

Abstract
Excitotoxicity, caused by exaggerated neuronal stimulation by Glutamate (Glu), is a major cause of neurodegeneration in brain ischemia. While we know that neurodegeneration is triggered by overstimulation of Glu-Receptors (GluRs), the subsequent mechanisms that lead to cellular demise remain controversial. Surprisingly, signaling downstream of GluRs can also activate neuroprotective pathways. The strongest evidence involves activation of the transcription factor cAMP Response Element Binding-protein (CREB), widely recognized for its importance in synaptic plasticity. Canonical views describe CREB as a phosphorylation-triggered transcription factor, where transcriptional activation involves CREB phosphorylation and association with CREB Binding Protein (CBP). However, given CREB’s ubiquitous cross-tissue expression, the multitude of cascades leading to CREB phosphorylation, and its ability to regulate thousands of genes, it remains unclear how CREB exerts closely-tailored, differential neuroprotective responses in excitotoxicity. A non-canonical, alternative cascade for activation of CREB-mediated transcription involves the CREB co-factor cAMP-regulated transcriptional co-activator (CRTC), and may be independent of CREB phosphorylation. To identify cascades that activate CREB in excitotoxicity we used a C. elegans model of neurodegeneration by excitotoxic necrosis. We demonstrated that CREB’s neuroprotective effect was conserved, and seemed most effective in neurons with moderate Glu exposure. We found that factors mediating canonical CREB activation were not involved. Instead, phosphorylation-independent CREB activation in nematode excitotoxic necrosis hinged on CRTC. CREB-mediated transcription that depends on CRTC, but not on CREB phosphorylation, might lead to expression of a specific subset of neuroprotective genes. Elucidating conserved mechanisms of excitotoxicity-specific CREB activation can help us focus on core neuroprotective programs in excitotoxicity.
Keywords: C. elegans, CREB, CRTC, Excitotoxicity, Neurodegeneration, Neuroprotection
Graphical Abstract
Excitotoxicity is a major cause of neurodegeneration in brain ischemia, and the transcription factor cAMP Response Element Binding-protein (CREB) mediates neuroprotection in excitotoxicity. Canonical views describe CREB as triggered by phosphorylation, but the wide use of this mechanism makes it unclear how CREB exerts specific neuroprotective responses. We use a model of excitotoxic necrosis in C. elegans, and show that a conserved mechanism in CREB activation in excitotoxic necrosis involves a CREB co-factor (CRTC) and is independent of CREB phosphorylation. Using this non-canonical mechanism of CREB activation might lead to expression of a specific subset of neuroprotective genes in excitotoxicity.
Introduction
Excitotoxic neurodegeneration is a leading cause of neuronal damage in brain ischemia, and a contributing factor in a range of progressive neurological diseases (Choi & Rothman 1990, Moskowitz et al. 2010, Lai et al. 2014, Tymianski 2014). Excitotoxicity is triggered by malfunction of Glutamate Transporters (GluTs) (Danbolt 2001), leading to the accumulation of Glutamate (Glu) in the synapse, overstimulation of Glu Receptors (GluRs) on postsynaptic neurons, and postsynaptic buildup of toxic Ca2+ concentrations. The exaggerated Ca2+ influx eventually causes extensive neurodegeneration, with morphology that spans the range from necrosis to apoptosis, correlated with the extent of the insult (Choi & Rothman 1990, Mehta et al. 2007, Moskowitz et al. 2010, Lai et al. 2014). Neuronal destruction in brain ischemia is progressive: Neuronal damage is most rapid and severe in the core of the stroke area, while the surrounding penumbra is initially only “stunned”. The fate of neurons in the penumbra (degeneration by necrosis, by apoptosis, or recovery) becomes apparent only at a later stage. The neurons in the penumbra area are therefore considered salvageable if appropriate therapy can be devised to protect them (Moskowitz et al. 2010). Although a plethora of intricate signaling cascades has been proposed to mediate the toxic effect of GluR hyperactivation (Mehta et al. 2007, Lai et al. 2011, Tymianski 2011, Fan et al. 2017), the contribution of each of these mechanisms to excitotoxicity in vivo is fiercely debated. Furthermore, a large number of clinical trials based on GluR antagonists or inhibitors of the proposed immediate downstream mechanisms have failed (Ikonomidou & Turski 2002, Tymianski 2014, Lai et al. 2014). These failures suggested that in the clinical setting, application of GluR antagonists “missed the boat”, as neurodestructive processes are already well underway by the time treatment is administered (Ikonomidou & Turski 2002).
However, these failures also emphasized a novel perspective: Surprisingly, in addition to its role in neurodegeneration, GluR activation also triggers neuroprotective signaling cascades that can mitigate neuronal demise. These neuroprotective effects were initially studied when protection was triggered at the same time of-, or even before- hyperstimulation of GluR (the latter being an example of preconditioning) (Meller et al. 2005, Gidday 2006, Hardingham & Bading 2010, Kitagawa 2012, Lai et al. 2014). In both cases, the transcription factor cAMP Response Element Binding protein (CREB) has a central role (Walton & Dragunow 2000, Ikonomidou & Turski 2002, Sakamoto et al. 2011, Lai et al. 2014). Importantly, subsequent studies showed that activating GluR- and CREB-mediated neuroprotection has beneficial effects even if triggered considerably after the onset of the excitotoxic insult, marking it with special clinical relevance (Papadia et al. 2005, Liu et al. 2007, Zhao et al. 2012).
CREB was originally identified as a bZIP-domain containing transcription factor (TF) that is widely found in many cell types, and is activated by phosphorylation on Ser133 (a residue located within a kinase-inducible transactivation domain, or KID domain) by the cAMP-dependent kinase PK-A (Montminy & Bilezikjian 1987, Yamamoto et al. 1988, Gonzalez & Montminy 1989, Goodman 1990). With time, CREB was marked as a prototypic example of a TF activated by phosphorylation by a range of kinases (Mayr & Montminy 2001, West et al. 2001, Lonze & Ginty 2002, Deisseroth et al. 2003). Other phosphorylation sites were also recognized in CREB, with the most critical of those (S142;S143) serving an inhibitory function (Mayr & Montminy 2001, Deisseroth & Tsien 2002, West et al. 2002, Lonze & Ginty 2002). CREB was further identified as a central TF in neuroscience, as it was found to mediate the changes in gene expression that occur in synaptic plasticity (Dash et al. 1990, Sheng et al. 1990, Kandel 2001, West et al. 2002, Deisseroth et al. 2003, Carlezon et al. 2005), a function that is conserved throughout evolution (Yin et al. 1995, Kandel 2001). Ca2+ was determined to be the central trigger of CREB phosphorylation and activation following synaptic activity (Sheng et al. 1990, Sheng et al. 1991, Deisseroth et al. 1996, Greer & Greenberg 2008), and a Histone Acetyl Transferase (HAT) protein called CREB Binding Protein (CBP) was found to bind the KID transactivation domain of phosphorylated CREB and mediate transcriptional activation (Chrivia et al. 1993). In the canonical view of CREB activation by synaptic activity (Figure 1A), Ca2+ influx through GluRs triggers the activation of Ca2+-Calmodulin (CaM)-dependent kinases that lead to phosphorylation of CREB on its transactivation domain, allowing it to recruit CBP and stimulate transcription. Ca2+-induced CREB phosphorylation is suggested to come from the moderate activation of the CREB-kinase CaMK-IV by CaM in the nucleus, an effect that is strongly augmented with the robust activation of CaMK-IV by the CaMK-IV-activating kinase CaMKK (Impey & Goodman 2001, Lonze & Ginty 2002, West et al. 2002, Wayman et al. 2008, Flavell & Greenberg 2008). Some studies suggest that there is special importance to a nuclear Ca2+ signal as the critical trigger for CaMK-IV activation (Bading 2013). The same mechanisms were also suggested to be at work to provide neuroprotection from excitotoxicity (Walton et al. 1999, Walton & Dragunow 2000, Mabuchi et al. 2001, Mantamadiotis et al. 2002, Hardingham et al. 2002, Ikonomidou & Turski 2002, Kitagawa 2007, Hardingham & Bading 2010, Sakamoto et al. 2011, Lai et al. 2014). Further studies identified CREB-targeted neuroprotective genes that are turned-on under conditions of canonical CREB activation, focusing on anti-apoptotic genes (Hardingham et al. 2002, Zhang et al. 2007, Zhang et al. 2009). However the target genes and mechanisms of CREB-mediated protection from excitotoxic necrosis remain under-explored.
Figure 1: Models of CREB activation by GluR activity leading to neuroprotection.

A) Canonical. B) Non Canonical.
Moreover, it is unclear how the widely-used canonical mode of CREB activation is able to specify a closely-tailored pattern of gene expression specific for excitotoxic neuroprotection. Indeed CREB is ubiquitously expressed in all tissues (Mayr & Montminy 2001), it can be stimulated by hundreds of different stimuli (Johannessen et al. 2004), and it regulates the expression of a total of 4,000–6,000 different genes (Zhang et al. 2005, Impey et al. 2004). Only a subset of these genes is suggested to be expressed in any given scenario (Cha-Molstad et al. 2004, Zhang et al. 2005). Wholesale hyperactivation of CREB is not a beneficial anti-excitotoxicity strategy, as it was found to cause mis-regulation of many genes, leading to extensive neurodegeneration of hippocampal neurons by excitotoxicity (probably due to exaggerated enhancement of neuronal activity) (Lopez de Armentia et al. 2007, Valor et al. 2010, Benito et al. 2011). Specificity in CREB’s action is believed to be conferred by the constellation of cellular conditions that trigger CREB activation in each case. This variability in activation modes might in turn give rise to diversification in the way CREB is activated and its interaction with other TFs (Lyons & West 2011).
