

Abstract
Aims
The aims of this study were to describe the pharmacokinetics of tacrolimus immediately after kidney transplantation, and to develop a clinical tool for selecting the best starting dose for each patient.
Methods
Data on tacrolimus exposure were collected for the first 3 months following renal transplantation. A population pharmacokinetic analysis was conducted using nonlinear mixed‐effects modelling. Demographic, clinical and genetic parameters were evaluated as covariates.
Results
A total of 4527 tacrolimus blood samples collected from 337 kidney transplant recipients were available. Data were best described using a two‐compartment model. The mean absorption rate was 3.6 h−1, clearance was 23.0 l h–1 (39% interindividual variability, IIV), central volume of distribution was 692 l (49% IIV) and the peripheral volume of distribution 5340 l (53% IIV). Interoccasion variability was added to clearance (14%). Higher body surface area (BSA), lower serum creatinine, younger age, higher albumin and lower haematocrit levels were identified as covariates enhancing tacrolimus clearance. Cytochrome P450 (CYP) 3A5 expressers had a significantly higher tacrolimus clearance (160%), whereas CYP3A4*22 carriers had a significantly lower clearance (80%). From these significant covariates, age, BSA, CYP3A4 and CYP3A5 genotype were incorporated in a second model to individualize the tacrolimus starting dose:
Both models were successfully internally and externally validated. A clinical trial was simulated to demonstrate the added value of the starting dose model.
Conclusions
For a good prediction of tacrolimus pharmacokinetics, age, BSA, CYP3A4 and CYP3A5 genotype are important covariates. These covariates explained 30% of the variability in CL/F. The model proved effective in calculating the optimal tacrolimus dose based on these parameters and can be used to individualize the tacrolimus dose in the early period after transplantation.
Keywords: cytochrome P450 enzymes, genetics and pharmacogenetics, immunosuppression Immunology, pharmacokinetics, population analysis, renal transplantation
What is Already Known about this Subject
Patients with low tacrolimus predose concentrations are at an increased risk for rejection while those with high predose concentrations are at a higher risk of toxicity. Achieving the therapeutic range is important, but it can take up to two weeks.
In clinical practice only 37% of patients are directly within the target range at first steady state.
Two externally validated models to predict the starting dose of tacrolimus have been published. One of these was prospectively tested and could not predict the tacrolimus exposure.
What this Study Adds
In the first 3 months post‐transplantation, age, albumin, body surface area, serum creatinine, CYP3A5 genotype, CYP3A4 genotype, haematocrit and lean bodyweight significantly influence the pharmacokinetics of tacrolimus in adult renal transplant recipients.
A separate model for the starting dose was developed:
The tacrolimus starting dose should be higher in CYP3A5 expressers, younger patients and those with a higher body surface area (BSA). It should be lower in patients carrying the CYP3A4*22 allele.
The starting dose model can be used to individualize the tacrolimus starting dose following kidney transplantation.
Introduction
Tacrolimus is the most used immunosuppressive drug to prevent acute rejection following renal transplantation 1. Short‐term kidney allograft survival has greatly improved with the use of immunosuppressive drug combination therapy 2, 3. However, prolonged use of immunosuppressive drugs leads to substantial toxicity, including increased rates of infections, hypertension, post‐transplant diabetes mellitus, neurotoxicity and nephrotoxicity 4, 5, 6, 7. These adverse events augment the limited long‐term renal allograft survival and the high cardiovascular morbidity and mortality of transplant recipients 8, 9. Rejection rates and most of the adverse events seem to be concentration related, with higher tacrolimus concentrations being related to toxicity and lower concentrations to an increased risk of acute rejection 10, 11.
The use of tacrolimus is hampered by its narrow therapeutic index with large intra‐ and interpatient variability in its pharmacokinetics that requires therapeutic drug monitoring (TDM) to individualize the dose to prevent toxicity and rejection 11. Multiple factors influence the clearance (CL) of tacrolimus, including cytochrome P450 (CYP) 3A genotype 12, 13, haematocrit 14, age 10, 15, bodyweight, ethnicity 16, 17 and drug–drug interactions 18. In routine clinical practice, the tacrolimus starting dose is based solely on bodyweight, even though the available evidence is scarce 19. Pharmacokinetic models have conflicting results demonstrating that bodyweight does 20, 21, 22, 23, 24 or does not 10, 25, 26, 27 have a statistically significant influence on the clearance of tacrolimus. Subsequent doses are adjusted by means of TDM, which limits the time a patient is exposed to concentrations outside the target range, although it may take up to 14 days to reach the target exposure 24. Therefore, patients are at an increased risk of sub‐ or supratherapeutic tacrolimus exposure during these first weeks after transplantation, and may have an increased risk of developing adverse events 28.
A population pharmacokinetic model with clinically relevant covariates may help predict an individual's tacrolimus pharmacokinetics and can be applied prior to the start of therapy to reach target exposure as soon as possible 29. To date, several models to predict the tacrolimus starting dose have been developed for adult 10, 12, 14, 20, 21, 22, 23, 26, 27, 30 and paediatric renal transplant recipients 31. Of these adult models, only two were successfully externally validated in an independent dataset 10, 12. One of these models was subsequently prospectively tested by another research group in a completely new population. Unfortunately, this model was unable to successfully predict tacrolimus exposure 32. The other externally validated model had several shortcomings, including flip‐flop kinetics, where the absorption constant is much slower than the elimination constant. Besides this, the external validation cohort had its limitations as only patients 1 month post‐transplant were included and only five were CYP3A4*22 carriers 12. The algorithm by Chen et al. was not externally validated but was prospectively tested in a randomized clinical trial in Chinese patients 22. Unfortunately, an algorithm designed for Asian patients cannot be extrapolated to Caucasian transplant populations.
The aim of the current study was to describe the population pharmacokinetics of twice‐daily, immediate‐release tacrolimus in the first 3 months following renal transplantation, and to develop a dosing algorithm for the starting dose. In contrast to previous studies, many covariates were tested (including CYP3A genotype, haematocrit and age), a rich database was used [for 100 patients a full area under the concentration vs. time curve (AUC) was available], and the model was extensively validated, both internally and externally. A separate starting dose model was developed.
Methods
Study design
The model building cohort consisted of a total of 337 patients. Of these patients, 237 were transplanted in the Erasmus MC and participated in a randomized–controlled clinical trial (RCT; Rotterdam cohort) 33. For these patients, additional pharmacokinetic data were retrospectively retrieved from the medical records. The Ethics Review Board of the Erasmus MC provided a waiver for the Medical Research Involving Human Subjects Act, for this study (Medical Ethical Review Board number 2017–1029).
The remaining 100 patients were transplanted in the Leiden University Medical Center (LUMC, Leiden cohort) 34. The inclusion criteria and patient demographics of these two cohorts have been described previously 33, 34. All clinical data were collected from 24 h before transplantation until 3 months post‐transplantation.
External validation of the pharmacokinetic model was performed on an independent dataset consisting of 304 adult renal transplant recipients (validation cohort). This cohort has been described previously 12. These patients were not included in the initial model building cohort.
Immunosuppression
All patients were treated with oral twice‐daily tacrolimus (Prograft, Astellas Pharma, Leiden, The Netherlands) in combination with mycophenolic acid. Tacrolimus doses were tailored using TDM. The tacrolimus predose concentration (C0) was measured for the first time on day 3 following transplantation in the Rotterdam cohort 33. In the Leiden cohort, blood samples were drawn before tacrolimus ingestion, and 1, 2, 3, 4, 5 and 6 h postingestion. This is routine clinical care in Leiden. In the Leiden cohort, tacrolimus concentrations for the pharmacokinetic curve were obtained at steady state, with a median of 2 weeks after transplantation. The validation cohort consisted of 304 patients, of whom seven participated in the Symphony‐Elite study. In this study, blood samples were drawn before tacrolimus ingestion, and 0.3, 0.7, 1.3, 2, 3, 4, 6, 8, 10 and 12 h postingestion 35. For the remaining 297 patients, only C0 was available 12.
In the Rotterdam cohort, the target tacrolimus C0 range was 10.0–15.0 ng ml–1 in week 1–2, 8.0–12.0 ng ml–1 in weeks 3–4, and 5.0–10.0 ng ml–1 after week 4 post transplantation. In the Leiden cohort, the target AUC was 210 ng h ml–1 with a corresponding C0 range of 10.0–15.0 ng ml–1 the first 6 weeks following transplantation. After week 6 post transplantation, the target AUC was 125 ng h ml–1 with a corresponding C0 range of 4.0–9.0 ng ml–1.
