

Abstract
A polymeric dual delivery nanoscale device (DDND) was designed for combined delivery of micro RNA (miR-345) and gemcitabine (GEM) to treat pancreatic cancer (PC). This temperature and pH-responsive pentablock copolymer system was able to restore miR-345, making xenograft tumors more susceptible to GEM, the standard therapy for PC. Restoration using DDND treatment results in sonic hedgehog signaling down regulation, which decreases desmoplasia, thereby resulting in improved GEM perfusion to the tumor and better therapeutic outcomes. The release of miR-345 and GEM could be tuned by using the DDND in the form of micelles or in the form of thermoreversible gels, based on polymer concentration. The DDNDs enabled miR-345 stability and sustained co-release of miR-345 and GEM, thereby facilitating dose-sparing use of GEM. Further, enhanced in vitro cellular uptake due to amphiphilic character, and endosomal escape because of the cationic end blocks led to efficient transfection with DDNDs. The combined DDND treatment enabled efficient reduction in cell viability of Capan-1 and CD18/HPAF cells in vitro compared with either GEM or miR-345 treatment alone. Mice carrying xenograft tumors treated with DDNDs carrying both miR-345 and GEM combination therapy displayed reduced tumor growth and less metastasis in distant organs compared to individual drug treatments. Immuno-histochemical analysis of the xenograft tissues revealed significant down regulation of desmoplastic reaction, SHH, Gli-1, MUC4, and Ki67 compared to control groups.
Keywords: miR-345, gemcitabine, nanoscale delivery, pancreatic cancer
1. Introduction
Dismal prognosis of pancreatic cancer (PC) patients [1] is due to late clinical presentation[2], early and aggressive local invasion, and resistance to conventional chemo-radiation therapies[3]. Additionally, PC is characterized by extensive desmoplasia, which is primarily driven by sonic hedgehog (SHH) signaling that plays an important role in tumor growth, metastasis[4], angiogenesis[5], and limits the delivery and efficacy of chemotherapy. SHH regulates the epithelial to mesenchymal transition (EMT), is involved in regulation of cancer stem cells, and contributes to drug resistance resulting in PC progression [6]. The current standard therapy for PC involves surgical resection followed by systemic gemcitabine (GEM) therapy, and provides symptomatic improvement in a small proportion of patients [7]. Moreover, GEM therapy also results in high toxicity, frequent resistance, and has not provided a significant improvement in the overall survival of PC patients with advanced disease [8, 9] [10]. Further, GEM treatment can result in up-regulation of SHH, known cancer stem cell (CSCs) markers (CD24, CD44, CD133, OCT4) and other genes (β-catenin, SNAIL, SLUG, TWIST, E-cadherin, and vimentin [11]) that facilitate GEM therapy resistance [12–14],[15–22]. Therefore, there is an urgent need to target SHH to improve GEM therapy.
Recently, the use of microRNAs (miRNAs) to target major cancer pathways is a potential therapeutic option for cancer treatment [16, 20, 21, 23] [24, 25] [26]. miRNAs provide several advantages over traditional therapeutics such as upstream target gene silencing as they direct sequence-specific degradation of target messenger RNAs (mRNA), acting catalytically prior to translation, reducing excess drug consumption and drug-associated side effects, and reducing costs [16, 20, 21]. Despite these practical benefits, the potential clinical application of miRNA therapy is currently limited by challenges related to delivery, stability, transport, diffusion, cellular entry, efficient release, endosomal escape, and activity in the cytoplasm [16, 17, 19–22, 27–34]. We showed that miRNA miR-345 is significantly decreased in PC tissues and PC cells [35]. Reintroducing this miRNA into PC cell lines resulted in reduced growth, motility, and invasion; with up-regulation of epithelial markers, and down-regulation of mesenchymal markers [35]. Further, the ectopic expression of miR-345 in CD18/HPAF-II and Capan-1 cells resulted in down-regulation of SHH, cMYC, XIAP; and up-regulation of cleaved caspase 3, cleaved caspase 7, PTCH 2, and PARP (unpublished data). Therefore, the development of a multifunctional miRNA (miR-345) and drug (gemcitabine) dual-delivery nanoscale device (DDND), could facilitate cellular entry and endosomal escape, while maintaining miRNA stability and provide sustained release and of both drugs as a new treatment for PC. However, targeted co-delivery of miR-345 and GEM using a single nanodelivery device is challenging due to varying physicochemical properties, drug release rates, and pharmacokinetics [22, 24, 28, 36–41].
There have been reports that seek to co-deliver miRNA/drug combinations using different polymers to address the limitations of existing systems such as co-loading of drug and miRNA, non-specific toxicity, and sustained miRNA/drug release [19, 22, 24, 28, 29, 36, 37, 42–50]. The most significant issues with these systems are the ineffective co-incorporation of miRNA/drug combinations, stability, and/or premature release of the miRNA/drug load. Our study involved the development of a temperature and pH responsive, cationic, amphiphilic, and bio-compatible PB co-polymer based DDND for in vitro and in vivo combined delivery of miR-345 and GEM for the treatment of PC. Our approach builds upon the promising results of our previous in vitro and in vivo studies [51–57], using the temperature responsive polymer Pluronic F127 (poly(ethyleneoxide)-block-poly(propyleneoxide)-block poly(ethyleneoxide) (PEO-PPO-PEO)) as middle blocks and pH responsive cationic (poly(2-diethylaminoethyl methacrylate)) (PDEAEM) end blocks. This novel multifunctional DDND design consists of amphiphilic Pluronic F127 blocks to enhance cellular uptake, while the protonatable tertiary amine groups of PDEAEM end blocks facilitate endosomal escape and efficient miR-345 release in the cytoplasm, providing dose-sparing effect [51, 57–61]. The miR-345/PB copolymer nanocomplex system is further self-assembled with GEM-encapsulated Pluronic F127 to provide shielding of excess charges, enhance cellular uptake, GEM loading and simultaneous miR-345/GEM release by temperature responsive micellization. At increased polymer concentrations, the DDND delivery system also undergoes physical thermoreversible gelation at physiological temperatures, allowing for a depot to be injected that dissolves slowly over time, providing controlled release of the miR-345 and GEM [40]. Using the DDND system, we show that restoration of miR-345 through the DDND treatment results in down-regulation of the SHH signaling pathway and leads to inhibition of desmoplasia, pancreatic stellate cells (PSCs) and cancer stem cells (CSCs), thus facilitating improved therapeutic outcome of GEM treatment in PC through its improved perfusion to the tumor. The results presented in this study help to enhance an understanding of the synergistic activity of a combined GEM and miR-345 mimic delivery and generate valuable information that significantly advances the goal of combining GEM and miR-345 mimic delivery for treatment of PC patients in the future.
2. Materials and Methods
2.1. Materials
Polymer synthesis materials were obtained from Sigma-Aldrich and Fisher Scientific and prepared as mentioned in our previous work [54]. Poly(ethylene glycol) diacrylate (PEG-DA) MW:4000 was purchased from Polysciences Inc. Photoinitiator 4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-propyl)ketone (Irgacure 2959) was obtained from BASF. CellTiter 96® Aqueous One Cell Proliferation Kit and Luciferase Assay System were purchased from Promega. Cell culture reagents: Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), 0.25% trypsin-EDTA and penicillin streptomycin (Pen-Strep) were purchased from Invitrogen. Lipofectamine and all Alexa Fluor 488 were obtained from Invitrogen. The pGWIZ-luc plasmid encoding the luciferase reporter gene was purified with the Qiagen HiSpeed® Plasmid Maxi Kit. Quant-iT™ RiboGreen® RNA Reagent was purchased from Invitrogen. Hsa-miR-345-5p mimic miRNA sequence GCUGACUCCUAGUCCAGGGCUC was obtained from Ambion.