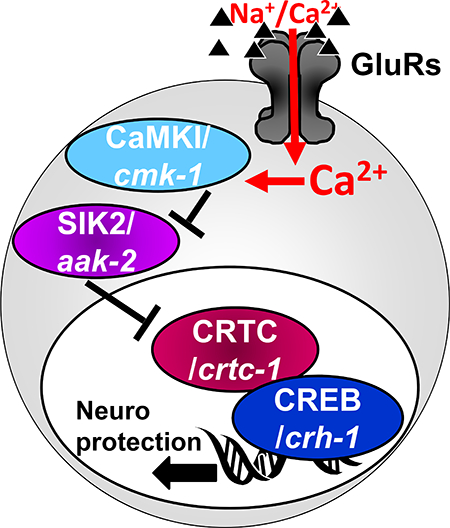
For instance, a subset of CREB triggering conditions leads to the recruitment members of the family of CREB co-factors called cAMP-regulated transcriptional co-activators (CRTCs), which shuttle between the cytoplasm and the nucleus, where they can activate the nucleus-resident CREB. Different CRTCs, (with CRTC1 being a brain-specific member of this TF family (Wu et al. 2006, Altarejos et al. 2008, Watts et al. 2011)), regulate a number of biological processes, including metabolism, cell transformation, memory and lifespan, and when disrupted cause age-related brain diseases and neurodegeneration (Altarejos & Montminy 2011, Saura & Cardinaux 2017, Parra-Damas et al. 2017a). Unlike CBP, CRTCs bind directly to CREB’s bZIP DNA-binding domain, to stabilize CREB’s interaction with the DNA (Iourgenko et al. 2003, Conkright et al. 2003, Screaton et al. 2004, Takemori et al. 2007, Altarejos & Montminy 2011). CRTC includes its own transactivation domain, adding to the capability of the complex to bind additional factors and activate transcription (Altarejos & Montminy 2011). The availability of CRTC in the nucleus depends on its own phosphorylation state (Figure 1B): When CRTC is unphosphorylated, it translocates into the nucleus and activates CREB. However, under resting conditions CRTC is prevented from entering the nucleus due to its phosphorylation by the Salt Induced Kinases (SIK1/2). In the non-canonical mode of CREB activation, Ca2+-triggered inhibition of SIK1/2 allows CRTC to enter the nucleus and activate CREB-mediated transcription, independently of CREB phosphorylation and CBP binding. Under these conditions, the CRTC::CREB complex associates instead with other HATs such as PCAF/KAT2 or KAT5 (Ravnskjaer et al. 2013, Clark et al. 2015) that mediate a different profile of histone acetylation (compared to pCREB::CBP) (Hirano et al. 2016, Uchida et al. 2017). Such non-canonical, CRTC-mediated activation of CREB-mediated transcription was observed in neurons under some conditions of synaptic plasticity (Kovacs et al. 2007, Ch’ng et al. 2012, Nonaka et al. 2014, Briand et al. 2015, Uchida et al. 2017, Parra-Damas et al. 2017b) and in Huntington’s disease (Jeong et al. 2012). One study further suggests that the non-canonical mode of CREB activation is also operational in excitotoxic neuroprotection, where SIK2 inhibition is achieved by Ca2+-mediated activation of cytoplasmic CaMK-I, which can phosphorylate and inhibit SIK2 (Sasaki et al. 2011). Therefore, while the prevalent view of CREB-mediated transcription in neuronal plasticity and neuroprotection is based on the canonical activation of transcription (by phosphorylation of CREB and its association with CBP), a few lines of evidence suggest instead that neuroprotection and some types of memory paradigms might be achieved by a non-canonical mode of activation. This non-canonical mechanism depends on the function of CaMK-I, SIK2, and CRTC, it is independent of CBP or CREB phosphorylation, and it might result in a different profile of histone acetylation (Altarejos & Montminy 2011, Sasaki et al. 2011, Ch’ng et al. 2012, Hirano et al. 2016, Uchida et al. 2017). Unlike the rather indiscriminately broad effect of hyperactivation of CREB, hyperactivation of CRTC seems to have a more focused and beneficial effect, and it is not reported to result in spontaneous neurodegeneration (Parra-Damas et al. 2014, Parra-Damas et al. 2017a). However, CRTC’s effect on induced excitotoxicity is under-investigated. Given the paucity of evidence in support of non-canonical activation of CREB in neuroprotection, it remains unclear if this mode of CREB activation is widely involved in reducing excitotoxic damage, and if it protects from apoptosis or necrosis.
These two mechanisms of CREB activation might contribute to different modes of gene activation and neuroprotection programs, depending on the exact conditions (such as the presence of apoptosis vs necrosis). However, while transcriptional programs for GluR-mediated protection from apoptosis have been studied before (Zhang et al. 2007, Zhang et al. 2009), evolutionarily conserved processes that promote neuronal survival in excitotoxic necrosis are understudied. Filling this gap might highlight the critical core of the neuroprotective pathway in this devastating form of neurodegeneration in excitotoxicity. We therefore turned to study this question in our model of excitotoxic necrosis in the nematode C. elegans (Mano & Driscoll 2009). In addition to the strong conservation of key cellular mechanisms (including cell death) (Lettre & Hengartner 2006), the powerful genetic (Brenner 1974) and neuroscience (White et al. 1986, Rankin 2002) tools available in this system make the elucidation of critical signaling cascades especially productive. Glutamate is a central neurotransmitter in the worm and is widely used to stimulate command interneurons through conserved GluRs (Brockie & Maricq 2006). GluRs mediate both basic signaling and synaptic plasticity (Rose & Rankin 2006, Rose et al. 2005, Rose et al. 2003, Emtage et al. 2009, Stetak et al. 2009). CREB (worm homolog name: CRH-1) and the components of its canonical activation cascade are also well conserved in the worm, and regulate learning and memory and synaptic plasticity (Kimura et al. 2002, Bates et al. 2006, Suo et al. 2006, Kauffman et al. 2010, Timbers & Rankin 2011, Nishida et al. 2011, Yu et al. 2014, Lakhina et al. 2015, Chen et al. 2016, Moss et al. 2016, Freytag et al. 2017, Nishijima & Maruyama 2017, Arey et al. 2018, Kaletsky et al. 2018). CREB/CRH-1 can be activated by phosphorylation (on a site homologous to Ser133) by the nematode’s combined CaMK I/IV homolog CMK-1, whose basal activity is greatly stimulated by CaMKK/CKK-1 (Kimura et al. 2002, Yu et al. 2014) (while the other CREB phosphorylation sites seen in mammals do not seem to be conserved in the nematode). Some studies also showed regulation of CREB/CRH-1 by KIN-1/KIN-2, the nematode homologs of the regulatory and catalytic subunits of cAMP dependent kinase PK-A (Rojo Romanos et al. 2017), which might determine the speed of developmental modifications in some neurons in the nerve cord (Yu et al. 2017). At least in some scenarios, (such as synapse development in the nematode) PK-A –regulated CREB/CRH-1 activity is independent of CRTC, further supporting the division of CREB functions into subgroups of target genes, only some of which requiring CRTC (Maeder et al. 2018). Similarly, CBP/CBP-1 cooperates with CREB/CRH-1 in many cells (Eastburn & Han 2005), and protects neurons from polyglutamine-induced neurodegeneration (Bates et al. 2006). SIK1/2 homologs are also present, including KIN-29 and AAK-2 (Lanjuin & Sengupta 2002, Savage-Dunn et al. 2003, Singaravelu et al. 2007, Apfeld et al. 2004, Mair et al. 2011). Similarly to SIK1/2 in mammals, AAK-2 modulates the nuclear translocation of CRTC/CRTC-1 to regulate CREB/CRH-1 –mediated transcription (and here too, CRTC-1 needs to be unphosphorylated in order to be preferentially localized to the nucleus) (Mair et al. 2011, Burkewitz et al. 2015).
In our model of nematode excitotoxicity (Mano & Driscoll 2009), knockout (KO) of the cardinal GluT gene glt-3 (Mano et al. 2007) in the nuIs5 sensitized background (Berger et al. 1998) causes the necrotic death of some of the neurons postsynaptic to Glu connections. This Glu-triggered neuronal necrosis is independent of canonical apoptosis (Tehrani et al. 2014), and it shows key features conserved in excitotoxicity, such as dependence on key Ca2+-permeable GluRs (Brockie & Maricq 2006), Ca2+ release from intracellular stores, involvement of DAPK (Del Rosario et al. 2015), and modulation by FoxO/DAF-16 (Tehrani et al. 2014). We therefore set out to address the controversy regarding the mechanism of CREB-mediated neuroprotection in excitotoxic necrosis, using a powerful genetic model where the most important core events in this process are likely to be conserved. In the current study we find that indeed CREB/CRH-1 has a neuroprotective role in neurons exposed to a moderate excitotoxic necrosis, and that the non-canonical mechanism of CREB activation is the one that is evolutionary conserved in neuroprotection from excitotoxic necrosis.
Methods
Strains
Strains were maintained at 20°C according to Brenner (Brenner 1974), and grown on MYOB agar plates (Church et al. 1995) seeded with OP50 (Stiernagle 2006). All the major new strains were constructed twice from independent crosses, and data was verified to be similar.
Strains used: WT:Bristol N2 (RRID:CGC_N2 (ancestral)); Excitotoxicity strain: ZB1102: glt-3(bz34) IV; nuIs5 V (RRID:CGC_ZB1102) (Mano & Driscoll 2009); crh-1: YT17: crh-1(tz2) III (RRID:CGC_YT17) (Kimura et al. 2002); age-1: TJ1052: age-1(hx546) II (RRID:CGC_TJ1052) (Friedman & Johnson 1988); cbp-1 hyperactivity: MH2430: cbp-1(ku258) III (RRID:CGC_MH2430) (Eastburn & Han 2005); cmk-1: VC220: cmk-1(ok287) IV; gkDf56 Y102A5C.36(gk3558) V (RRID:CGC_VC220) (Satterlee et al. 2004); aak-2: RB754: aak-2(ok524) X (RRID:CGC_RB754) (Apfeld et al. 2004); crtc-1: crtc-1(tm2869) I (Mair et al. 2011); rol-6: HE1006: rol-6(su1006) II (RRID:CGC_HE1006); WT CRTC labeled with tdTOMATO: AGD418: uthIs205[Pcrtc-1::crtc-1::tdTOMATO::unc-54 3’UTR; rol-6(su1006)] (RRID:CGC_AGD418) (Mair et al. 2011); Unphosphorylatable CRTC: Extrachromosomal, AGD466: uthEx222[Pcrtc-1::crtc-1 (S76A, S179A)::tdTOMATO::unc-54 3’UTR; rol-6(su1006)]; Integrated, WBM55: uthIs226[Pcrtc-1::crtc-1(S76A, S179A)::tdTOMATO::unc-54 3’UTR + rol-6(su1006)] (RRID:CGC_WBM55) (Burkewitz et al. 2015); Glutamatergic behavioral negative control: VM1268: nmr-1(ak4) II; glr-2(ak10) glr-1(ky176) III. Some strains were obtained from C. elegans Genetic Center (CGC), Japanese National Bioresource Project (NBRP) or from the original creators. Strains created in this study: crh-1 in excitotoxicity (by crossing YT17 and ZB1102) IMN36: crh-1(tz2) III; glt-3(bz34) IV; nuIs5 V; age-1 in excitotoxicity (by crossing TJ1052 and ZB1102) IMN37: age-1(hx546) II; glt-3(bz34) IV; nuIs5 V; age-1 and crh-1 epistasis in excitotoxicity (by crossing IMN36 and IMN37) IMN38: age-1(hx546) II; crh-1(tz2) III; glt-3(bz34) IV; nuIs5 V; cbp in excitotoxicity (by crossing MH2430 and ZB1102) IMN39: cbp-1(ku528) III; glt-3(bz34) IV; nuIs5 V; cmk-1 in excitotoxicity (by crossing VC220 and ZB1102) IMN40: cmk-1(ok287) IV; glt-3(bz34) IV; nuIs5 V aak-2 in excitotoxicity (by crossing RB754 and ZB1102) IMN41: glt-3(bz34) IV; nuIs5 V; aak-2(ok524) X; crtc in excitotoxicity (by crossing crtc-1(tm2869) I and ZB1102) IMN42: crtc-1(tm2869) I; glt-3(bz34) IV; nuIs5 V; crtc & aak-2 epistasis in excitotoxicity (by crossing IMN41 and IMN42) IMN43: crtc-1(tm2869) I; glt-3(bz34) IV; nuIs5 V; aak-2(ok524) X; roller phenotype control in excitotoxicity (by crossing HE1006 and ZB1102) IMN44 rol-6(su1006) II; glt-3(bz34) IV; nuIs5 V; WT CRTC overexpression in excitotoxicity (by crossing AGD418 and ZB1102): IMN45 glt-3(bz34) IV; nuIs5 V; uthIs205[Pcrtc-1::crtc-1::tdTOMATO::unc-54 3’UTR; rol-6(su1006)]; unphosphorylatable CRTC overexpression in excitotoxicity (by crossing AGD466 or WBM55 with ZB1102): Extrachromosomal, IMN46: glt-3(bz34) IV; nuIs5 V; uthEx222[Pcrtc-1::crtc-1 (S76S, S179A)::tdTOMATO::unc-54 3’UTR; rol-6(su1006)]; Integrated, IMN49: glt-3(bz34) IV; nuIs5 V; uthIs226 [Pcrtc-1::crtc-1 (S76S, S179A)::tdTOMATO::unc-54 3’UTR; rol-6(su1006)]; WT CREB rescue: IMN47 crh-1(tz2) III; glt-3(bz34) IV; nuIs5 V; Ex[Pglr-1::crh-1 cDNA::dsRed; Pmec-4::RFP]; Phosphorylation mutant CREB rescue IMN48: crh-1(tz2) III; glt-3(bz34) IV; nuIs5 V; Ex[Pglr-1::S29A crh-1 cDNA::dsRed; Pmec-4::GFP].
