

Abstract
Background
In wine fermentation starter cultures, the blending of non-Saccharomyces yeast with Saccharomyces cerevisiae to improve the complexity of wine has become common practice, but data regarding the impact of co-cultivation on yeast physiology and on genetic and metabolic regulation remain limited. Here we describe a transcriptomic analysis of mixed fermentations of Saccharomyces cerevisiae and Lachancea thermotolerans. The fermentations were carried out in carefully controlled environmental conditions in a bioreactor to reduce transcriptomic responses that would be due to factors other than the presence of the second species.
Results
The transcriptomic data revealed that both yeast species showed a clear response to the presence of the other. Affected genes primarily belonged to two groups: genes whose expression can be linked to the competition for certain trace elements such as copper and iron, as well as genes required for cell wall structure and integrity. Furthermore, the data revealed divergent transcriptional responses with regard to carbon metabolism in response to anoxic conditions.
Conclusions
The results suggest that the mixed fermentation created a more competitive and stressful environment for the two species than single strain fermentations independently from total biomass, i.e. competition between cells of the same species is less stressful, or may present a different set of challenges, than interspecies competition. The changes in cell wall and adhesion properties encoding genes suggest that the adjustment of physical contact between cells may play a direct role in the response to the presence of competing species.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5511-x) contains supplementary material, which is available to authorized users.
Keywords: Transcriptome, RNA-seq, Wine fermentation, Yeast interactions, Lachancea thermotolerans, Saccharomyces cerevisiae, Mixed-cultures, Anoxia
Background
During crushing of grapes, grape juice is exposed to air and becomes saturated with oxygen. Oxygen may also be discretely introduced into the wine fermentation process at various stages in amounts ranging from 2 mg L− 1 to 6 mg L− 1 depending on the method employed [1, 2]. Most of the oxygen is rapidly consumed by yeast cells, and concomitant CO2 production has a negative impact on oxygen dissolution, creating an anaerobic environment [1]. Several yeast species that constitute the wine fermentation community including members of the genera Saccharomyces, Torulaspora, Hanseniaspora, Lachancea, Pichia, Candida and Starmerella are facultative anaerobes that can grow and survive under these conditions. However, most of the non-Saccharomyces yeasts have high oxygen demands and low fermentation capacity compared to Saccharomyces species, particularly Saccharomyces cerevisiae [3–5]. Consequently, such species are used in mixed cultures with S. cerevisiae to ensure complete conversion of grape sugars to ethanol, CO2 and other metabolites that constitute the wine fermentation bouquet. This practice has gained tremendous interest in the global wine industry over the past decade, mainly due to improved wine flavour and aroma complexity, as well as potential reduction in ethanol levels, resulting from the underlying yeast-yeast interactions [6–12]. Consequently, understanding the interaction between Saccharomyces and non-Saccharomyces yeasts has become a central focus of ecological and of wine-related research. The nature of some of the ecological interactions between two yeast species have been previously evaluated. The data show that in mixed fermentation of S. cerevisiae and non-Saccharomyces yeasts, S. cerevisiae displays antagonistic interaction towards non-Saccharomyces yeasts such as Torulaspora delbrueckii, Hanseniaspora guilliermondii, Lachancea thermotolerans and Kluyveromyces lactis [13–15]. Early studies revealed that presence of S. cerevisiae cells at a high concentration causes cellular death in T. delbrueckii and L. thermotolerans [13]. These studies were subsequently followed by pioneering work attributing this antagonism to direct cell-cell contact as well as the production of antimicrobial peptides by S. cerevisiae [15, 16]. The data strongly suggest the existence of specific physical and metabolic interactions between yeast species, but do not provide any insights about the molecular mechanism behind such interactions, and little is known about the molecular factors influencing the response of any yeast species to the presence of another species. Such studies are challenging because of the complexity of multispecies systems and of ecological interactions. In particular, very few investigations have thus far been published reporting genome-wide data sets for such interactions, and most of these studies have primarily been reporting on the response of S. cerevisiae to the presence of another species. For instance, DNA microarray-based transcriptome analyses and mass spectrometry-based proteome analyses have been used to study the interaction between yeast and bacteria as well as between S. cerevisiae and non-Saccharomyces yeasts under oenological conditions [17–24]. Furthermore, these studies have usually relied on batch fermentation conditions. Such conditions make it difficult to differentiate the relative impact of the continuous changes in media composition from the specific response of one yeast species to the presence of the other.