2.2. Methods
2.2.1. Preparation of DDND
The pentablock (PB) copolymers were synthesized by atom transfer radical polymerization (ATRP) and characterized as described in our previous work [54]. miR-345/PB copolymer polyplexes at various N/P ratios (N/P: molar ratios of nitrogen (N) in the pentablock copolymer to phosphate (P) in miRNA) were prepared by adding appropriate quantities of PB copolymer (8 mg/mL stock) in 0.5 M HEPES buffer at pH 7.0 to miR-345 solution (10 pmol) as reported previously [59], Gemcitabine (GEM) encapsulated PluronicF127 was prepared as described [57], The GEM encapsulation efficiency was expressed as the ratio of the initial drug amount to the amount of encapsulated drug, which was found to be about 70%. For DDND formulations, GEM-encapsulated Pluronic F127 (10 mg/mL in 0.5 M HEPES Buffer at pH 7.0) was added to the miR-345/PB copolymer polyplexes at the same volume, to result in a weight ratio of 5:1 with regard to the corresponding PB to provide co-loading. GEM/Pluronic F127 and miR-345/PB copolymer polyplexes were then incubated together for 30 min at room temperature for nanoscale self-assembly to form the DDND micelles through hydrophobic interaction. Using the method, ~100% self-assembly was observed without loss of GEM. For the in vitro and in vivo tests, the DDND micelle formulations were used throughout the study. However, for the release studies, DDND gels were prepared to demonstrate the controlled release properties of these thermoreversibly gelling systems. The DDNDs can be injected as liquid and can form a solid gel at physiological temperatures at certain concentrations, and act like a drug depot providing controlled release [57]. In order to prepare DDND gels, the polyplex solution was further assembled with appropriate amount of Pluronic F127 at 4 °C to make the final Pluronic F127 concentration ~20 wt.%.
2.2.2. Characterization of DDND
The electrophoretic mobility of polyplex formations was characterized on a 1% (w/v) agarose gel containing 0.5 μg/mL ethidium bromide for 30 min at 100 V in 1×Tris/borate/EDTA (TBE) buffer. The samples were loaded in wells and polyplex electrophoretic mobility was visualized and image capture using a UV-trans illuminator. The serum and RNase stability of designed DDND systems at different N/P ratios was also tested using agarose gel electrophoresis after 72 h incubation at 37 °C in culture media containing 10% serum and 0.25% RNase. The size and zeta potential values and long-term stability in serum-containing cell culture media of the designed DDNDs were determined using dynamic light scattering (DLS).
2.2.3. Release Experiments
GEM and miR-345 release from DDNDs (in the form of micelles as well as gels) was evaluated as described in our previous study [57], The DDNDs were injected into the concave surface of molded polyethylene glycol-diacrylate (PEG-Da) gels mimicking extracellular matrix and were placed in 6-well plate transwell inserts filled with PBS at pH 7.4 as release media. The plate was then placed on an incubator shaker (100 rpm at 37°C) and samples were collected at predetermined times. The concentration of miR-345 was determined using the Quant-iT™ Ribo Green RNA reagent by following the manufacturer’s protocol. The amount of released GEM was measured using high performance liquid chromatography (HPLC) as described elsewhere [62].
2.2.4. Cell Culture
Pancreatic cancer cell lines Capan-1 and CD18/HPAF-II, were grown in DMEM supplemented with 10% (v/v) FBS and 1% Pen-Strep at 37 °C in a 5% CO2 humidifed atmosphere. Cells were subcultured approximately every 2–3 days.
2.2.5. Cell Viability Assay
To evaluate the in vitro efficiency of the DDNDs to kill Capan-1 and CD18/HPAF-II pancreatic cells, a cell viability was assessed. Briefly, Capan-1 and CD18/HPAF-II cells were plated at a density of 2×104 cells per well in 96-well plates and were incubated for 24 h at 37 °C in a 5% CO2 humidified atmosphere. The described N/P ratio DDNDs, (10 μL of DDND solution having 10 pmol miR-345 and 3μg GEM) along with the negative and positive controls, were added to the wells at vaious doses. After 24 h of incubation, cell viability was tested using the CellTiter 96® Aqueous One Proliferation Kit following manufacturer’s protocol.
2.2.6. Cellular Uptake, Intracellular Distribution and Endosomal Escape of DDNDs
The cellular uptake, intracellular distribution and endosomal escape capabilities of the DDNDs in Capan-1 and CD18/HPAF-II cells were qualitatively evaluated by confocal microscopy. For this purpose, the DDNDs were prepared using the Alexa Fluor 488 dye-attached PB copolymers, which were prepared following the same procedure mentioned previously [59], Capan-1 and CD18/HPAF-II cells were plated at a density of 2 × 105 cells per well in cell culture petri dishes and were incubated 24 h at 37 °C in a 5% CO2 humidified atmosphere. Then, the dye labeled DDNDs were added to the wells and incubated with the cells for 24 h under same conditions. For live cell imaging, the lysosomes and nucleus were stained with Lysotracker Red and Hoechst respectively, as described [59].
2.2.7. Transfection Efficiency of DDNDs
The transfection efficiency of the developed DDNDs was tested as described in our previous work [57], Capan-1 and CD18/HPAF-II cells were seeded at a cell density of 1×104 cells per well in 96-well plates and were incubated overnight. Transfections were carried out by adding DDNDs prepared at different N/P ratios to contain: 0.6 μg of luciferase expressing pGWIZ-luc plasmid, 10 pmol miR-345, and 3 μg GEM per well. Cells were allowed to incubate with the polyplexes for 6 h, at which time the media was changed to remove any excess polyplexes. The cells were incubated for an additional 42 h before they were lysed and analyzed for luciferase activity according to the Luciferase Assay System protocol with a Veritas Microplate Luminometer. Lipofectamine was used as positive control at an N/P ratio of 8, according to the manufacturer’s protocol.
2.2.8. In vivo Studies
In the present study, animal experiments were carried out in accordance with the United States Public Health Service “Guidelines for the Care and Use of Laboratory Animals” under an approved protocol by the University of Nebraska Medical Center’s Institutional Animal Care and Use Committee (IACUC). Six to eight-week-old athymic nude mice (n=12 for each group) of either sex were obtained from ENVIGO and maintained in pathogen-free conditions for two weeks for acclimatization. CD18/HPAF-II PC cells labeled with luciferase (0.5×106), were suspended in 50 μM phosphate-buffered saline (PBS) and were orthotopically implanted into the mice pancreata to establish xenograft tumors. Ten days post-implantation, mice were imaged with the IVIS spectrum (Perkin Elmer, USA) machine following luciferase intraperitoneal (i/p) injection of D-luciferin (100 μM of 15mg/ml in PBS). Based on luciferase expression, mice were randomized into four groups (n=12 animals/group) for treatment, Group 1: DDND vehicle control; Group 2: miR-345 alone; Group 3: DDND with GEM alone; and Group 4: DDNDs with GEM + miR-345. The treatment schedules and doses were set up as follows; DDNDs were administered i/p twice a week for 4 weeks by 100 μL DDND solution having 10 pmol miR-345 and 3μg GEM. During the course of treatment, D-luciferin was i/p injected to detect/monitor tumor growth in mice non-invasively using the IVIS Spectrum imaging system, at days 0, 1,8, 15 and 22 (study endpoint) of treatment. The animals were sacrificed by asphyxiation with CO2 followed by cervical dislocation at the study endpoint. Tumors were removed and weighed. Vital organs were carefully observed for metastatic lesions prior to collection. Half of each primary tumor or metastatic tumor was flash frozen and the other half was fixed in 10% buffered formalin for immune-histochemistry and protein analysis.
2.2.9. Immuno-histochemicai Analysis
The histological changes associated with DDND treatments were assessed with respective control group. Tumor specimens were fixed in 10% neutral buffered formalin solution for 48 hours prior to embedding in paraffin. Pancreatic and metastatic tumor tissues were sectioned at 5μm thickness and stained with hematoxylin and eosin. Immuno-histochemical analysis was performed on primary tumor sections from each treatment group for SHH, GLI1, Trichrome, MUC4, and KI67 antibodies with established protocols [37, 63].
2.2.10. Statistical Analysis
The significant differences between the groups for the in vitro experiments were evaluated using ANOVA analysis by Tukey’s method with a 95% confidence interval. The results are presented as the average ± standard deviation calculated from at least three independent experiments. The Student t-test was used to determine statistical differences between control and treatment groups for in vivo experiments. A p-value less than 0.05 was considered to be statistically significant. Error bars represent the calculated standard error of means (SEM).
3. Results
The molecular weight of the synthesized PB copolymer used to form polyplexes for miR-345 and GEM delivery, was estimated to be 18 kDa with a polydispersity index of 1.2, which was similar to our previous reports [54, 57, 59], Gel electrophoresis indicated that complete complex formation occurred in DDND systems with N/P ratios above 20, which provided the miR-345 both protection and stability, while preventing the premature release (Figure 1).
