


Abstract
SMYD3 is a methylase previously linked to cancer cell invasion and migration. Here we show that SMYD3 favors TGFβ-induced epithelial–mesenchymal transition (EMT) in mammary epithelial cells, promoting mesenchymal and EMT transcription factors expression. SMYD3 directly interacts with SMAD3 but it is unnecessary for SMAD2/3 phosphorylation and nuclear translocation. Conversely, SMYD3 is indispensable for SMAD3 direct association to EMT genes regulatory regions. Accordingly, SMYD3 knockdown or its pharmacological blockade with the BCI121 inhibitor dramatically reduce TGFβ-induced SMAD3 association to the chromatin. Remarkably, BCI121 treatment attenuates mesenchymal genes transcription in the mesenchymal-like MDA-MB-231 cell line and reduces their invasive ability in vivo, in a zebrafish xenograft model. In addition, clinical datasets analysis revealed that higher SMYD3 levels are linked to a less favorable prognosis in claudin-low breast cancers and to a reduced metastasis free survival in breast cancer patients. Overall, our data point at SMYD3 as a pivotal SMAD3 cofactor that promotes TGFβ-dependent mesenchymal gene expression and cell migration in breast cancer, and support SMYD3 as a promising pharmacological target for anti-cancer therapy.
INTRODUCTION
A crucial event occurring in the metastatic process is the epithelial–mesenchymal transition (EMT), which endows tumor cells with the ability to trans-differentiate from epithelial to mesenchymal-like cells and to acquire the capability to leave the primary tumor mass and disseminate at distant sites (1,2). EMT has been extensively linked to both metastatic progression of cancer (1,3) and acquisition of stem-cell properties (4,5), strengthening the hypothesis that reprogramming from epithelial to mesenchymal phenotype leads to acquired migration and self-renewal abilities, which foster the formation of secondary tumors at distant sites.
Specific extracellular signals activate the EMT and promote epithelial cells reprogramming towards a mesenchymal phenotype (6). During this process, epithelial features such as cell-cell adhesion, polarity and lack of motility are lost, and cells acquire mesenchymal characteristics, including motility and ability to invade. In breast cancer progression, EMT is triggered and maintained by various extracellular signals promoting either autocrine or paracrine stimuli. Among these signals, the transforming growth beta (TGFβ) signaling pathway plays a crucial role (1,3,7). Evolution to invasive and metastatic forms of breast cancer correlates with the activation of EMT (6,8,9) and with increased levels of TGF-β1 in plasma of breast cancer patients and at the invasive front in human breast cancer tissue sections (10,11).
TGFβ signaling pathway activation results in SMAD2 and SMAD3 phosphorylation, their association with SMAD4, and translocation to the nucleus, where SMAD2/3 promote transcriptional activation of EMT-inducing transcription factors (EMT-TFs): Snail1, Slug, ZEB1/2, Twist1/2 and Sox4 (12,13). SMAD2/3/4 transcription factors orchestrate the epithelial cell reprogramming in concert with epigenetic factors that regulate epigenome plasticity and coordinate the expression of molecular signatures associated with the EMT program (14,15). Different DNA-binding partners confer SMADs target gene selectivity and influence the recruitment of transcriptional coactivators or co-repressors. SMADs can bind to acetyltransferases as p300 and P/CAF, but also histone deacetylases and chromatin repressors, which contribute to target genes regulation (8,16).
Expression profiles analysis of human breast cancers allowed the identification of at least five different molecular subtypes (luminal A, luminal B, Her2+, basal and claudin-low) (17,18). Among these subtypes, claudin-low tumors express characteristic mesenchymal genes, and the EMT signature is particularly enriched in this set of tumors, which associate with poorer prognosis (19,20).
The methylase SMYD3 was reported to promote cancer cells proliferation and invasion, and to be over-expressed in several cancers, e.g. colorectal, pancreatic, liver and breast cancers, and was also identified in gene signatures of metastatic pancreatic cancer cells (21–25). SMYD3 has been reported to regulate the metalloprotease MMP9 and cancer invasion (26–29). Recently, Sarris et al. (23) described that SMYD3 is recruited at chromatin regulatory regions of proliferation and EMT genes, in mouse models of liver and colon cancers and the authors depict SMYD3 as a transcriptional ‘potentiator’ for proliferation and EMT genes, during cancer progression (23).
Nevertheless, although SMYD3 oncogenic function is well established, the molecular mechanism through which it promotes tumor cells migration and invasion has not been fully described yet. To elucidate the underlying dynamics of SMYD3-mediated regulation of EMT, we employed breast cancer cells as a model system and investigated SMYD3 link with the TGFβ/SMADs signaling pathway. Here, we report that SMYD3 is indispensable for SMAD3 mediated regulation of target genes, in TGFβ treated breast cancer cells. SMYD3 blockade with the BCI121 inhibitor reduced cell motility, both in cell cultures and in an in vivo model of zebrafish xenograft. Our study provides novel insight in TGFβ-induced transcriptional activation and it supports SMYD3 as a promising therapeutic target for cells that undergo EMT.
MATERIALS AND METHODS
Cell cultures and reagents
NMuMG, MCF10A and MDAMB231 cell lines were purchased from the American Type Culture Collection, grown in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin. MCF10A cell line was grown in DMEM F-12 supplemented with 5% HS, 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin, 100 U/ml penicillin and 100 mg/ml streptomycin. Cells were grown in a humidified incubator with 5% CO2 at 37°C. Cells were starved in serum free growth medium for 12 h and then they were fed with fresh medium containing FBS and 5 ng/ml TGFβ.
TGFβ was reconstituted in 10 mM citric acid (pH 3.0) to a final concentration of 0.1 mg/ml, then further diluted in PBS containing 0.1% BSA to a final concentration of 0.01 mg/ml and stored at –20°C. BCI121 (Innovamol, Italy) was dissolved in dimethyl sulfoxide (DMSO) and stored at –20°C. Unless differently described, BCI121 was used at a final concentration of 10 μM.
All cell lines were periodically tested for mycoplasma with MycoAlert Mycoplasma detection kit (Euroclone, Italy). All cell lines were fed every 48/72 h, for a maximum number of 30 passages. mSMYD3 expression plasmid was purchased from Origene (PS100001).
Cell proliferation and wound healing assays
Cells growth was determined with a Bürker chamber, counting cells after 48 or 72 h of BCI121 or DMSO exposure.
Wound healing assays was performed using Dish Culture–Inserts (Ibidi). 50 000 cells per well were plated and dish were incubated at 37°C and 5% CO2. After 24 h, the Culture-Insert was removed and medium was changed. Pictures were taken at time 0 and 16 h, to evaluate migration ability. The ‘wounded’ area was manually selected (blue lines) and quantified with ImageJ.
RNA tsolation and real time PCR (qRT-PCR)
Total RNA was extracted using TRI reagent (Sigma) according to the manufacturer's instruction. cDNA was synthesized from RNA (1μg) using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystem). qRT-PCR was performed in triplicate using SYBR Green PCR Master Mix (Bio-Rad) or 2X Xtra Master Mix (GeneSpin) on a CFX Connect Real-Time PCR Detection System (Bio-Rad). The qRT-PCR reactions were normalized using GAPDH as housekeeping gene and relative quantification was done using the ddCT method. List of primers used in this study can be found in supplementary methods.
RNA interference and retroviral infections
siRNAs targeting human SMYD3 (5′-GAUUGAAGAUUUGAUUCUA-3′) were synthetized by Eurofins Genomics, and SMAD2/3 siRNAs were purchased from Santa Cruz Biotechnology (sc-37238). siRNAs were transfected (150nM) with Lipofectamine 2000 according to the manufacturer's instructions. Scrambled siRNA (5′-GCGUUGCUCGGAUCAGAAA-3′) was used as negative control. shRNAs used for retroviral/lentiviral infections and siRNA transfection in NMuMG cells were previously described (30). Retroviral and lentiviral infections were performed as in (31). Retrovirus carrying full-length hSMYD3 or SMYD3 mutants were previously described (30).