Laboratory analysis
Genotyping for CYP3A5*3 and CYP3A4*22 was performed as described previously 33, 34. Deviations from Hardy–Weinberg equilibrium were tested using the χ2 goodness‐of‐fit (GOF) test. Tacrolimus concentrations in the Rotterdam cohort were analysed in whole‐blood samples using two different immunoassays: the antibody‐conjugated magnetic immunoassay (ACMIA) and the enzyme multiplied technique (EMIT), as described previously 33. The lower limits of quantification were 1.5 ng ml–1 (ACMIA) and 2.0 ng ml–1 (EMIT). The upper limit of quantification was 30.0 ng ml–1. Tacrolimus concentrations in the Leiden cohort were measured using a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) method 34. The lower limit of quantification was 1.0 ng ml–1 and the upper limit of quantification 50.0 ng ml–1. In the validation cohort, samples were measured using a validated LC–MS/MS 12.
Population pharmacokinetic modelling
Pharmacokinetic analysis was conducted by nonlinear mixed‐effects modelling using NONMEM version 7.2 (FOCE+I, ICON Development Solutions, Ellicott City, MD, USA) and PsN version 4.6.0. Pirana software was used as an interface between NONMEM, R (version 3.2.2) and Xpose (version 4).
Base model development
One‐ and two‐compartment models were considered based on visual inspection of the data and a review of the literature. Typical values for lag‐time (tlag), absorption rate constant (ka), central volume of distribution (V1), peripheral volume of distribution (V2), CL and intercompartmental clearance (Q) were estimated. As bioavailability (F) could not be estimated, F was fixed to 1 and certain values were estimated as ratios: CL/F, Q/F, V 1 /F and V 2 /F. Interindividual variability (IIV) and interoccasion variability (IOV) were modelled for each pharmacokinetic parameter using an exponential model. An occasion was defined as the measurement of a C0. Residual variability was incorporated as an additive and proportional error for immunoassay, and as a proportional error for LC–MS/MS. For all model parameters for which IIV was estimated, shrinkage was calculated. A shrinkage value below 25% was considered acceptable 36. Minimum objective function values (OFVs, P < 0.01), parameter precision, error estimates, shrinkage value and visual inspection of the GOF plots were considered for model selection.
Covariate model development
Covariates were selected based on their known or theoretical relationships with tacrolimus pharmacokinetics and theoretical plausibility. The following demographic, clinical and genetic characteristics were evaluated as potential model covariates: weight, height, time post‐transplant, sex, age, ethnicity, haematocrit, creatinine, estimated glomerular filtration rate (Cockcroft–Gault and Modification of Diet in Renal Disease), aspartate aminotransferase, albumin, C‐reactive protein, total protein, bilirubin, CYP3A4 genotype, CYP3A5 genotype, combination of CYP3A4 and CYP3A5 (as described previously 12), ABCB1 (previously known as multidrug resistance‐1) genotype 3435C > T polymorphism, P‐450 oxidoreductase*28 (POR) genotype, comedication known to interact with tacrolimus (calcium channel blockers, glucocorticoids), glucocorticoid dose, primary kidney disease, number of previous kidney transplantations, renal replacement therapy prior to transplantation (pre‐emptive, haemodialysis or peritoneal dialysis), delayed graft function, human leucocyte antigen mismatches, panel reactive antibodies, body mass index, lean body weight (LBW), ideal bodyweight, fat mass and body surface area.
First, the relationship between IIV and covariates was investigated graphically. Covariates with a visually apparent relationship and a clinically plausible relationship with the pharmacokinetic parameter were univariately added to the model. Covariates included in previously published population pharmacokinetic models were also univariately added to the model, regardless of the visually apparent relationship. A univariate analysis was performed to determine which covariates improved the model (P < 0.05). The stepwise covariate modelling with forward inclusion‐backward elimination method was used 37. Covariates that significantly improved the model (P < 0.05, i.e. decrease in OFV of 3.84) were added to the full model. A backward elimination process with a stricter statistical criterion was then performed (P < 0.01, i.e. increase in OFV of 6.64). A shark plot was generated for each covariate for case‐deletion diagnostics.
Internal model evaluation
The model was validated using a prediction corrected visual predictive check (VPC) by simulating 500 datasets, and a normalized prediction distribution errors (NPDE) analysis (1000 simulations). The VPC was stratified for the covariates included in the final model.
Simulations were performed with the final model with different values of the covariate to evaluate the effect of significant covariates. All simulated patients received 0.2 mg kg–1 divided into two equal doses. Concentration–time profiles were simulated for 1000 patients for each included covariate. All other parameters were fixed to the median.
External model evaluation
An independent dataset consisting of 340 adults treated with the same immunosuppressive regimen was used for external validation using a VPC. The VPC was prediction corrected and stratified for the covariates included in the final model.
Statistical analyses other than those mentioned above, were performed using SPSS version 23 (SPSS Inc., Chicago, IL, USA). Data on patients' baseline characteristics are presented as median value and range for continuous variables.
Starting dose model
The final model was used to develop a model for the starting dose of tacrolimus after kidney transplantation. Each significant covariate in the final model was evaluated if it was clinically relevant, feasible to use, and if it significantly influenced the starting dose of tacrolimus. The starting dose model was then validated using the techniques mentioned in sections Internal Model Evaluation and External Model Evaluation.
Simulation trial
To demonstrate the added value of the starting dose model, a clinical trial was simulated using the patient characteristics of those included in the model building cohort. Each patient was given the standard bodyweight dose and a dose based on the starting dose model calculated using equation (3). For each patient, the C0 and AUC were simulated 1000 times at day 10 post transplantation.
Results
A total of 337 patients were included in the model building group. Patient characteristics are presented in Table 1. From these patients, 4527 blood samples were collected and analysed for tacrolimus concentrations (range 1.6–96.0 ng ml–1). A quarter of the blood samples in the model building groups was drawn the first week following transplantation. In total, three samples (0.07%) in the Rotterdam cohort were below the lower limit of quantification of the immunoassay and were discarded. A total of 40 samples (0.88%) in the Rotterdam cohort were above the upper limit of quantification. These samples were estimated by NONMEM and after every critical model building step checked if the estimate was plausible. The allele frequencies of the tested single‐nucleotide polymorphisms are depicted in Table 1. There was no deviation from the Hardy–Weinberg equilibrium.
Table 1.
