

Abstract
Iron is an essential nutrient for all living organisms and plays a vital role in many fundamental biochemical processes, such as oxygen transport, energy metabolism, and DNA synthesis. Due to its capability to produce free radicals, iron has deleterious effects and thus, its level needs to be tightly controlled in the body. Deregulation of iron metabolism is known to cause diseases, including anemia by iron deficiency and hereditary hemochromatosis by iron overload. Interestingly, dysregulated iron metabolism occurs frequently in tumor cells and contributes to tumorigenesis. In this review, we will discuss the role of p53 tumor suppressor in iron homeostasis.
Keywords: Iron homeostasis, tumor suppression, mitochondrial iron homeostasis, p53, IRP1/2, FDXR
P53 tumor suppressor plays a critical role in maintaining the genome integrity. Recent studies show that p53 is instrumental in regulating iron homeostasis by modulating several iron regulators. Some iron regulators also form a feedback regulatory loop with p53. Thus, the crosstalk between p53 and the iron regulators may shed a light on the mechanisms by which iron metabolism is altered in cancers.
Introduction
Iron is essential for many cellular processes including cell growth and proliferation. The most important feature of iron is that it mediates electron transfer by interchanging between ferrous (Fe(II), Fe2+) and ferric (Fe(III), Fe3+) states. Iron acts as an electron donor in the ferrous state but as an electron acceptor in the ferric state. Consequently, iron plays a vital role in many fundamental biochemical activities, such as oxygen transport, energy metabolism, and DNA synthesis [1]. However, iron can be highly toxic due to its capability to participate in Fenton reaction. Ferrous iron donates an electron in a reaction with hydrogen peroxide to generate the hydroxyl radical, a reactive oxygen species (ROS), which leads to oxidative stress, lipid peroxidation, and DNA damage. Thus, the uptake, storage, and usage of iron need to be tightly controlled. Indeed, all organisms have developed sophisticated mechanisms to modulate iron homeostasis.
Systemic iron homeostasis
Circulating iron in the body is controlled by four main cell types: enterocytes in the duodenum to absorb dietary iron; erythrocyte precursors in the bone marrow to incorporate iron into hemoglobin; macrophages in the liver, spleen, and bone marrow to recycle iron; and hepatocytes in the liver to store iron. The control of circulating iron is mediated by factors that can sense the iron level and subsequently, regulate the expression of genes necessary for iron homeostasis. One of the major regulatory mechanisms is the hepcidin-ferroportin (FPN) axis (Fig. 1). FPN is a transmembrane protein that allows iron to be transported out of cells and into the bloodstream. High levels of FPN are found in enterocytes, macrophages, and hepatocytes. The role of FPN in iron export has been demonstrated in mouse models. Loss of FPN leads to embryonic lethality in mice, suggesting a critical role of FPN in development [2]. Interestingly, conditional deletion of FPN in mouse intestine leads to accumulation of iron in duodenal enterocytes and these mice become severely anemic within weeks [2]. FPN-mediated iron efflux is negatively controlled by hepcidin, a 25-amino acid peptide hormone produced by hepatocytes [3, 4]. Circulating hepcidin inhibits iron export from macrophages, enterocytes, and hepatocytes and thereby regulates the amount of iron in the bloodstream [5–7]. Mechanistically, hepcidin binds to the extracellular domain of FPN and induces its endocytosis and subsequent degradation [5, 8, 9], leading to reduced export of cellular iron. The importance of the hepcidin-FPN axis in systemic iron homeostasis is exemplified by several iron disorders. For example, decreased FPN activity, led by high levels of hepcidin, is found in iron-restriction syndromes, including iron-refractory iron deficiency anemia (IRIDA) [10] or anemia of inflammation [11], in which iron accumulates in recycling macrophages and enterocytes, but may become insufficient in other tissues. By contrast, hyperactive FPN, usually as a result of hepcidin deficiency, is found in hereditary hemochromatosis [12] or β-thalassemia intermedia [13], in which excessive iron absorption and toxic iron deposition are found in hepatocytes and other parenchymal cells, but relative iron depletion occurs in macrophages.
Figure 1.

Systemic iron homeostasis is controlled by the hepcidin-FPN axis. The peptide hormone hepcidin negatively regulates the expression of ferroportin (FPN), the major cellular iron exporter responsible for transporting iron from cell into plasma.