Cell extracts and immunoblot analysis
Cells were collected and homogenized in RIPA lysis buffer (50 mM Tris–HCl pH 7.4, 0,5% sodium deoxycholate, 0,1% SDS, 250 mM NaCl and 1% NP40) supplemented with protease and phosphatase inhibitors (Sigma). Homogenates were solubilised in Laemmli Sample buffer and 30 μg proteins were separated on 8%, 10% or 12% SDS-PAGE, and transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer System (BioRad). Membranes were blocked with 5% nonfat dry milk in PBS/0.1% Tween and incubated with primary antibody, overnight. Employed antibodies were: E-caderin (Cell Signaling, 24E10), Occludin (Santa Cruz, sc-5562), Fibronectin (Santa Cruz, sc-9068), SMYD3 (GeneTex, GTX121945), SMYD3 (Cell Signaling, D2Q4V), Snail1 (Cell Signaling, C15D3), Actin (Santa Cruz, sc-8432), p-SMAD2/3 (Santa Cruz, sc-11763), Vimentin (Santa Cruz, sc-6260), SMAD4 (Santa Cruz, sc-7966), histone H3 (Santa Cruz, sc-10809), Flag (Sigma, F3165), HA (Santa Cruz, sc-805), Snail2 (Santa Cruz, sc-166476), Lamin A/C (Cell Signaling, 4C11), c-myc (Santa Cruz, sc-789), AKT1/2/3 (Santa Cruz, sc-8312), p-AKT1/2/3 (Santa Cruz, sc-7985-R), ERK (Santa Cruz, sc-94), p-ERK (Santa Cruz, sc-sc-7383), GAPDH (Santa Cruz, sc-32233). Nuclear and cytoplasmic extracts were performed with Nuclear Extract Kit (Active Motif) or according to the procedure described in (32). Images derived from Western blot were quantified using ImageJ (NIH, MD, USA) software.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed as previously described (33). MCF10A and NMuMG cells were treated with 5 ng/ml TFGβ for 6 hrs before formaldehyde crosslinking. 2μg of SMYD3 (NovusBio, NBP1-79393) and SMAD2/3 (Cell Signaling, D7G7) antibodies were used in MCF10A cells, SMYD3 (GeneTex, GTX121945), SMAD3 (Abcam, ab28379) RNAPolII (GeneTex), H3K4me3 (Millipore, 05-745), H3K27me3 (Active Motif, 39536), H3K9me3 (Active Motif, 39161), H3K9Ac (Millipore, 06-942). List of primers used in this study can be found in supplementary methods.
Recombinant proteins
GST-SMYD3 was expressed in Escherichia coli TUNER cells, previously transfected with the Chaperone pKJE7 plasmid (Takara), by induction with 0.1 mM IPTG O/N at 4°C. Recombinant GST-SMYD3 was purified using Pierce Glutathione Superflow Agarose resin (Thermo Scientific) and eluted with 125 mM Tris–HCl, 150 mM NaCl, 10 mM reduced glutathione, 0.2% Triton X100, pH 8.0. GST tag was removed with PreScission Protease (GE Healthcare), according to the manufacturer's instructions.
Immunoprecipitation
Whole cell extracts were obtained from HEK293T transfected cells or NMuMG cells collected and homogenized in IP lysis buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 5 mM MgCl2, 2 mM EDTA, 10% glycerol, 0.1% NP40) supplemented with protease and phosphatase inhibitors (Sigma, Italy). Cytoplasmic/nuclear fractionation was performed as in (32) and 500 μg of both extracts was employed for immunoprecipitation.
Protein contents of all the samples were determined by the Bradford's method. 500 μg of whole extract were incubated with Myc (Millipore, 05-419), SMYD3 (Santa Cruz, sc-49519X), normal IgG (Santa Cruz, sc-2027, sc-2028) at 4°C for 2 h and Protein G Agarose beads (Pierce, Italy) were added for 45 additional minutes. IP for Flag were performed with Flag-M2 affinity gel (Sigma, A2220). For IP with recombinant proteins, 200ng of GST-SMAD3 (Sigma, Italy) and SMYD3 were incubated with 2μg of anti-SMYD3 antibody (Santa Cruz, sc-49519X) or with goat IgG (Santa Cruz, sc-20208) in IP Buffer (50 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1% NP40, pH 8.0) and incubated at 4°C for 2 h and Protein G Agarose beads (Pierce, Italy) were added for 45 additional minutes. Immuno-complexes were washed 3 times, resolved by SDS-PAGE and analyzed by Western blot with SMAD2/3 (Cell Signaling, D7G7), HA (Santa Cruz, sc-805), Flag (Santa Cruz, sc-807), SMYD3 (GeneTex, GTX121945).
Immunofluorescence
Cells seeded on coverslips were washed with PBS, fixed in methanol and blocked with 3% BSA in 1XPBS 0.02% Triton for 1 h at room temperature. Samples were then probed overnight with Paxillin (Santa Cruz, 5574), ZO1 (Santa Cruz 10804), N-cadherin (Santa Cruz, 7939), Phalloidin (Santa Cruz, sc-363795), p-SMAD3 (Cell Signaling, C25A9). After three 5 min washes with PBS, cells were incubated at room temperature with rabbit or mouse Alexa Fluor secondary antibodies for 1 h. Cells were then washed three times with PBS/Triton for 5 min, coverslips were mounted on slides with Fluoroshield with DAPI (Sigma). The samples were examined with a fluorescence microscope (Zeiss). Pictures of staining were obtained using an AxioCam (Carl Zeiss Vision).
Zebrafish care and tumor xenograft
Tg(fli1a:EGFP)y1 transgenic embryos, collected by natural spawning, were staged and raised at 28°C in fish water (Instant Ocean, 0.1% methylene blue), according to National (Italian D.lgs 26/2014) and European laws (2010/63/EU and 86/609/EEC). Dechorionated embryos at 48 h postfertilization hpf were anesthetized with 0.04 mg/ml of tricaine (Sigma-Aldrich). Tumor cells, labeled with a red fluorescent dye for cell viability (CellTracker™ CM-DiI, Invitrogen) and resuspended in PBS, were implanted in the sub-peridermal space, close to the sub-intestinal vessels (SIV) plexus, of 48 hpf zebrafish embryos (100 cells for each embryo). After cell implantation, embryos showing cells into the yolk sac and/or in the vasculature embryos were excluded from further analyses. Correctly grafted embryos were split in two experimental groups that were treated with 50 μM BCI121 and the vehicle alone (DMSO). BCI121 and DMSO were diluted directly into the fish water. Treated and control embryos were incubated at 32°C. At 48 h post injection (hpi), the presence of circulating grafted cells into the trunk/tail region was evaluated through a fluorescence stereomicroscope (Leica DM6000B equipped with LAS Leica imaging software). The spread of injected tumor cells throughout the embryo was quantified in both 50 μM BCI121-treated and control embryos by the ‘Analyze Particle’ plugin of Fiji software. Data from two independent experiments were pooled. Statistical significance was calculated with unpaired Student's t-test. *P < 0.05.
Clinical datasets analysis
The website FireBrowse (http://firebrowse.org) was used to interrogate the TGCA database for the average SMYD3 expression in breast invasive carcinomas versus normal tissue.
The website cBioPortal (http://www.cbioportal.org) was used for meta-analysis (34,35). The Metabric dataset contains data from 2509 patients with breast cancer, and includes Illumina v3 RNA microarray and clinical data with a follow-up period up to 350 months (36,37). Only 1866 patients were included in the present study, because they displayed all the parameters considered. Patients were stratified based on SMYD3 z-score and quartiles were identified as SMYD3 high (q1: SMYD3 z-score ≥ 0.998. n = 468) and SMYD3 low (q3 SMYD3 z-score ≤ –0.327. n = 467). SMYD3 expression levels were correlated with EMT genes transcript levels by Pearson correlation analysis. Estimated probability of overall survival and distant metastasis free survival was calculated with the Kaplan-Meier method.
NKI295 dataset was downloaded from http://ccb.nki.nl/data/ (38). Correlation analysis in the NKI295 set was limited to the available data for EMT-related target probes. The subgroup of patients with BRCA1 mutations was excluded from the analysis.
Statistical analysis
Statistical significance of the results was analyzed using the unpaired Student's t-tail test, one-way Anova followed by Tukey post-test, two-way Anova followed by Bonferroni post-test using GraphPad (Prism6). Survival curves were analyzed by Log-rank (Mantel-Cox) Test. Correlations were analyzed by Pearson test. *P < 0.05, **P < 0.01 and ***P < 0.001 were considered statistically significant.
RESULTS
SMYD3 is required for efficient TGFβ-induced EMT
Our analysis of the Human Cancer Genome Atlas (TCGA) database (34,35) revealed that SMYD3 transcripts are over-expressed in a variety of cancers, and amplification frequency measured by RNA transcript levels is especially apparent in several breast cancers cohorts (Supplementary Figure S1A).