Figure 1.
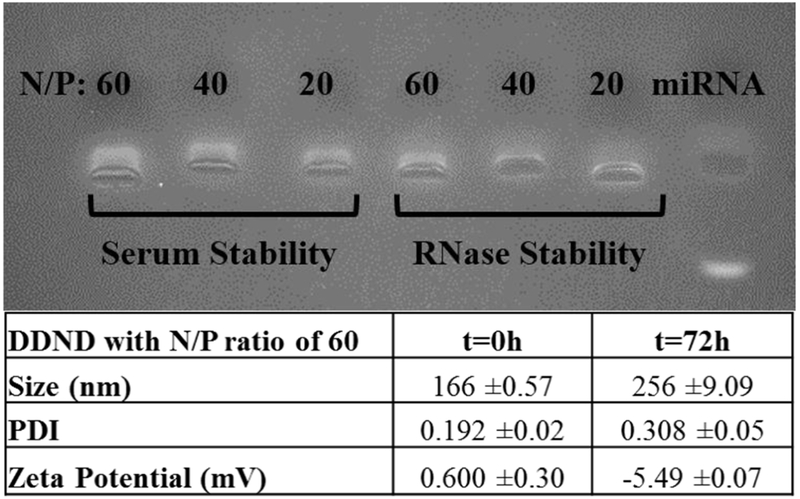
Stability of DDND systems at different N/P ratios in media containing 10% serum and 0.25% RNase enzyme. Incubation time: 72h. Size and zeta potential stability of the DDND system with N/P ratio of 60 in 10% serum containing cell culture media.
The serum and RNase stability of the miR-345 in the DDND systems was also tested for 72h (Figure 1) and our results indicated that DDNDs with N/P ratios of 20, 40 and 60 showed good miR-345 protection and stability against serum and RNase enzymes by not showing any significant signs of miR-345 degradation, disassembly or dissociation in the presence of RNase or serum proteins due to the strong electrostatic complexation and self-assembly. However, a slight decrease in miR-345 intensity on the agarose gel was observed in the DDNDs with N/P ratios 20 and 40, indicating a slight miR-345 degradation in the presence of serum proteins and RNase, which was not observed in the DDNDs with a N/P ratio of 60. Considering effective miR-345 protection, size and zeta potential stability was evaluated particularly for DDNDs with an N/P ratio of 60 (Figure 1). The results indicated that DDND with an N/P ratio of 60 possesses almost neutral charge due to the Pluronic F127 surface self-assembly and average cumulative size of 166 nm in PBS. Following the 72h incubation in serum-containing cell culture media, the size of the 60 N/P DDND increased to 257 nm, with a low polydispersity index value; and the zeta potential changed to −5.49 mV, which could be due to the attachment of cell culture media components (Figure 1) [59]. The enhanced stability of the DDNDs is attributed to the hydrophilic PEO chains of Pluronic F127, which play a vital role in providing shielding and stealth effects, and the strong electrostatic condensation of miR-345 in the polyplex matrix, which minimizes its external interactions [52, 57, 59]. In addition, the responsive PB copolymer-based systems also show enhanced stability compared to liposomal systems due to the ability to tailor the ratio of the blocks with different features [64, 65]. Although the strong electrostatic complexation and self-assembly enables efficient protection and stability, it may prevent the efficient release of the payload at the site of action [66]. Our results indicated that the DDND systems (both as micelles and gels) were able to provide simultaneous release of miR-345 and GEM over 5 days, providing efficient stability, protection, and preventing premature release (Figure 2).
Figure 2.
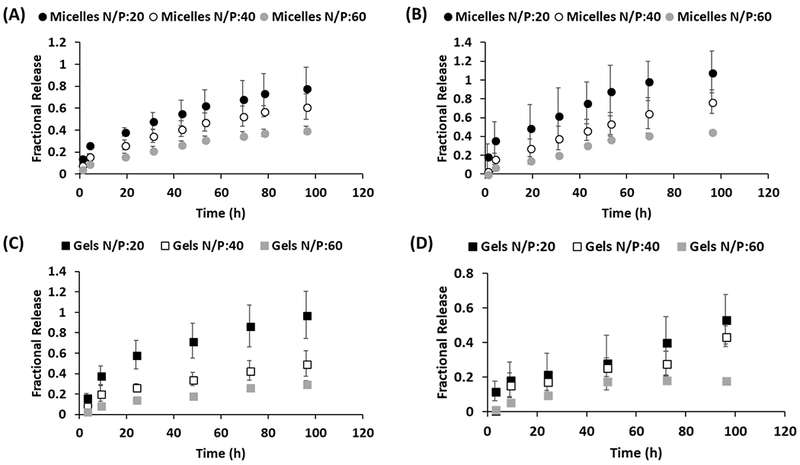
Release profiles of (A) Gemcitabine and (B) miR-345 from DDND micelles with N/P ratios of 20, 40 and 60. Release profiles of (C) Gemcitabine and (D) miR-345 from DDND gels with N/P ratios of 20, 40 and 60.
It was also observed that the increase in N/P ratio from 20 to 60 resulted in slower release of miR-345 and GEM for both micelles and gels. ~90% of the initially loaded miR-345 and GEM were released within 5 days from the micelles with N/P ratio of 20. The PB copolymer system is able to complex the miR-345 because of its cationic blocks, and can simultaneously undergo thermoreversible gelation at physiological temperatures (at polymer concentrations greater than 20%) to form gels that provide sustained release of the GEM and miR-345 slowly over time [57]. The release of GEM from the outer Pluronic F127 layer was observed to be less controlled due to it is small molecular size and high hydrophilicity. The gels provided more controlled release with slower rates compared to the micelles. The gels with N/P ratio of 60 released ~20% of initially loaded miR-345 and GEM within 5 days, which can be considered a favorable release timeframe for positive clinical outcomes [67]. Therefore, it is possible to tune the controlled release characteristics of the DDNDs by changing N/P ratio or creating micelles/gels.
Capan-1 and CD18/HPAF-II cells were exposed to DDNDs with N/P ratios of 60, 40 and 20 (Figure 3) to determine PC cell line viability in vitro. The DDNDs with N/P ratios of 60 (miR-345 and GEM co-loaded) showed the greatest effect with ~85% and 80% reduction in cell viability for Capan-1 and CD18/HPAF-II cells respectively (Figure 3A and 3B).
Figure 3.
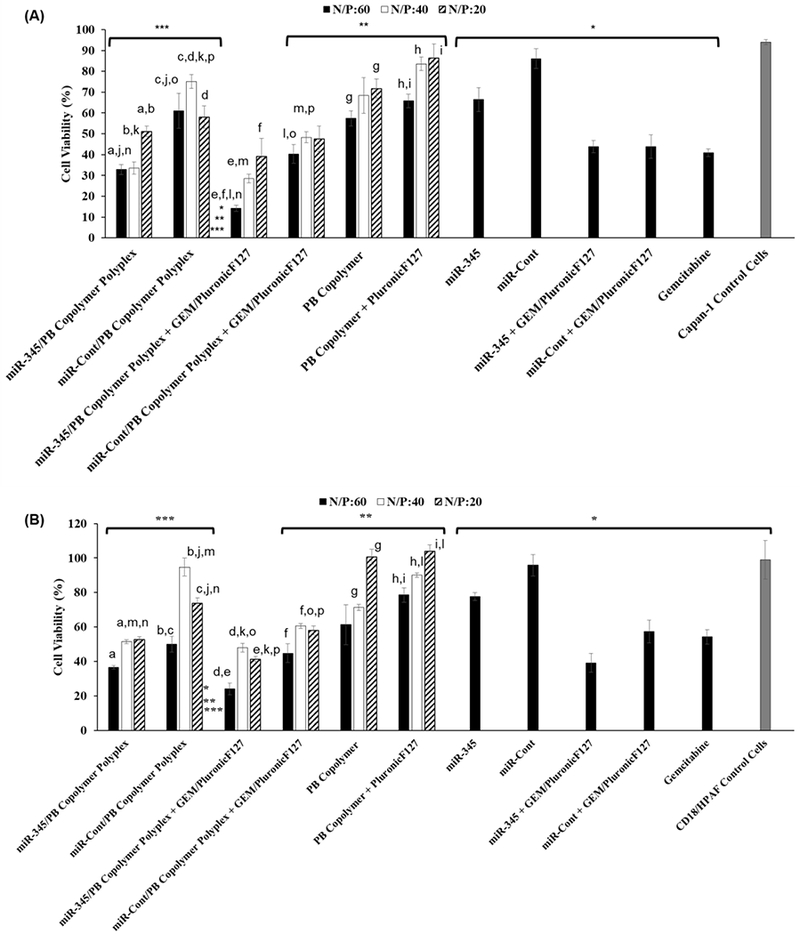
Effect of DDND systems with N/P ratios of 60, 40 and 20 on (A) Capan-1 and (B) CD18/HPAF PC cell viability. Cell Density: 1×104 cell/well. miRNA Dose: 10 pmol. Gemcitabine dose: 3 μg/well. Incubation time: 72h. The same letters represent statistically significant differences between cases (p<0.05).*, ** and *** represent the statistically significant differences of the DDND system with N/P ratios of 60 against all controls (p<0.05).