All strains were confirmed homozygous using PCR. Transgenes expressing green or red fluorescence were followed by microscopy. All key strains were derived by two independent crosses, and the effects were scored in each sub-strain. The magnitude of the effect and its significance were verified to be similar, and then the data from the separate sub-strains was pooled.
Overall Quantification of Neurodegeneration.
Overall neurodegeneration levels was quantified in large number of animals using an inverted scope (AxioZeiss) and Nomarski Differential Interference Contrast (DIC). We mounted agar chunks from freshly growing nematode culture plates on a cover slip (without anesthetics), scanned for animals at random, identified their developmental stage (by the shape of the uterus), and counted the number of necrotic neurons in each animal (necrotic neurons appear as vacuolar-looking structures), as previously described (see more details below) (Mano & Driscoll 2009, Tehrani et al. 2014, Del Rosario et al. 2015). The data was confirmed in independent lines (data for separate strains is not shown). Data collection was blinded in terms of the identity of the strain, and animal samples were random as they disperse on the surface of the culture plate from which the chunk is taken for evaluation of neurodegeneration. At least 30 animals were tested for each strain, at each developmental stage. Larger numbers of animals were assessed for neurodegeneration in the L3 developmental stage, since this is when degeneration reaches its peak, and is thus the most informative stage. The range of number of animals assessed for neurodegeneration in each subgroup is indicated by the n= # in the figures. The average number of degenerating head neurons per animal (in each of the indicated developmental stage) is depicted on the graphs in figures 2,3,5,6,7,and 9. Normally, new combination mutants will be compared to a concurrently grown culture of the excitotoxicity strain glt-3;nuIs5. In figure 9, since the CRTC-1 expressing strains from Mair & Dillin have a co-injection marker of rol-6, we compare the new combination strains to rol-6;glt-3;nuIs5. This precaution is taken because in some cases of necrosis examined by the Driscoll lab, such as that caused by mec-4(d), the large necrotic vacuoles might be affected by the mechanics of animal rolling.
Figure 2: CREB/CRH-1 has a conserved role in neuroprotection.

Left panel - DIC images: Vacuole-like structures in the nematode’s head indicate necrotic neurodegeneration in glt-3;nuIs5 (top image) and crh-1;glt-3;nuIs5 (bottom image) animals. Filled red arrows indicate location of necrotic neurons in this or immediately adjacent focal plane; Empty red arrows indicate detection of necrotic neurons in other focal planes. In these and subsequent images: anterior is left, dorsal is up. Right panel - bar graph: Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to a similar strain where CREB/CRH-1 is eliminated (crh-1;glt-3;nuIs5). In this and all subsequent bar graphs: Error bars represent SEM of the number of degenerating head neurons per animal. One-way ANOVAs were performed at each life stage. Asterisks represent statistical significance of the difference between the indicated groups, where * indicates p≤0.05; ** indicates p≤0.01; *** indicates p≤0.001, n.s. denotes non-significant difference between groups. At least 30 animals were scored for each strain, at each developmental stage. The range of the number of animals scored is given as n. Except for extremely rare cases, N2 WT animals do not show necrotic vacuoles (Mano & Driscoll 2009). No necrotic corpses are seen in crh-1 mutants (or other mutants described below) in the absence of excitotoxic conditions (not shown).
Figure 3: CREB/CRH-1 and the Insulin/IIS pathway regulate neuroprotection in separate pathways.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5), excitotoxicity with CREB KO (crh-1;glt-3;nuIs5), excitotoxicity with IIS neuroprotective mutation (age-1;glt-3;nuIs5), and excitotoxicity with a combination of CREB KO and IIS modification (age-1;crh-1;glt-3;nuIs5). All statistical comparisons, at each life-stage: one-way ANOVAs, post hoc Tukey-HSD test, ***p≤0.001. For clarity, only differences between crh-1;glt-3;nuIs5, and age-1;glt-3;nuIs5, and age-1;crh-1;glt-3;nuIs5 are highlighted.
Figure 5: Canonical activator, CBP-1, has no effect on excitotoxic necrosis.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to a similar strain carrying also a hyperactivating mutation in CBP (cbp-1;glt-3;nuIs5). The difference between the two strains is significant (by one-way ANOVA) only in L1 (p≤0.001). However, this difference in L1 might be influenced by cbp-1’s pleiotropic effects, given its “bag-of-worms” phenotype.
Figure 6: Non-canonical CREB modulators affect excitotoxic necrosis.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to a similar strain where a mediator of the non canonical pathway is eliminated: B) Testing the CaMK-I/IV homolog CMK-1 (cmk-1;glt-3;nuIs5); C) Testing the SIK homolog AAK-2 (glt-3;nuIs5;aak-2); D) Testing the CREB cofactor CRTC homolog CRTC-1 (crtc-1;glt-3;nuIs5). All statistical comparisons, at each life-stage used one-way ANOVAs, where * indicates p≤0.05; ** indicates p≤0.01; *** indicates p≤0.001.
Figure 7: SIK/AAK-2 and CRTC-1 work in the same pathway.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to similar strain where SIK/AAK-2 is eliminated (glt-3;nuIs5;aak-2), or CRTC-1 is eliminated (crtc-1;glt-3;nuIs5), or both AAK-2 and CRTC-1 are eliminated (crtc-1;glt-3;nuIs5;aak-2). All statistical comparisons, at each life stage: one-way ANOVA, post hoc Tukey-HSD * indicates p≤0.05; ** indicates p≤0.01; *** indicates p≤0.001. n.s. denotes not significant. At least 30 animals were tested for each strain, at each developmental stage.
Figure 9: Overexpression of either WT or hyperactive CRTC-1 protect from excitotoxicity to a similar extent.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to a similar strain carrying also an integrated transgenic construct expressing cDNAs encoding either WT CRTC-1 or S76A,S179A non-phosphorylatable mutant CRTC-1. Since the integrated strains for WT or hyperactive CRTC-1 overexpression were prepared (by the Dillin & Mair labs) with a rol-6 transgenic marker (a mutation that causes the nematodes to roll around their heads ceaselessly, leading to unusual mechanical forces on the head), we included this transgenic marker also in our control line (rol-6, glt-3;nuIs5). Statistical comparisons, at each lifestage: one-way ANOVAs, post hoc Tukey, ** indicates p≤0.01; *** indicates p≤0.001. For clarity, only significant differences are highlighted.
WT animals or animals carrying the crh-1 mutation alone (or any of the other CREB-related mutations discussed below) without the excitotoxicity background (glt-3;nuIs5) almost never show any necrotic neurodegeneration at any developmental stage (not shown). The nematode excitotoxicity strain (glt-3;nuIs5) combines a KO of the centrally important GluT gene glt-3 with the sensitizing transgenic modification nuIs5 (Mano & Driscoll 2009). The nuIs5 transgene puts ~30 identified neurons postsynaptic to Glu connections at risk of neurodegeneration by expressing a hyperactive Gαs and GFP under the promoter of the GluR subunit glr-1 (Berger et al. 1998). Combining the nuIs5 sensitized background with the GluT KO mutation glt-3(bz34) gives rise to seemingly stochastic- (but see below), and GluR-dependent- necrosis of some of these at-risk neurons. We quantify the extent of necrosis by observing animals at different developmental stages and counting the number of swollen neurons in the head (by screening through a large number of live animals in a mixed population, identifying developmental stage and counting vacuole-like structures in each animal using DIC optics). Typically, excitotoxic neurodegeneration peaks at the L3 developmental stage (coinciding with the maturation of Glu signaling) at the level of ~4.5 head neurons/animal (Mano & Driscoll 2009). Previous studies of neuronal necrosis in C. elegans suggest that necrotic corps are removed within few hours by classic engulfment mechanisms and do not linger between developmental stages (Chung et al. 2000). Our unpublished data (Idrizi & Mano) supports that excitotoxic necrotic corpses are also removed by this mechanism.
Identification of Specific Degenerating Neurons
Identification of the specific neurons that die is a much more meticulous process than overall quantification of degeneration levels, and was done in a smaller group of animals. To identify the specific neurons dying in some of these strains (Figure 4) we imaged L3 animals from strains ZB1102 and IMN32 with Nomarski DIC and fluorescence microscopy (AxioZeiss) with the 63X objective; We mounted L3 animals on agarose gel pads (2%) microscope slides with a drop of M9 buffer. Z-stacks were acquired using Metamorph Software (RRID:SCR_002368). Some animals were treated with 10mM NaN3 and showed the same degeneration pattern as animals that were analyzed without the use of NaN3. DIC and GFP images were examined independently and were merged in ImageJ (imagej.nih.gov) (RRID:SCR_003070). To identify the degenerating neuron, the location of the swollen- (as seen in DIC) and GFP-labeled- cell body and the shape of the neuron’s processes (seen by GFP fluorescence) were compared to those of glr-1–expressing neurons (Brockie & Maricq 2006, Altun et al. 2002–2018). Usually, the GFP signal partially persists in the neurons as they swell-up and go through degeneration, and disappears only in the later stages of cellular demise (at which stage we will see a large non-fluorescent cell corpse in a spot typically occupied by a glr-1 –expressing neuron. We therefore used the persistent GFP label, as well as recording non-labeled neurons, to analyze the identity of degenerating neurons in a representative group of L3 animals. nuIs5 alone showed low background levels of neurodegeneration (which are GluR-independent (Mano & Driscoll 2009)) that was evenly distributed between the different glr-1 –expressing neurons (data not shown).
Figure 4: Neurodegeneration shows variability, but also possible patterns of neuronal vulnerability.