Cellular iron homeostasis
Cellular iron homeostasis is regulated via iron uptake, trafficking and export (Fig. 2). These processes are tightly controlled by iron regulatory protein 1 (IRP1) and IRP2 (also known as ACO1 and IREB2, respectively) [14, 15]. IRPs are RNA-binding proteins and interact with conserved cis-regulatory hairpin structures known as IREs (iron-responsive elements), which are present in the 5’ or 3’untranslated regions (UTRs) of target mRNAs. Generally, IRPs inhibit the translation of mRNAs that contain an IRE in their 5’UTRs and increase the stability of mRNAs that contain an IRE in their 3’UTRs. For example, IRPs suppress the translation of FTH1, FTL, and FPN1 mRNAs via binding to an IRE in their 5’UTRs. By contrast, IRPs stabilize mRNAs of TFR1 and DMT1 via binding to an IRE in their 3’UTRs [16, 17]. Interestingly, the activity of IRPs is controlled by iron. When intracellular levels of iron are low, IRPs are active and stabilize mRNAs for proteins involved in iron uptake but repress the translation of mRNAs for proteins involved in iron storage and export. In iron-replete cells, IRP1 functions as an aconitase but not a RNA-binding protein whereas IRP2 undergoes iron-dependent degradation. As a result, both IRPs are inactive and thus unable to increase the level of proteins involved in iron uptake or decrease the level of proteins involved in iron storage and export. The biological significance of IRPs in regulating cellular iron homeostasis has been confirmed in mouse models. Genetic ablation of both IRPs in the mouse leads to embryonic lethality [18, 19], suggesting a critical role of iron homeostasis during development. However, mice deficient in IRP1 or IRP2 are viable and fertile, indicating a redundant function between these two IRPs. Notably, IRP2 deficiency leads to abnormal body iron distribution, a mild microcytic anemia, and neurodegeneration [20, 21]. IRP1-KO mice were initially found to be asymptomatic, suggesting that IRP1 is not necessary in iron metabolism [22]. Later, it was found that IRP1 plays an essential role in regulation of systemic iron homeostasis and erythropoiesis [23, 24]. Additionally, it was found that overexpression of IRP1 reduces, whereas overexpression of IRP2 promotes, tumor growth in vivo [25–28]. These data suggest that IRP1 and IRP2 are not functionally redundant and may have different impacts in the content of cancer.
Figure 2.

Cellular and mitochondrial iron homeostasis. The cellular iron storage, uptake and export is controlled by iron regulatory proteins (IRP1/2). Mitochondria are a major site for ISC (iron sulfur cluster) protein synthesis as well as heme synthesis. The regulators of mitochondrial homeostasis include Mitoferrin (for iron delivery), FtMt (ferritin mitochondrial, for iron storage) and ABCB7 (iron transporter).
Mitochondrial Iron homeostasis
Mitochondria play a central role in energy production, oxygen transport, and deoxynucleotide synthesis. Mitochondria are also essential for iron metabolism (Fig. 2). Intracellular iron can be transported to mitochondria, where it is utilized to synthesize essential cofactors for a number of proteins. Indeed, the mitochondrion is the only site where heme is synthesized. Heme is a prosthetic group for proteins involved in cellular respiration, oxygen transport/storage, and enzymatic functions [29], such as hemoglobin, myoglobin, and cytochrome c. A multi-step reaction is necessary for heme biogenesis, which is catalyzed by eight enzymes [30, 31]. The first rate-limiting step occurs in mitochondria and is mediated by δ-aminolevulinic acid synthase (ALAS1/2) to form δ-aminolevulinic acid (ALA). ALA is exported to the cytosol and converted to coproporphyrinogen III (CPgenIII), which is then transported back to mitochondria and converted to protoporphyrin IX (PPIX). The final step is mediated by ferrochelatase (FECH), which inserts ferrous iron into PPIX to form heme. Defective heme synthesis is associated with sideroblastic anemia [32], in which erythroblasts cannot make enough hemoglobin due to disrupted heme production. Consistently, patients with sideroblastic anemia were found to have hereditary gene mutations in ALAS2 and FECH, both of which are known to be required for heme synthesis [33, 34].
Mitochondria are the major site to produce iron sulfur clusters (ISCs), which are cofactors essential for proteins involved in mitochondrial respiration, DNA replication/repair, and enzymatic functions [35, 36]. The initial stage of ISC assembly is carried out by a multimeric protein complex [37, 38]: 1) cysteine desulfurase (NFS1) which provides sulfur by removing it from cysteine residues; 2) ISD11, a binding partner to stabilize NFS1; 3) ISCU (iron-sulfur cluster assembly enzyme), a scaffold protein that supplies the backbone structure to synthesize the nascent Fe-S cluster; and 4) Frataxin that supplies the source of iron [39]. In addition, ferredoxin reductase (FDXR) and ferredoxins 1 and 2 (FDX1/2) provide an electron that reduces sulfane to sulfide in order to achieve an appropriate electronic configuration for a given ISC [40]. Once the nascent Fe-S cluster is formed, it is transferred to recipient apo-proteins, thereby converting them to their holo-forms. Importantly, genetic mutations of the genes involved in ISC biosynthesis have been identified in neurodegenerative and metabolic diseases associated with mitochondrial iron overload [38]. For example, reduced expression of frataxin due to homozygous unstable GAA trinucleotide expansion in the FXN gene can result in Friedreich’s ataxia (FA), an autosomal recessive disease characterized by severe neurodegeneration and cardiomyopathy [41]. Consistent with this, mice deficient in frataxin are embryonically lethal whereas conditional loss of frataxin in neuron/cardiac muscle phenocopies the pathophysiological and biochemical features of the human FA [42]. Mutation of NFS1 leads to infantile mitochondrial complex II/III deficiency, an autosomal recessive disease characterized by hypotonia, lactic acidemia, and multisystem organ failure [43]. Moreover, loss of function of ISCU due to splicing mutation leads to ISCU myopathy, a disease leading to severe exercise intolerance in skeletal muscles [44, 45]. Furthermore, biallelic mutations in FDX2 are found in patients with mitochondrial muscle myopathy [46]. Recently, two groups discovered that FDXR mutations are found in patients with auditory neuropathy, optic atrophy, and other neurological signs of mitochondriopathy [47, 48].