Patient characteristics
| Model building group 1 (n = 237) | Model building group 2 (n = 100) | Model validation group (n = 304) | |
|---|---|---|---|
| Recipient sex | |||
| Male | 148 (62.4%) | 56 (56.0%) | 200 (65.8%) |
| Age of recipient (years) | 58.5 (19.4–79.4) | 54.0 (15.0–77.0) | 52.0 (17.0–83.0) |
| Ethnicity | |||
| Caucasian | 186 (78.4%) | 78 (78.0%) | 304 (100%) |
| Asian | 23 (9.7%) | 8 (8.0%) | 0 (0%) |
| African descent | 23 (9.7%) | 1 (1.0%) | 0 (0%) |
| Other | 5 (2.1%) | 13 (13.0%) | 0 (0%) |
| Bodyweight (kg) * | 79.4 (37.6–132.0) | 74.0 (40.0–114.0) | 68.0 (40.0–106.0) |
| Height (cm) * | 183 (145–203) | 172 (141–195) | 166 (145–190) |
| Body mass index (kg m –2 ) | 25.8 (17.2–42.2) | 24.8 (15.6–38.2) | 24.7 (16.3–44.1) |
| Body surface area (m 2 ) | 2.03 (1.24–2.66) | 1.90 (1.33–2.48) | 1.78 (1.18–2.36) |
| Ideal bodyweight (kg) | 68.3 (46.8–89.8) | 65.3 (41.7–83.9) | 60.9 (45.7–80.2) |
| Lean bodyweight (kg) | 64.0 (33.1–85.3) | 55.9 (33.6–81.7) | 51.4 (34.9–76.3) |
| Fat mass (kg) | 21.7 (12.0–44.0) | 26.0 (14.1–49.5) | 25.5 (11.3–50.2) |
| Laboratory measurements * | |||
| Haematocrit (l l –1 ) | 0.34 (0.15–0.80) | 0.34 (0.24–0.45) | 0.33 (0.18–0.59) |
| Creatinine (μmol l –1 ) | 137 (38–1885) | 124 (62–920) | 139 (47–1284) |
| ASAT (U l –1 ) | 21 (<5–662) | Unknown | Unknown |
| Albumin (g l –1 ) | 42 (12–57) | Unknown | Unknown |
| Bilirubin (μmol l –1 ) | 6 (<2–305) | Unknown | Unknown |
| Total protein (g l –1 ) | 64 (23–86) | Unknown | Unknown |
| CRP (mg l –1 ) | 11 (<0.3–320) | Unknown | Unknown |
| Genotype | |||
| CYP3A4 | |||
| * 1 | 205 (86.5%) | 91 (91.0%) | 275 (90.5%) |
| * 22 | 22 (9.3%) | 9 (9.0%) | 29 (9.5%) |
| Unknown | 10 (4.2%) | 0 (0%) | 0 (0%) |
| CYP3A5 | |||
| * 1/ * 1 | 9 (3.8%) | 4 (4.0%) | 0 (0%) |
| * 1/ * 3 | 56 (23.6%) | 17 (17.0%) | 49 (16.1%) |
| * 3/ * 3 | 172 (72.6%) | 76 (76.0%) | 255 (83.9%) |
| * 3/ * 6 | 0 (0%) | 3 (3.0%) | 0 (0%) |
| ABCB1 3435C > T | |||
| CC | 55 (24.3%) | Unknown | Unknown |
| CT | 111 (49.1%) | Unknown | Unknown |
| TT | 60 (26.5%) | Unknown | Unknown |
| POR * 28 | |||
| CC | 128 (56.4%) | Unknown | Unknown |
| CT | 78 (34.4%) | Unknown | Unknown |
| TT | 21 (9.3%) | Unknown | Unknown |
| Primary diagnosis | |||
| Diabetic nephropathy | 48 (20.3%) | 21 (21.0%) | 16 (5%) |
| Polycystic kidney disease | 39 (16.5%) | 15 (15.0%) | 36 (12%) |
| Glomerulonephritis | 44 (18.6%) | 15 (15.0%) | 97 (32%) |
| Hypertensive nephropathy | 42 (17.7%) | 15 (15.0%) | 16 (5%) |
| Reflux/chronic pyelonephritis | 23 (9.7%) | 3 (3.0%) | 0 (0%) |
| Other | 20 (8.4%) | 26 (26.0%) | 51 (17%) |
| Unknown | 21 (8.9%) | 5 (5.0%) | 88 (29%) |
| Number of kidney transplantations | |||
| 1 | 218 (92.0%) | Unknown | 243 (80%) |
| 2 | 16 (6.8%) | Unknown | 49 (16%) |
| 3 or more | 3 (1.3%) | Unknown | 12 (4%) |
| RRT prior to transplantation | |||
| Haemodialysis | 90 (38.0%) | Unknown | Unknown |
| Peritoneal dialysis | 44 (18.6%) | Unknown | Unknown |
| Pre‐emptive | 102 (43.0%) | Unknown | Unknown |
| Delayed graft function | |||
| Yes | 11 (4.6%) | Unknown | Unknown |
| No | 224 (94.5%) | Unknown | Unknown |
| Unknown | 2 (0.8%) | Unknown | Unknown |
| Co‐medication | |||
| Calcium channel blockers | Unknown | Unknown | |
| Amlodipine | 25 (10.5%) | ||
| Nifedipine | 44 (18.6%) | ||
| Barnidipine | 2 (0.8%) | ||
| Time of tacrolimus concentration measurement (days after transplantation) | 23.7 (0.7–99.9) | 7.2 (3–100) | 30 (6–97.5) |
| Distribution of tacrolimus samples | |||
| Total samples | 3661 | 866 | 1334 |
| 0–7 days post‐transplantation | 734 (20.0%) | 359 (41.5%) | 287 (21.5%) |
| 8–14 days post‐transplantation | 642 (17.5%) | 244 (28.2%) | 60 (4.5%) |
| 15–30 days post‐transplantation | 722 (19.7%) | 113 (13.0%) | 604 (45.3%) |
| 31–100 days post‐transplantation | 1563 (42.7%) | 150 (17.3%) | 383 (28.7%) |
Model building group 1 consists of patients transplanted in the Erasmus MC (Rotterdam cohort). Model building group 2 consists of patients transplanted in the LUMC (Leiden cohort)
ASAT, aspartate aminotransferase; CRP, C‐reactive protein; CYP, cytochrome P450; POR*28, P‐450 oxidoreductase*28; RRT, renal replacement therapy
Presented as median and range over a 3‐month period for continuous variables
Base model
The data were best described by a two‐compartment model with first order absorption. Including IIV on CL/F, V 1 /F, V 2 /F and Q/F significantly improved the model fit. The OFV decreased further, and parameter precision, error estimates and GOF plots improved after introduction of IOV on CL/F. Building the different analytical techniques for tacrolimus into the residual error model improved the base model. The residual error was described with a combined additive and proportional error model for the immunoassay measured concentrations, and with a separate proportional error model for the LC–MS/MS measured concentrations. Parameter estimates of the base model, final model and simulation model are presented in Table 2.
Table 2.
Parameter estimates of the base model, final model and bootstrap analysis
| Parameter | Base model (RSE %) [shrinkage] | Final model (RSE %) [shrinkage] | Starting dose model (RSE %) [shrinkage] |
|---|---|---|---|
| t lag (h) | 0.29 (17) | 0.38 (49) | 0.39 (12) |
| k a (l h –1 ) | 3.26 (19) | 3.58 (40) | 3.70 (13) |
| CL/F (l h –1 ) | 25.9 (3) | 23.0 (3) | 22.5 (3) |
| V 1 /F (l) | 655 (7) | 692 (8) | 685 (5) |
| Q/F (l h –1 ) | 10.5 (7) | 11.6 (10) | 10.6 (6) |
| V 2 /F (l) | 6320 (14) | 5340 (22) | 6590 (14) |
| Covariate effect on CL | |||
| CYP3A5*1 | ‐ | 1.63 (15) | 1.62 (14) |
| CYP3A4*22 | ‐ | 0.80 (32) | 0.81 (36) |
| Haematocrit (l l –1 ) | ‐ | −0.76 (11) | ‐ |
| Creatinine (μmol l –1 ) | ‐ | −0.14 (26) | ‐ |
| Albumin (g l –1 ) | ‐ | 0.43 (30) | ‐ |
| Age (years) | ‐ | −0.43 (19) | −0.50 (15) |
| BSA (m 2 ) | ‐ | 0.88 (24) | 0.72 (29) |
| Covariate effect on V 1 | |||
| Lean bodyweight (kg) | ‐ | 1.52 (20) | ‐ |
| IIV (%) | |||
| CL/F | 46.3 (5) 10 | 38.6 (6) 8 | 39.4 (6) 10 |
| V 1 /F | 50.2 (11) 19 | 49.2 (7) 25 | 54.0 (11) 19 |
| V 2 /F | 52.3 (14) 38 | 53.0 (16) 39 | 53.7 (13) 40 |
| Q/F | 79.6 (12) 29 | 78.7 (11) 28 | 79.6 (11) 29 |
| IOV (%) | |||
| CL/F | 15.1 (9) | 13.6 (10) | 14.6 (9) |
| Residual variability | |||
| Proportional (%) | |||
| Immunoassay | 16.6 (6) 26 | 17.7 (7) 22 | 16.9 (6) 25 |
| LC–MS/MS | 24.7 (5) 12 | 24.5 (5) 12 | 24.4 (5) 12 |
| Additive Immunoassay (μg l –1 ) | 1.02 (9) 26 | 0.88 (13) 22 | 1.02 (10) 25 |
CL, clearance; CYP, cytochrome P450; F, bioavailability of oral tacrolimus; IIV, interindividual variability; IOV, interoccasion variability; Ka, absorption rate constant; LC–MS/MS, liquid chromatography–tandem mass spectrometry; OFV, objective function value; Q, intercompartmental clearance of tacrolimus; RSE, residual standard error; tlag, lag time; V1, central compartment for tacrolimus; V2, peripheral compartment for tacrolimus
Covariate analysis
The base two‐compartment model with IOV on CL/F was used as a reference for the covariate analysis. After graphical analysis, the univariate analysis resulted in seven significant covariates correlated with CL/F. The covariates were added in the following order: haematocrit (dOFV 94.0), CYP3A5 genotype (dOFV 74.0), albumin (dOFV 71.9), creatinine (dOFV 36.0), age (dOFV 32.7), CYP3A4 genotype (dOFV 8.2) and body surface area (BSA; dOFV 19.2). Based on graphical analysis there was no difference in the effect on CL/F between CYP3A5 expressers (CYP3A5*1/*1 and CYP3A5*1/*3). LBW significantly influenced V 1 /F (dOFV 24.3). After forward inclusion‐backward elimination (stepwise covariate modelling method) 37, the covariates remained in the final model. Equation (1) describes the final model for estimation of tacrolimus CL/F (l h–1) in the first 3 months post‐transplant:
| (1) |
The NONMEM control stream for the analysis has been included in the Supporting Information Data S1.