In addition to its role in cancer cells proliferation, SMYD3 was shown to regulate EMT genes (23). Since EMT is causatively linked to distant metastases for epithelial cancers including breast cancer, we initially tested SMYD3 role in EMT employing mouse mammary gland epithelial (NMuMG) cells, which can be induced to undergo EMT when treated with TGFβ. We knocked down SMYD3 levels by Sh-interference, using a retrovirus carrying a short-hairpin sequence targeting SMYD3. As expected, following TGFβ treatment, ShScramble NMuMG cells morphology acquired a mesenchymal phenotype (Figure 1A, upper panels), accompanied by the progressive down-regulation of epithelial transcripts E-cadherin and Claudin6 (Supplementary Figure S2A). Remarkably, ShSMYD3 cells maintained a more epithelial morphology (Figure 1A, lower panel) and the epithelial transcripts E-Cadherin and Claudin6 failed to decrease (Supplementary Figure S2A). Immunoblot analysis revealed that E-Cadherin and Occludin protein levels were maintained at higher levels following TGFβ stimulation, when compared to ShScramble cells at 96 h of TGFβ treatment (Figure 1B). Moreover, ShSMYD3 silencing reduced TGFβ induced upregulation of the mesenchymal markers Fibronectin1 and N-cadherin, when compared to ShScramble cells (Figure 1B and Supplementary Figure S2A). Snail1, which plays a critical role in the transcriptional repression of epithelial genes and transition to a mesenchymal phenotype during EMT (39), failed to be upregulated both at the transcriptional and protein level in ShSMYD3 cells (Figure 1C). Moreover, AKT and ERK phosphorylation levels, which were shown to be affected by SMYD3 depletion in previous reports (25,40,41), remained unchanged in our model system (Supplementary Figure S2B).
Figure 1.

SMYD3 depletion impairs TGFβ-induced EMT in NMuMG and MCF10A cells. (A) Brightfield images of shSMYD3 or shScramble NMuMG cells treated with 10 ng/ml TGFβ or vehicle for 24 h. Scale bar = 10 μm. (B) Western blot analysis of epithelial (E-Cadherin and Occludin) and mesenchymal (N-Cadherin and Fibronectin) genes in shSMYD3 or shScramble NMuMG cells treated with 10 ng/ml TGFβ for 0, 48, 72 and 96 h. GAPDH serves as a loading control. Normalized band intensity in immunoblots is reported above signals. (C) Upper panel: Snail1 transcript levels were quantified by qRT-PCR, after 0, 6, 12 h of TGFβ treatment. Data represent means±SD. Statistical analysis was performed with one-way ANOVA, followed by post-hoc Tukey test. n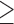 3, * P ≤ 0.05. Lower panel: Western blot to evaluate Snail1 protein levels at 0, 6 12, 24 h following TGFβ treatment. β-actin was used as loading control. Normalized band intensity in immunoblots is reported above signals. (D) immunofluorescence microscopy of mesenchymal and epithelial markers in shSMYD3 or shScramble NMuMG cells. Left panels show untreated cells and the right panel shows cells treated with 10 ng/ml TGFβ for 24 h. Epithelial (ZO-1) and mesenchymal (N-Cadherin) markers were analyzed. Paxillin was used to detect focal adhesion plaques, and phalloidin to visualize the actin cytoskeleton. Scale bar = 20 μm. (E) MCF10A cells were treated with 5 ng/ml TGFβ for 72 h and mRNA levels of epithelial (Occludin) and mesenchymal genes (MMP9, Vimentin, N-Cadherin and Fibronectin) were determined by quantitative real-time PCR analysis, relative to GAPDH. Data represent means ± SD of nFold relative to a SiScramble untreated sample. Statistical analysis was performed with unpaired Student's t test. n
3, * P ≤ 0.05. Lower panel: Western blot to evaluate Snail1 protein levels at 0, 6 12, 24 h following TGFβ treatment. β-actin was used as loading control. Normalized band intensity in immunoblots is reported above signals. (D) immunofluorescence microscopy of mesenchymal and epithelial markers in shSMYD3 or shScramble NMuMG cells. Left panels show untreated cells and the right panel shows cells treated with 10 ng/ml TGFβ for 24 h. Epithelial (ZO-1) and mesenchymal (N-Cadherin) markers were analyzed. Paxillin was used to detect focal adhesion plaques, and phalloidin to visualize the actin cytoskeleton. Scale bar = 20 μm. (E) MCF10A cells were treated with 5 ng/ml TGFβ for 72 h and mRNA levels of epithelial (Occludin) and mesenchymal genes (MMP9, Vimentin, N-Cadherin and Fibronectin) were determined by quantitative real-time PCR analysis, relative to GAPDH. Data represent means ± SD of nFold relative to a SiScramble untreated sample. Statistical analysis was performed with unpaired Student's t test. n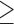 3, * P ≤ 0.05, ** P ≤ 0.01. (F) MCF10A cells were incubated with Scramble or SMYD3 siRNAs for 24 h, and then treated with 5 ng/ml TGFβ for 6 h. mRNA levels of EMT-TFs was determined by quantitative real-time PCR analysis, relative to GAPDH. Data represent means±SD of nFold relative to a SiScramble untreated sample. Statistical analysis was performed with unpaired Student's t test. n
3, * P ≤ 0.05, ** P ≤ 0.01. (F) MCF10A cells were incubated with Scramble or SMYD3 siRNAs for 24 h, and then treated with 5 ng/ml TGFβ for 6 h. mRNA levels of EMT-TFs was determined by quantitative real-time PCR analysis, relative to GAPDH. Data represent means±SD of nFold relative to a SiScramble untreated sample. Statistical analysis was performed with unpaired Student's t test. n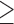 3, * P ≤ 0.05, ** P ≤ 0.01. (G) Rescue experiment in MCF10A cells knocked down for SMYD3 by siRNAs and co-transfected with a plasmid carrying the mSMYD3 cDNA. After 24 h, cells were treated with 5 ng/ml TGFβ for 72 h. Normalized band intensity in immunoblots is reported above signals. (H) Migration percentage was measured by wound healing assay in siScramble, siSMYD3 and mSMYD3-reconstituted siSMYD3 MCF10A cells treated with TGFβ (5 ng/ml) for 16 h. Statistical analysis was calculated with unpaired Student's t-test. n
3, * P ≤ 0.05, ** P ≤ 0.01. (G) Rescue experiment in MCF10A cells knocked down for SMYD3 by siRNAs and co-transfected with a plasmid carrying the mSMYD3 cDNA. After 24 h, cells were treated with 5 ng/ml TGFβ for 72 h. Normalized band intensity in immunoblots is reported above signals. (H) Migration percentage was measured by wound healing assay in siScramble, siSMYD3 and mSMYD3-reconstituted siSMYD3 MCF10A cells treated with TGFβ (5 ng/ml) for 16 h. Statistical analysis was calculated with unpaired Student's t-test. n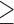 3, * P ≤ 0.05. Scale bar = 10 μm.
3, * P ≤ 0.05. Scale bar = 10 μm.
F-actin and Paxillin immunostaining revealed that SMYD3 depletion did not affect the epithelial morphology of NMuMG cells in the absence of TGFβ. Following TGFβ treatment, instead, expression of the epithelial marker ZO-1 was retained at the cell membrane of SMYD3-depleted cells, and the signal for the mesenchymal marker N-cadherin was barely detectable in ShSMYD3 cells (Figure 1D). TGFβ induced stress fibers and focal adhesion formation, revealed by F-actin and paxillin staining respectively, were almost absent in ShSMYD3 cells and revealed reduced remodeling of cytoskeleton, when compared to ShScramble TGFβ-treated cells (Figure 1D).
Similar results were obtained in immortalized benign MCF10A human mammary epithelial cells, in which the cuboidal cobblestone epithelial cells switch to a fibroblastoid morphology following TGFβ treatment (Supplementary Figure S2C, top panel). SMYD3 depletion by RNA interference led to a persistency of the epithelial morphology, upon TGFβ treatment (Supplementary Figure S2C, bottom panel); this lack of morphological change was accompanied by a delay in mesenchymal marker expression (N-cadherin, Vimentin, Fibronectin1 and MMP9) in TGFβ-treated SMYD3-depleted cells (Figure 1E). In addition, when SMYD3-deficient and control cells were exposed to TGFβ for 6 h, mRNA levels of a subset of EMT-TFs (Snail1, Snail2, Sox4, Twist1) failed to be upregulated in SMYD3-knockdown cells (Figure 1F). Concurrently, mRNA and protein levels of the epithelial marker Occludin were maintained at higher level in SMYD3-depleted cells, when compared to control scramble interfered cells (Figure 1E). Protein levels of the mesenchymal markers Snail1, Fibronectin and N-cadherin decreased in SMYD3-depleted MCF10A cells and their expression was rescued by reconstitution with a mouse SMYD3 protein, which promoted the upregulation of mesenchymal markers and led to a reduction of Occludin levels (Figure 1G). Likewise, in ShSMYD3 NMuMG cells, the reconstitution with the hSMYD3 protein (Supplementary Figure S2D) promoted the mesenchymal transcripts expression and reduced Occludin transcript levels (Supplementary Figure S2E). Both in NMuMG and MCF10A cells, SMYD3 transcript levels tended to slightly increase following TGFβ administration (Supplementary Figure S2F, G). Nonetheless, SMYD3 protein levels were not significantly affected by TGFβ treatment in our NMuMG model (Figure 1B).