The corresponding control treatments, consisted of the same amount of PB copolymer self-assembled with Pluronic F127 alone (without miR-345/GEM/both) to form the DDNDs at the different N/P ratios, and did not show significant toxicity in both cell lines. It was observed that PluronicF127 self-assembly provided a shielding effect, stability and reduction in toxicity commonly seen with synthetic cationic polymers [57]. Many cationic polymers have cytotoxicity issues, but our PB copolymer system can be tailored to optimize complexation and transfection efficiency while minimizing cytotoxicity, by simply changing the ratio of the cationic blocks to the non-ionic Pluronic F217 blocks [57, 59]. Moreover, the scrambled miRNA control, either alone or loaded in DDNDs, showed no effect on both cell lines (Figure 3). GEM alone or entrapped in Pluronic F127 showed significant cell death (~60% for Capan-1 and 45% for CD18/HPAF-II), which was higher than that observed with the miR-345 alone (~30% for Capan-1 and 10% for CD18/HPAF-II). The miR-345, directly mixed with GEM/PluronicF127 assembly without PB copolymer complexation, also did not provide enhanced effect on cell viability compared to the GEM/PluronicF127 assembly alone in both cell lines (Figure 3). On the other hand, when miR-345 was complexed with PB copolymer and further self-assembled with GEM/PluronicF127 to form miR-345/PB Copolymer Polyplex + GEM/PluronicF127 (Figure 3), it showed effective destruction of PC cell lines upon its treatment through the miR-345 and GEM synergism (~85% reduction in cell viability for Capan-1 and 80% reduction in cell viability for CD18/HPAF-II) compared with control groups. These results demonstrate that an efficient miR-345 restoration in combination with GEM therapy provided highly significant inhibition of growth in PC cell lines through induction of apoptosis [35, 36, 68]. In most treatments, the DDND with N/P ratio of 60 showed a more pronounced effect on cell viability as compared to the DDNDs with N/P ratios of 40 and 20 in both cell lines. This could be attributed to the stability, protection, and efficient simultaneous release of miR-345 and GEM resulting from the synergistic advantages of different blocks for this DDND [57, 59].
Based on the observed stability, simultaneous extended release, and cell viability results, the DDND system with a N/P ratio of 60 was selected for the remaining in vitro and in vivo experiments. The designed DDND system with N/P ratio of 60 provided efficient cellular entry and endosomal escape in Capan-1 and CD18/HPAF-II cells (Figure 4).
Figure 4.
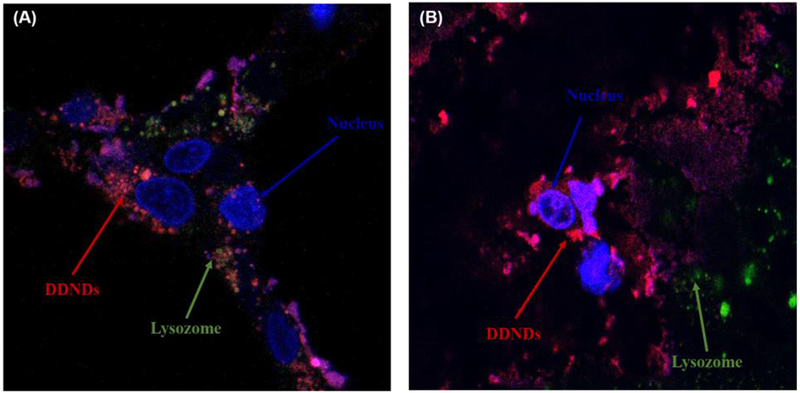
Confocal images of DDND systems prepared at N/P ratio of 60 using dye attached PB copolymer in (A) Capan-1 and (B) CD18/HPAF cells. Incubation time: 24 h. Red: DDND stained by Alexaflour647, Green: lysosome stained by Lysotracker Green, Blue: nucleus stained by Hoechst dye.
The enhanced cellular uptake was due to the presence hydrophobic PPO groups, which are capable of promoting cell membrane internalization through thermoreversible micellization and hydrophobic interactions [51, 52, 57–59, 69]. On the other hand, the PDEAEM end-blocks enhanced the endosomal escape through protonated tertiary amine groups by pH buffering (Figure 4) [57, 59, 61]. Quantitative analysis indicated that approximately ~82% of DDNDs managed to escape from the endosome through proton sponge effect in Capan-1 cells while ~85% of the DDNDs escaped from the endosome in CD18/HPAF-II cells.
Following efficient cellular entry and endosomal escape, the DDNDs were able to provide efficient transfection as depicted in Figure 5.
Figure 5.
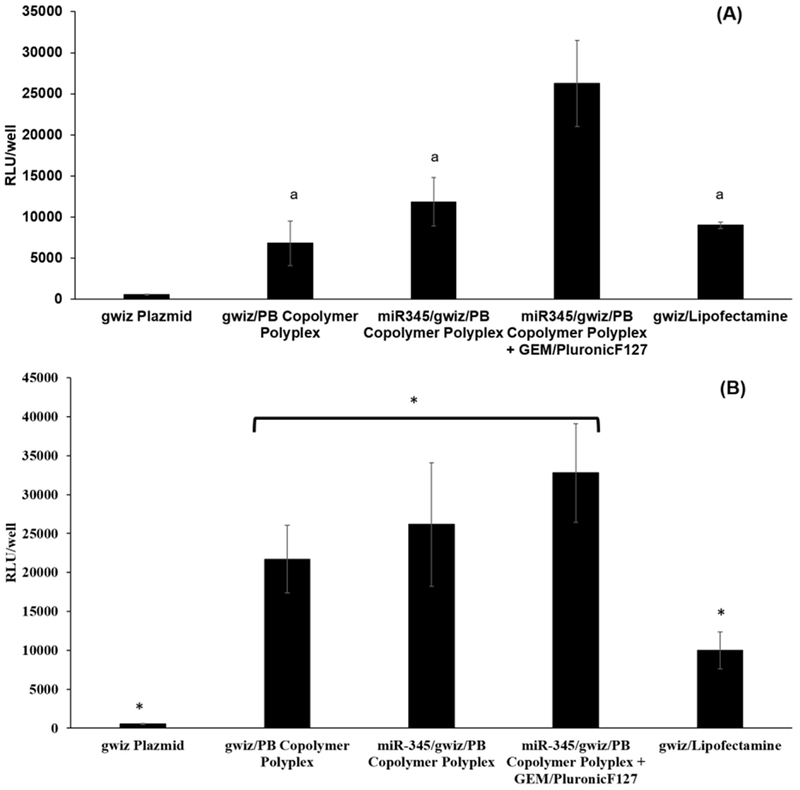
Transfection of DDNDs prepared at N/P ratio of 60 in (A) Capan-1 and (B) CD18/HPAF cells. Luciferase expressing gwiz plasmid complexed with Lipofectamine was used as positive control. Luciferase expressing gwiz plasmid alone or complexed with PB copolymers in the presence or absence of GEM/Pluronic F127 self-assembly were used as controls. * represents statistically significant differences (p<0.05) while same letters represent statistically insignificant difference (p>0.05).