Evaluation of the final model
All estimates were within the limits, given the criteria as defined in the Methods section, with the exception of shrinkage for V2 and Q. The population and individual predictions were evenly distributed around the line of unity. The conditional weighted residuals were normally distributed. (Figure 1). The median and variability of the C0 fell mostly within the corresponding simulations as shown in the VPCs, with concentrations in simulations slightly lower than the measured concentrations approximately 2.5–4 h after dose (Figure 2A). NPDE analysis showed adequate predictive ability with distribution of the NPDEs within an acceptable deviation from a normal distribution (Supporting Information Data S2). Evaluation of the individual's influence on a change in OFV by shark plot showed that 73% of patients had a decrease in OFV with the final model compared with the base model. In the external validation, the median of the observed data was close to the lower bound of the simulated data in the second half of the curve. However, for an external validation in clinical data, the median was acceptably described (Figure 2B). Unfortunately, we had no albumin levels at our disposal in the external validation cohort and therefore we fixed the albumin concentration to the population albumin median in the external validation.
Figure 1.
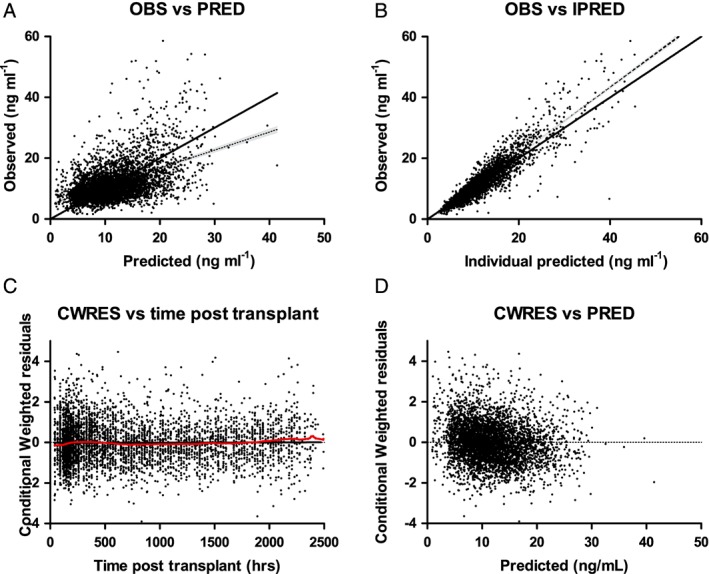
Goodness‐of‐fit plots of the final model. (A) DV plotted against PRED. (B) DV plotted against IPRED. (C) The correlation of CWRES with the time after the tacrolimus dose. (D) The correlation of CWRES with PRED. The line represents the line of identity. CWRES, conditional weighted residuals; DV, observed concentrations; IPRED, individual predicted concentration; OBS, observed concentration; PRED, predicted concentration
Figure 2.
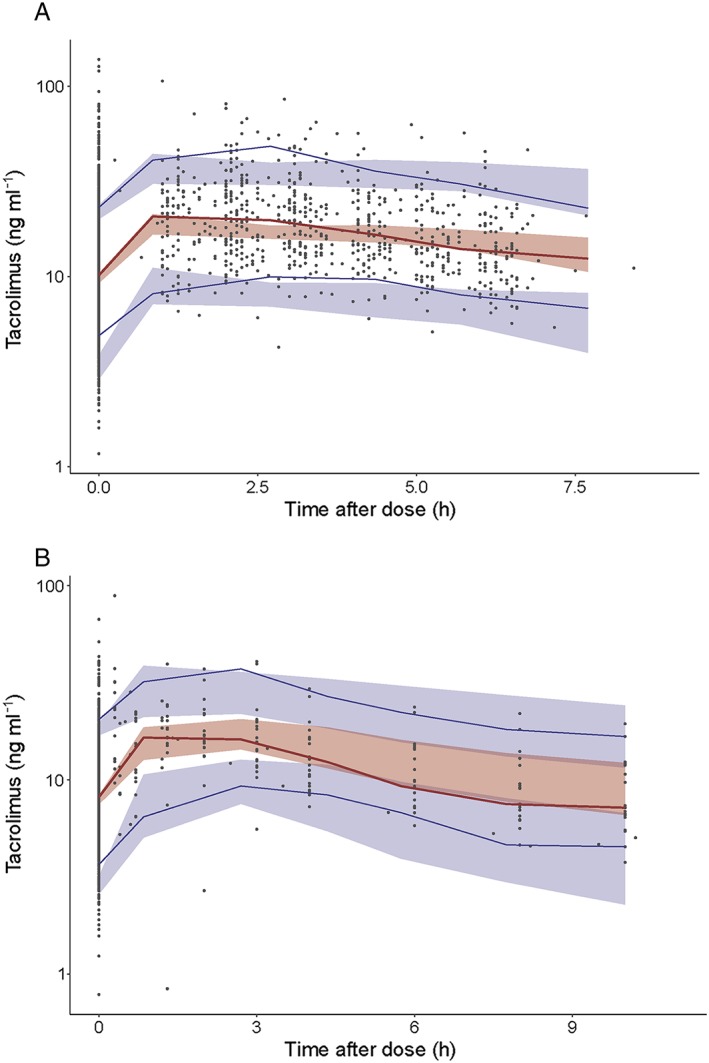
Prediction‐corrected visual predictive check (VPC) showing how well the average trend of the observations (red line) and how well the variability of the observed data (blue lines) fall within the model simulated (n = 500) average trend (red shaded area) and the model simulated variability (blue shaded areas) represented as 95% confidence interval. The average and the variability of the observed data both fall within the corresponding simulations. (A) Prediction‐corrected VPC of the final model (internal dataset). (B) Prediction‐corrected VPC of the final model (external dataset)
Simulations
The effects of the significant covariates on CL/F and are shown in Figure 3. Based on the final model, CYP3A5 expressers had a 1.6 times higher CL/F. Patients carrying the CYP3A4*22 allele had a 0.8 times lower CL/F. An increase in age from 25 to 65 years resulted in a 34% lower CL/F, whereas a decrease in BSA from 2.25 to 1.5 resulted in a 43% lower CL/F. In total, these covariates explained 30% of the variability in CL/F of tacrolimus.
Figure 3.
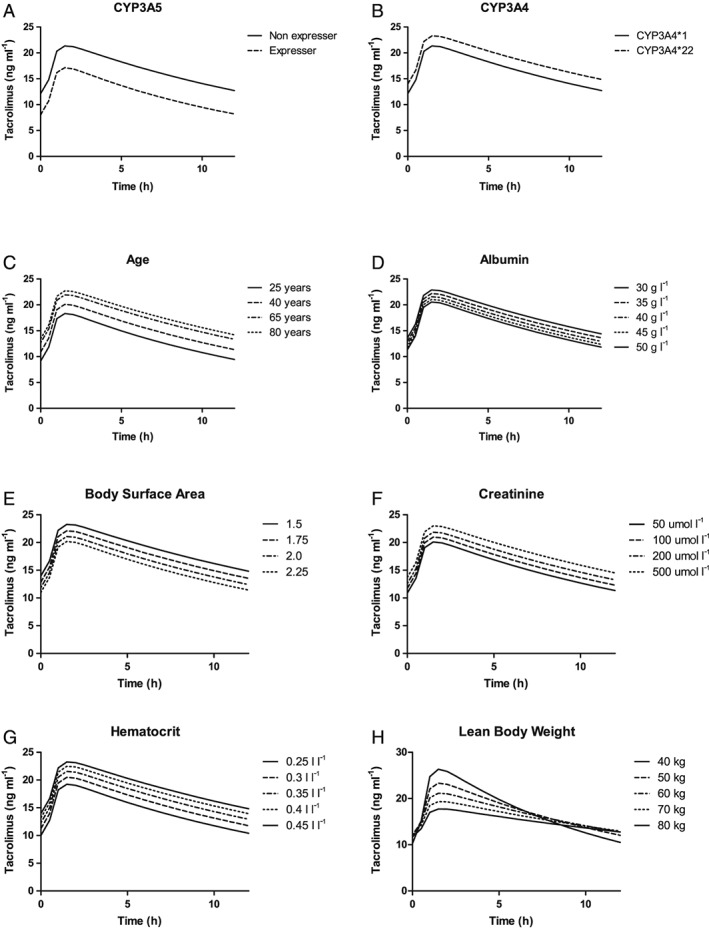
Simulated plasma profiles of tacrolimus at first steady state after transplantation. (A) Simulated plasma profiles of tacrolimus for CYP3A5 nonexpressers (CYP3A5*3/*3) and CYP3A5 expressers (CYP3A5*1/*1 or CYP3A5*1/*3). (B) Simulated plasma profiles of tacrolimus for patients carrying the CYP3A4*1 allele and the CYP3A4*22 allele. (C) Simulated plasma profiles of tacrolimus for patients aged 25, 40, 65 and 80 years. (D) Simulated plasma profiles of tacrolimus for patients with albumin levels of 30, 35, 40, 45 and 50 g l–1. (E) Simulated plasma profiles of tacrolimus for patients with a BSA of 1.5, 1.75, 2 and 2.25 m2. (F) Simulated plasma profiles of tacrolimus for patients with creatinine concentrations of 50, 100, 200 and 500 μmol l–1. (G) Simulated plasma profiles of tacrolimus for patients with haematocrit levels of 0.25, 0.3, 0.35, 0.4 and 0.45 l l–1. (H) Simulated plasma profiles of tacrolimus for patients with an LBW of 40, 50, 60, 70 and 80 kg. BSA, body surface area; CYP, cytochrome P450; LBW, lean body weight
Starting dose model
The final model was used to develop a model for the starting dose of tacrolimus after kidney transplantation. Time after transplantation was not a significant covariate, therefore the starting dose model was based on the same data as the final model. As in clinical practice C0 is commonly used, and CL is the main parameter that influences C0, only those covariates significantly influencing CL/F were included in the starting dose model. The last measured albumin, serum creatinine and haematocrit before transplantation did not significantly influence the CL/F, and because these parameters change substantially after transplantation, they were also not incorporated in the starting dose model. Equation (2) describes the estimation of tacrolimus CL/F (l h–1) right after transplantation:
| (2) |
The median and variability of the C0 fell mostly within the corresponding simulations as shown in the VPCs, demonstrating the good predictive performance in the internal validation (Figure 4A). In the external validation, both the median and variability were adequately described (Figure 4B).
