Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res, 652, 169–173. [DOI] [PubMed] [Google Scholar]
- Deretic V, Jiang S and Dupont N (2012) Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends in cell biology, 22, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H and Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci U S A, 101, 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC and Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A and Pulliam L (2014) Exosomes as mediators of neuroinflammation. J Neuroinflammation, 11, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie LN, Abiraman K, Plyler ES et al. (2016) Attenuation of PKR-like ER Kinase (PERK) Signaling Selectively Controls Endoplasmic Reticulum Stress-induced Inflammation Without Compromising Immunological Responses. J Biol Chem, 291, 15830–15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LD, Yin Y, Attarwala IY, Begum G, Deng J, Yan HQ, Dixon CE and Sun D (2015) Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagi R, Kumagai T, Nishi H, Kawakami T, Miyata T, Fujita T and Nangaku M (2008) Preconditioning with endoplasmic reticulum stress ameliorates mesangioproliferative glomerulonephritis. J Am Soc Nephrol, 19, 915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski C and Callewaert DM (1983) An enzyme-release assay for natural cytotoxicity. J Immunol Methods, 64, 313–320. [DOI] [PubMed] [Google Scholar]
- Li Y, Guo Y, Tang J, Jiang J and Chen Z (2014) New insights into the roles of CHOP-induced apoptosis in ER stress. Acta biochimica et biophysica Sinica, 46, 629–640. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W and Popko B (2009) Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci, 12, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M and Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem, 266, 17067–17071. [PubMed] [Google Scholar]
- Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF and Zanetti M (2011) Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci U S A, 108, 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & development, 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares GP, Hughes KJ, Jaimes KF, Salvatori AS, Rhodes CJ and Corbett JA (2010) AMP-activated protein kinase attenuates nitric oxide-induced beta-cell death. J Biol Chem, 285, 3191–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA and Benveniste EN (2014) PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol Cell Biol, 34, 3911–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollereau B, Manie S and Napoletano F (2014) Getting the better of ER stress. J Cell Commun Signal, 8, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M, Hino S, Saito A et al. (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol, 26, 9220–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racek T, Buhlmann S, Rust F, Knoll S, Alla V and Putzer BM (2008) Transcriptional repression of the prosurvival endoplasmic reticulum chaperone GRP78/BIP by E2F1. J Biol Chem, 283, 34305–34314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, Willert K, Lin JH and Zanetti M (2017) Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci Signal, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D and Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol, 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Rutkowski D, Arnold S, Miller C et al. (2006a) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol, 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN et al. (2006b) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol, 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders GC, Horigan V, Tout AC and Windl O (2007) Identification of a proteinase K resistant protein for use as an internal positive control marker in PrP Western blotting. Res Vet Sci, 83, 157–164. [DOI] [PubMed] [Google Scholar]
- Schinzel R and Dillin A (2015) Endocrine aspects of organelle stress—cell non-autonomous signaling of mitochondria and the ER. Curr Opin Cell Biol, 33, 102–110. [DOI] [PubMed] [Google Scholar]
- Song B, Scheuner D, Ron D, Pennathur S and Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. The Journal of clinical investigation, 118, 3378–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenkle NT, Sims SG, Sanchez CL and Meares GP (2017) Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener, 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I and Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol, 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Berendzen KM and Dillin A (2014) Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat Rev Mol Cell Biol, 15, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC and Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell, 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AG, Gray E, Heman-Ackah SM, Mäger I, Talbot K, Andaloussi SE, Wood MJ and Turner MR (2016) Extracellular vesicles in neurodegenerative disease - pathogenesis to biomarkers. Nat Rev Neurol, 12, 346–357. [DOI] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Bateman JF and Cheah KSE (2010) In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci, 123, 2145–2154. [DOI] [PubMed] [Google Scholar]
- Walter P and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Williams KW, Liu T, Kong X et al. (2014) Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab, 20, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti M, Rodvold JJ and Mahadevan NR (2016) The evolving paradigm of cell-nonautonomous UPR-based regulation of immunity by cancer cells. Oncogene, 35, 269–278. [DOI] [PubMed] [Google Scholar]
- Zhang H, Yue Y, Sun T, Wu X and Xiong S (2017) Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3-induced viral myocarditis. Sci Rep, 7, 42162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH and Kaufman RJ (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell, 124, 587–599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sets are available from the corresponding author on request.