Iron homeostasis and cancer
Deregulation of iron metabolism has been implicated in several types of cancer with abnormal iron uptake, utilization and storage [49]. Cancer cells often have an increased expression of iron importers and decreased expression of iron exporters. For example, FPN expression is reduced in breast cancer along with increased levels of the labile iron pool and enhanced tumor growth [50]. Similarly, FPN was found to be decreased in prostate cancer and ovarian cancer [51, 52]. By contrast, hepcidin, the negative regulator of FPN, was found to be increased in patients diagnosed with prostate cancer, breast cancer, hepatocellular carcinoma, and ovarian cancer [53–56]. Additionally, ferritin, TfR, and STEAP3, a metalloreductase that converts iron from an insoluble ferric (Fe3+) to a soluble ferrous (Fe2+) form, were found to be highly expressed in cancer cells to increase iron intake [57–59]. Recently, it was found that NFS1, a regulator of ISC assembly, promotes lung cancer by regulating iron homeostasis [60]. It is suggested that iron promotes tumor formation as an essential growth factor. Indeed, iron is necessary for the function of many enzymes involved in DNA synthesis and cell cycle [61]. By contrast, iron depletion inhibits cell proliferation by decreasing cyclin D1 and Cdk2 expression [62, 63]. Additionally, iron can function as a tumor promoter by generating reactive oxygen species via the Fenton reaction. This reaction not only damages lipids and proteins, but also causes oxidative damage to DNA, including DNA base modifications and DNA strand breaks [64, 65], which can be mutagenic [66, 67].
The crosstalk between tumor suppressor p53 and iron regulators
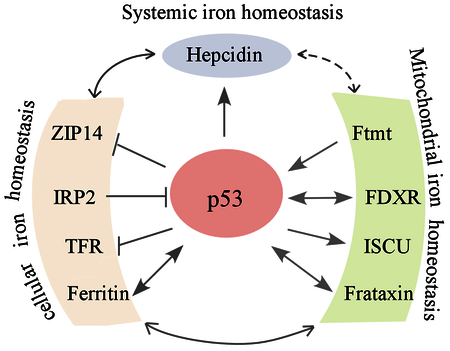
The tumor suppressor p53 is often referred to as the “guardian of the genome” [68, 69]. p53 is the most commonly mutated gene in human cancer and loss of p53 is known to play a central role in tumor development [70]. The importance of p53 in tumor suppression is underscored by its ability as a transcription factor to regulate a series of target genes necessary for cell survival and death [71]. Recent studies suggest that p53 plays a role in iron homeostasis and conversely, p53 is also regulated by several key regulators in iron metabolism (Fig. 3).
Figure 3.

Crosstalk between p53 and iron regulators. The crosstalk between p53 and various iron regulators involving systemic, cellular and mitochondrial iron homeostasis.
p53 expression is modulated by both iron overload and iron deficiency.
Upon treatment with iron chelators, p53 expression is increased and then induces growth inhibition by activating its targets such as p21 [72–75]. The enhanced expression of p53 by iron chelators is likely due to the increased p53 protein stability by hypoxia-inducible factor 1 alpha (HIF1α) [76]. HIF1α is activated in response to iron-deprived conditions [77, 78]. Conversely, p53 expression is decreased upon exposure to excess iron or iron overload via heme-p53 interaction [79]. Consistently, p53 expression is decreased in Hfe−/− mice that exhibit hereditary hemochromatosis, an iron overload disease [80]. Mechanistically, heme interacts with p53 DNA-binding domain, which promotes p53 export out of the nucleus through CRM1 and subsequently, p53 degradation [79]. However, p53 is also found to be upregulated upon exposure to excess iron but downregulated upon iron depletion via MDM2 [81]. It is not clear why two opposing observations were obtained in response to iron alterations. One possibility is that different cell types, e.g., cancer cells [72–75] vs. hepatocytes [81], were used in these studies. Thus, a systemic study is warranted to determine how p53 expression is altered by iron chelation or depletion.
p53 plays a role in iron homeostasis by modulating iron regulators.