A) Frequency of degeneration of identified neurons in the original excitotoxicity strain (glt-3;nuIs5) is compared to degeneration frequencies in a similar strain where CREB is eliminated (crh-1;glt-3;nuIs5). The X axis lists the categories of glr-1 expressing neurons according to previously established data (Brockie & Maricq 2006). Degenerating cells were identified by the location of their cell bodies and morphology of their processes. Degeneration frequencies show trends, but the high variability prevents affirmation of significance. The background neurodegeneration in nuIs5 alone (which is GluR independent) (Mano & Driscoll 2009) is equally distributed at a very low level without any preference to neuronal types (not shown). B) Data grouped from A, combining data for sensory neurons, motor neurons, and interneurons. ** indicates p≤0.01; *** indicates p≤0.001.
Fluorescence Confocal Microscopy to Study CRTC-1 Expression.
To study the cellular expression on CRTC-1 we mounted L3 animals on agarose gel pads (2%) microscope slides with a drop of M9 buffer. 10mM sodium azide (NaN3, (amresco® CAS # 26628–22-8) was added to immobilize worms for CRTC localization. Strains AGD418 (Mair et al. 2011), IMN41, and IMN49 were imaged with Zeiss LSM880 (RRID:SCR_015963), using a 63X objective lens with GaAsP detection; Z-stacks were obtained with the multi-dimensional acquisition tool using Zen Black 2015 2.1 SP2 software. Images were examined using Fiji (http://fiji.sc) (RRID:SCR_002285) and ImageJ (imagej.nih.gov) (RRID:SCR_003070).
Molecular Biology
Two pENTR Gateway vectors, one expressing wildtype CREB/crh-1 cDNA and a the other expressing CREB/crh-1 cDNA with a single point mutation making a phosphorylation mutant (S29A) were a gift from Hidehito Kuroyanagi (Kimura et al. 2002). In addition, Vector KP#889 (Pglr-1::dsRed, a gift from the Kaplan and the Juo labs) (Kowalski et al. 2011)), was used as the destination vector. Plasmid expressing Pmec-4::GFP and Pmec-4::mCherry (Gifts from Driscoll lab (Royal et al. 2005, Toth et al. 2012)) were used as coinjection markers. WT crh-1 and S29A crh-1 cDNAs were inserted at the BamHI site of KP#889 vector to create a final vectors: Pglr-1::crh-1 cDNA(WT)::dsRed and Pglr-1::crh-1 cDNA(S29A)::dsRed. Final plasmids were confirmed with sequencing using a 5’ CTTCGTCTCGGTCACTTCACTTCG primer for the KP#889 promoter of glr-1. Plasmids were injected into IMN36 crh-1;glt-3;nuIs5 worms (L4 or Young Adult) using standard protocols. Two independent lines were maintained for each wildtype and mutant strains.
Behavioral assays
Locomotion assays (duration of spontaneous forward mobility) (Brockie et al. 2001b) and nose touch assays (NOT) (Kaplan & Horvitz 1993, Hart et al. 1995) were performed blindly, in three independent trials.
Statistics
We calculated averages and SEM of the number of degenerating head neurons per animal (in each of the indicated developmental stage) (figures 2,3,5,6,7, and 9). Degeneration frequency of identified neurons (figure 4) was calculated as the sum of neurons (individual or by type) divided by the total possible (individual or type) number of neurons dying. Statistical analysis was carried out using SPSS v20.0.0 (IBM) (RRID:SCR_002865). Means were compared using one-way ANOVAs/Welch’s ANOVA. Levene’s tests were performed for each set of data to assess the equality of variances. Shapiro-Wilk test was carried out to test for normality; there was a mix of normality and non-normality as determined by the Shapiro-Wilk test. We performed post-hoc Tukey, Games-Howell, or Bonferroni tests, depending upon passing normality and results of the homogeneity tests. No statistical methods were employed to predetermine the sample size, and no active randomization of the samples was performed in the study. We also performed additional statistical analysis using two way ANOVA to examine the main and interaction effects of genotype and developmental stage (see Supplementary Statistical Analysis).
Results
The neuroprotective role of CREB/CRH-1 is conserved in nematode excitotoxicity.
We tested the effect of CREB in nematode excitotoxicity by introducing a standard knockout (KO) allele of the CREB homolog gene (crh-1(tz2)III, missing the bZIP domain) (Kimura et al. 2002) (a strain which does not show neurodegeneration) into our excitotoxicity strain (glt-3(bz34) IV; nuIs5 V) (Mano & Driscoll 2009). Here and in all subsequent crosses we obtained two independent mutant combination lines, we analyzed them separately, verified that the effect of mutant combination in the independently derives strains was similar in magnitude and significance, and then pooled the data to calculate overall averages and determine statistical significance. We observe that adding the CREB/crh-1 ko to the excitotoxicity strain (glt-3;nuIs5) caused a dramatic surge in the extent of necrotic neurodegeneration, increasing the average number of dying head neurons per animal at L3 from 4.5 to above 8 (Figure 2) (ANOVA at L3: F(1,208)= 332.595, p=0.000, with similar significance in other life stages.
Two way ANOVA analysis of this and subsequent experiments yielded similar significance and conclusions as the one‐way ANOVA for genotype effects at each developmental stage, but also revealed a significant main effect of Developmental Stage and a significant interaction of genotype x developmental stage (see supplementary statistical analysis). These observations indicate that the number of degenerating neurons is not the same at each stage, and that the effect of genotype on neuron degeneration may not be the same at each stage. Nonetheless, these results suggest that the presence of fully functional CREB/CRH-1 in wild type (WT) animals protects some of the at-risk neurons from excitotoxic necrosis, and the absence of CREB/CRH-1 causes more of these at-risk neurons to die. We therefore conclude that the neuroprotective effect of CREB/CRH-1 in excitotoxic necrosis is conserved.
The neuroprotective effect of CREB/CRH-1 is independent of neuroprotection by the Insulin/IGF-1 Signaling (IIS) cascade.
Our previous studies on the regulation of excitotoxicity in nematodes and in mouse spinal cultures showed that excitotoxic neuroprotection is modulated by the Insulin/IGF-1 Signaling (IIS) cascade (Mojsilovic-Petrovic et al. 2009, Tehrani et al. 2014). Components of this cascade, and especially Akt, have also been suggested by others to mediate cross-talk with the CREB activation cascades (Soderling 1999, Walton & Dragunow 2000, Yuan & Yankner 2000). To determine if the neuroprotective effect of CREB is mediated by the same pathway as the IIS cascade we used genetic epistasis with a mutation in age-1, encoding the PI3 Kinase that is responsible for the activation of Akt. We have previously shown that the age-1(hx546)II mutation causes increased neuroprotection in nematode excitotoxicity (Mojsilovic-Petrovic et al. 2009). If age-1’s effect is mediated through CREB, then CREB/crh-1 ko should block the neuroprotective effect of the age-1 mutation, and the age-1;crh-1 combination should have the same neurodestructive effect as crh-1 ko alone. In contrast, if age-1 works in a pathway that is independent CREB, then the combination of the neuroprotective age-1 mutation and the neurodestructive crh-1 mutation should result in an intermediate level of neurodegeneration. Our data shows (Figure 3) that the combination strain age-1;crh-1;glt-3;nuIs5 has moderate levels of neurodegeneration, which is between those seen for age-1;glt-3;nuIs5 and crh-1;glt-3;nuIs5. We obtained a similar result using the PI3-kinase chemical inhibitor LY294002 in crh-1;glt-3;nuIs5 animals (not shown). These results demonstrate that IIS and CREB consist of two independent signaling cascades that separately modulate necrotic neurodegeneration in nematode excitotoxicity. We therefore wanted to learn more on CREB’s mechanism of action.
The neuroprotection provided by CREB/CRH-1 in nematode excitotoxicity is more apparent in interneurons.
To gain further insight into the process of neurodegeneration and neuroprotection we looked in more detail at the dying neurons and tried to identify them. Previously, our initial evaluation suggested that the overall pattern of necrosis of the at-risk neurons in the parental excitotoxicity strain (glt-3;nuIs5) is generally stochastic, so that each animal presents a different constellation of dying neurons (based on the rough location of the vacuole-like structures). However, more recently we suspected that the effect of CREB/CRH-1 might be preferentially pronounced in a specific subset of these at-risk neurons, which requires a more detailed analysis. To identify the specific neurons dying in excitotoxicity and neuroprotection, we take advantage of the GFP fluorescence of at-risk neurons in the excitotoxicity strain (glt-3;nuIs5), and compare neuron location and the structure of its processes to the documented features of glr-1 –expressing neurons (Brockie et al. 2001a, Brockie & Maricq 2006, Altun et al. 2002–2018).
We found that both in the regular excitotoxicity strain (glt-3;nuIs5) and in the excitotoxicity plus CREB-KO strain (crh-1;glt-3;nuIs5) the vast majority of swollen degenerating neurons are labeled, if weakly, with GFP. Based on their location, the very few swollen neurons that do not show easily detectible GFP label could be at-risk neurons where GFP has degraded. Excitotoxic neurodegeneration in our model, therefore, seems cell-autonomous. We noticed, however, that even though there is great variability in the probability of each neuron to die, there are also overall patterns that can be distinguished (Figure 4). While the large variability in degeneration levels prevents us from making conclusions on specific neurons (Figure 4A), combining the neurons into groups (according to their role, as defined in WormAtlas.org, Figure 4B) provides sufficient basis for determining the difference in degeneration-levels to be statistically significant. Under normal excitotoxic conditions (glt-3;nuIs5), most of the neurons that die fall into the categories of the sensory neurons of the URY group, and the motor neurons of the RMD and SMD groups. In this original glt-3;nuIs5 excitotoxicity strain, command interneurons such as AIB, AVA, AVB, AVD, and AVE die at lower frequencies. We further observe that the effect of CREB KO (in fold, comparing neurodegeneration levels with and without CREB) is particularly pronounced in the command interneurons (p<0.001) (Figure 4B). This difference in vulnerability can be based on a difference in the neuron’s exposure to the excitotoxic insult (e.g., difference in extracellular Glu concentrations, or a difference in the levels of expression of GluRs), or based on a difference in resilience of the postsynaptic neuron to a given insult. Since the command interneurons are relatively resilient while seemingly being exposed to intense Glu insult (as they are reported to have particularly high levels of GluRs (Brockie et al. 2001a)), we considered other mechanisms for differential vulnerability. To gain more insight into the mechanism of resilience, we study the pathway that allows CREB/CRH-1 to confer neuroprotection.
Key components of the canonical cascade for CREB/CRH-1 activation do not contribute significantly to neuroprotection in nematode excitotoxicity.