Since TGFβ treatment promotes cell migration in epithelial breast cancer cells (2), we evaluated SMYD3 impact on cell migration through a wound-healing assay. We observed that SMYD3 knockdown reduced TGFβ-induced migration in NMuMG and MCF10A cells and that SMYD3 reconstitution rescued the defective ability to migrate in both cell types (Supplementary Figure S2H–J and Figure 1H).
Overall, these data indicate that SMYD3 plays a role in regulating EMT-TFs and mesenchymal genes transcription and modulates TGFβ-induced cell migration in human and mouse mammary cells.
SMYD3 pharmacological blockade prevents EMT
SMYD3 function was recently challenged by the small inhibitor BCI121 (41). Thus, in a parallel set of experiments, SMYD3 was pharmacologically blocked, by treating both NMuMG and MCF10A cells with increasing concentration of the SMYD3 inhibitor BCI121. Both cell lines were initially treated with different doses of BCI121 for 3 h and subsequently co-treated with TGFβ for additional 48 h. In NMuMG cells, increasing doses of BCI121 progressively reduced TGFβ-induced mesenchymal genes upregulation and epithelial genes downregulation. At the 10 μM BCI121 dose, TGFβ treated NMuMG cells failed to induce Fibronectin and N-cadherin mesenchymal transcripts and to down-regulate mRNA levels of the epithelial genes E-cadherin and Claudin6. A lower BCI121 dose (5 μM) was sufficient to mask TGFβ-mediated effects on Claudin6 and Fibronectin transcript levels (Figure 2A). Likewise, SMYD3 pharmacological blockade in MCF10A cells resulted in a modest increase in the mesenchymal transcript N-cadherin following TGFβ treatment, at the 5 μM BCI121 dose, and MMP9 expression was abrogated at the 10 and 50 μM doses (Figure 2B). In agreement with BCI121 ability to temper TGFβ-induced effects, we also observed reduced migration ability in BCI121-treated NMuMG and MCF10A cells (Figure 2C and D).
Figure 2.

SMYD3 blockade with the BCI121 inhibitor hampers TGFβ-induced EMT. (A) Epithelial (E-Cadherin, Claudin-6) and mesenchymal (N-Cadherin and Fibronectin) transcripts were quantified by quantitative real-time PCR analysis, relative to GAPDH. NMuMG cells were treated with 10 ng/ml TGFβ in the presence of 0, 1, 5 10 and 50 μM BCI121, for 48 h. Statistical analysis was performed with one-way ANOVA, followed by post-hoc Tukey test. Data represent means±SD. n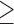 3, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (B) N-Cadherin and MMP9 transcript levels were quantified by quantitative real-time PCR normalized to GAPDH in MCF10A cells treated with 5 ng/ml TGFβ in the presence of 0, 1, 5 10 and 50 μM BCI121 for 48 h. Data represent means±SD. Statistical analysis was performed with one-way ANOVA, followed by post-hoc Tukey test. n
3, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (B) N-Cadherin and MMP9 transcript levels were quantified by quantitative real-time PCR normalized to GAPDH in MCF10A cells treated with 5 ng/ml TGFβ in the presence of 0, 1, 5 10 and 50 μM BCI121 for 48 h. Data represent means±SD. Statistical analysis was performed with one-way ANOVA, followed by post-hoc Tukey test. n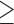 3, * P ≤ 0.05, ** P ≤ 0.01, ***P ≤ 0.001. (C, D) Wound healing assay on NMuMG and MCF10A cells treated with 10μM BCI121 and 5 and 10 ng/ml TGFβ respectively, for 0 and 16 h. Migration percentage is reported in the right panel. Statistical significance was calculated with paired Student's t-test. * P ≤ 0.05, *** P ≤ 0.001. Scale bar = 10 μm.
3, * P ≤ 0.05, ** P ≤ 0.01, ***P ≤ 0.001. (C, D) Wound healing assay on NMuMG and MCF10A cells treated with 10μM BCI121 and 5 and 10 ng/ml TGFβ respectively, for 0 and 16 h. Migration percentage is reported in the right panel. Statistical significance was calculated with paired Student's t-test. * P ≤ 0.05, *** P ≤ 0.001. Scale bar = 10 μm.
As previously reported for other cell lines, 72 h BCI121 treatment led to decreased cell viability (41), both in NMuMG and MCF10A cells. With the exception of the 50 μM dose in TGFβ treated NMuMG cells, BCI121 did not significantly affect cell growth at 48 h treatment, suggesting that the inhibitor effects on EMT were largely independent on its impact on cell proliferation at earlier stages (Supplementary Figures S3A and S3B). Likewise, SMYD3 genetic depletion did not affect cell growth throughout TGFβ treatment, both in NMuMG and MCF10A cells (Supplementary Figure S3C and D).
Overall, these data suggest that SMYD3 pharmacological blockade prevents the full activation of TGFβ induced EMT and reduces cell migration.
SMYD3 directly interacts with SMAD3
Transcriptional regulation of TGFβ modulated genes is largely controlled by SMAD2/3 transcription factors during EMT (39). Therefore, we first checked whether SMYD3 interacted with SMAD3 in HEK293T cells, transfected with Flag-SMYD3 and HA-SMAD3 plasmids.
Immunoprecipitation experiments revealed that over-expressed SMYD3 was able to associate to SMAD3, in the absence of TGFβ stimulation (Figure 3A). Remarkably, exogenously expressed SMAD2 and SMAD4 were also able to associate with SMYD3 in HEK293T cells (Supplementary Figure S4A and B). Next, we determined whether SMYD3 was able to interact with endogenous SMAD2/3 and performed immunoprecipitation experiments using NMuMG extracts. These experiments indicated that SMDA3/SMYD3 association occurs both in untreated and TGFβ-treated cells and that it is not affected by the presence of the TGFβ inhibitor SB43152 in growth medium (Figure 3B). Immnuoprecipitation experiments with recombinant GST-SMAD3 and SMYD3 proteins revealed a direct association between the two proteins in vitro (Figure 3C).
Figure 3.

SMYD3 directly interacts with SMAD3. (A) Western-blot analysis of over-expressed Flag-SMYD3 and HA-SMAD3 following co-immunoprecipitation with anti-Flag antibody. (B) Co-immunoprecipitation analysis of endogenous SMYD3 and SMAD3 in NMuMG cells. Whole cell extract were prepared from NMuMG cells after starvation, from cells treated with the TGFβ-signaling inhibitor SB431542, or starved and treated with 10 ng/ml TGFβ for 30 and 60 minutes. IP was performed with antibodies raised against SMYD3. (C) in vitro co-immunoprecipitation of GST-SMAD3 and SMYD3 was performed with SMYD3 antibodies. Immunoblot was performed with anti-SMAD2/3 and anti-SMYD3 antibodies. (D) Co-immunoprecipitation with anti-Myc antibody in whole cell extracts of HEK293T cells over-expressing Myc-SMYD3 and Flag-SMAD3 WT or Flag-tagged SMAD3 domains MH1, L-MH2 and MH2. Lower panel represents a schematic representation of SMAD3 mutants. (E) Co-immunoprecipitation with anti-HA antibody of HEK293T cells over-expressing HA-SMAD3 and Flag-SMYD3 WT or Flag-tagged SMYD3 mutants 1–219, 219–428, 111–428, 1–380. Lower panel represents a schematic representation of SMYD3 mutant. (F) ΔΔNMuMG cells were transduced with human full-length or SMYD3 mutants by retroviral infection and than knocked down for endogenous SMYD3. 24 h after SMYD3 depletion, cells were treated with 10ng/ml TGFβ for 12 h (Snail1) or 48 h (Fibronectin). Snail1 and Fibronectin transcripts were quantified by quantitative real-time PCR analysis, relative to GAPDH. nFold is relative to siScramble untreated cells. SiS refers to SiSMYD3 cells. Statistical analysis was performed with unpaired Student's t-test. Data represent means ± SD. n = 3, *P ≤ 0.05, **P ≤ 0.01.
To define the protein domains involved in the interaction, we performed immunoprecipitation experiments in HEK293T cells over-expressing SMAD3 deletion mutants with full-length SMYD3 and found that the MH2 domain is required for SMYD3 association (Figure 3D and Supplementary Figure S4C). Co-immunoprecipitation experiments with full-length HA-SMAD3 and Flag-SMYD3 deletion mutants revealed that mutant SMYD3 encompassing aa 219–428 and aa 111–428 associate with SMAD3 as the full-length protein. Conversely SMYD3 proteins lacking the C-terminal region (SMYD3_1–219 and SMYD3_1–380) lost the ability to interact with SMAD3 (Figure 3E and Supplementary Figure S4D).