C57BL/6 mice fed a high-iron diet show a decrease in p53 protein levels in the liver [79]. Consistent with this, loss of p53 leads to increased levels of serum iron in mice fed with excess dietary iron [82]. As a master regulator, p53 is likely to regulate expression of key iron sensors to control intracellular iron pool. Indeed, the promoter of HAMP, the gene encoding hepcidin, contains a putative p53-responsive element and can be activated by p53 but decreased when p53 is silenced [83]. It is suggested that an increased level of hepcidin by p53 plays a role in the pathogenesis of anemia, the most common haematological abnormality in cancer patients, whose p53 levels are often increased during cancer treatment. Additionally, it was found that ISCU is a target of p53 and the increased expression of ISCU by p53 protects cells from iron overload [82]. Thus, ISCU serves as a mediator of p53 to maintain the intracellular iron pool [82], consistent with the study that p53 is capable of increasing ferritin, but decreasing TFR1, expression [72]. Intriguingly, p53 appears to increase ferritin expression via a posttranscriptional mechanism [72]. However, another study showed that p53 is recruited by NF-Y to the H ferritin promoter and subsequently, represses ferritin expression [84]. Moreover, the metal transporter ZIP14 was identified as a p53-regulated protein [85]. Knockdown of endogenous p53 increased ZIP14 expression and subsequently, cellular non-transferrin-bound iron (NTBI) uptake. Interestingly, p53 appears to modulate ZIP14 expression through interaction with ZIP14, subsequently preventing ZIP14 from degradation.
p53 plays a role in mitochondrial iron homeostasis.
p53 is found to modulate several mitochondrial proteins involved in iron metabolism. For example, ferredoxin reductase (FDXR) is identified as a target of p53 [86, 87]. FDXR is the sole human ferredoxin reductase involved in the biosynthesis of ISCs and heme. Mice deficient in FDXR are embryonically lethal, likely due to iron overload in developing embryos [88]. It is suggested that p53-mediated FDXR expression plays a role in mitochondrial iron homeostasis and subsequently, modulates ISC or heme synthesis [89]. In addition, frataxin, a key regulator of ISC synthesis, is regulated by p53 [90, 91]. Notably, frataxin expression was found to be associated with p53 status and the promoter of the FXN gene contains a putative p53-responsive element [90]. Nevertheless, the biological significance of p53-mediated frataxin expression remains to be elucidated.
Multiple feedback loops between p53 and iron regulators.
Feedback regulation is involved in many cellular processes. Since p53 regulates several key iron regulators, it is likely that some of these regulators in turn modulate p53 expression. Indeed, our laboratory showed that FDXR is necessary for p53 expression [88]. Specifically, we showed that loss of FDXR reduces, whereas ectopic expression of FDXR increases, p53 expression. Mechanistically, FDXR signals through ferredoxin 2, a substrate of FDXR, which subsequently activates IRP2 to decrease p53 mRNA translation. As a result, mice heterozygous in Fdxr had a short life span and were prone to spontaneous tumors and liver abnormalities [88]. In addition, knockdown of frataxin induces cell death in human astrocytes, which is accompanied with a significant up-regulation of p53 and p21 [92]. Similarly, it was found that silencing of frataxin expression triggers p53-dependent apoptosis in human neuron-like cells [93]. Consistent with this, high levels of p53 protein were expressed in B cells derived from Friedreich’s ataxia patients, suggesting that frataxin deficiency activates p53 expression [94]. Moreover, ferritin, an iron storage protein, was found to activate p53 expression under oxidative stress [95]. Specifically, ferritin binds to p53 and subsequently, increases the transcriptional activity of p53, which is independent of the ferroxidase activity of ferritin. Consistent with this, knockdown of ferritin alleviates induction of p53 target genes upon treatment with hydrogen peroxide [95]. Similarly, it was found that mitochondrial ferritin (Ftmt), a mitochondrial iron storage protein, activates p53 expression along with its target p21, leading to growth suppression [96].
p53 and ferropotosis
Ferroptosis is an iron-mediated, caspase-independent, cell death pathway that requires the accumulation of lipid hydroperoxides [97] and recent studies suggest that ferroptosis may be a new option for cancer therapy [98]. Recently, it was found that ferropotosis is a crucial component of p53-mediated tumor suppression [99]. p53 induces ferroptosis at least in part via transcriptional repression of SLC7A11, a component of system xc−, a cystine/glutamate antiporter [99]. Additionally, p53 mediates ferroptosis by activating GLS2 and SAT1 [100–102]. Intriguingly, it was found that p53 inhibits ferroptosis by inhibiting dipeptidyl-peptidase-4 (DPP4) activity in a transcription-independent manner in human colorectal cancer cell lines [103]. In another study, it was found that p53 decreases system xc− activity, and simultaneously reduces the sensitivity of cells to metabolic stress-mediated ferroptosis [104]. These apparently opposing observations are likely due to different settings. For example, basal p53 was found to induce ferroptosis [100–102], whereas stress-induced p53 was found to inhibit ferroptosis [103, 105]. Nevertheless, a comprehensive understanding of how p53 modulates ferroptosis is needed, which would be beneficial for developing cancer therapies targeting ferroptosis.