Figure 4.
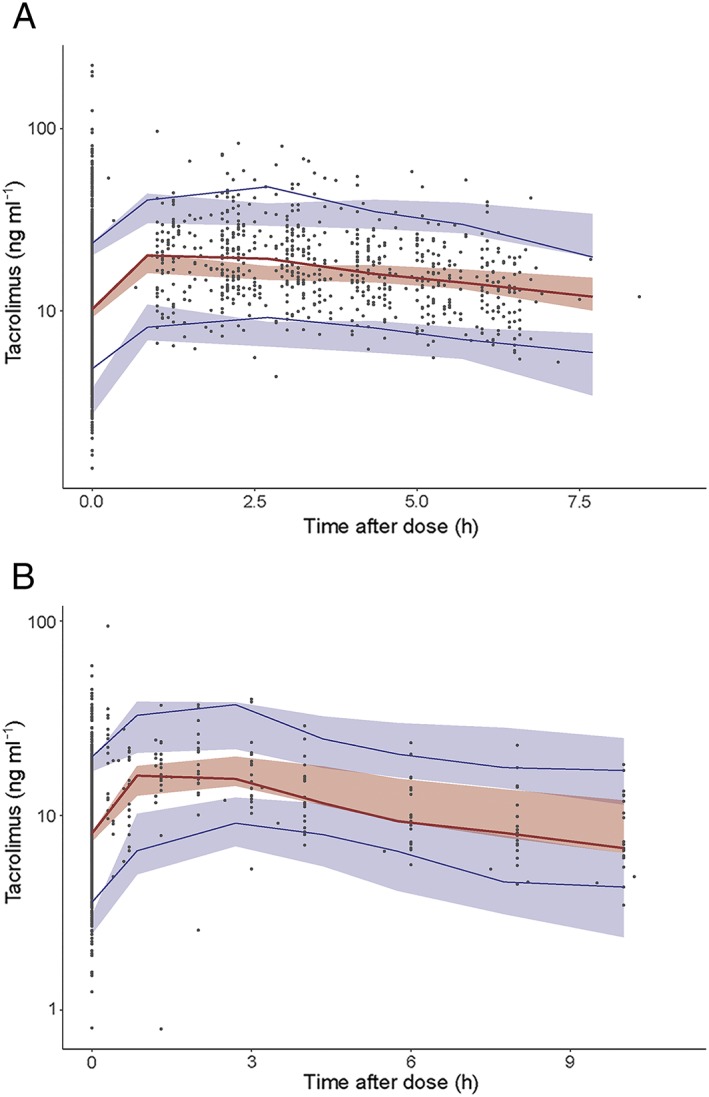
Prediction‐corrected visual predictive check (VPC) of the starting dose model. (A) Prediction‐corrected VPC of the starting dose model (internal dataset). (B) Prediction‐corrected VPC of the starting dose model (external dataset)
The required dose can be calculated using the equation: Dose = CL/F * AUC. In our study, a tacrolimus C0 of 10 ng ml–1 corresponded with an AUC0‐12h of 222 ng h ml–1, 12.5 ng ml–1 with 277 ng h ml–1, and 15 ng ml–1 with 332 ng h ml–1. This leads to equation (3) for a target C0 of 10 ng ml–1 based on a twice daily dose:
| (3) |
The NONMEM control stream for the analysis is shown in the Supporting Information Data S1.
Simulation trial
The results are shown in Figure 5. In the standard bodyweight‐based group, 26.1% were on target (10–15 ng ml–1) 41 vs. 33.0% in the model‐based group. In the bodyweight‐based group, 44.5% were above target compared with 36.8% in the model‐based group. The median tacrolimus C0 in the bodyweight‐based dose group was 13.9 ng ml–1, and in the model‐based dose group 12.9 ng ml–1. There were fewer extreme concentrations in the model‐based dose group, with 5.2% markedly subtherapeutic (<5 ng ml–1) compared to 7.2% in the bodyweight‐based group. In the model‐based dose group, 15.6% were markedly supratherapeutic (>20 ng ml–1) compared to 24.6% in the bodyweight‐based group.
Figure 5.
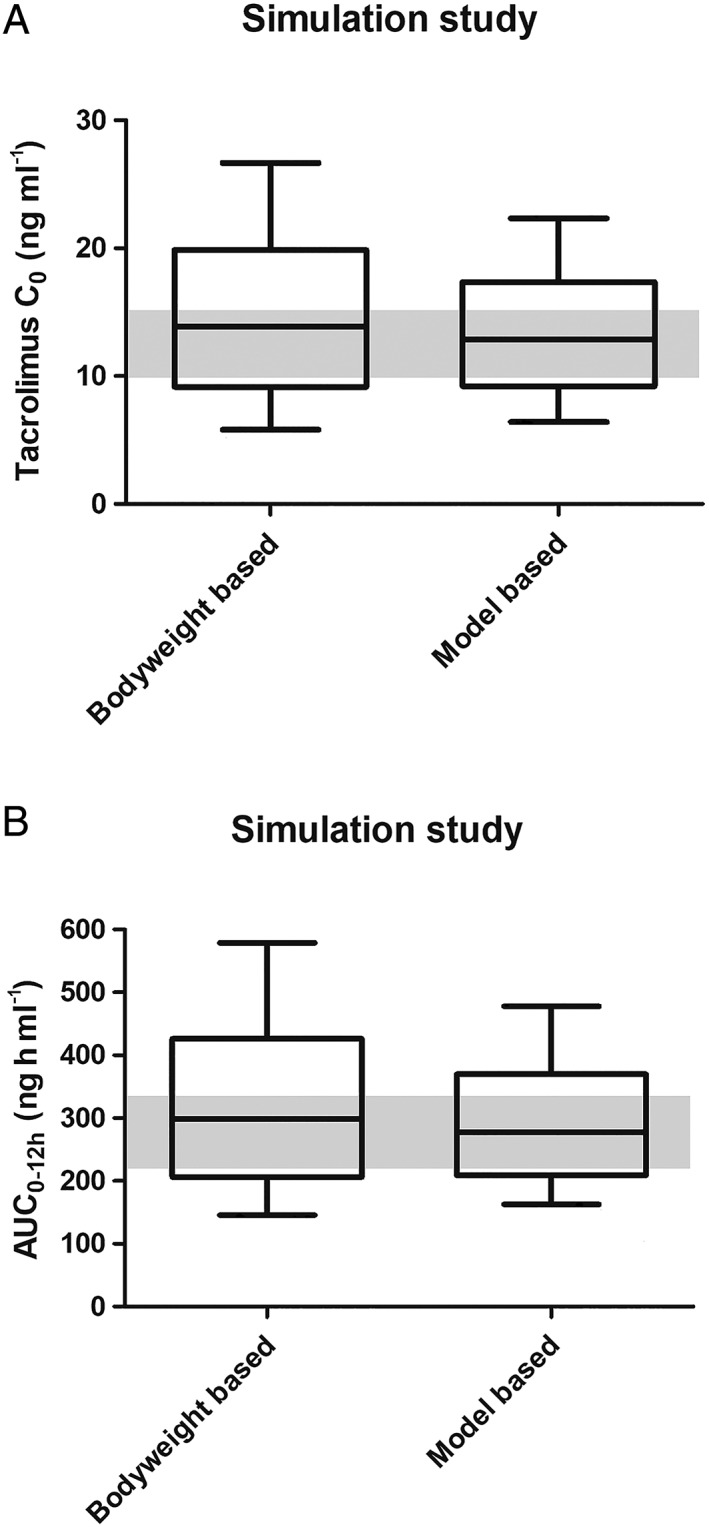
Boxplot with 10–90 percentile whiskers comparing simulations of the standard bodyweight‐based dose and a dose based on the starting dose model. (A) Simulated predose concentrations. The median tacrolimus C0 in the bodyweight‐based dose group was 13.9 ng ml–1, and in the model‐based dose group 12.9 ng ml–1. (B) Simulated AUCs. The median tacrolimus AUC in the bodyweight‐based dose group was 298.5 ng h ml–1, and in the model‐based dose group 277.9 ng h ml–1. AUC, area under the curve
Discussion
This study demonstrates that multiple clinical (albumin, creatinine, haematocrit), demographic (age, BSA, LBW), and genetic (CYP3A4 and CYP3A5 genotype) factors significantly influence the pharmacokinetics of tacrolimus in the first 3 months following renal transplantation. Together, these covariates explained 30% of the total variability in tacrolimus CL/F. A model for the starting dose was developed incorporating CYP3A5 genotype, CYP3A4 genotype, age and BSA. A simulation showed that more patients were on target when the starting dose proposed by the model was used compared with the standard bodyweight‐based dose group (33.0% vs. 26.1%).