We next asked if the canonical mode of CREB activation by phosphorylation is involved in nematode excitotoxicity. Surprisingly, we have recently found that the CaMKK homolog CKK-1 (Eto et al. 1999), which is needed for a large potentiation of phospho-CREB –mediated transcription (Kimura et al. 2002, Yu et al. 2014, Moss et al. 2016), has no effect in nematode excitotoxicity (using the ckk-1(ok1033) III allele in the glt-3;nuIs5 background) (Del Rosario et al. 2015). The effect of CaMK-IV in the nucleus is harder to decipher in the nematode, since the functions of both the cytoplasmic CaMK-I and the nuclear CaMK-IV are mediated in the nematode by a single nematode homolog, CMK-1 (Yu et al. 2014, Schild et al. 2014), which is expressed throughout the nervous system. The function of phosphorylation-dependent CREB partner CBP (which is also widely expressed in neurons (Hunt-Newbury et al. 2007)) can be studied in the worm using a gain-of-function allele of the cbp-1 gene (cbp-1(ku258) III), encoding a CBP with a seven-fold increase in HAT activity (Eastburn & Han 2005, Moss et al. 2016). Similarly to the lack of effect of CaMKK/CKK-1, we find that extensive elevation of CBP/CBP-1 activity has no effect in nematode excitotoxicity (Figure 5). The lack of effect of ckk-1 and cbp-1 argues strongly against the involvement of canonical CREB activation in the neuroprotective effect of CREB in nematode excitotoxicity.
The non-canonical mediators of CREB/CRH-1 activation are central to neuroprotection in nematode excitotoxicity.
Examining non-canonical mechanisms for CREB involvement in nematode excitotoxicity, we turned to test the CaMK-I/SIK/CRTC axis (Figure 6A). As mentioned above, the function of both CaMK-I and CaMK-IV is mediated in the worm by a single gene, cmk-1. In both canonical and non-canonical modes of CREB activation, the effect of WT CMK-1 is predicted to potentiate neuroprotection (either by directly activating CREB, or by suppressing the inhibitor of it co-factor). Indeed, elimination of CMK-1 (using cmk-1(ok287) IV in the excitotoxicity strain glt-3;nuIs5) enhances neurodegeneration (equivalent to suppressing neuroprotection) (Figure 6B, L3 ANOVA: F(1,68)=35.375, p=0.000). Since this result cannot differentiate between the two modes of CREB activation, we advanced further down the pathway and examined the role of the SIK1/2 homolog AAK-2. We found that introducing the aak-2(ok524)X mutation to the excitotoxicity strain (glt-3;nuIs5) causes a pronounced decrease in neurodegeneration (equivalent to increasing neuroprotection) (Figure 6C, L3 ANOVA: F(1,138)=50.798, p=0.000). This observation is in line with a role for WT AAK-2 as a potentiator of excitotoxicity, and in line with the non-canonical mechanism of CREB activation. We finally checked the effect of the defining component of the non-canonical mechanism of CREB activation, CRTC. Elimination of CRTC (using the crtc-1(tm2869)I allele) had an extensive enhancing effect on the level of neurodegeneration, similar to the effect of eliminating CREB/CRH-1 itself (Figure 6D, L3 ANOVA: F(1,138)=50.798, p=0.000). Put together, these results suggest that the components of the non-canonical pathway for CREB activation are involved in neuroprotection from excitotoxicity. However, these results, in-and-of-themselves, do not provide information on whether they work together in a combined pathway, and on the hierarchical organization of the cascade.
CRTC/CRTC-1 acts downstream of SIK2/AAK-2 to regulate neuroprotection in nematode excitotoxicity.
AAK-2 has been previously shown to phosphorylate CRTC-1, thus determining its cytoplasmic vs. nuclear distribution (Mair et al. 2011, Sasaki et al. 2011, Burkewitz et al. 2014, Altarejos & Montminy 2011, Burkewitz et al. 2015, Escoubas et al. 2017). However, AAK-2 has also been shown to regulate the activity of FoxO/DAF-16 to modulate levels of neurodegeneration (Greer et al. 2007, Tullet et al. 2014, Vazquez-Manrique et al. 2016). Since we have previously demonstrated that the IIS cascade and FoxO/DAF-16 regulate neuroprotection in nematode excitotoxicity (Mojsilovic-Petrovic et al. 2009, Tehrani et al. 2014), it becomes unclear if the effect of AAK-2 seen here is mediated by the CRTC-1/CRH-1 CREB cascade. We therefore used epistasis to test if SIK/AAK-2 and CRTC-1 act in the same pathway to regulate neuroprotection, by combining both the aak-2 mutation and the crtc-1 mutation in the excitotoxicity strain (glt-3;nuIs5) (Figure 7). Our results show that the neurodestructive effect of crtc-1 ko on nematode excitotoxicity remains intact in the absence of aak-2 (whereas absence of aak-2 alone is neuroprotective). These observations indicate that AAK-2 works in the same non-canonical CREB activation pathway as CRTC-1, and that CRTC-1 acts downstream of AAK-2, as suggested before for other effects of the AAK-2/CRTC-1/CRH-1 axis (Mair et al. 2011). We next wanted to see if other aspects of CRTC-1’s function are also in effect in nematode excitotoxicity.
Our data is in line with the model of phosphorylation-dependent translocation of CRTC-1 between the cytoplasm and the nucleus in nematode excitotoxicity.
Another prediction of the proposal that CRTC-1 is responsible for CREB activation in nematode excitotoxicity is that the hyperactivation of postsynaptic neurons causes CRTC to translocate into the nucleus of the at-risk neurons to mediate their protection, in accordance with previously suggested models (Mair et al. 2011, Burkewitz et al. 2015, Ch’ng et al. 2012). Trying to gain visual access to this process we take advantage of a CRTC-1 labeled with red fluorescence developed by Mair & Dillin (Mair et al. 2011, Burkewitz et al. 2015) and used it to monitor its translocation into the nucleus. We used a chromosomally-integrated transgene of CRTC-1::tdTOMATO expressed from its native promoter, and combined it with our excitotoxicity strain (glt-3;nuIs5), where at-risk neurons are labeled with Pglr-1::GFP (Figure 8). We observe that CRTC-1 is widely expressed in many neurons, and in basal conditions (in WT background) it is excluded from the nucleus (confocal image in Figure 8, left, top panel). However, when we combined this CRTC-1 reporter with the excitotoxicity strain (glt-3;nuIs5), we noticed that in cells exposed to excitotoxicity (labeled with green) CRTC-1 is no longer excluded from the nucleus (Figure 8, left, bottom three panels). This suggests that similarly to the effect of synaptic activity or ischemia on mammalian CRTC (Sasaki et al. 2011, Ch’ng et al. 2012), exposure to the excitotoxic insult causes nematode CRTC-1 to equilibrate into the nucleus. In contrast, a chromosomally integrate construct expressing the unphosphorylatable CRTC-1 (Burkewitz et al. 2015) (CRTC-1(S76A,S179A)::tdTOMATO expressed from its native promoter) is evenly expressed in neurons and does not show nuclear exclusion (Figure 8, right, top panel). Combining this constitutively active CRTC with the excitotoxicity strain (glt-3;nuIs5) had pleiotropic effects, as many animals seemed paralyzed and arrested in L1. Examining the expression of the hyperactive CRTC-1(S76A,S179A) in animals that managed to progress to L3, we see that the hyperactive CRTC-1 is now expressed in tight puncta in many cells, and that its expression in at-risk neurons is similar to its expression in other locations (Figure 8, right, bottom three panels). We therefore conclude that WT CRTC-1 equilibrates to the nucleus upon exposure of at-risk neurons to the excitotoxic insult, and that unphosphorylatable CRTC-1 equilibrates to the nucleus in all expressing neurons. We also note that combining hyperactive CRTC-1 with excitotoxicity seems to have pleiotropic effects. Though pleiotropic effects of neuronally expressed hyperactive CRTC-1 outside the nervous system were reported before (Burkewitz et al. 2015), we do not know why such effects seem more pronounced in the excitotoxicity strain (glt-3;nuIs5).
Figure 8: CRTC-1 nuclear localization depends on the presence of excitotoxic insult and its phosphorylation sites.

Epifluorescence confocal images showing expression of CRTC-1 (from integrated constructs uthIs205[Pcrtc-1::crtc-1::tdTOMATO] and uthIs226[Pcrtc-1::crtc-1(S76S, S179A)::tdTOMATO]) in the head neurons (red) compared to the location of GLR-1 expressing neurons (green). Top panels: overexpression of either WT (left) or hyperactive (right) CRTC-1 in a WT background. Bottom three panels: overexpression of either WT (left) or hyperactive (right) CRTC-1 in the excitotoxicity background (glt-3;nuIs5). The images show the head area of representative animals. Dorsal is up, anterior is left, dashed line and the label “ph” mark the approximate area of the pharynx. Scale bar represents 10 μm.
We further asked if over-expression of WT or hyperactive CRTC in these strains modulates neuroprotection in nematode excitotoxicity. We used the integrated form of WT CRTC-1, and both the extrachromosomal (Supplementary Figure 1) and integrated (Figure 9) forms of hyperactive CRTC-1 (CRTC-1(S76A,S179A)::tdTOMATO). We note that all these strains also express endogenous CRTC-1 from their genome, thus potentially limiting the supplemental effect of additional CRTC-1. We observe that overexpression of either WT or hyperactive CRTC-1 reduced neurodegeneration to similar levels (Figure 9 & Supplementary Figure 1), suggesting that there is a ceiling effect to neuroprotection by CRTC-1 overexpression.
CREB/CRH-1 activity in nematode excitotoxicity is cell autonomous and independent of its phosphorylation.
To firmly establish that the neuroprotection provided by CREB in nematode excitotoxicity is turned on via the phosphorylation-independent, non-canonical pathway, we used a phosphorylation-deficient mutant version of CREB (Kimura et al. 2002). The conserved site of phosphorylation in the transactivation / KID domain of nematode CREB/CRH-1 has been determined to be Ser29, and previous studies showed that an S29A mutant construct was unable to give rise to CaMKIV/CMK-1 or CaMKK/CKK-1 -induced transcription by CRH-1. The use of the S133/S29 phosphorylation site in nematode CRH-1 has been confirmed by several additional studies (Kauffman et al. 2010, Chen et al. 2016, Freytag et al. 2017). In contrast, the sequence of other CREB regulatory phosphorylation sites used in mammals (Mayr & Montminy 2001, Deisseroth & Tsien 2002, West et al. 2002, Lonze & Ginty 2002, Altarejos & Montminy 2011) is not conserved in nematode CRH-1 (not shown; we do not exclude the possibility that non-typical phosphorylation sites in CRH-1 might be recognized in the future, but these have not been demonstrated so far). We compared the ability of WT and S29A mutant crh-1 cDNA to rescue the effect of crh-1 ko and reduce neurodegeneration towards more normal levels. To address the question of cell autonomous/non-autonomous effect of CREB in our model, we expressed the WT and mutant crh-1 cDNA under the glr-1 promoter (i.e., in the at-risk neurons). Both transgenes were expressed as non-integrated extrachromosomal constructs. We observe that both WT and S29A non-phosphorylatable CREB/CRH-1 can rescue the neurodegeneration (partially, as expected from a non-integrated construct), indicating a cell-autonomous effect. Importantly, the two versions of CREB/CRH-1 rescue neurodegeneration to the same extent (Figure 10). We note that the similarity in partial effect between WT and S29A mutant CREB (Figure 10) is likely to be different from the similarity in effect seen with WT and mutant CRTC (Figure 9), which we interpreted as a ceiling effect. This is because the extensive overexpression of transgenic CRTC-1 was on the background of endogenously-expressed WT version of the protein, whereas here the moderate expression of transgenic WT or mutant CRH-1 is in the absence of endogenous protein. The ability of S29A non-phosphorylatable CREB/CRH-1 to provide protection to the same extent as WT CREB/CRH-1 indicates that CREB/CRH-1 does not need to be phosphorylated (at least not in the canonical phosphorylation site in its transactivation domain) to provide neuroprotection. Together, these results firmly establish that CREB-mediated neuroprotection in nematode excitotoxicity is achieved cell autonomously and is independent of CREB activation by canonical phosphorylation in its transactivation domain.