Conclusions and future directions
Emerging evidence suggests that dysregulation of iron metabolism contributes to tumorigenesis and many iron regulatory genes are found to be altered in cancers. Generally, cancer cells require high amount of iron for proliferation and thus, are vulnerable to iron deficiency. Thus, iron chelators are currently being tested for the treatment of several types of cancers including solid tumors and blood cancers [106–109]. For example, deferoxamine showed anti-tumor effect in patients with advanced HCC [110, 111]. In addition, iron chelator triapine showed promising results for patients with stage IB2-IIIB cervical cancer [112] and is currently being advanced to a phase II randomized trial for treatment of advanced cervical and vaginal cancers [113]. It is suggested that the anti-tumor effect of iron chelator is at least in part via activation of p53 or its downstream target, p21 [74]. Interestingly, p53 is not only necessary for inducing growth suppression upon iron depletion but also necessary for the maintenance of the intracellular iron pool. Thus, many questions remain unanswered. First, p53 is mutated in more than half of human cancers. Interestingly, it was found that ISCU expression was significantly lower in hepatocellular carcinoma tissues with mutant p53 as compared to those with wild-type p53 [82], suggesting a role of mutant p53 in deregulating iron homeostasis. Therefore, the role of mutant p53 in iron metabolism needs to be elucidated. Second, the genetic status of the p53 gene in tumors need to be determined when iron chelating agents are considered as a therapeutic strategy. Accordingly, the anti-tumor effect of iron chelator needs to be carefully examined in tumors with a mutant p53. Third, as p53 appears to modulate expression of genes involving in both systemic and cellular iron homeostasis, further studies are needed to determine how p53 cooperates with these iron regulators to modulate iron metabolism, which may shed a light to the role of p53 in diseases associated with aberrant iron homeostasis. Forth, several regulatory loops exist between p53 and mitochondrial iron regulators, such as FDXR-p53 and Frataxin-p53 loops. For example, FDXR is required for both wild-type and mutant p53 expression, thus, targeting FDXR may provide an ideal approach for tumors bearing a mutant p53 but not for the ones with wild-type p53. Thus, understanding the role of these feedback loops in cancer is required to target these loops as a cancer therapeutic strategy.
Aknowledgements:
This work is supported in part by NIH grants (CA224433, CA076069, CA081237, and CA195828)
Abbreviations:
- IRP1
iron regulatory protein 1
- IRP2
iron regulatory protein 2
- IRE
iron-responsive elements
- UTR
untranslated region
- ALA
δ-aminolevulinic acid
- ALAS1
δ-aminolevulinic acid synthase 1
- CPgenIII
coproporphyrinogen III
- PPIX
protoporphyrin IX
- ISC
iron sulfur cluster
- FDXR
ferredoxin reductase
- FDX1/2
ferredoxins
- FA
Friedreich’s ataxia
- Ftmt
mitochondrial ferritin
- FPN
ferroportin
References
- 1.Loreal O, et al. , Iron, hepcidin, and the metal connection. Front Pharmacol, 2014. 5: p. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donovan A, et al. , The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab, 2005. 1(3): p. 191–200. [DOI] [PubMed] [Google Scholar]
- 3.Park CH, et al. , Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem, 2001. 276(11): p. 7806–10. [DOI] [PubMed] [Google Scholar]
- 4.Krause A, et al. , LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett, 2000. 480(2–3): p. 147–50. [DOI] [PubMed] [Google Scholar]
- 5.Knutson MD, et al. , Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A, 2005. 102(5): p. 1324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laftah AH, et al. , Effect of hepcidin on intestinal iron absorption in mice. Blood, 2004. 103(10): p. 3940–4. [DOI] [PubMed] [Google Scholar]
- 7.Ramey G, et al. , Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica, 2010. 95(3): p. 501–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemeth E, et al. , Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science, 2004. 306(5704): p. 2090–3. [DOI] [PubMed] [Google Scholar]
- 9.Delaby C, et al. , Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood, 2005. 106(12): p. 3979–84. [DOI] [PubMed] [Google Scholar]
- 10.De Falco L, et al. , Iron refractory iron deficiency anemia. Haematologica, 2013. 98(6): p. 845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraenkel PG, Anemia of Inflammation: A Review. Med Clin North Am, 2017. 101(2): p. 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pietrangelo A, Hereditary hemochromatosis--a new look at an old disease. N Engl J Med, 2004. 350(23): p. 2383–97. [DOI] [PubMed] [Google Scholar]
- 13.Origa R, beta-Thalassemia. Genet Med, 2017. 19(6): p. 609–619. [DOI] [PubMed] [Google Scholar]
- 14.Anderson CP, et al. , Mammalian iron metabolism and its control by iron regulatory proteins. Biochim Biophys Acta, 2012. 1823(9): p. 1468–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenstein RS, Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr, 2000. 20: p. 627–62. [DOI] [PubMed] [Google Scholar]