In this study, CYP3A5 expressers required a 1.6‐fold higher tacrolimus dose than CYP3A5 nonexpressers. This is in line with previous research 42, 43, 44, 45, 46. Patients carrying the CYP3A4*22 allele required 20% less tacrolimus than the CYP3A4*1 carriers independent of CYP3A5 genotype status, confirming findings from previous research 7, 12, 47, 48, 49. Given the wide availability of TDM, genotyping patients for CYP3A is most useful prior to initiation of tacrolimus therapy. Two RCTs demonstrated that optimization of the initial tacrolimus dose using CYP3A5 genetic testing does not improve clinical outcomes when TDM is performed 24, 33. However, this model is more sophisticated than basing the dose solely on bodyweight and CYP3A5 genotype. For example, it has been suggested that the CYP3A4*22 allele should be included in the Clinical Pharmacogenetics Consortium guidelines when considering a Caucasian population 42, 50.
As approximately 70–80% of tacrolimus is distributed in erythrocytes, low haematocrit will reduce the whole‐blood concentrations of tacrolimus 51. We found in our study that patients with a lower haematocrit had higher CL/F. This underlines previous findings 12, 14, 23, 26, 27, 52, 53. The unbound concentration of tacrolimus is pharmacologically active. Haematocrit does not influence the unbound fraction. However, low albumin concentrations will increase the unbound fraction 52. In contrast to what we expected, patients with hypoalbuminaemia had a lower tacrolimus CL/F. We did not find a similar effect on V1, which one would expect if the correlation were due to protein binding. A possible explanation could be that the reduced CL/F is caused by an underlying inflammatory response, as described previously 54. Hypoalbuminaemia can be an expression of inflammation, which can result in reduced CYP3A activity 55, 56, 57. Unfortunately, no C‐reactive protein levels were available to test this hypothesis. Patients with lower serum creatinine concentrations had an increased CL/F. Tacrolimus undergoes almost no renal elimination and therefore the explanation for the observed association remains unclear. Some studies have reported a significant correlation between serum creatinine and tacrolimus CL 58, 59, whereas others found no such effect 39, 60, 61. Research has shown that CYP3A5 expresser genotype is associated with a greater extent of renal tacrolimus metabolism and a lower apparent urinary tacrolimus CL compared with subjects having the CYP3A5*3/*3 genotype. This is indicative of substantial intrarenal CYP3A5‐dependent tacrolimus metabolism. Patients with poor renal function, and especially patients with delayed graft function, may therefore have a lower tacrolimus CL 62. It is unclear whether this is caused by decreased intrinsic metabolic capacity of the kidney or is an indirect effect of uraemic toxins on hepatic metabolism 40.
Younger patients had an increased tacrolimus CL compared with older patients. A few years ago, Jacobson et al. examined age‐related changes in the metabolism of tacrolimus and nicely demonstrated that older patients (>65 years) had significantly higher weight‐normalized tacrolimus C0 than younger patients (<34 years) 38. Other developed pharmacokinetic models have found a similar effect 10, 12, 63. Research has shown that basing the tacrolimus starting dose solely on bodyweight, will result in overexposure in a considerable proportion of patients 19. BSA is a better indicator of metabolic mass than bodyweight because it is less affected by abnormal adipose mass. In both cohorts of the model building group this correlation between CL and BSA was seen. To the best of our knowledge this is the first pharmacokinetic model to incorporate BSA as a covariate.
In the prediction‐corrected VPCs, the median and variability of the observations fell for the biggest part within the corresponding 95% prediction intervals of the simulations. However, approximately 2.5–4 h postingestion the simulations were slightly lower than the observations. This is explained by the relatively small proportion of patients with an AUC at our disposal (19%). Furthermore, the aim of this study was to develop a pharmacokinetic model for the starting dose of tacrolimus. Therefore, we chose to not describe the absorption with an over‐parametrized transit compartment model.
The main strength of this study is the extensive validation of both models. The models were validated both internally and externally with clinical data using VPCs, and an NPDE was performed. Another strength of the study is the large number of patients included, and the high proportion of patients for whom an AUC was available. Furthermore, the Rotterdam data were of high quality as they were collected in a large RCT, rather than routinely collected clinical data. Another strength is the usage of data collected in four different centres. The final strength of the study is that a separate model was developed to predict the starting dose of tacrolimus.
The main limitation of the current study is that in the model building cohort, three different analytical techniques were used (ACMIA and EMIT in model building group 1, and LC–MS/MS in model building group 2). However, to solve this issue, the residual error model was coded in such a way that it calculates separate residuals errors for the two different bioanalytical techniques. Furthermore, albumin concentrations were not available in the external validation cohort and therefore we could not validate the model for this parameter. The final limitation is the relatively large proportion of C0 (81%) in the model building group. However, in clinical practice tacrolimus is usually dosed based on C0, rather than AUC. Furthermore, population pharmacokinetics using nonlinear mixed effect modelling is the optimal method to handle unevenly distributed data.
The next step is to prospectively test the starting dose model in a pilot study. We have received approval from the ERB and have started dosing patients based on the starting dose algorithm presented in this manuscript 64. If this is successful, the final step to show the additional value of a model based starting dose would be to prospectively test the developed models in an RCT. The starting dose in the experimental arm of such a trial should be adjusted using the starting dose model, with subsequent dose adjustments based on the final model which includes all significant covariates.
Conclusion
The population pharmacokinetics of tacrolimus during the first 3 months following renal transplantation was adequately described using the models presented in this article. CYP3A5 expressers and CYP3A4*1 homozygotes had a higher tacrolimus CL/F. Higher BSA, lower creatinine, younger age, higher albumin and lower haematocrit also resulted in higher tacrolimus CL/F. In total, these covariates explained 30% of the variability in CL/F. By combining these clinical, demographic and genetic parameters, an individualized model has been developed that accurately estimates the tacrolimus CL and which can be used clinically to calculate the starting dose and posterior dose adjustments. The tacrolimus starting dose should be increased to 160% in individuals carrying the CYP3A5*1 allele, whereas it should be reduced to 80% in patients carrying the CYP3A4*22 allele.
Competing Interests
D.A.H. has received lecture and consulting fees from Astellas Pharma and Chiesi Farmaceutici Spa, as well as grant support (paid to his institution) from Astellas Pharma, Chiesi Farmaceutici Spa, and Bristol Myers‐Squibb. T.v.G. has received lecture and consulting fees from Astellas Pharma, Roche Diagnostics and Chiesi Farmaceutici Spa, as well as grant support (paid to his institution) from Astellas Pharma and Chiesi Farmaceutici Spa. D.J.A.R.M. has received lecture fees from Astellas Pharma and Chiesi Farmaceutici, as well as grant support (paid to his institution) from Astellas Pharma and Chiesi Farmaceutici. B.C.M.d.W. has received grant support from Astellas Pharma. The other authors have no competing interests to declare.