The DDND system with an N/P ratio of 60 provided better transfection efficiency compared to the control cases (gwiz alone, gwiz/PB copolymer complex and miR-345/gwiz/PB copolymer complex) for Capan-1 cells (Figure 5A), whereas the difference was insignificant for CD18/HPAF-II cells except against gwiz alone control (Figure 5B). DDNDs also showed significantly higher transfection (~2.5 fold higher in Capan-1 (Figure 5A) and ~3 fold higher in CD18/HPAF-II (Figure 5B)) than a commercially available agent, Lipofectamine 2000 (Figure 5). The DDND system containing a self-assembled Pluronic F127 layer provided further enhanced transfection compared to the miR-345/PB copolymer polyplexes, as mentioned in our previous work on delivery of plasmid DNA [57]. Besides, the stable structure and the novel design of the DDND also has a positive effect on cellular uptake, endosomal escape and transfection efficiency.
Based on our optimized in vitro results, we sought to evaluate the in vivo therapeutic potential of the DDND system with N/P ratio of 60 on tumorigenicity and metastasis. Athymic nude mice carrying CD-18/HPAF-II xenograft tumors were treated with DDNDs loaded with both GEM and miR-345 alone or in combination, administered i/p twice a week for 4 weeks. Non-invasive live animal IVIS spectrum imaging revealed significant tumor growth inhibition in the GEM+miR-345 mice compared to other treatment groups (Figure 6A). The average tumor weight was substantially decreased due to the synergistic effect of GEM and miR-345 with respect to the GEM and miR-345 alone control groups (Figure 6B). These large differences in the tumor weight (1.2g vs. ~0.3g) reflect the fact that more aggressive tumor growth occurs in pancreas of the control mice as compared to other treatment groups; further, similar to our in vitro results, we also observed synergism when GEM and miR-345 were combined for in vivo treatment.
Figure 6.
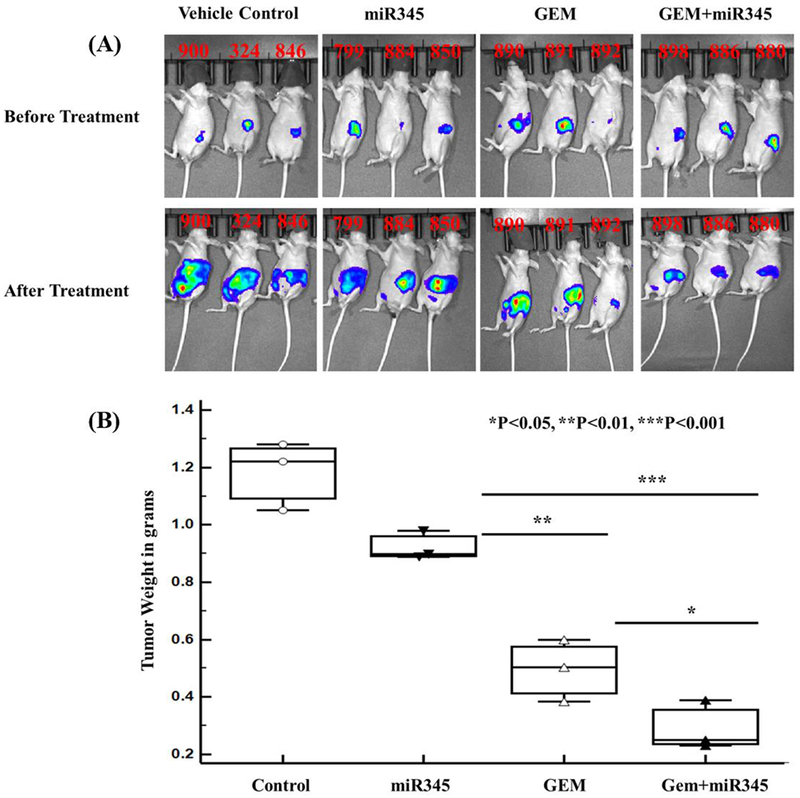
Therapeutic potential of nanoparticle carrying GEM + miR-345 mimics on orthotopically grown PC tumors in athymic nude mice. (A) Luciferase live animal IVIS imaging to monitor growth of tumors during the treatment period (B) Tumor weights at study endpoints.
We investigated the effect of miR-345 and GEM-loaded DDNDs on histopathology and local, distant metastatic lesions in vivo after treating athymic nude mice carrying xenograft tumors. Metastatic lesions at distant organs were significantly reduced in animals treated with DDNDs loaded with miR-345 and GEM as compared to all other groups (Figure 7B). We observed a significantly higher incidence of metastasis in distant organs like liver, spleen, kidney, ovary, diaphragm, and intestine in the control group (Figure 7C, example control group metastases) compared to the group with DDNDs loaded with GEM + miR-345 (Figure 7B). Histopathological changes were evaluated in the primary pancreatic tumor tissues and metastatic lesions isolated from distant organs from all treatment groups. H&E staining of excised mouse primary pancreatic tissue sections (Figure 7A) showed that significant reduction in desmoplasia in primary tumors of animals treated with DDND loaded with GEM + miR-345 compared with other miR-345 and control groups. Tumor metastasis to distant organs was clearly inhibited in mice treated with DDNDs loaded GEM + miR-345. Further, H&E staining also revealed that presence of micrometastatic lesions in the distant organs of mice treated with vehicle control (polymer treated mice) compared with mice treated with DDND loaded with miR-345/GEM alone/in combination.
Figure 7.
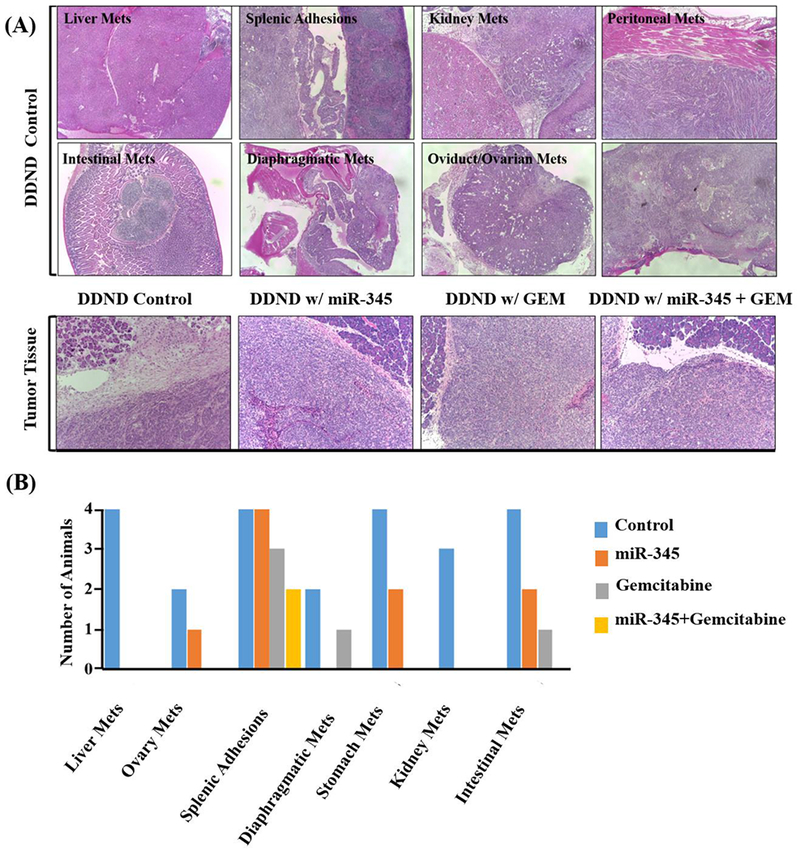
Effect of DDNDs loaded with miR-345 and GEM on tumorigenicity and metastasis (A) Changes in distant organs with metastatic lesions and pancreatic tumor tissue histology as a result of DDND mediated delivery of miR-345 and GEM. The first and second rows are the light microscopy images (100×) of H&E stained metastatic lesions from the control group. The third row shows H&E stained primary pancreatic tissue sections for each treatment group. (B) The number of animals showing incidence of metastasis at distant organs in the various treatment groups.