To investigate whether SMYD3 C-terminal region is relevant to the modulation of TGFβ induced genes, we reconstituted SMYD3-depleted NMuMG cells with the full-length human SMYD3, hSMYD3_1–380, hSMYD3_111–428 deletion mutants and the methylation defective hSMYD3_ΔEEL (Supplementary Figure S4E). Following TGFβ treatment, Snail1 and Fibronectin transcript levels were rescued by SMYD3 full-length, hSMYD3_111–428 and hSMYD3_ΔEEL over-expression, in SMYD3-depleted cells (Figure 3F). The hSMYD3_1–380 mutant, instead, was unable to promote Snail1 and Fibronectin expression in ShSMYD3 NMuMG cells (Figure 3F).
Furthermore, we tested whether SMYD3 methylase activity was functionally indispensable for SMYD3-mediated EMT regulation. We evaluated the ability of SMYD3 catalytically defective mutant (SMYD3-ΔEEL) to promote EMT, when compared to wild type SMYD3 (SMYD3-WT). Both SMYD3-WT and the inactive SMYD3-ΔEEL mutant promoted upregulation of the mesenchymal genes N-cadherin and Vimentin, and equally reduced E-cadherin and Occludin protein levels, as well as membrane-associated ZO-1 (Supplementary Figures S4F and S4G). These results are in agreement with previously reported data (23) and they suggest that SMYD3-mediated EMT genes regulation is independent on SMYD3 methylation activity.
Overall, these data suggest that SMYD3 directly interact with SMAD3 through its C-terminal domain, and this region is crucial for EMT genes modulation, independently of SMYD3 methylation activity.
SMYD3 is indispensable for SMAD3 association to the chromatin
SMYD3 protein localized both in the cytoplasm and nucleus of TGFβ-treated NMuMG cells, and was more abundant in the former compartment (Supplementary Figure S5A). Following TGFβ treatment, SMAD3 primarily localizes in the nucleus, where SMYD3/SMAD3 interaction was detectable, pointing to a potential role of this interaction in nuclear functions (Supplementary Figure S5A).
SMYD3 critical role in EMT regulation (23) (Figures 1–3) prompted us to investigate SMYD3 ability to directly associate to mesenchymal gene regulatory regions. ChIP assays in TGFβ treated MCF10A cells showed that SMYD3 is recruited at the Snail1, Sox4, MMP9 and Vimentin regulatory regions. To provide further mechanistic insight, we assayed SMAD2/3 recruitment at these mesenchymal target genes in SMYD3 knockdown and control ShScramble cells. Chromatin immunoprecipitation (ChIP) assays revealed that SMAD2/3 engagement at mesenchymal genes was significantly reduced by SMYD3 depletion (Figure 4A and Supplementary Figure S5B).
Figure 4.

SMYD3 assists SMAD3 recruitment to the chromatin. (A) SMAD2/3 and SMYD3 association to regulatory regions of EMT markers was analyzed by ChIP qPCR, in ShScramble and ShSMYD3 MCF10A cells treated with 5ng/ml TGFβ for 24 h. Data are expressed as fold enrichment relative to ShScramble cells and represent means±SD. Statistical analysis was performed with Student's t test. n = 4, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (B) SMAD2/3 and SMYD3 association to EMT markers was analyzed by ChIP qPCR, in MCF10A cells co-treated with 5ng/ml TGFβ and 10 μM BCI121 for 24 h. Data are expressed as fold enrichment relative to control cells. Data represent means±SD. Statistical analysis was performed with unpaired Student's t test. n = 4, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (C) immunofluorescence microscopy of shSMYD3 or shScramble NMuMG cells treated with 10 ng/ml TGFβ for 2 h. The staining for the phosphorylated SMAD3 (P-SMAD3) (left panel), for DAPI (center panel) and the merge of the two stainings (right panel) are shown. Scale bar = 10 μm. (D) Western-blot analysis of P-SMAD3 and SMAD2/3 levels in shSMYD3 or shScramble NMuMG cells, treated with 10 ng/ml TGFβ for for 30 min. Relative band intensity, normalized over total SMAD2/3 proteins, is indicated. (E) Western-blot analysis of cytoplasmic (C) and nuclear (N) P-SMAD3 in shSMYD3 or shScramble NMuMG cells treated with 10 ng/ml TGFβ for 30 min. GAPDH was used as cytoplasmic marker, histone H3 as nuclear marker. (F) ChIP assay was performed to evaluate SMYD3, SMAD2/3, RNAPolII, H3K9me3, H3K9Ac, H3K27me3 occupancy at the Snail1 promoter, in NMuMG cells treated with 10ng/ml TGFβ for 6 h. Data are normalized on IgG and represent means ± SD. Statistical analysis was performed with Student's t test. n = 3, * P ≤ 0.05, ** P ≤ 0.01. (G) ChIP assay was performed to evaluate SMYD3 and SMAD2/3 occupancy at the Snail1 promoter, in NMuMG cells obtained as in Figure 3F, treated with 10ng/ml TGFβ for 6 h. SiS refers to SiSMYD3 cells. Data are normalized on IgG and represent means±SD. Statistical analysis was performed with unpaired Student's t test. n = 3, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
BCI121 is predicted to bind the substrate binding pocket, and the inner part of the lysine channel, forming hydrogen bonds with SMYD3 Ser202 and Tyr239 (41). Thus, by interacting with the histone lysine-binding pocket, BCI121 exerts a dual function on SMYD3, both inhibiting its catalytic activity and preventing its recruitment to the chromatin (41). Thus, as a second approach we performed ChIP assays with BCI121-treated MCF10A cells, in which BCI121 treatment did not prevent SMAD2/3 interaction with SMYD3 (Supplementary Figure S5C). As expected, BCI121 administration prevented SMYD3 occupancy at these regulatory regions. In addition, SMAD2/3 recruitment was also dramatically decreased at regulatory regions of MMP9, Snail1, Sox4, and Vimentin (Figure 4B and Supplementary Figure S5D).
Remarkably, both immunoblot and immunofluorescence experiments revealed that TGFβ dependent SMAD2/3 phosphorylation was unaffected by SMYD3 knockdown (Figure 4C and 4D). Likewise, SMAD2/3 nuclear translocation (Figure 4C and E) and SMAD4 interaction (Supplementary Figure S5E) were not influenced by SMYD3 reduction, suggesting that SMYD3 depletion did not impair TGFβ induced SMAD2/3 nuclear localization.
ChIP assays in NMuMG cells, confirmed a reduction in SMAD2/3 and SMYD3 occupancy at the Snail1 promoter, in ShSMYD3 TGFβ-treated cells compared to ShScramble cells. Additionally, histone marks analysis at Snail1 regulatory regions revealed that SMYD3 depletion was accompanied by a local decrease in histone H3K9ac and H3K4me3. Conversely, repressive histone marks H3K27me3 and H3K9me3 occupancy was comparable in ShSMYD3 and ShScramble cells. Concurrently, RNA Polymerase II recruitment was reduced at Snail1 promoter in ShSMYD3 NMuMG cells (Figure 4F and Supplementary Figure S5F), in agreement with Snail1 gene reduced transcription (Figure 1C).
Taken together, these findings suggest that SMYD3 directly takes part in the transcriptional regulation of a subset of mesenchymal genes, and that its engagement correlates with SMAD2/3 and RNAPolII recruitment at regulatory regions.
We repeated SMYD3 and SMAD3 ChIP-assays in SMYD3-depleted NMuMG cells that were reconstituted with full-length hSMYD3, with the methylation defective mutant (hSMYD3_ΔEEL) and with the N-term or C-term deletion mutants (hSMYD3_111–428 and hSMYD3_1–380). All SMYD3 mutant forms were able to associate to the Snail1 promoter. ChIP-assays with the SMAD3 antibody revealed that the hSMYD3_1–380 mutant failed to recruit SMAD3 to the Snail1 promoter (Figure 4G and Supplementary Figure S5I), in agreement with Snail1 transcript levels reported in Figure 3F. To further dissect SMYD3/SMAD3 interplay, we evaluated SMYD3 ability to associate EMT-genes in SMAD3 knocked-down MCF10A cells, by ChIP assay. SMAD3 was necessary for SMYD3 recruitment, suggesting that both proteins are required to promote EMT-genes transcription (Supplementary Figures S5G and H).
Overall, SMYD3 appears to promote EMT-TFs and mesenchymal genes transcription by favoring SMAD2/3 chromatin stabilization, in a methylation independent manner.
SMYD3 inhibition decreases MDA-MB-231 mesenchymal phenotype and cells migration ability
We next asked whether SMYD3 knockdown or pharmacological blockade could attenuate the mesenchymal phenotype, in a metastatic mesenchymal-like breast cancer cell line. We therefore treated human MDA-MB-231 cells with siSMYD3 RNAs or BCI121 and assessed whether SMYD3 blockade could reduce mesenchymal gene transcription and promote epithelial markers expression. SMYD3-depleted cells displayed higher levels of E-cadherin and a reduced expression of mesenchymal markers (Fibronectin, Vimentin, Snail1) (Figure 5A). In addition, we treated MDA-MB-231 cells with two doses of BCI121 (10 and 50 μM) for 48 h and found that SMYD3 inhibition increased the expression of epithelial markers Occludin and Claudin6 and reduced the expression of mesenchymal markers Snail2, Fibronectin and N-cadherin (Figure 5B). Snail1, Snail2, Vimentin and Fibronectin protein levels also decreased following BCI121 administration (Figure 5C). Functionally, BCI121 treatment decreased MDA-MB-231 ability to migrate in vitro, as shown by wound-healing assays (Figure 5D).