- 16.Muckenthaler MU, Galy B, and Hentze MW, Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr, 2008. 28: p. 197–213. [DOI] [PubMed] [Google Scholar]
- 17.Recalcati S, Minotti G, and Cairo G, Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal, 2010. 13(10): p. 1593–616. [DOI] [PubMed] [Google Scholar]
- 18.Smith SR, et al. , Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol Dis, 2006. 36(2): p. 283–7. [DOI] [PubMed] [Google Scholar]
- 19.Galy B, et al. , Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab, 2008. 7(1): p. 79–85. [DOI] [PubMed] [Google Scholar]
- 20.Cooperman SS, et al. , Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood, 2005. 106(3): p. 1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galy B, et al. , Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2). Blood, 2005. 106(7): p. 2580–9. [DOI] [PubMed] [Google Scholar]
- 22.Meyron-Holtz EG, et al. , Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J, 2004. 23(2): p. 386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh MC, et al. , Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha. Cell Metab, 2013. 17(2): p. 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson SA, et al. , The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab, 2013. 17(2): p. 282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maffettone C, et al. , Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS One, 2010. 5(4): p. e10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G, et al. , Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis, 2007. 28(4): p. 785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, et al. , Insights on regulation and function of the iron regulatory protein 1 (IRP1). Hemoglobin, 2008. 32(1–2): p. 109–15. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, et al. , IRP2 regulates breast tumor growth. Cancer Res, 2014. 74(2): p. 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poulos TL, Heme enzyme structure and function. Chem Rev, 2014. 114(7): p. 3919–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Severance S and Hamza I, Trafficking of heme and porphyrins in metazoa. Chem Rev, 2009. 109(10): p. 4596–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schultz IJ, et al. , Iron and porphyrin trafficking in heme biogenesis. J Biol Chem, 2010. 285(35): p. 26753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bottomley SS and Fleming MD, Sideroblastic anemia: diagnosis and management. Hematol Oncol Clin North Am, 2014. 28(4): p. 653–70, v. [DOI] [PubMed] [Google Scholar]
- 33.Cotter PD, Rucknagel DL, and Bishop DF, X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood, 1994. 84(11): p. 3915–24. [PubMed] [Google Scholar]
- 34.Caudill JS, et al. , Congenital sideroblastic anemia associated with germline polymorphisms reducing expression of FECH. Haematologica, 2008. 93(10): p. 1582–4. [DOI] [PubMed] [Google Scholar]
- 35.Beinert H, Holm RH, and Munck E, Iron-sulfur clusters: nature’s modular, multipurpose structures. Science, 1997. 277(5326): p. 653–9. [DOI] [PubMed] [Google Scholar]
- 36.Stehling O and Lill R, The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol, 2013. 5(8): p. a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bandyopadhyay S, Chandramouli K, and Johnson MK, Iron-sulfur cluster biosynthesis. Biochem Soc Trans, 2008. 36(Pt 6): p. 1112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouault TA, Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech, 2012. 5(2): p. 155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stemmler TL, et al. , Frataxin and mitochondrial FeS cluster biogenesis. J Biol Chem, 2010. 285(35): p. 26737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, et al. , Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim Biophys Acta, 2012. 1823(2): p. 484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campuzano V, et al. , Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 1996. 271(5254): p. 1423–7. [DOI] [PubMed] [Google Scholar]
- 42.Puccio H, et al. , Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet, 2001. 27(2): p. 181–6. [DOI] [PubMed] [Google Scholar]
- 43.Farhan SM, et al. , Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol Genet Genomic Med, 2014. 2(1): p. 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mochel F, et al. , Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet, 2008. 82(3): p. 652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsson A, et al. , Myopathy with lactic acidosis is linked to chromosome 12q23.3–24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet, 2008. 17(11): p. 1666–72. [DOI] [PubMed] [Google Scholar]