Our hypothesis was that the restoration of miR-345 through DDND treatment would result in down regulation of the SHH signaling pathway and lead to inhibition of desmoplasia, pancreatic stellate cells (PSCs) and cancer stem cells (CSCs). This in turn would lead to an improved therapeutic outcome of GEM in PC through improving its perfusion to tumor. Our immunohistochemical analysis in primary tumors from each treatment group (Figure 8) indicated that there was significant down-regulation of SHH (sonic hedgehog protein) and its downsteam effector GLI1 in the tumor tissues of mice treated with DDNDs loaded with GEM+ miR-345 compared to GEM alone or the untreated DDND control group. As expected, a reduction in SHH and GLI1 was also observed in the miR-345 treated group. Pancreatic ductal adenocarcinoma (PDAC) is characterized by an abundant desmoplastic stroma that promotes tumor formation, invasion, and metastasis. In addition, desmoplastic stroma creates an additional physical barrier that prevents perfusion of chemotherapy and thus leads to increased chemoresistance [70]. Masson trichrome staining of tumor tissues showed high stromal density in control group and mice treated with DDND loaded with miR-345 compared to other treatment groups; with the combined GEM + miR-345 treatment providing a significantly reduced stromal density (Figure 8, third row).
Figure 8.
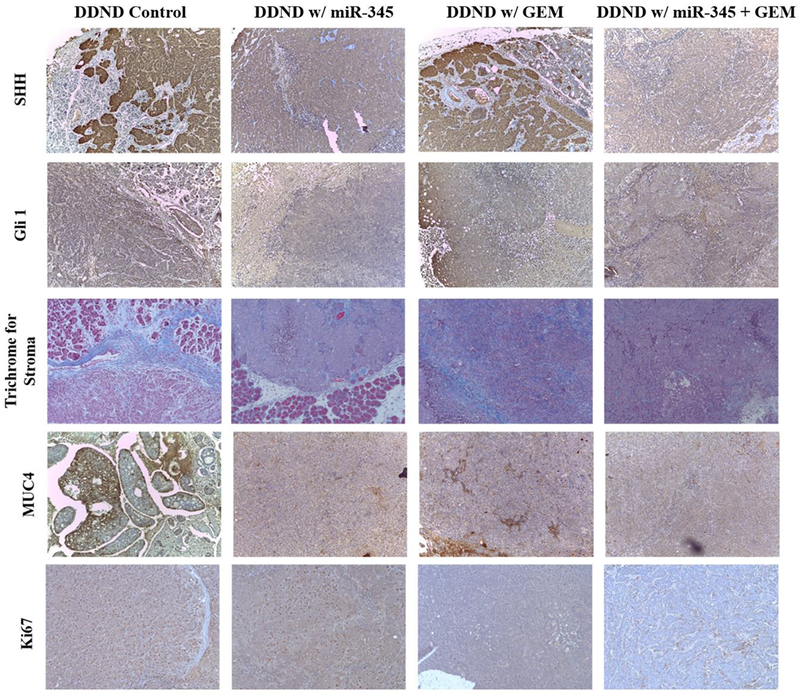
Analysis of tumor samples for SHH, Glil, Trichrome, MUC4, and Ki-67 staining for each treatment groups.
Our in silico target prediction and microarray data from miR-345 over expression in PC cells, revealed that miR-345 potentially targets MUC4 which is highly overexpressed in human PC and has a role in the progression and metastasis of PC cells [63]. MUC4 is undetectable in the normal pancreas, whereas its expression increases progressively with the advancement of PC where it plays an active role in progression and metastasis [63]. Our results demonstrated that restoration of miR-345 by means of DDNDs loaded with miR-345 alone or in combination (GEM and miR-345) treatment resulted in down-regulation of MUC4 as compared to other groups (Figure 8, fourth row). In addition, our immune-histochemical analysis of tumor sections with KI67 (proliferation marker) revealed that the mice treated with DDNDs loaded with miR-345 and GEM display lower KI67 staining as compared to other treatment groups which indicates these are less proliferative tumors (Figure 8, last row).
4. Discussion
The sophisticated DDND design and stimuli responsive features of the PB copolymers had a significant influence on these promising in vitro and in vivo results. Previous studies aimed to co-delivering miRNA/drug combinations using various polymers to address the limitations of existing systems include ineffective co-loading of drug and miRNA, non-specific toxicity, stability, premature miRNA/drug release, and multidrug resistance etc. [19, 22, 24, 28, 29, 36, 37, 42]. For instance, similar approaches using other polymeric delivery systems have shown much lower drug loadings (~5-10%) with short-term stability in serum for only 12-24 h, and sustained release of 80% of loaded drug over 2 days[28],[24]. Despite the limitations of these systems in terms of loading, stability, and premature release, they were able to show a reversal in chemoresistance to gemcitabine, inhibition of migration, invasion and tumor cell proliferation (up to ~20-40% cell in MIA PaCa-2 and Capan-1 PC cells) with an accompanying increased in apoptosis [28],[24] in vitro.
In our study, we were able to obtain ~70% GEM and ~100% miR-345 loading. In the micelle formulations (for N/P ratio of 60), ~40% of the initial miR-345 and GEM total dose released in a sustained manner over 5 days while this was reduced to ~20% for gel formulations providing a slower and more controlled release. The size of our 60 N/P system is 166 nm in cell culture media supplemented with 10% serum and showed extended serum stability over 72 h. A comparison of transfection efficiency showed a ~2.5 and ~3-fold higher cellular uptake as compared to the commercially available Lipofectamine 2000, was also achieved in Capan-1 and CD18/HPAF-II cells respectively. Our design achieved the same or a better reduction in cell viability of Capan-1 and MIA PaCa-2 cells with a lower miR-345 and GEM doses (10 μL of DDND solution having 10 pmol miR-345 and 3μg GEM) as compared to other in vitro studies [28],[24]. Using the CD18/HPAF-II in vivo pancreatic orthotopic tumor model, a decrease in the expression of SHH, GLI1 and KI67 was observed when the combined treatment was administered. These studies further revealed that the combination therapy showed significant inhibition of tumor growth accompanied by significant decrease in metastasis in vivo. Therefore, our in vivo results also revealed that we achieved a significant decrease in the tumor weight, from 1.2 g to 0.1g, as well as significant decrease in overexpressed proteins, through the injection of DDNDs (i/p twice weekly for 4 weeks by 100 μL DDND solution having 10 pmol miR-345 and 3μg GEM). Although the injection regimens are slightly different from the other mentioned studies, the applied miR-345 dose is significantly lower. In addition, compared to the actually applied dose of GEM in humans with pancreatic cancer (1000 mg/m2 body surface area) our applied dose is almost 4-fold lower when we calculate it in terms of dose per average body surface area of a mouse (which is 3 μg / 0.0066 m2 = ~0.45 mg/m2). These evaluations indicated the superiority of our DDND design and materials over other attempts and the development of a clinically applicable dose regimen. The design strategy, mainly based on PB copolymers electrostatically complexed with miR-345 and subsequently self-assembled with GEM encapsulated Pluronic F127 layers, provided an extended stability and sustained co-release as thereby allowed for dose-sparing use of GEM [61, 69]. Amphiphilic Pluronic F127 blocks enhanced cellular uptake via thermoreversible micellization while the protonatable tertiary amine groups of PDEAEM end blocks facilitated endosomal escape via the proton sponge effect [51, 57–61]. Further, self-assembly by GEM encapsulated Pluronic F127 also provided shielding of excess charges, enhanced cellular uptake, GEM loading and simultaneous miR-345/GEM release by temperature responsive micellization. Thus, the promising in vitro and in vivo results presented in this study help to enhance our understanding of mechanisms underlying combined delivery – as well as generate valuable information regarding the synergistic activity of combining GEM with a miR-345 mimic that significantly advances the goal of an improvement upon GEM treatment for PC patients. Additional studies are needed, but this work incrementally advances nano-delivery in relevant PC models to develop novel combination treatments for lethal pancreatic cancer.