Figure 5.

SMYD3 knock down or pharmacological blockade tempers MDA-MB-231 mesenchymal phenotype and migration ability. (A) SMYD3 was knocked down by siRNA transfection in MDA-MB-231 cells, total extracts were collected after 48 h and assayed for epithelial (E-cad) or mesenchymal (Fibronectin, Vimentin and Snail1) protein levels by immunoblot. GAPDH was used as a loading control. nFold is relative to DMSO treated cells. (B) MDA-MB-231 cells were treated with increasing doses of BCI121 (0, 10 or 50 μM) for 48 h. Epithelial (Occludin, Claudin6) or mesenchymal (Snail2, N-cadherin and Fibronectin) marker mRNA levels were measured by qRT-PCR. GAPDH was used as housekeeping gene. Data represent means±SD. Statistical analysis was performed with 1 way ANOVA, followed by post-hoc Tukey test n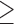 3, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (C) MDA-MB-231 cells were treated with increasing doses of BCI121 (0, 10 or 50 μM) for 72 h and whole cell extracts were assayed for mesenchymal (Snail1, Snail2, Vimentin and Fibronectin) protein levels by immunoblot. GAPDH was used as a loading control. (D) Wound-healing assay on MDA-MB-231 cells treated with 10 μM BCI121 for 0 and 16 h. Migration percentage is reported in the right panel. Statistical significance was calculated with Student's t-test. *** P ≤ 0.001. Scale bar = 10μm. (E) in vivo migration assay in zebrafish xenografts. 1 × 102 MDA-MB-231 cells were injected in the subepidermal pocket of zebrafish embryos at 48 hpf. Embryos were treated with 50 μM BCI-121 or DMSO, for 48 h, n = 14 DMSO embryos, n = 12 BCI121 embryos. (F) Cells migrating in the tail, heart and head were counted in the two groups, 48 hpi. Scale bar = 200μm. Statistical analysis was performed with unpaired Student's t-test. * P ≤ 0.05. Normalized band intensity in immunoblots is reported above signals.
3, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. (C) MDA-MB-231 cells were treated with increasing doses of BCI121 (0, 10 or 50 μM) for 72 h and whole cell extracts were assayed for mesenchymal (Snail1, Snail2, Vimentin and Fibronectin) protein levels by immunoblot. GAPDH was used as a loading control. (D) Wound-healing assay on MDA-MB-231 cells treated with 10 μM BCI121 for 0 and 16 h. Migration percentage is reported in the right panel. Statistical significance was calculated with Student's t-test. *** P ≤ 0.001. Scale bar = 10μm. (E) in vivo migration assay in zebrafish xenografts. 1 × 102 MDA-MB-231 cells were injected in the subepidermal pocket of zebrafish embryos at 48 hpf. Embryos were treated with 50 μM BCI-121 or DMSO, for 48 h, n = 14 DMSO embryos, n = 12 BCI121 embryos. (F) Cells migrating in the tail, heart and head were counted in the two groups, 48 hpi. Scale bar = 200μm. Statistical analysis was performed with unpaired Student's t-test. * P ≤ 0.05. Normalized band intensity in immunoblots is reported above signals.
MDA-MB-231 cells have been employed to evaluate in vivo migration, in zebrafish xenografts. MDA-MB-231 cells were injected in the sub-peridermal space, close to the sub-intestinal vessels (SIV) plexus, in zebrafish embryos at 48 h post fertilization (hpf). After cells injection, embryos were treated either with 50 μM BCI121 or DMSO for 48 h. We then evaluated the number of single cells that invaded the zebrafish body, either in the tail, heart or head, at 2 days post-injection (dpi). We found that the number of invading MDA-MB-231 cells was significantly reduced in zebrafish embryos treated with BCI121, when compared to DMSO treated embryos (Figure 5E and F).
Overall, these results indicate that BCI121 treatment decreases MDA-MB-231 cells mesenchymal signature and it reduces their invasion ability, both in vitro and in vivo.
SMYD3 is highly expressed in human breast cancers and correlates with increased metastasis
To assess whether our findings could be related to the pathogenesis of human breast cancer and metastasis formation, we studied SMYD3 expression in one of the publicly available data sets in the TCGA repository (Supplementary Figure S1). In the Metabric cohort (35, 36) SMYD3 was altered in 24.1% of the 2501 cases (Supplementary Figure S1).
Analysis of RNA-seq expression profiles of breast invasive carcinomas obtained from the TCGA database and analyzed with the Firebrowse portal showed that SMYD3 expression was significantly enhanced in breast cancer tumors (1093 tumor samples) when compared to healthy tissue (112 normal samples) (Figure 6A). Remarkably, we could not point out any specific SMYD3 enrichment in relation to any breast cancer molecular subtype or in relation to different tumor stages (Supplementary Figure S6A and B). We next focused on EMT signature genes and interrogated expression data from 1394 tumor samples obtained from the Metabric dataset to investigate whether SMYD3 transcript levels were associated with EMT genes expression. We stratified patients for SMYD3 levels (Supplementary Figure S6C), and found that Snail2, Zeb1, Zeb2, Twist1, Twist2, N-cadherin, Fibronectin and Vimentin were significantly upregulated in high SMYD3 tumors compared to the low SMYD3 group (Supplementary Figure S6D). We observed a positive correlation between SMYD3 and mesenchymal genes expression (Snail2, Zeb1, Zeb2, Fibronectin, N-Cadherin and Twist1) in the Metabric dataset (Supplementary Figure S6E). To confirm our findings, we extended our analysis to the NKI295 breast cancer dataset (42), and confirmed a positive correlation between SMYD3 and Snail2, N-cadherin and Fibronectin expression (Supplementary Figure S6F).
Figure 6.

Prognostic value of SMYD3 in breast cancer. (A) Box plots showing SMYD3 expression levels in normal and tumor breast tissues. Analysis was performed on 1098 breast cancer tissues and 112 normal tissues on TCGA data, downloaded through Firebrowse. *** P < 0.0001, unpaired Student's t test. (B) Correlation between SMYD3 and the mesenchymal markers Snail2, ZEB1, ZEB2, TWIST1, Fibronectin and N-Cadherin expression, calculated on claudin-low tumors (Metabric), by Pearson correlation analysis. Pearsons’ coefficient tests were performed to assess statistical significance. (C) Claudin-low tumors (Metabric) were stratified for SMYD3 expression (high vs low) and Snail2, ZEB1, ZEB2, TWIST1, TWIST2, Vimentin, Fibronectin mRNA levels were compared in SMYD3 low and SMYD3 high tumors. Statistical significance was calculated with unpaired Student's t test. *P ≤ 0.05, **P ≤ 0.001, ***P ≤ 0.0001. (D) Kaplan–Meier plots of survival probability of claudin-low patients stratified by SMYD3 expression in the Metabric dataset. P value calculated by log rank test. (E) Kaplan–Meier plots of distant metastasis-free survival of patients with grade 3 tumors, stratified by SMYD3 expression levels. P value was calculated by log rank test.
Core-EMT signature genes are highly expressed in claudin-low breast cancers, when compared to other molecular breast cancers subtypes (20). We therefore queried for a correlation between SMYD3 expression and a subset of core-EMT genes in claudin-low tumors, within the Metabric dataset. Our analysis confirmed a positive correlation between SMYD3 expression and EMT-TFs (Snail2, Zeb2, Zeb1, Twist1), as well as mesenchymal gene transcripts (Fibronectin, N-Cadherin) and the degree of correlation was more robust in claudin-low subtype than in the whole Metabric cohort (Figure 6B and Supplementary Figure S6E). The basal subtype of breast cancers differ from claudin-low tumors for a lower expression of EMT-signature transcripts and TGFβ activation pathway (20). Remarkably, the correlation between mesenchymal markers and SMYD3 transcript levels was less evident in the basal tumors subgroup than in claudin-low tumors (Figure S7A).
Transcript levels for Snail2, ZEB1, ZEB2, Twist1, Vimentin and Fibronectin were significantly higher in claudin-low tumors over-expressing SMYD3 compared to the SMYD3 low tumors (Figure 6C), and the differences between the two groups were more prominent than in the analysis on the entire dataset and the basal subtype (Figure 6C, Supplementary Figures S6D and S7B).