Supporting information
Data S1 Example NONMEM control stream
Data S2 Normalized prediction distribution error (NPDE) plot for the starting dose model showing NPDE quantiles
Andrews, L. M. , Hesselink, D. A. , van Schaik, R. H. N. , van Gelder, T. , de Fijter, J. W. , Lloberas, N. , Elens, L. , Moes, D. J. A. R. , and de Winter, B. C. M. (2019) A population pharmacokinetic model to predict the individual starting dose of tacrolimus in adult renal transplant recipients. Br J Clin Pharmacol, 85: 601–615. 10.1111/bcp.13838.
References
- 1. Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group . KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant 2009; 9 (Suppl. 3): S1–S155. [DOI] [PubMed] [Google Scholar]
- 2. Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Eng J Med 2000; 342: 605–612. [DOI] [PubMed] [Google Scholar]
- 3. Meier‐Kriesche HU, Li S, Gruessner RW, Fung JJ, Bustami RT, Barr ML, et al Immunosuppression: evolution in practice and trends, 1994‐2004. Am J Transplant 2006; 6: 1111–1131. [DOI] [PubMed] [Google Scholar]
- 4. Burckart GJ, Liu XI. Pharmacogenetics in transplant patients: can it predict pharmacokinetics and pharmacodynamics? Ther Drug Monit 2006; 28: 23–30. [DOI] [PubMed] [Google Scholar]
- 5. Hesselink DA, van Schaik RH, van Agteren M, de Fijter JW, Hartmann A, Zeier M, et al CYP3A5 genotype is not associated with a higher risk of acute rejection in tacrolimus‐treated renal transplant recipients. Pharmacogenet Genomics 2008; 18: 339–348. [DOI] [PubMed] [Google Scholar]
- 6. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 2009; 4: 481–508. [DOI] [PubMed] [Google Scholar]
- 7. Hesselink DA, Bouamar R, Elens L, van Schaik RHN, van Gelder T. The role of pharmacogenetics in the disposition of and response to tacrolimus in solid organ transplantation. Clin Pharmacokinet 2014; 53: 123–139. [DOI] [PubMed] [Google Scholar]
- 8. Hesselink DA, Hoorn EJ. Improving long‐term outcomes of kidney transplantation: The pressure is on. Neth J Med 2014; 72: 248–250. [PubMed] [Google Scholar]
- 9. Lamb KE, Lodhi S, Meier‐Kriesche HU. Long‐term renal allograft survival in the United States: a critical reappraisal. Am J Transplant 2011; 11: 450–462. [DOI] [PubMed] [Google Scholar]
- 10. Passey C, Birnbaum AK, Brundage RC, Oetting WS, Israni AK, Jacobson PA. Dosing equation for tacrolimus using genetic variants and clinical factors. Br J Clin Pharmacol 2011; 72: 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet 2004; 43: 623–653. [DOI] [PubMed] [Google Scholar]
- 12. Andreu F, Colom H, Elens L, van Gelder T, van Schaik RHN, Hesselink DA, et al A new CYP3A5*3 and CYP3A4*22 cluster influencing tacrolimus target concentrations: a population approach. Clin Pharmacokinet 2017; 56: 963–975. [DOI] [PubMed] [Google Scholar]
- 13. van Gelder T, van Schaik RH, Hesselink DA. Pharmacogenetics and immunosuppressive drugs in solid organ transplantation. Nat Rev Nephrol 2014; 10: 725–731. [DOI] [PubMed] [Google Scholar]
- 14. Storset E, Holford N, Midtvedt K, Bremer S, Bergan S, Åsberg A. Importance of hematocrit for a tacrolimus target concentration strategy. Eur J Clin Pharmacol 2014; 70: 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang JT, de Winter BC, Hesselink DA, Sombogaard F, Wang LL, van Gelder T. The pharmacokinetics and pharmacodynamics of mycophenolate mofetil in younger and elderly renal transplant recipients. Br J Clin Pharmacol 2017; 83: 812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang JT, Andrews LM, van Gelder T, Shi YY, van Schaik RHN, Wang LL, et al Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: recent developments and ethnic considerations. Expert Opin Drug Metab Toxicol 2016; 12: 555–565. [DOI] [PubMed] [Google Scholar]
- 17. Oetting WS, Schladt DP, Guan W, Miller MB, Remmel RP, Dorr C, et al Genomewide association study of tacrolimus concentrations in African American kidney transplant recipients identifies multiple CYP3A5 alleles. Am J Transplant 2016; 16: 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Gelder T. Drug interactions with tacrolimus. Drug Saf 2002; 25: 707–712. [DOI] [PubMed] [Google Scholar]
- 19. Andrews LM, de Winter BC, Tang JT, Shuker N, Bouamar R, van Schaik RHN, et al Overweight kidney transplant recipients are at risk of being overdosed following standard bodyweight‐based tacrolimus starting dose. Transplant Direct 2017; 3: e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han N, Yun HY, Hong JY, Kim IW, Ji E, Hong SH, et al Prediction of the tacrolimus population pharmacokinetic parameters according to CYP3A5 genotype and clinical factors using NONMEM in adult kidney transplant recipients. Eur J Clin Pharmacol 2013; 69: 53–63. [DOI] [PubMed] [Google Scholar]
- 21. Bergmann TK, Hennig S, Barraclough KA, Isbel NM, Staatz CE. Population pharmacokinetics of tacrolimus in adult kidney transplant patients: impact of CYP3A5 genotype on starting dose. Ther Drug Monit 2014; 36: 62–70. [DOI] [PubMed] [Google Scholar]
- 22. Chen SY, Li JL, Meng FH, Wang XD, Liu T, Li J, et al Individualization of tacrolimus dosage basing on cytochrome P450 3A5 polymorphism – a prospective, randomized, controlled study. Clin Transplant 2013; 27: E272–E281. [DOI] [PubMed] [Google Scholar]
- 23. Golubovic B, Vucicevic K, Radivojevic D, Kovačević SV, Prostran M, Miljković B. Total plasma protein effect on tacrolimus elimination in kidney transplant patients – population pharmacokinetic approach. Eur J Pharm Sci 2014; 52: 34–40. [DOI] [PubMed] [Google Scholar]
- 24. Thervet E, Loriot MA, Barbier S, Buchler M, Ficheux M, Choukroun G, et al Optimization of initial tacrolimus dose using pharmacogenetic testing. Clin Pharmacol Ther 2010; 87: 721–726. [DOI] [PubMed] [Google Scholar]
- 25. Press RR, Ploeger BA, den Hartigh J, van der Straaten T, van Pelt J, Danhof M, et al Explaining variability in tacrolimus pharmacokinetics to optimize early exposure in adult kidney transplant recipients. Ther Drug Monit 2009; 31: 187–197. [DOI] [PubMed] [Google Scholar]
- 26. Asberg A, Midtvedt K, van Guilder M, Størset E, Bremer S, Bergan S, et al Inclusion of CYP3A5 genotyping in a nonparametric population model improves dosing of tacrolimus early after transplantation. Transpl Int 2013; 26: 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zuo XC, Ng CM, Barrett JS, Luo AJ, Zhang BK, Deng CH, et al Effects of CYP3A4 and CYP3A5 polymorphisms on tacrolimus pharmacokinetics in Chinese adult renal transplant recipients: a population pharmacokinetic analysis. Pharmacogenet Genomics 2013; 23: 251–261. [DOI] [PubMed] [Google Scholar]
- 28. MacPhee IA, Fredericks S, Tai T, Syrris P, Carter ND, Johnston A, et al The influence of pharmacogenetics on the time to achieve target tacrolimus concentrations after kidney transplantation. Am J Transplant 2004; 4: 914–919. [DOI] [PubMed] [Google Scholar]
- 29. Andrews LM, Riva N, de Winter BC, Hesselink DA, de Wildt SN, Cransberg K, et al Dosing algorithms for initiation of immunosuppressive drugs in solid organ transplant recipients. Expert Opin Drug Metab Toxicol 2015; 11: 921–936. [DOI] [PubMed] [Google Scholar]
- 30. Antignac M, Barrou B, Farinotti R, Lechat P, Urien S. Population pharmacokinetics and bioavailability of tacrolimus in kidney transplant patients. Br J Clin Pharmacol 2007; 64: 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andrews LM, Hesselink DA, van Gelder T, Koch BCP, Cornelissen EAM, Brüggemann RJM, et al A population pharmacokinetic model to predict the individual starting dose of tacrolimus following pediatric renal transplantation. Clin Pharmacokinet 2018; 57: 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boughton O, Borgulya G, Cecconi M, Fredericks S, Moreton‐Clack M, MacPhee IAM. A published pharmacogenetic algorithm was poorly predictive of tacrolimus clearance in an independent cohort of renal transplant recipients. Br J Clin Pharmacol 2013; 76: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shuker N, Bouamar R, van Schaik RH, Clahsen‐van Groningen MC, Damman J, Baan CC, et al A randomized controlled trial comparing the efficacy of CYP3A5 genotype‐based with bodyweight‐based tacrolimus dosing after living donor kidney transplantation. Am J Transplant 2016; 16: 2085–2096. [DOI] [PubMed] [Google Scholar]