Acknowledgements
SKM is very grateful to have had outstanding mentors, Prof. Nicholas Peppas and Prof. Robert Langer, for her doctoral and postdoctoral research respectively. As giants in the biomaterials and controlled release fields to whom this special issue is jointly dedicated to, SKM would like to thank them for inspiring her as well as several generations of biomaterials researchers, for leading by example and for creating an outstanding body of work that helped define the field. The authors in this manuscript were, in parts, supported by the following grants from the National Institutes of Health (RO1 CA183459, RO1 CA183459, R01GM113166, and SPORE P50CA127297). SKM is also grateful to the Carol Vohs Johnson Chair for support. We also thank Mrs. Kavita Mallya, for her invaluable technical support for this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2016, CA: a cancer journal for clinicians, 66 (2016) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Matsuno S, Egawa S, Fukuyama S, Motoi F, Sunamura M, Isaji S, Imaizumi T, Okada S, Kato H, Suda K, Nakao A, Hiraoka T, Hosotani R, Takeda K, Pancreatic Cancer Registry in Japan: 20 years of experience, Pancreas, 28 (2004) 219–230. [DOI] [PubMed] [Google Scholar]
- [3].Sultana A, Tudur Smith C, Cunningham D, Starling N, Neoptolemos JP, Ghaneh P, Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses, British Journal of Cancer, 99 (2008) 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, Ouellette MM, Hollingsworth MA, Sonic hedgehog promotes desmoplasia in pancreatic cancer, Clinical cancer research : an official journal of the American Association for Cancer Research, 14 (2008) 5995–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bailey JM, Mohr AM, Hollingsworth MA, Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer, Oncogene, 28 (2009) 3513–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee CJ, Li C, Simeone DM, Human Pancreatic Cancer Stem Cells: Implications for How We Treat Pancreatic Cancer, Translational Oncology, 1 (2008) 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD, Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial, Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 15 (1997) 2403–2413. [DOI] [PubMed] [Google Scholar]
- [8].Ciliberto D, Botta C, Correale P, Rossi M, Caraglia M, Tassone P, Tagliaferri P, Role of gemcitabine-based combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomised trials, European journal of cancer (Oxford, England : 1990), 49 (2013) 593–603. [DOI] [PubMed] [Google Scholar]
- [9].Bae YH, Park K, Targeted drug delivery to tumors: myths, reality and possibility, J Control Release, 153 (2011) 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Conroy T , Desseigne F , Ychou M , Bouché O , Guimbaud R , Bécouarn Y , Adenis A , Raoul J-L , Gourgou-Bourgade S , de la Fouchardière C , Bennouna J , Bachet J-B , Khemissa-Akouz F , Péré-Vergé D , Delbaldo C , Assenat E , Chauffert B , Michel P , Montoto-Grillot C , Ducreux M FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer, New England Journal of Medicine, 364 (2011) 1817–1825. [DOI] [PubMed] [Google Scholar]
- [11].Quint K, Tonigold M, Di Fazio P, Montalbano R, Lingelbach S, Ruckert F, Alinger B, Ocker M, Neureiter D, Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition, International journal of oncology, 41 (2012) 2093–2102. [DOI] [PubMed] [Google Scholar]
- [12].Huang ZQ, Saluja AK, Dudeja V, Vickers SM, Buchsbaum DJ, Molecular Targeted Approaches for Treatment of Pancreatic Cancer, Current pharmaceutical design, 17 (2011) 2221–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Spadi R, Brusa F, Ponzetti A, Chiappino I, Birocco N, Ciuffreda L, Satolli MA, Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians, World Journal of Clinical Oncology, 7 (2016) 27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu X, Zhang Y, Chen C, Yao Q, Li M, Targeted Drug Delivery in Pancreatic Cancer, Biochimica et biophysica acta, 1805 (2010) 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li F, Mahato RI, MicroRNAs and Drug Resistance in Prostate Cancers, Molecular pharmaceutics, 11 (2014) 2539–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Y, Sarkar FH, MicroRNA Targeted Therapeutic Approach for Pancreatic Cancer, International Journal of Biological Sciences, 12 (2016) 326–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, Zarone MR, Gulla A, Tagliaferri P, Tassone P, Caraglia M, Mir-34: A New Weapon Against Cancer?, Molecular Therapy-Nucleic Acids, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pramanik D, Campbell NR, Karikari C, Chivukula R, Kent OA, Mendell JT, Maitra A, Restitution of Tumor Suppressor MicroRNAs Using a Systemic Nanovector Inhibits Pancreatic Cancer Growth in Mice, Molecular Cancer Therapeutics, 10 (2011) 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P, Targeting miR-21 for the Therapy of Pancreatic Cancer, Molecular Therapy, 21 (2013) 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Simonson B, Das S, MicroRNA Therapeutics: the Next Magic Bullet?, Mini-Reviews in Medicinal Chemistry, 15 (2015) 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Z, Rao DD, Senzer N, Nemunaitis J, RNA Interference and Cancer Therapy, Pharm. Res, 28 (2011) 2983–2995. [DOI] [PubMed] [Google Scholar]
- [22].Yanes RE, Lu J, Tamanoi F, Nanoparticle-Based Delivery of siRNA and miRNA for Cancer Therapy, in: Guo F, Tamanoi F (Eds.) Enzymes, Vol 32: Eukaryotic Rnases and Their Partners in Rna Degradation and Biogenesis, Pt B2012, pp. 185–203. [Google Scholar]
- [23].Wu Y, Crawford M, Mao Y, Lee RJ, Davis IC, Elton TS, Lee LJ, Nana-Sinkam SP, Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer, Molecular Therapy - Nucleic Acids, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kumar V, Mondal G, Slavik P, Rachagani S, Batra SK, Mahato RI, Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer, Mol Pharm, 12 (2015) 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Devulapally R, Sekar NM, Sekar TV, Foygel K, Massoud TF, Willmann JK, Paulmurugan R, Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy, ACS nano, 9 (2015) 2290–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Luna JM, Scheel TKH, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, Jacobson IM, Rice CM, Darnell RB, Hepatitis C virus RNA functionally sequesters miR-122, Cell, 160 (2015) 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li W, Lebrun DG, Li M, The expression and functions of microRNAs in pancreatic adenocarcinoma and hepatocellular carcinoma, Chinese journal of cancer, 30 (2011) 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mittal A, Chitkara D, Behrman SW, Mahato RI, Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer, Biomaterials, 35 (2014) 7077–7087. [DOI] [PubMed] [Google Scholar]
- [29].Shi M, Cui J, Xie K, Signaling of MiRNAs-FOXM1 in Cancer and Potential Targeted Therapy, Current Drug Targets, 14 (2013) 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tutar Y, miRNA and Cancer; Computational and Experimental Approaches, Current Pharmaceutical Biotechnology, 15 (2014) 429–429. [DOI] [PubMed] [Google Scholar]
- [31].Uz M, Altinkaya SA, Mallapragada SK, Stimuli responsive polymer-based strategies for polynucleotide delivery, Journal of Materials Research, (2017) 1–24. [Google Scholar]
- [32].Kaczmarek JC, Kowalski PS, Anderson DG, Advances in the delivery of RNA therapeutics: from concept to clinical reality, Genome Medicine, 9 (2017) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pannier AK, Shea LD, Controlled release systems for DNA delivery, Molecular therapy : the journal of the American Society of Gene Therapy, 10 (2004) 19–26. [DOI] [PubMed] [Google Scholar]
- [34].Ramos J, Rege K, Poly(aminoether)–Gold Nanorod Assemblies for shRNA Plasmid-Induced Gene Silencing, Molecular pharmaceutics, 10 (2013) 4107–4119. [DOI] [PubMed] [Google Scholar]
- [35].Srivastava SK, Bhardwaj A, Arora S, Tyagi N, Singh S, Andrews J, McClellan S, Wang B, Singh AP, MicroRNA-345 induces apoptosis in pancreatic cancer cells through potentiation of caspase-dependent and -independent pathways, Br J Cancer, 113 (2015) 660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dai X, Tan C, Combination of microRNA therapeutics with small-molecule anticancer drugs: Mechanism of action and co-delivery nanocarriers, Advanced Drug Delivery Reviews, 81 (2015) 184–197. [DOI] [PubMed] [Google Scholar]