SMYD3 higher expression levels did not correlate with a poorer prognosis in the Metabric cohort as a whole or in the basal subtype subgroup of patients (Supplementary Figure S7C and D); however, SMYD3 high patients had a significantly lower survival probability in the claudin-low tumor subgroup, when compared to the claudin-low SMYD3 low patients’ group (Figure 6D).
To further investigate SMYD3 prognostic value, we performed a parallel analysis in different clinical microarray datasets from 1354 breast tumors (43). We first focused our analysis to an advanced tumor stage, and found that distant metastasis free survival was significantly higher for patients with low SMYD3 grade 3 tumors, independently of lymph node status and systemic treatment they underwent (n = 458, P value 0.022) (Figure 6E). Likewise, SMYD3 high levels in grade 1 lymph node negative tumors, in patients that didn’t undergo chemotherapy or endocrine therapy, displayed a significantly lower probability of distant metastasis free survival (n = 94, P = 0.031) (Supplementary Figure S7E).
Overall, clinical dataset analysis indicates that SMYD3 levels positively correlate with EMT-TFs and mesenchymal gene expression in breast tumors. Higher SMYD3 levels in breast tumors associate with reduced metastasis free survival, and with a worst overall survival probability in claudin-low tumors.
DISCUSSION
Results presented in this paper highlight SMYD3 nuclear functions in regulating EMT-TFs and mesenchymal gene expression, in the context of TGFβ stimulation. We describe a direct TGFβ-independent interaction between SMYD3 and SMAD3, and a functional interplay between SMYD3 and SMAD3 in EMT genes transcriptional regulation. We propose that SMYD3 is indispensable for TGFβ-induced SMAD3 recruitment at chromatin regulatory regions of mesenchymal targets and EMT-TFs. Remarkably, SMAD3 is likewise necessary for SMYD3 association, suggesting that both factors are required for transcriptional competence and that SMYD3 may stabilize SMAD3-orchestrated complex and potentiate transcriptional activation.
Several lines of evidence previously linked SMYD3 to migration and invasion, in different cell lines 22,24. Our findings confirm this indication in TGFβ-induced EMT and provide a molecular mechanism that supports recently reported in vivo findings (23), highlighting a crucial role for SMYD3 in promoting EMT genes activation. Sarris et al. (23) showed by ChIP-Seq assay that SMYD3 occupies several regulatory regions in liver tumors and revealed that SMYD3 association to a subset of proliferation and EMT loci promotes their transcriptional activation. We propose that SMYD3 cooperates with SMAD3 at EMT genes, driving mesenchymal and EMT-TFs transcription. Following TGFβ-mediated stimulation, SMAD2 and SMAD3 are activated, translocate to the nucleus and bind to different transcription factors, co-activators or co-repressors thus achieving higher affinity and selectivity for target genes regulatory regions (16). In different cell types SMAD3 occupancy is directed by cell-type-specific master transcription factors, which are responsible for orchestrating the cell-type-specific effects of TGFβ signaling (44). Therefore, cell-type-specific effects of TGFβ signaling are in large part determined by SMAD2/3 interactions with transcription factors and cofactors and they can vary in a cell/tissue-specific manner. In this scenario, we propose that SMYD3 takes part in the orchestrated SMAD3 recruitment at a subset of mesenchymal and EMT-TFs genes, following TGFβ stimulation. SMAD3 interacts with SMYD3 through the MH2 domain, which is responsible for association to various transcription factors and cofactors, for the interaction between SMAD proteins and that is also crucial for transcriptional activation (45). SMYD3 interaction domain, instead, encompasses the SMYD3 C-terminal region, which was previously reported to be involved in HSP90 (46) and H3K4me3 interaction in vitro (23).
Our ChIP data show that SMYD3 depletion does not affect distribution of the repressive histone marks H3K9me3 and H3K27me3 at the Snail1 promoter. These findings are in agreement with the concept that SMYD3 is recruited at open-chromatin regions (23) and suggest that SMYD3/SMAD2/3 independent mechanisms are involved in H3K9me3 and H3K27me3 removal (47). Conversely, SMYD3 promotes H3K4me3 and H3K9Ac marks deposition at the Snai1 promoter, as well as SMAD3 and RNAPolII recruitment, and transcriptional activation. These data suggest that SMYD3 association to the Snail1 gene is subsequent to repressive histone marks removal and that SMYD3 association promotes transcription through SMAD2/3 complex stabilization and RNAPolII recruitment, at EMT loci. SMAD3 was previously reported to mediate COMPASS recruitment and precede H3K4 trimethylation at the Snail1 promoter, in TGFβ-treated DU145 cells (48). Additionally, SMAD3 interacts with p300/CBP and TGFβ treatment promotes acetylation of SMAD2/3 target genes (49,50), further suggesting that SMAD2/3 association to the chromatin is required to promote H3K4me3 and H3K9Ac. Additionally, SMYD3 associates with the RNAPolII complex (30,51), and this interaction may favor the transcriptional machinery recruitment at EMT genes.
Remarkably, SMYD3-mediated regulation of EMT is independent of its catalytic activity, suggesting that SMYD3 co-activator functions are not mediated by SMYD3 methylation activity at the mesenchymal/EMT-TFs genes. Likewise, SMYD3 knockdown did not lead to ERK and AKT phosphorylation modulation (25,40,41), indicating that SMYD3 cytosolic and methylation-dependent mechanisms are finely tuned and operate differently in different context.
In the patho-physiological context, patients’ data on survival and metastasis free survival support the experimentally described SMYD3 role in promoting EMT. SMYD3 mRNA levels are upregulated in breast cancers compared to normal breast tissues, but SMYD3 transcripts appear to be upregulated in all breast cancer subtypes. SMYD3 also promotes transcriptional activation of proliferation genes and its augmented levels may confer an advantage for tumor growth in different breast cancer subtypes. Additionally, SMYD3 was shown to regulate HER2 dimerization through direct methylation (52), suggesting that in different breast cancer subtypes SMYD3 may provide selective growth advantages, through distinct mechanisms.
Remarkably, we were unable to observe a significant correlation between Snail1 and SMYD3 expression in our bioinformatics analysis of breast tumor datasets. Although Snail1 transcriptional upregulation is known to occur following TGFβ stimulation, experimental models of breast cancer have shown that Snail1 transient expression in the primary tumor is sufficient to increase lung metastasis (53) and that TGFβ signaling is a transient and spatially restricted event, suggesting that Snail1 plays a crucial role in EMT initiation, while other EMT-TFs participate in EMT maintenance (53,54).
Claudin-low breast cancer cells were shown to be more likely than other breast cancer subtypes to promote the breast tumor initiating cancer cell (BTICs) population in response to TGFβ (55). We propose that in TGFβ sensitive cells, such as claudin-low cancer cells, SMYD3 may promote EMT genes activation through its interplay with SMAD3. Recent evidences show that EMT-associated markers are highly expressed in cancer stem cells and promote their self-renewal (4). Remarkably, BTICs are enriched within the claudin-low molecular subtype of breast cancer, where TGFβ/SMAD signaling promote BTICs expansion (55). Further investigations will reveal whether SMYD3 likewise promotes BTICs stemness in claudin-low cells.