- 46.Spiegel R, et al. , Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur J Hum Genet, 2014. 22(7): p. 902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng Y, et al. , Biallelic mutations in the ferredoxin reductase gene cause novel mitochondriopathy with optic atrophy. Hum Mol Genet, 2017. 26(24): p. 4937–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paul A, et al. , FDXR Mutations Cause Sensorial Neuropathies and Expand the Spectrum of Mitochondrial Fe-S-Synthesis Diseases. Am J Hum Genet, 2017. 101(4): p. 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torti SV and Torti FM, Iron and cancer: more ore to be mined. Nat Rev Cancer, 2013. 13(5): p. 342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinnix ZK, et al. , Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med, 2010. 2(43): p. 43ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basuli D, et al. , Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene, 2017. 36(29): p. 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tesfay L, et al. , Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res, 2015. 75(11): p. 2254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanno T, et al. , Hepcidin, anaemia, and prostate cancer. BJU Int, 2011. 107(4): p. 678–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamy PJ, Durigova A, and Jacot W, Iron homeostasis and anemia markers in early breast cancer. Clin Chim Acta, 2014. 434: p. 34–40. [DOI] [PubMed] [Google Scholar]
- 55.Kijima H, et al. , Expression of hepcidin mRNA is uniformly suppressed in hepatocellular carcinoma. BMC Cancer, 2008. 8: p. 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kali A, Charles MV, and Seetharam RS, Hepcidin - A novel biomarker with changing trends. Pharmacogn Rev, 2015. 9(17): p. 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Isobe T, et al. , Human STEAP3 maintains tumor growth under hypoferric condition. Exp Cell Res, 2011. 317(18): p. 2582–91. [DOI] [PubMed] [Google Scholar]
- 58.Jeong SM, Hwang S, and Seong RH, Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem Biophys Res Commun, 2016. 471(3): p. 373–9. [DOI] [PubMed] [Google Scholar]
- 59.Schonberg DL, et al. , Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell, 2015. 28(4): p. 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alvarez SW, et al. , NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature, 2017. 551(7682): p. 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muckenthaler MU, et al. , A Red Carpet for Iron Metabolism. Cell, 2017. 168(3): p. 344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Le NT and Richardson DR, The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta, 2002. 1603(1): p. 31–46. [DOI] [PubMed] [Google Scholar]
- 63.Nurtjahja-Tjendraputra E, et al. , Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood, 2007. 109(9): p. 4045–54. [DOI] [PubMed] [Google Scholar]
- 64.Inoue S and Kawanishi S, Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res, 1987. 47(24 Pt 1): p. 6522–7. [PubMed] [Google Scholar]
- 65.Dizdaroglu M, et al. , Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch Biochem Biophys, 1991. 285(2): p. 317–24. [DOI] [PubMed] [Google Scholar]
- 66.Dizdaroglu M and Jaruga P, Mechanisms of free radical-induced damage to DNA. Free Radic Res, 2012. 46(4): p. 382–419. [DOI] [PubMed] [Google Scholar]
- 67.Dizdaroglu M, et al. , Free radical-induced damage to DNA: mechanisms and measurement. Free Radic Biol Med, 2002. 32(11): p. 1102–15. [DOI] [PubMed] [Google Scholar]
- 68.Vogelstein B, Lane D, and Levine AJ, Surfing the p53 network. Nature, 2000. 408(6810): p. 307–10. [DOI] [PubMed] [Google Scholar]
- 69.Vousden KH and Prives C, Blinded by the Light: The Growing Complexity of p53. Cell, 2009. 137(3): p. 413–31. [DOI] [PubMed] [Google Scholar]
- 70.Kandoth C, et al. , Mutational landscape and significance across 12 major cancer types. Nature, 2013. 502(7471): p. 333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harms K, Nozell S, and Chen X, The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci, 2004. 61(7–8): p. 822–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang F, et al. , Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem, 2008. 283(49): p. 33911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saletta F, et al. , Iron chelator-mediated alterations in gene expression: identification of novel iron-regulated molecules that are molecular targets of hypoxia-inducible factor-1 alpha and p53. Mol Pharmacol, 2010. 77(3): p. 443–58. [DOI] [PubMed] [Google Scholar]
- 74.Liang SX and Richardson DR, The effect of potent iron chelators on the regulation of p53: examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis, 2003. 24(10): p. 1601–14. [DOI] [PubMed] [Google Scholar]
- 75.Kim BM, et al. , Desferrioxamine (DFX) has genotoxic effects on cultured human lymphocytes and induces the p53-mediated damage response. Toxicology, 2007. 229(3): p. 226–35. [DOI] [PubMed] [Google Scholar]
- 76.An WG, et al. , Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature, 1998. 392(6674): p. 405–8. [DOI] [PubMed] [Google Scholar]
- 77.Peyssonnaux C, et al. , Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest, 2007. 117(7): p. 1926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peyssonnaux C, Nizet V, and Johnson RS, Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle, 2008. 7(1): p. 28–32. [DOI] [PubMed] [Google Scholar]
- 79.Shen J, et al. , Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep, 2014. 7(1): p. 180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bahram S, et al. , Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc Natl Acad Sci U S A, 1999. 96(23): p. 13312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dongiovanni P, et al. , Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol, 2010. 176(2): p. 1006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Funauchi Y, et al. , Regulation of iron homeostasis by the p53-ISCU pathway. Sci Rep, 2015. 5: p. 16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weizer-Stern O, et al. , Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol, 2007. 138(2): p. 253–62. [DOI] [PubMed] [Google Scholar]