- 34. Moes DJ, Swen JJ, den Hartigh J, van der Straaten T, van der Heide JJH, Sanders JS, et al Effect of CYP3A4*22, CYP3A5*3, and CYP3A Combined genotypes on cyclosporine, everolimus, and tacrolimus pharmacokinetics in renal transplantation. CPT Pharmacometrics Syst Pharmacol 2014; 3: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ekberg H, Tedesco‐Silva H, Demirbas A, Vítko Š, Nashan B, Gürkan A, et al Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007; 357: 2562–2575. [DOI] [PubMed] [Google Scholar]
- 36. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther 2007; 82: 17–20. [DOI] [PubMed] [Google Scholar]
- 37. Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res 1998; 15: 1463–1468. [DOI] [PubMed] [Google Scholar]
- 38. Jacobson PA, Schladt D, Oetting WS, Leduc R, Guan W, Matas AJ, et al Lower calcineurin inhibitor doses in older compared to younger kidney transplant recipients yield similar troughs. Am J Transplant 2012; 12: 3326–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sam WJ, Tham LS, Holmes MJ, Aw M, Quak SH, Lee KH, et al Population pharmacokinetics of tacrolimus in whole blood and plasma in Asian liver transplant patients. Clin Pharmacokinet 2006; 45: 59–75. [DOI] [PubMed] [Google Scholar]
- 40. Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J Am Soc Nephrol 2006; 17: 2363–2367. [DOI] [PubMed] [Google Scholar]
- 41. Jusko WJ, Thomson AW, Fung J, McMaster P, Wong SH, Zylber‐Katz E, et al Consensus document: therapeutic monitoring of tacrolimus (FK‐506). Ther Drug Monit 1995; 17: 606–614. [DOI] [PubMed] [Google Scholar]
- 42. Birdwell KA, Decker B, Barbarino JM, Peterson JF, Stein CM, Sadee W, et al Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther 2015; 98: 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Macphee IA, Fredericks S, Tai T, Syrris P, Carter ND, Johnston A, et al Tacrolimus pharmacogenetics: polymorphisms associated with expression of cytochrome p4503A5 and P‐glycoprotein correlate with dose requirement. Transplantation 2002; 74: 1486–1489. [DOI] [PubMed] [Google Scholar]
- 44. Picard N, Bergan S, Marquet P, van Gelder T, Wallemacq P, Hesselink DA, et al Pharmacogenetic biomarkers predictive of the pharmacokinetics and pharmacodynamics of immunosuppressive drugs. Ther Drug Monit 2016; 38 (Suppl. 1): S57–S69. [DOI] [PubMed] [Google Scholar]
- 45. Andrews LM, De Winter BC, Van Gelder T, Hesselink DA. Consideration of the ethnic prevalence of genotypes in the clinical use of tacrolimus. Pharmacogenomics 2016; 17: 1737–1740. [DOI] [PubMed] [Google Scholar]
- 46. Moes DJ, van der Bent SA, Swen JJ, van der Straaten T, Inderson A, Olofsen E, et al Population pharmacokinetics and pharmacogenetics of once daily tacrolimus formulation in stable liver transplant recipients. Eur J Clin Pharmacol 2016; 72: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elens L, van Schaik RH, Panin N, de Meyer M, Wallemacq P, Lison D, et al Effect of a new functional CYP3A4 polymorphism on calcineurin inhibitors' dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenomics 2011; 12: 1383–1396. [DOI] [PubMed] [Google Scholar]
- 48. Lloberas N, Elens L, Llaudo I, Padullés A, van Gelder T, Hesselink DA, et al The combination of CYP3A4*22 and CYP3A5*3 single‐nucleotide polymorphisms determines tacrolimus dose requirement after kidney transplantation. Pharmacogenet Genomics 2017; 27: 313–322. [DOI] [PubMed] [Google Scholar]
- 49. Woillard JB, Mourad M, Neely M, Capron A, van Schaik RH, van Gelder T, et al Tacrolimus updated guidelines through popPK modeling: how to benefit more from CYP3A pre‐emptive genotyping prior to kidney transplantation. Front Pharmacol 2017; 8: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Elens L, Haufroid V. Genotype‐based tacrolimus dosing guidelines: with or without CYP3A4*22? Pharmacogenomics 2017; 18: 1473–1480. [DOI] [PubMed] [Google Scholar]
- 51. Venkataramanan R, Swaminathan A, Prasad T, Jain A, Zuckerman S, Warty V, et al Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet 1995; 29: 404–430. [DOI] [PubMed] [Google Scholar]
- 52. Størset E, Holford N, Hennig S, Bergmann TK, Bergan S, Bremer S, et al Improved prediction of tacrolimus concentrations early after kidney transplantation using theory‐based pharmacokinetic modelling. Br J Clin Pharmacol 2014; 78: 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Woillard JB, de Winter BC, Kamar N, Marquet P, Rostaing L, Rousseau A. Population pharmacokinetic model and Bayesian estimator for two tacrolimus formulations – twice daily Prograf and once daily Advagraf. Br J Clin Pharmacol 2011; 71: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Franken LG, Masman AD, de Winter BCM, Baar FPM, Tibboel D, van Gelder T, et al Hypoalbuminaemia and decreased midazolam clearance in terminally ill adult patients, an inflammatory effect? Br J Clin Pharmacol 2017; 83: 1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harvey RD, Morgan ET. Cancer, inflammation, and therapy: effects on cytochrome p450‐mediated drug metabolism and implications for novel immunotherapeutic agents. Clin Pharmacol Ther 2014; 96: 449–457. [DOI] [PubMed] [Google Scholar]
- 56. Rivory LP, Slaviero KA, Clarke SJ. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute‐phase response. Br J Cancer 2002; 87: 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Slaviero KA, Clarke SJ, Rivory LP. Inflammatory response: an unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol 2003; 4: 224–232. [DOI] [PubMed] [Google Scholar]
- 58. Fukatsu S, Yano I, Igarashi T, Hashida T, Takayanagi K, Saito H, et al Population pharmacokinetics of tacrolimus in adult recipients receiving living‐donor liver transplantation. Eur J Clin Pharmacol 2001; 57: 479–484. [DOI] [PubMed] [Google Scholar]
- 59. Jacobson P, Ng J, Ratanatharathorn V, Uberti J, Brundage RC. Factors affecting the pharmacokinetics of tacrolimus (FK506) in hematopoietic cell transplant (HCT) patients. Bone Marrow Transplant 2001; 28: 753–758. [DOI] [PubMed] [Google Scholar]
- 60. Gruber SA, Hewitt JM, Sorenson AL, Barber DL, Bowers L, Rynders G, et al Pharmacokinetics of FK506 after intravenous and oral administration in patients awaiting renal transplantation. J Clin Pharmacol 1994; 34: 859–864. [DOI] [PubMed] [Google Scholar]
- 61. Staatz CE, Willis C, Taylor PJ, Lynch SV, Tett SE. Toward better outcomes with tacrolimus therapy: population pharmacokinetics and individualized dosage prediction in adult liver transplantation. Liver Transpl 2003; 9: 130–137. [DOI] [PubMed] [Google Scholar]
- 62. Zheng S, Tasnif Y, Hebert MF, Davis CL, Shitara Y, Calamia JC, et al Measurement and compartmental modeling of the effect of CYP3A5 gene variation on systemic and intrarenal tacrolimus disposition. Clin Pharmacol Ther 2012; 92: 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ogasawara K, Chitnis SD, Gohh RY, Christians U, Akhlaghi F. Multidrug resistance‐associated protein 2 (MRP2/ABCC2) haplotypes significantly affect the pharmacokinetics of tacrolimus in kidney transplant recipients. Clin Pharmacokinet 2013; 52: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Available at http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=7568 (last accessed 8 January 2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Example NONMEM control stream
Data S2 Normalized prediction distribution error (NPDE) plot for the starting dose model showing NPDE quantiles