Figure 10: CREB’s ability to protect from excitotoxic necrosis is independent of its phosphorylation status.

Average number of degenerating head neurons per animal in different developmental stages, comparing the original excitotoxicity strain (glt-3;nuIs5) to a similar strain carrying also extrachromosomal non-integrated transgenic constructs expressing cDNA encoding either WT or a S29A (non-phosphorylatable) mutant CRH-1 ( crh-1;glt-3;nuIs5;Ex[Pglr-1::crh-1] and crh-1;glt-3;nuIs5;Ex[Pglr-1:: crh-1(S29A)] ). At each life stage, means were compared using one-way ANOVAs, post-hoc Tukey test (*p≤0.05, **p≤0.01,***p≤0.001).
Discussion
In this study we establish that the non-canonical mode of CREB activation mediates protection from necrotic neurodegeneration in nematode excitotoxicity. Our data shows that the neuroprotective function of CREB in excitotoxic necrosis (Figure 2) is conserved over a large evolutionary distance, suggesting that it forms a critical core biochemical cascade in neuroscience. We also show that the effect of the IIS cascade on neuroprotection is separate from the effect of CREB (Figure 3).
Unlike other forms of cell death in C. elegans (Lettre & Hengartner 2006), the pattern of cell death in excitotoxic necrosis is not stereotypic, showing considerable variability (Figure 4A). Nonetheless, it also shows a clear pattern of susceptibility and resilience: under basal excitotoxic conditions, most of the dying neurons are the URY sensory neurons and the RMD & SMD motorneurons; under these conditions, the command interneurons are mostly protected, and they degenerate in large numbers only when CREB/CRH-1 is eliminated (Figure 4B). It is not immediately clear what causes the special vulnerability of the RMD and SMD neurons to excitotoxic neurodegeneration in the WT background, and what is the source of CREB’s ability to protect the command interneurons. The resilience of the command interneurons (when CREB is present) is in stark contrast to the fact that they express the highest levels of all GluRs (Brockie et al. 2001a), arguing against postsynaptic susceptibility that is based on the expression level of the GluRs or their potential modification (also supported by Supplementary Figure 2). One possible presynaptic explanation to the differential susceptibility is that the sensitive neurons receive more/stronger Glu synaptic inputs. In C. elegans, the number of synaptic inputs is indicative of the strength of signaling between neurons (Gray et al. 2005, Leinwand & Chalasani 2013). However, analyzing publicly available data on the number of Glu synapses that each of these neurons receives (White et al. 1986, Jarrell et al. 2012) (using WormWeb.org & WormWiring.org) (Supplementary Table 1) shows no correlation between the number of incoming Glu connections and the tendency of the neurons to die by excitotoxicity (see RMDs vs. AVA, or SMDs vs. AVB, AVD, and AVE). One possibly important correlation to susceptibility to neurodegeneration is the location of these synapses in the nerve ring: the synapses from the glutamatergic neuron RIA onto SMDs and RMDs are located in the inner-most aspect of the nerve ring, close to the space between the pharyngeal muscle and the nerve ring (Supplementary Figure 3 and WormWiring.org), a compartment washed by body fluids (Altun et al. 2002–2018) whose Glu content is regulated by glt-3 (Mano et al. 2007). It therefore seems that the neurons most susceptible to excitotoxic necrosis in our model are those whose who have synapses that are most exposed to elevation in ambient Glu concentrations in body fluids as caused by KO of glt-3. It therefore seems that CREB/CRH-1 has the greatest neuroprotective capacity to make a difference in viability fate in a “penumbra-like” region of moderate Glu insult experienced by the command interneurons.
We further asked how CREB is activated in nematode excitotoxicity. We demonstrate that canonical mediators of CREB activation and function, such as CaMKK/CKK-1 and CBP/CBP-1 have little to no role in excitotoxic necrosis in the nematode (Figure 5 and our previous studies (Del Rosario et al. 2015)). We further demonstrate that the mediators of the non-canonical route have a very strong effect on CREB’s activation (Figure 6), and that the cascade is arranged in the same order (Figure 7) as the one suggested in mammals (Altarejos & Montminy 2011, Sasaki et al. 2011). Our data shows that neuroprotection hinges on CRTC-1, which is widely expressed in the nematode’s nervous system (Figure 8). Interestingly we show that overexpressed WT CRTC-1 equilibrates to the nucleus of at-risk neurons, which are exposed to the excitotoxic insult (Figure 8, left panels), similarly to the effect seen in mammals (Sasaki et al. 2011, Ch’ng et al. 2012). Though mutant hyperactive CRTC-1 seems equilibrated to the nucleus in all expressing cells, the pleiotropic effects seen when it is combined with the excitotoxicity condition (Figure 8, right panels) might be detrimental and prevent further protection. Together with the fact that this overexpression of WT or mutant CRTC-1 is done in the background of endogenously expressed WT CRTC-1, it is not surprising that CRTC-1 seems to have a ceiling neuroprotective effect (Figure 9).
The phosphorylation of CREB in the transactivation domain has been the cornerstone of suggested mechanisms of CREB activity for many years (Mayr & Montminy 2001, West et al. 2001, Lonze & Ginty 2002, Deisseroth et al. 2003). It serves both as a crossroad for many signaling pathways, and, together with the phosphorylation-dependent binding of the histone acetyl transferase (HAT) CBP, as a main mechanism for transcriptional activation. How does CREB activation by phosphorylation and CBP association confer neuroprotection remains unclear. Moreover, reports that this mode of CREB activation confers neuronal hyper excitability (Soriano et al. 2006, Dong et al. 2006, Yiu et al. 2014, Lisman et al. 2018) and cause neurodegeneration (Lopez de Armentia et al. 2007, Valor et al. 2010, Benito et al. 2011) suggest that indiscriminative hyperactivation of CREB is unlikely to be productive and a neuroprotective approach in excitotoxicity. A number of recent studies have shown that in some important cases the non-canonical pathway is at play, involving CREB stabilization on the DNA by CRTC. This non-canonical pathway might have more restricted-, yet beneficial- effects (Sasaki et al. 2011, Nonaka et al. 2014, Parra-Damas et al. 2017a). Importantly, our studies in nematode excitotoxicity support that this non-canonical, CRTC-dependent, and pCREB::CBP -independent pathway of CREB activation has a conserved role in neuroprotection. Nonetheless, our CRTC-1 overexpression experiment suggest that CRTC also has pleiotropic effects, and more accurate mitigation of excitotoxicity might require further analysis of CREB::CRTC’s targets. Indeed, this pathway has the potential to activate a separate sub-set of CREB targets compared with pCREB::CBP, since CRTC has its own transactivation domain, and could recruit other HATs with other histone acetylation profiles (Hirano et al. 2016, Uchida et al. 2017, Uchida & Shumyatsky 2018a, Uchida & Shumyatsky 2018b). The non-canonical pathway for CREB activation might therefore direct the differential expression of target genes that were not studied before. Our findings do not negate the significance of canonical CREB activation in excitotoxicity that leads to apoptosis. However, necrotic neurodegeneration, which is much less understood, is a leading mediator of neurodegeneration in brain ischemia (including in the non-immediate stages of neurodegeneration, when neurons are “stunned” and need to choose their survival fate). Together with the recent findings on CRTC’s involvement in learning, memory, and neurodegenerative & psychiatric diseases (Saura & Cardinaux 2017, Uchida & Shumyatsky 2018b), these notions focus further attention on non-canonical targets of CREB that might provide accurate tools for neuroprotection. Finding the right handles that might tip the balance towards scenario-specific CREB activation, and identifying the most critical neuroprotective genes associated with this specific mode of CREB activity, will be crucial for the future development of therapeutic interventions in brain ischemia.
--Human subjects -- Involves human subjects: If yes: Informed consent & ethics approval achieved: => if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods. ARRIVE guidelines have been followed: Yes => if No or if it is a Review or Editorial, skip complete sentence => if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Supplementary Material
Acknowledgments, Conflict of interest disclosure
We would like to thank the Li lab (especially A. Alexander and J.-S. Yang) and the Emerson lab for help with molecular biology and microinjections. We would like to thank all members of the Mano lab, A. Alexander, and A. Khan for technical support and critical reading of this manuscript. We would also like to thank M. Driscoll and H. Kuroyanagi for gift of plasmids. We thank the Caenorhabditis Genetic Center at the Univ. of Minnesota (which is funded by NIH Office of Research Infrastructure Programs, P40 OD010440), and the Japanese National Bioresource Project (NBRP, Ministry of Education, Culture, Science, Sports and Technology, Japan) for providing strains. This project was supported by The Alexandrine and Alexander L. Sinsheimer Fund (P60134 to IM), the American Heart Association (16GRNT31500004 to IM), and NIH/NINDS (NS096687 to IM). The Mano lab is also supported by NIH/NINDS (NS098350 to IM), and by an institutional RCMI grant (G12RR003060–26 to CCNY).
Abbreviations:
- CaMK
Ca2+/Calmodulin -dependent Kinase
- CBP:
CREB Binding Protein
- CRTC
cAMP-regulated transcriptional co-activator
- CREB
cAMP Response Element Binding protein
- Glu
L-Glutamate
- GluRs
Glutamate Receptors
- SIK
Salt Induced Kinase
- TF
Transcription Factor
Footnotes
The authors declare no conflict of interest.
A preprint version of this manuscript was posted in BioRxiv at https://www.biorxiv.org/content/early/2018/02/07/261420
This study was not pre-registered with clinicaltrials.gov (or similar) and did not require institutional approval.
References
- Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, Sawchenko PE and Montminy M (2008) The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat Med, 14, 1112–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY and Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol, 12, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun ZF, Herndon LA, Crocker C, Lints R and Hall DH (2002-2018) WormAtlas.org.