- [37].Kumar V, Mondal G, Dutta R, Mahato RI, Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis, Biomaterials, 76 (2016) 144–156. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Y, Wang Z, Gemeinhart RA, Progress in MicroRNA Delivery, Journal of controlled release : official journal of the Controlled Release Society, 172 (2013) 962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen Y, Gao D-Y, Huang L, In vivo delivery of miRNAs for cancer therapy: Challenges and strategies, Advanced Drug Delivery Reviews, 81 (2015) 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gao ZG, Tian L, Hu J, Park IS, Bae YH, Prevention of metastasis in a 4T1 murine breast cancer model by doxorubicin carried by folate conjugated pH sensitive polymeric micelles, J Control Release, 152 (2011) 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, Wang AZ, Sokolsky-Papkov M, Kabanov AV, Drug Combination Synergy in Worm-like Polymeric Micelles Improves Treatment Outcome for Small Cell and Non-Small Cell Lung Cancer, ACS nano, 12 (2018) 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Arora S, Swaminathan SK, Kirtane A, Srivastava SK, Bhardwaj A, Singh S, Panyam J, Singh AP, ynthesis, characterization, and evaluation of poly (D, L-lactide-co-glycolide)-based nanoformulation of miRNA-150: potential implications for pancreatic cancer therapy, International Journal of Nanomedicine, 9 (2014) 2933–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu C, Wang P, Zhang J, Tian H, Park K, Chen X, Pulmonary Codelivery of Doxorubicin and siRNA by pH-Sensitive Nanoparticles for Therapy of Metastatic Lung Cancer, Small (Weinheim an der Bergstrasse, Germany), 11 (2015) 4321–4333. [DOI] [PubMed] [Google Scholar]
- [44].Popilski H, Abtew E, Schwendeman S, Domb A, Stepensky D, Efficacy of paclitaxel/dexamethasone intra-tumoral delivery in treating orthotopic mouse breast cancer, J Control Release, 279 (2018) 1–7. [DOI] [PubMed] [Google Scholar]
- [45].Riehle R, Pattni B, Jhaveri A, Kulkarni A, Thakur G, Degterev A, Torchilin V, Combination Nanopreparations of a Novel Proapoptotic Drug - NCL-240, TRAIL and siRNA, Pharm Res, 33 (2016) 1587–1601. [DOI] [PubMed] [Google Scholar]
- [46].Sarisozen C, Pan J, Dutta I, Torchilin VP, Polymers in the co-delivery of siRNA and anticancer drugs to treat multidrug-resistant tumors, Journal of Pharmaceutical Investigation, 47 (2017) 37–49. [Google Scholar]
- [47].Salzano G, Costa Daniel F, Sarisozen C, Luther E, Mattheolabakis G, Dhargalkar Pooja P, Torchilin Vladimir P, Mixed Nanosized Polymeric Micelles as Promoter of Doxorubicin and miRNA‐34a Co‐Delivery Triggered by Dual Stimuli in Tumor Tissue, Small (Weinheim an der Bergstrasse, Germany), 12 (2016) 4837–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cho H, Lai TC, Kwon GS, Poly(ethylene glycol)-block-poly(ε-caprolactone) micelles for combination drug delivery: Evaluation of paclitaxel, cyclopamine and gossypol in intraperitoneal xenograft models of ovarian cancer, Journal of Controlled Release, 166 (2013) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK, Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer, Journal of Controlled Release, 171 (2013) 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lee E, Oh C, Kim I-S, Kwon IC, Kim S, Co-delivery of chemosensitizing siRNA and an anticancer agent via multiple monocomplexation-induced hydrophobic association, Journal of Controlled Release, 210 (2015) 105–114. [DOI] [PubMed] [Google Scholar]
- [51].Agarwal A, Unfer R, Mallapragada SK, Novel cationic pentablock copolymers as non-viral vectors for gene therapy, J. Control. Release, 103 (2005) 245–258. [DOI] [PubMed] [Google Scholar]
- [52].Agarwal A, Vilensky R, Stockdale A, Talmon Y, Unfer RC, Mallapragada SK, Colloidally stable novel copolymeric system for gene delivery in complete growth media, J. Control. Release, 121 (2007) 28–37. [DOI] [PubMed] [Google Scholar]
- [53].Determan MD, Cox JP, Mallapragada SK, Drug release from pH-responsive thermogelling pentablock copolymers, J. Biomed. Mater. Res. Part A, 81A (2007) 326–333. [DOI] [PubMed] [Google Scholar]
- [54].Determan MD, Cox JP, Seifert S, Thiyagarajan P, Mallapragada SK, Synthesis and characterization of temperature and pH-responsive pentablock copolymers, Polymer, 46 (2005) 6933–6946. [Google Scholar]
- [55].Mallapragada SK, Anderson BC, Ieee, Ieee, Ieee, Design and synthesis of novel pH and temperature sensitive copolymers for injectable delivery, Second Joint Embs-Bmes Conference 2002, Vols 1-3, Conference Proceedings: Bioengineering - Integrative Methodologies, New Technologies, Ieee, New York, 2002, pp. 486–487. [Google Scholar]
- [56].Peleshanko S, Anderson K, Goodman MD, Determan MD, Mallapragada SK, Tsukruk VV, Interfacial behavior of the thermo-responsive pentablock copolymer, Abstr. Pap. Am. Chem. Soc, 233 (2007) 1. [DOI] [PubMed] [Google Scholar]
- [57].Zhang BQ, Jia F, Fleming MQ, Mallapragada SK, Injectable self-assembled block copolymers for sustained gene and drug co-delivery: An in vitro study, Int. J. Pharm, 427 (2012) 88–96. [DOI] [PubMed] [Google Scholar]
- [58].Agarwal A, Unfer RC, Mallapragada SK, Dual-role self-assembling nanoplexes for efficient gene transfection and sustained gene delivery, Biomaterials, 29 (2008) 607–617. [DOI] [PubMed] [Google Scholar]
- [59].Uz M, Mallapragada SK, Altinkaya SA, Responsive pentablock copolymers for siRNA delivery, RSC Advances, 5 (2015) 43515–43527. [Google Scholar]
- [60].Zhang BQ, Kanapathipillai M, Bisso P, Mallapragada S, Novel Pentablock Copolymers for Selective Gene Delivery to Cancer Cells, Pharm. Res, 26 (2009) 700–713. [DOI] [PubMed] [Google Scholar]
- [61].Zhang BQ, Mallapragada S, The mechanism of selective transfection mediated by pentablock copolymers; Part II: Nuclear entry and endosomal escape, Acta Biomater., 7 (2011) 1580–1587. [DOI] [PubMed] [Google Scholar]
- [62].Singh R, Shakya AK, Naik R, Shalan N, Stability-Indicating HPLC Determination of Gemcitabine in Pharmaceutical Formulations, International Journal of Analytical Chemistry, 2015 (2015) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rachagani S, Torres MP, Kumar S, Haridas D, Baine M, Macha MA, Kaur S, Ponnusamy MP, Dey P, Seshacharyulu P, Johansson SL, Jain M, Wagner KU, Batra SK, Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy, Journal of hematology & oncology, 5 (2012) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liechty WB, Kryscio DR, Slaughter BV, Peppas NA, Polymers for Drug Delivery Systems, Annual review of chemical and biomolecular engineering, 1 (2010) 149–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S, Advances and Challenges of Liposome Assisted Drug Delivery, Frontiers in Pharmacology, 6 (2015) 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Varkouhi AK, Mountrichas G, Schiffelers RM, Lammers T, Storm G, Pispas S, Hennink WE, Polyplexes based on cationic polymers with strong nucleic acid binding properties, Eur. J. Pharm. Sci, 45 (2012) 459–466. [DOI] [PubMed] [Google Scholar]
- [67].Kim ST, Jang K-T, Lee SJ, Jang H-L, Lee J, Park SH, Park YS, Lim HY, Kang WK, Park JO, Tumour shrinkage at 6 weeks predicts favorable clinical outcomes in a phase III study of gemcitabine and oxaliplatin with or without erlotinib for advanced biliary tract cancer, BMC Cancer, 15 (2015) 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Srivastava SK, Bhardwaj A, Arora S, Singh S, McClellan S, Wang B, Grizzle WE, Singh AP, miR-345 downregulation is associated with apoptosis resistance in pancreatic cancer: therapeutic implications, Cancer Research, 72 (2012). [Google Scholar]
- [69].Zhang BQ, Mallapragada S, The mechanism of selective transfection mediated by pentablock copolymers; Part I: Investigation of cellular uptake, Acta Biomater., 7 (2011) 1570–1579. [DOI] [PubMed] [Google Scholar]
- [70].Wang LM, Silva MA, D’Costa Z, Bockelmann R, Soonawalla Z, Liu S, O’Neill E, Mukherjee S, McKenna WG, Muschel R, Fokas E, The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma, Oncotarget, 7 (2016) 4183–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]