- 84.Faniello MC, et al. , p53-mediated downregulation of H ferritin promoter transcriptional efficiency via NF-Y. Int J Biochem Cell Biol, 2008. 40(10): p. 2110–9. [DOI] [PubMed] [Google Scholar]
- 85.Zhao N, et al. , The Tumor Suppressor, P53, Decreases the Metal Transporter, ZIP14. Nutrients, 2017. 9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hwang PM, et al. , Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med, 2001. 7(10): p. 1111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu G and Chen X, The ferredoxin reductase gene is regulated by the p53 family and sensitizes cells to oxidative stress-induced apoptosis. Oncogene, 2002. 21(47): p. 7195–204. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Y, et al. , Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sheftel AD, et al. , Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A, 2010. 107(26): p. 11775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shimizu R, et al. , p53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene, 2014. 551(1): p. 79–85. [DOI] [PubMed] [Google Scholar]
- 91.Sawamoto M, et al. , The p53-dependent expression of frataxin controls 5-aminolevulinic acid-induced accumulation of protoporphyrin IX and photo-damage in cancerous cells. Photochem Photobiol, 2013. 89(1): p. 163–72. [DOI] [PubMed] [Google Scholar]
- 92.Loria F and Diaz-Nido J, Frataxin knockdown in human astrocytes triggers cell death and the release of factors that cause neuronal toxicity. Neurobiol Dis, 2015. 76: p. 1–12. [DOI] [PubMed] [Google Scholar]
- 93.Palomo GM, et al. , Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum Mol Genet, 2011. 20(14): p. 2807–22. [DOI] [PubMed] [Google Scholar]
- 94.Guccini I, et al. , Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis, 2011. 2: p. e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee JH, et al. , Ferritin binds and activates p53 under oxidative stress. Biochem Biophys Res Commun, 2009. 389(3): p. 399–404. [DOI] [PubMed] [Google Scholar]
- 96.Shi ZH, et al. , Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell Mol Life Sci, 2015. 72(5): p. 983–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dixon SJ, et al. , Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 2012. 149(5): p. 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shen Z, et al. , Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv Mater, 2018. 30(12): p. e1704007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang L, et al. , Ferroptosis as a p53-mediated activity during tumour suppression. Nature, 2015. 520(7545): p. 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gao M, et al. , Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell, 2015. 59(2): p. 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ou Y, et al. , Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A, 2016. 113(44): p. E6806–E6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jennis M, et al. , An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev, 2016. 30(8): p. 918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie Y, et al. , The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep, 2017. 20(7): p. 1692–1704. [DOI] [PubMed] [Google Scholar]
- 104.Tarangelo A, et al. , p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Reports, 2018. 22(3): p. 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tarangelo A, et al. , p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep, 2018. 22(3): p. 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Corce V, et al. , Recent advances in cancer treatment by iron chelators. Bioorg Med Chem Lett, 2016. 26(2): p. 251–256. [DOI] [PubMed] [Google Scholar]
- 107.Yu Y, et al. , Iron chelators for the treatment of cancer. Curr Med Chem, 2012. 19(17): p. 2689–702. [DOI] [PubMed] [Google Scholar]
- 108.Kalinowski DS and Richardson DR, The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev, 2005. 57(4): p. 547–83. [DOI] [PubMed] [Google Scholar]
- 109.Richardson DR, Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol, 2002. 42(3): p. 267–81. [DOI] [PubMed] [Google Scholar]
- 110.Yamasaki T, Saeki I, and Sakaida I, Efficacy of iron chelator deferoxamine for hepatic arterial infusion chemotherapy in advanced hepatocellular carcinoma patients refractory to current treatments. Hepatol Int, 2014. 8 Suppl 2: p. 492–8. [DOI] [PubMed] [Google Scholar]
- 111.Yamasaki T, Terai S, and Sakaida I, Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med, 2011. 365(6): p. 576–8. [DOI] [PubMed] [Google Scholar]
- 112.Kunos CA and Sherertz TM, Long-Term Disease Control with Triapine-Based Radiochemotherapy for Patients with Stage IB2-IIIB Cervical Cancer. Front Oncol, 2014. 4: p. 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kunos CA, et al. , Radiochemotherapy plus 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in advanced-stage cervical and vaginal cancers. Gynecol Oncol, 2013. 130(1): p. 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]