- Apfeld J, O’Connor G, McDonagh T, DiStefano PS and Curtis R (2004) The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev, 18, 3004–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arey RN, Stein GM, Kaletsky R, Kauffman A and Murphy CT (2018) Activation of Galphaq Signaling Enhances Memory Consolidation and Slows Cognitive Decline. Neuron, 98, 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2013) Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci. [DOI] [PubMed] [Google Scholar]
- Bates EA, Victor M, Jones AK, Shi Y and Hart AC (2006) Differential Contributions of Caenorhabditis elegans Histone Deacetylases to Huntingtin Polyglutamine Toxicity. J. Neurosci, 26, 2830–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Valor LM, Jimenez-Minchan M, Huber W and Barco A (2011) cAMP Response Element-Binding Protein Is a Primary Hub of Activity-Driven Neuronal Gene Expression. J Neurosci, 31, 18237–18250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AJ, Hart AC and Kaplan JM (1998) Galphas-induced neurodegeneration in Caenorhabditis elegans. J Neurosci, 18, 2871–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Lee BG, Lelay J, Kaestner KH and Blendy JA (2015) Serine 133 phosphorylation is not required for hippocampal CREB-mediated transcription and behavior. Learning & Memory, 22, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Madsen DM, Zheng Y, Mellem J and Maricq AV (2001a) Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J Neurosci, 21, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ and Maricq AV (2006) Ionotropic glutamate receptors: genetics, behavior and electrophysiology. In: WormBook, (The C. elegans Research Community ed.). www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Mellem JE, Hills T, Madsen DM and Maricq AV (2001b) The C. elegans glutamate receptor subunit NMR-1 is required for slow NMDA-activated currents that regulate reversal frequency during locomotion. Neuron, 31, 617–630. [DOI] [PubMed] [Google Scholar]
- Burkewitz K, Morantte I, Weir HJM et al. (2015) Neuronal CRTC-1 governs systemic mitochondrial metabolism and lifespan via a catecholamine signal. Cell, 160, 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Zhang Y and Mair, William B (2014) AMPK at the Nexus of Energetics and Aging. Cell Metabolism, 20, 10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr., Duman RS and Nestler EJ (2005) The many faces of CREB. Trends Neurosci, 28, 436–445. [DOI] [PubMed] [Google Scholar]
- Ch’ng TH, Uzgil B, Lin P, Avliyakulov NK, O’Dell TJ and Martin KC (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell, 150, 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S and Goodman RH (2004) Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci U S A, 101, 13572–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Chen HJ, Tseng WC, Hsu JM, Huang TT, Chen CH and Pan CL (2016) A C. elegans Thermosensory Circuit Regulates Longevity through crh-1/CREB-Dependent flp-6 Neuropeptide Signaling. Dev Cell, 39, 209–223. [DOI] [PubMed] [Google Scholar]
- Choi DW and Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci, 13, 171–182. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR and Goodman RH (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Chung S, Gumienny TL, Hengartner MO and Driscoll M (2000) A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol, 2, 931–937. [DOI] [PubMed] [Google Scholar]
- Church DL, Guan KL and Lambie EJ (1995) Three genes of the MAP kinase cascade, mek-2, mpk-1//sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development, 121, 2525–2535. [DOI] [PubMed] [Google Scholar]
- Clark M, Kumar GS, Marcum R, Luo Q, Zhang Y and Radhakrishnan I (2015) Molecular Basis for the Mechanism of Constitutive CBP/p300 Coactivator Recruitment by CRTC1-MAML2 and its Implications in cAMP Signaling. Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB and Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell, 12, 413–423. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol, 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Dash PK, Hochner B and Kandel ER (1990) Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature, 345, 718–721. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H and Tsien RW (1996) Signaling from Synapse to Nucleus: Postsynaptic CREB Phosphorylation during Multiple Forms of Hippocampal Synaptic Plasticity. Neuron, 16, 89–101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H and Tsien RW (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol, 13, 354–365. [DOI] [PubMed] [Google Scholar]
- Deisseroth K and Tsien RW (2002) Dynamic multiphosphorylation passwords for activity-dependent gene expression. Neuron, 34, 179–182. [DOI] [PubMed] [Google Scholar]
- Del Rosario JS, Feldmann KG, Ahmed T et al. (2015) Death Associated Protein Kinase (DAPK) –Mediated Neurodegenerative Mechanisms in Nematode Excitotoxicity. BMC Neuroscience, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ and Malenka RC (2006) CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci, 9, 475–477. [DOI] [PubMed] [Google Scholar]
- Eastburn DJ and Han M (2005) A gain-of-function allele of cbp-1, the Caenorhabditis elegans ortholog of the mammalian CBP/p300 gene, causes an increase in histone acetyltransferase activity and antagonism of activated Ras. Mol Cell Biol, 25, 9427–9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emtage L, Chang H, Tiver R and Rongo C (2009) MAGI-1 modulates AMPA receptor synaptic localization and behavioral plasticity in response to prior experience. PLoS One, 4, e4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas CC, Silva-Garcia CG and Mair WB (2017) Deregulation of CRTCs in Aging and Age-Related Disease Risk. Trends Genet, 33, 303–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Takahashi N, Kimura Y, Masuho Y, Arai K, Muramatsu MA and Tokumitsu H (1999) Ca(2+)/Calmodulin-dependent protein kinase cascade in Caenorhabditis elegans. Implication in transcriptional activation. J Biol Chem, 274, 22556–22562. [DOI] [PubMed] [Google Scholar]
- Fan J, Dawson TM and Dawson VL (2017) Cell Death Mechanisms of Neurodegeneration. Adv Neurobiol, 15, 403–425. [DOI] [PubMed] [Google Scholar]
- Flavell SW and Greenberg ME (2008) Signaling Mechanisms Linking Neuronal Activity to Gene Expression and Plasticity of the Nervous System. Annual Review of Neuroscience, 31, 563–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag V, Probst S, Hadziselimovic N et al. (2017) Genome-wide temporal expression profiling in C. elegans identifies a core gene set related to long-term memory. J Neurosci, 37, 6661–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB and Johnson TE (1988) A Mutation in the age-1 Gene in Caenorhabditis elegans Lengthens Life and Reduces Hermaphrodite Fertility. Genetics, 118, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM (2006) Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci, 7, 437–448. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA and Montminy MR (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell, 59, 675–680. [DOI] [PubMed] [Google Scholar]
- Goodman RH (1990) Regulation of neuropeptide gene expression. Annu Rev Neurosci, 13, 111–127. [DOI] [PubMed] [Google Scholar]
- Gray JM, Hill JJ and Bargmann CI (2005) A circuit for navigation in Caenorhabditis elegans. Proc Natl Acad Sci U S A, 102, 3184–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP and Brunet A (2007) An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol, 17, 1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL and Greenberg ME (2008) From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron, 59, 846–860. [DOI] [PubMed] [Google Scholar]
- Hardingham GE and Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci, 11, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y and Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci, 5, 405–414. [DOI] [PubMed] [Google Scholar]
- Hart AC, Sims S and Kaplan JM (1995) Synaptic code for sensory modalities revealed by C. elegans GLR-1 glutamate receptor. Nature, 378, 82–85. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Ihara K, Masuda T et al. (2016) Shifting transcriptional machinery is required for long-term memory maintenance and modification in Drosophila mushroom bodies. Nat Commun, 7, 13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R et al. (2007) High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol, 5, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C and Turski L (2002) Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet neurology, 1, 383–386. [DOI] [PubMed] [Google Scholar]
- Impey S and Goodman RH (2001) CREB signaling--timing is everything. Sci STKE, 2001, pe1. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H et al. (2004) Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell, 119, 1041–1054. [DOI] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C et al. (2003) Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A, 100, 12147–12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrell TA, Wang Y, Bloniarz AE, Brittin CA, Xu M, Thomson JN, Albertson DG, Hall DH and Emmons SW (2012) The Connectome of a Decision-Making Neural Network. Science, 337, 437–444. [DOI] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L et al. (2012) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med, 18, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP and Moens U (2004) What turns CREB on? Cellular Signalling, 16, 1211–1227. [DOI] [PubMed] [Google Scholar]
- Kaletsky R, Yao V, Williams A, Runnels AM, Tadych A, Zhou S, Troyanskaya OG and Murphy CT (2018) Transcriptome analysis of adult Caenorhabditis elegans cells reveals tissue-specific gene and isoform expression. PLoS Genet, 14, e1007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science, 294, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Kaplan JM and Horvitz HR (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc Natl Acad Sci U S A, 90, 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AL, Ashraf JM, Corces-Zimmerman MR, Landis JN and Murphy CT (2010) Insulin Signaling and Dietary Restriction Differentially Influence the Decline of Learning and Memory with Age. PLoS Biol, 8, e1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Corcoran EE, Eto K et al. (2002) A CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep, 3, 962–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K (2007) CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J, 274, 3210–3217. [DOI] [PubMed] [Google Scholar]
- Kitagawa K (2012) Ischemic tolerance in the brain: endogenous adaptive machinery against ischemic stress. J Neurosci Res, 90, 1043–1054. [DOI] [PubMed] [Google Scholar]
- Kovacs KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O and Cardinaux JR (2007) TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A, 104, 4700–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski JR, Dahlberg CL and Juo P (2011) The Deubiquitinating Enzyme USP-46 Negatively Regulates the Degradation of Glutamate Receptors to Control Their Abundance in the Ventral Nerve Cord of Caenorhabditis elegans. J. Neurosci, 31, 1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Shyu WC and Wang YT (2011) Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med, 17, 266–275. [DOI] [PubMed] [Google Scholar]
- Lai TW, Zhang S and Wang YT (2014) Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Progress in Neurobiology, 115. [DOI] [PubMed] [Google Scholar]
- Lakhina V, Arey, Rachel N, Kaletsky R, Kauffman A, Stein G, Keyes W, Xu D and Murphy, Coleen T (2015) Genome-wide Functional Analysis of CREB/Long-Term Memory-Dependent Transcription Reveals Distinct Basal and Memory Gene Expression Programs. Neuron, 85, 330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanjuin A and Sengupta P (2002) Regulation of chemosensory receptor expression and sensory signaling by the KIN-29 Ser/Thr kinase. Neuron, 33, 369–381. [DOI] [PubMed] [Google Scholar]
- Leinwand SG and Chalasani SH (2013) Neuropeptide signaling remodels chemosensory circuit composition in Caenorhabditis elegans. Nat Neurosci, 16, 1461–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G and Hengartner MO (2006) Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol, 7, 97–108. [DOI] [PubMed] [Google Scholar]
- Lisman J, Cooper K, Sehgal M and Silva AJ (2018) Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat Neurosci, 21, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M et al. (2007) NMDA Receptor Subunits Have Differential Roles in Mediating Excitotoxic Neuronal Death Both In Vitro and In Vivo. J. Neurosci, 27, 2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE and Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron, 35, 605–623. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER and Barco A (2007) cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci, 27, 13909–13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR and West AE (2011) Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol, 94, 259–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K et al. (2001) Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci, 21, 9204–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder CI, Kim JI, Liang X et al. (2018) The THO Complex Coordinates Transcripts for Synapse Development and Dopamine Neuron Survival. Cell, 174, 1436–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues APC, Manning G, Montminy M, Shaw RJ and Dillin A (2011) Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature, 470, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano I and Driscoll M (2009) C. elegans Glutamate Transporter Deletion Induces AMPA-Receptor/Adenylyl Cyclase 9-Dependent Excitotoxicity. J Neurochem, 108, 1373–1384. [DOI] [PubMed] [Google Scholar]