Overall, our data further support SMYD3 as a promising pharmacological target for anti-cancer therapy.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr S. Simone (University of Bari, Italy) and Dr A. DelRio (CNR.ISOF, Bologna, Italy) for helpful discussions, Dr G. Pavesi in our Department for support with the bioinformatic analysis and Dr E. Garattini (Istituto Mario Negri, Milan) for helpful advice.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Trideo grant from Italian Association for Cancer Research/Fondazione Cariplo (to G.C.); Telethon and Worldwide Cancer Research (to G.C., in part); R. Fittipaldi was supported by a FIRC fellowship. Funding for open access charge: Project funding (to G.C.) Trideo AIRC/Fondazione Cariplo grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P.. EMT: 2016. Cell. 2016; 166:21–45. [DOI] [PubMed] [Google Scholar]
- 2. Imamura T., Hikita A., Inoue Y.. The roles of TGF-β signaling in carcinogenesis and breast cancer metastasis. Breast Cancer. 2012; 19:118–124. [DOI] [PubMed] [Google Scholar]
- 3. Thiery J.P. epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002; 2:442–454. [DOI] [PubMed] [Google Scholar]
- 4. Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M. et al. . The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheel C., Weinberg R.A.. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012; 22:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalluri R., Weinberg R.A.. The basics of epithelial–mesenchymal transition. J. Clin. Invest. 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barcellos-Hoff M.H., Akhurst R.J.. Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. 2009; 11:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. López-Novoa J.M., Nieto M.A.. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009; 1:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thiery J.P., Acloque H., Huang R.Y., Nieto M.A.. epithelial–mesenchymal transitions in development and disease. Cell. 2009; 139:871–890. [DOI] [PubMed] [Google Scholar]
- 10. Dalal B.I., Keown P.A., Greenberg A.H.. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am. J. Pathol. 1993; 143:381–389. [PMC free article] [PubMed] [Google Scholar]
- 11. Chod J., Zavadova E., Halaska M.J., Strnad P., Fucikova T., Rob L.. Preoperative transforming growth factor-beta 1 (TGF-beta 1) plasma levels in operable breast cancer patients. Eur. J. Gynaecol. Oncol. 2008; 29:613–616. [PubMed] [Google Scholar]
- 12. Budi E.H., Duan D., Derynck R.. Transforming growth Factor-β receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017; 27:658–672. [DOI] [PubMed] [Google Scholar]
- 13. Macias M.J., Martin-Malpartida P., Massagué J.. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015; 40:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nieto M.A. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013; 342:1234850. [DOI] [PubMed] [Google Scholar]
- 15. Tam W.L., Weinberg R.A.. The epigenetics of epithelial–mesenchymal plasticity in cancer. Nat. Med. 2013; 19:1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Massagué J., Seoane J., Wotton D.. Smad transcription factors. Genes Dev. 2005; 19:2783–2810. [DOI] [PubMed] [Google Scholar]
- 17. Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sørlie T., Perou C.M., Tibshirani R., Aas T., Geisler S., Johnsen H., Hastie T., Eisen M.B., van de Rijn M., Jeffrey S.S. et al. . Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prat A., Parker J.S., Karginova O., Fan C., Livasy C., Herschkowitz J.I., He X., Perou C.M.. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010; 12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sabatier R., Finetti P., Guille A., Adelaide J., Chaffanet M., Viens P., Birnbaum D., Bertucci F.. Claudin-low breast cancers: Clinical, pathological, molecular and prognostic characterization. Mol. Cancer. 2014; 13:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura T., Fidler I.J., Coombes K.R.. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res. 2007; 67:139–148. [DOI] [PubMed] [Google Scholar]
- 22. Hamamoto R., Silva F.P., Tsuge M., Nishidate T., Katagiri T., Nakamura Y., Furukawa Y.. Enhanced SMYD3 expression is essential for the growth of breast cancer cells. Cancer Sci. 2006; 97:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarris M.E., Moulos P., Haroniti A., Giakountis A., Talianidis I.. Smyd3 is a transcriptional potentiator of multiple Cancer-Promoting genes and required for liver and colon cancer development. Cancer Cell. 2016; 29:354–366. [DOI] [PubMed] [Google Scholar]
- 24. Giakountis A., Moulos P., Sarris M.E., Hatzis P., Talianidis I.. Smyd3-associated regulatory pathways in cancer. Semin. Cancer Biol. 2017; 42:70–80. [DOI] [PubMed] [Google Scholar]
- 25. Mazur P.K., Reynoird N., Khatri P., Jansen P.W., Wilkinson A.W., Liu S., Barbash O., Van Aller G.S., Huddleston M., Dhanak D. et al. . SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014; 510:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cock-Rada A.M., Medjkane S., Janski N., Yousfi N., Perichon M., Chaussepied M., Chluba J., Langsley G., Weitzman J.B.. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012; 72:810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang S.Z., Luo X.G., Shen J., Zou J.N., Lu Y.H., Xi T.. Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008; 41:294–299. [DOI] [PubMed] [Google Scholar]
- 28. Zou J.N., Wang S.Z., Yang J.S., Luo X.G., Xie J.H., Xi T.. Knockdown of SMYD3 by RNA interference down-regulates c-Met expression and inhibits cells migration and invasion induced by HGF. Cancer Lett. 2009; 280:78–85. [DOI] [PubMed] [Google Scholar]
- 29. Zeng B., Li Z., Chen R., Guo N., Zhou J., Zhou Q., Lin Q., Cheng D., Liao Q., Zheng L. et al. . Epigenetic regulation of miR-124 by hepatitis C virus core protein promotes migration and invasion of intrahepatic cholangiocarcinoma cells by targeting SMYD3. FEBS Lett. 2012; 586:3271–3278. [DOI] [PubMed] [Google Scholar]
- 30. Proserpio V., Fittipaldi R., Ryall J.G., Sartorelli V., Caretti G.. The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 2013; 27:1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caretti G., Di Padova M., Micales B., Lyons G.E., Sartorelli V.. The polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004; 18:2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gagnon K.T., Li L., Chu Y., Janowski B.A., Corey D.R.. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014; 6:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Segatto M., Fittipaldi R., Pin F., Sartori R., Dae Ko K., Zare H., Fenizia C., Zanchettin G., Pierobon E.S., Hatakeyama S. et al. . Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat. Commun. 2017; 8:1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. et al. . Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. et al. . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pereira B., Chin S.F., Rueda O.M., Vollan H.K., Provenzano E., Bardwell H.A., Pugh M., Jones L., Russell R., Sammut S.J. et al. . The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016; 7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y. et al. . The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van de Vijver M.J., He Y.D., van’t Veer L.J., Dai H., Hart A.A., Voskuil D.W., Schreiber G.J., Peterse J.L., Roberts C., Marton M.J. et al. . A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002; 347:1999–2009. [DOI] [PubMed] [Google Scholar]
- 39. Vincent T., Neve E.P., Johnson J.R., Kukalev A., Rojo F., Albanell J., Pietras K., Virtanen I., Philipson L., Leopold P.L. et al. . A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial–mesenchymal transition. Nat. Cell Biol. 2009; 11:943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshioka Y., Suzuki T., Matsuo Y., Nakakido M., Tsurita G., Simone C., Watanabe T., Dohmae N., Nakamura Y., Hamamoto R.. SMYD3-mediated lysine methylation in the PH domain is critical for activation of AKT1. Oncotarget. 2016; 7:75023–75037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peserico A., Germani A., Sanese P., Barbosa A.J., di Virgilio V., Fittipaldi R., Fabini E., Bertucci C., Varchi G., Moyer M.P. et al. . A SMYD3 Small-Molecule inhibitor impairing cancer cell growth. J. Cell. Physiol. 2015; 230:2447–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van 't Veer L.J., Dai H., van de Vijver M.J., He Y.D., Hart A.A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveen A.T. et al. . Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415:530–536. [DOI] [PubMed] [Google Scholar]
- 43. Györffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z.. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010; 123:725–731. [DOI] [PubMed] [Google Scholar]
- 44. Mullen A.C., Orlando D.A., Newman J.J., Lovén J., Kumar R.M., Bilodeau S., Reddy J., Guenther M.G., DeKoter R.P., Young R.A.. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011; 147:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Massagué J., Blain S.W., Lo R.S.. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000; 103:295–309. [DOI] [PubMed] [Google Scholar]
- 46. Brown M.A., Foreman K., Harriss J., Das C., Zhu L., Edwards M., Shaaban S., Tucker H.. C-terminal domain of SMYD3 serves as a unique HSP90-regulated motif in oncogenesis. Oncotarget. 2015; 6:4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ramadoss S., Chen X., Wang C.Y.. Histone demethylase KDM6B promotes epithelial–mesenchymal transition. J. Biol. Chem. 2012; 287:44508–44517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li D., Sun H., Sun W.J., Bao H.B., Si S.H., Fan J.L., Lin P., Cui R.J., Pan Y.J., Wen S.M. et al. . Role of RbBP5 and H3K4me3 in the vicinity of Snail transcription start site during epithelial–mesenchymal transition in prostate cancer cell. Oncotarget. 2016; 7:65553–65567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan H., Reddy M.A., Sun G., Lanting L., Wang M., Kato M., Natarajan R.. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am. J. Physiol. Renal. Physiol. 2013; 304:F601–F613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mishra V.K., Subramaniam M., Kari V., Pitel K.S., Baumgart S.J., Naylor R.M., Nagarajan S., Wegwitz F., Ellenrieder V., Hawse J.R. et al. . Krüppel-like transcription factor KLF10 suppresses TGFβ-Induced Epithelial-to-Mesenchymal transition via a negative feedback mechanism. Cancer Res. 2017; 77:2387–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F.P., Li M., Yagyu R., Nakamura Y.. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004; 6:731–740. [DOI] [PubMed] [Google Scholar]
- 52. Yoshioka Y., Suzuki T., Matsuo Y., Tsurita G., Watanabe T., Dohmae N., Nakamura Y., Hamamoto R.. Protein lysine methyltransferase SMYD3 is involved in tumorigenesis through regulation of HER2 homodimerization. Cancer Med. 2017; 6:1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tran H.D., Luitel K., Kim M., Zhang K., Longmore G.D., Tran D.D.. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014; 74:6330–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Giampieri S., Manning C., Hooper S., Jones L., Hill C.S., Sahai E.. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009; 11:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bruna A., Greenwood W., Le Quesne J., Teschendorff A., Miranda-Saavedra D., Rueda O.M., Sandoval J.L., Vidakovic A.T., Saadi A., Pharoah P. et al. . TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat. Commun. 2012; 3:1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.