- Mano I, Straud S and Driscoll M (2007) Caenorhabditis elegans Glutamate Transporters Influence Synaptic Function and Behavior at Sites Distant from the Synapse. J Biol Chem, 282, 34412–34419. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC et al. (2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet, 31, 47–54. [DOI] [PubMed] [Google Scholar]
- Mayr B and Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol, 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Mehta SL, Manhas N and Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev, 54, 34–66. [DOI] [PubMed] [Google Scholar]
- Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, Henshall DC and Simon RP (2005) CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab, 25, 234–246. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Nedelsky N, Boccitto M et al. (2009) FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J Neurosci, 29, 8236–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy MR and Bilezikjian LM (1987) Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature, 328, 175–178. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH and Iadecola C (2010) The Science of Stroke: Mechanisms in Search of Treatments. Neuron, 67, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss BJ, Park L, Dahlberg CL and Juo P (2016) The CaM Kinase CMK-1 Mediates a Negative Feedback Mechanism Coupling the C. elegans Glutamate Receptor GLR-1 with Its Own Transcription. PLoS Genet, 12, e1006180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Sugi T, Nonomura M and Mori I (2011) Identification of the AFD neuron as the site of action of the CREB protein in Caenorhabditis elegans thermotaxis. EMBO Rep, 12, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima S and Maruyama IN (2017) Appetitive Olfactory Learning and Long-Term Associative Memory in Caenorhabditis elegans. Front Behav Neurosci, 11, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka M, Kim R, Fukushima H et al. (2014) Region-specific activation of CRTC1-CREB signaling mediates long-term fear memory. Neuron, 84, 92–106. [DOI] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H and Hardingham GE (2005) Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci, 25, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra-Damas A, Chen M, Enriquez-Barreto L et al. (2017a) CRTC1 Function During Memory Encoding Is Disrupted in Neurodegeneration. Biol Psychiatry, 81, 111–123. [DOI] [PubMed] [Google Scholar]
- Parra-Damas A, Rubio-Ferrarons L, Shen J and Saura CA (2017b) CRTC1 mediates preferential transcription at neuronal activity-regulated CRE/TATA promoters. Scientific reports, 7, 18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra-Damas A, Valero J, Chen M, España J, Martín E, Ferrer I, Rodríguez-Alvarez J and Saura CA (2014) Crtc1 Activates a Transcriptional Program Deregulated at Early Alzheimer’s Disease-Related Stages. J Neurosci, 34, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin CH (2002) From gene to identified neuron to behaviour in Caenorhabditis elegans. Nat Rev Genet, 3, 622–630. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Hogan MF, Lackey D, Tora L, Dent SY, Olefsky J and Montminy M (2013) Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest, 123, 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo Romanos T, Petersen JG and Pocock R (2017) Control of Neuropeptide Expression by Parallel Activity-dependent Pathways in Caenorhabditis elegans. Scientific reports, 7, 38734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, Kaun KR, Chen SH and Rankin CH (2003) GLR-1, a non-NMDA glutamate receptor homolog, is critical for long-term memory in Caenorhabditis elegans. J Neurosci, 23, 9595–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK and Rankin CH (2006) Blocking Memory Reconsolidation Reverses Memory-Associated Changes in Glutamate Receptor Expression. J. Neurosci, 26, 11582–11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, Sangha S, Rai S, Norman KR and Rankin CH (2005) Decreased Sensory Stimulation Reduces Behavioral Responding, Retards Development, and Alters Neuronal Connectivity in Caenorhabditis elegans. J. Neurosci, 25, 7159–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal DC, Bianchi L, Royal MA, Lizzio M Jr., Mukherjee G, Nunez YO and Driscoll M (2005) Temperature-sensitive mutant of the Caenorhabditis elegans neurotoxic MEC-4(d) DEG/ENaC channel identifies a site required for trafficking or surface maintenance. J Biol Chem, 280, 41976–41986. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Karelina K and Obrietan K (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem, 116, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Takemori H, Yagita Y et al. (2011) SIK2 Is a Key Regulator for Neuronal Survival after Ischemia via TORC1-CREB. Neuron, 69, 106–119. [DOI] [PubMed] [Google Scholar]
- Satterlee JS, Ryu WS and Sengupta P (2004) The CMK-1 CaMKI and the TAX-4 Cyclic nucleotide-gated channel regulate thermosensory neuron gene expression and function in C. elegans. Curr Biol, 14, 62–68. [DOI] [PubMed] [Google Scholar]
- Saura CA and Cardinaux JR (2017) Emerging Roles of CREB-Regulated Transcription Coactivators in Brain Physiology and Pathology. Trends Neurosci. [DOI] [PubMed] [Google Scholar]
- Savage-Dunn C, Maduzia LL, Zimmerman CM, Roberts AF, Cohen S, Tokarz R and Padgett RW (2003) Genetic screen for small body size mutants in C. elegans reveals many TGFbeta pathway components. Genesis, 35, 239–247. [DOI] [PubMed] [Google Scholar]
- Schild LC, Zbinden L, Bell HW, Yu YV, Sengupta P, Goodman MB and Glauser DA (2014) The balance between cytoplasmic and nuclear CaM kinase-1 signaling controls the operating range of noxious heat avoidance. Neuron, 84, 983–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y et al. (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell, 119, 61–74. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G and Greenberg ME (1990) Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron, 4, 571–582. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA and Greenberg ME (1991) CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science, 252, 1427–1430. [DOI] [PubMed] [Google Scholar]
- Singaravelu G, Song H-O, Ji YJ, Jee C, Park B-J and Ahnn J (2007) Calcineurin interacts with KIN-29, a Ser/Thr kinase, in Caenorhabditis elegans. Biochemical and Biophysical Research Communications, 352, 29–35. [DOI] [PubMed] [Google Scholar]
- Soderling TR (1999) The Ca2+–calmodulin-dependent protein kinase cascade. Trends in Biochemical Sciences, 24, 232–236. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H and Hardingham GE (2006) Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci, 26, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetak A, Horndli F, Maricq AV, van den Heuvel S and Hajnal A (2009) Neuron-specific regulation of associative learning and memory by MAGI-1 in C. elegans. PLoS One, 4, e6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T (2006) Maintenance of C. elegans. WormBook, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suo S, Kimura Y and Van Tol HHM (2006) Starvation Induces cAMP Response Element-Binding Protein-Dependent Gene Expression through Octopamine-Gq Signaling in Caenorhabditis elegans. J. Neurosci, 26, 10082–10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori H, Kajimura J and Okamoto M (2007) TORC-SIK cascade regulates CREB activity through the basic leucine zipper domain. FEBS J, 274, 3202–3209. [DOI] [PubMed] [Google Scholar]
- Tehrani N, Del Rosario J, Dominguez M, Kalb R and Mano I (2014) The Insulin/IGF Signaling Regulators Cytohesin/GRP-1 and PIP5K/PPK-1 Modulate Susceptibility to Excitotoxicity in C. elegans. PLoS ONE, 9, e113060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timbers TA and Rankin CH (2011) Tap withdrawal circuit interneurons require CREB for long-term habituation in Caenorhabditis elegans. Behav Neurosci, 125, 560–566. [DOI] [PubMed] [Google Scholar]
- Toth ML, Melentijevic I, Shah L et al. (2012) Neurite Sprouting and Synapse Deterioration in the Aging Caenorhabditis elegans Nervous System. J Neurosci, 32, 8778–8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullet JMA, Araiz C, Sanders MJ et al. (2014) DAF-16/FoxO Directly Regulates an Atypical AMP-Activated Protein Kinase Gamma Isoform to Mediate the Effects of Insulin/IGF-1 Signaling on Aging in Caenorhabditis elegans. PLoS Genet, 10, e1004109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M (2011) Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci, 14, 1369–1373. [DOI] [PubMed] [Google Scholar]
- Tymianski M (2014) Stroke in 2013: Disappointments and advances in acute stroke intervention. Nat Rev Neurol, 10, 66–68. [DOI] [PubMed] [Google Scholar]
- Uchida S and Shumyatsky GP (2018a) Epigenetic regulation of Fgf1 transcription by CRTC1 and memory enhancement. Brain Res Bull. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S and Shumyatsky GP (2018b) Synaptically Localized Transcriptional Regulators in Memory Formation. Neuroscience, 370, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Teubner BJW, Hevi C et al. (2017) CRTC1 Nuclear Translocation Following Learning Modulates Memory Strength via Exchange of Chromatin Remodeling Complexes on the Fgf1 Gene. Cell Reports, 18, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor LM, Jancic D, Lujan R and Barco A (2010) Ultrastructural and transcriptional profiling of neuropathological misregulation of CREB function. Cell Death Differ, 17, 1636–1644. [DOI] [PubMed] [Google Scholar]
- Vazquez-Manrique RP, Farina F, Cambon K, Sequedo MD, Parker AJ, Millan JM, Weiss A, Deglon N and Neri C (2016) AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Human molecular genetics, 25, 1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ and Dragunow M (1999) CREB phosphorylation promotes nerve cell survival. J Neurochem, 73, 1836–1842. [PubMed] [Google Scholar]
- Walton MR and Dragunow M (2000) Is CREB a key to neuronal survival? Trends in neurosciences, 23, 48–53. [DOI] [PubMed] [Google Scholar]
- Watts AG, Sanchez-Watts G, Liu Y and Aguilera G (2011) The distribution of messenger RNAs encoding the three isoforms of the transducer of regulated cAMP responsive element binding protein activity in the rat forebrain. J Neuroendocrinol, 23, 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva AJ and Soderling TR (2008) Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron, 59, 914–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X and Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A, 98, 11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC and Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci, 3, 921–931. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN and Brenner S (1986) The structure of the nervous system of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond, 314, 1–340. [DOI] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y et al. (2006) Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci U S A, 103, 14379–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Jarrell TA, Wang Y, Cook SJ, Hall DH and Emmons SW (2013) Computer assisted assembly of connectomes from electron micrographs: application to Caenorhabditis elegans. PLoS One, 8, e54050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KK, Gonzalez GA, Biggs WH 3rd and Montminy MR (1988) Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature, 334, 494–498. [DOI] [PubMed] [Google Scholar]
- Yin JC, Del Vecchio M, Zhou H and Tully T (1995) CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell, 81, 107–115. [DOI] [PubMed] [Google Scholar]
- Yiu AP, Mercaldo V, Yan C et al. (2014) Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron, 83, 722–735. [DOI] [PubMed] [Google Scholar]
- Yu B, Wang X, Wei S, Fu T, Dzakah EE, Waqas A, Walthall WW and Shan G (2017) Convergent Transcriptional Programs Regulate cAMP Levels in C. elegans GABAergic Motor Neurons. Dev Cell. [DOI] [PubMed] [Google Scholar]
- Yu YV, Bell HW, Glauser DA, Van Hooser SD, Goodman MB and Sengupta P (2014) CaMKI-dependent regulation of sensory gene expression mediates experience-dependent plasticity in the operating range of a thermosensory neuron. Neuron, 84, 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J and Yankner BA (2000) Apoptosis in the nervous system. Nature, 407, 802–809. [DOI] [PubMed] [Google Scholar]
- Zhang S-J, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, Descombes P and Bading H (2007) Decoding NMDA Receptor Signaling: Identification of Genomic Programs Specifying Neuronal Survival and Death. Neuron, 53, 549–562. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P and Bading H (2009) Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet, 5, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH et al. (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A, 102, 4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ren C, Chen X and Shen J (2012) From rapid to delayed and remote postconditioning: the evolving concept of ischemic postconditioning in brain ischemia. Curr Drug Targets, 13, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.