In the current study, we evaluated the transcriptomic response of L. thermotolerans and S. cerevisiae in mixed fermentations when compared to single strain cultures in the same environmental conditions. We selected L. thermotolerans as a non-Saccharomyces wine yeast as that yeast has already been commercialised for use in mixed starter fermentations. Mixed culture fermentation with L. thermotolerans are known for leading to enhanced concentration of higher alcohols (particularly 2-phenylethanol), l-lactic acid, glycerol and esters, while in some conditions also resulting in lower ethanol wines [10]. The genome of this yeast has been sequenced and the genome sequence has been partially annotated. As demonstrated in previous studies [3, 5], oxygen enhances the growth and persistence of L. thermotolerans in mixed starter fermentations. To better characterise the molecular nature of the interactions, we used a controlled bioreactor system that allowed maintenance of two species in fermentation with continuous in-flow and out-flow of medium. The conditions were set to ensure that the total biomass of mixed and single species fermentations, and the environmental factors that strongly impact gene transcription in fermentative conditions such as nutrient availability, oxygen, ethanol and hexose concentrations, were maintained at similar levels in all fermentations. These settings should restrict the transcriptomic response to factors linked to the presence of a second species.
Results
Optimisation of fermentation conditions
Multispecies interaction studies at the molecular, transcriptomic or proteomic level face significant challenges. Indeed, when such studies are carried out in standard batch fermentation conditions, both species continuously modify gene expression to respond to the dynamic environment. Furthermore, population evolution leads to continuous change in the level of mutual exposure. In such conditions, any specific transcriptional response of one species to the presence of the other species will be hidden within a broader transcriptional response to changes in the environment. To overcome this problem, and to focus the investigation on the transcriptomic signature of the interaction between species, a system with similarity to a chemostat was optimised. The specific growth rate of S. cerevisiae and L. thermotolerans monocultures under anaerobic conditions, was found to be 0.2 h− 1 at a dilution rate of 0.1 h− 1 and 0.17 h− 1 at a dilution rate of 0.075 h− 1, respectively, and similar cell concentrations were obtained (Table 1). In order to avoid a washout of L. thermotolerans in mixed fermentations, the cultures were co-inoculated and cultivated in batch for 30 h, and then switched to continuous mode at a dilution rate of 0.1 h− 1 for the anaerobic fermentation. In contrast, under aerobic conditions, the two yeasts displayed similar specific growth rates and reached comparable cell concentrations under the same dilution rate (Table 1). Consequently, the same dilution rate could be applied in the mixed culture fermentations. The aim of this optimisation was to ensure similar population densities and similar growth medium composition in both monocultures and mixed culture fermentations. As illustrated in Fig. 1, under these optimised conditions, the species display similar growth rates in single and mixed fermentations, and after 48 h of continuous culture, the total number of cells, as well as the sugar concentrations were similar in all the fermentations. The viable counts obtained from samples used for RNA extraction and expression analysis, showed that the cell concentrations of both species were ≈ 108 CFU mL− 1 in both mono- and mixed-cultures (Table 1).
Table 1.
A summary of the dilutions rates applied to maintain similar cell concentrations, as well as the chemical fermentation parameters at the time of sampling for transcriptome analysis
| Fermentations | Dilution rate h− 1 | CFU mL− 1 at 48 h | Sugar concentration at 48 h (g L− 1) | Glycerol concentration at 48 h (g L− 1) | μMax at exponential phase (h− 1) |
|---|---|---|---|---|---|
| L. thermotolerans-AN | 0.075 | 2.1E+ 08 | 68.0 | 2.92 | 0.17 |
| S. cerevisiae-AN | 0.10 | 1.2E+ 08 | 62.5 | 2.40 | 0.20 |
| Mixed-AN | 0.10 | Sc- 1.1E+ 08 Lt- 8.6E+ 07 |
59.6 | 3.14 | |
| S. cerevisiae-AR | 0.125 | 1.0E+ 08 | 60.0 | 1.09 | 0.23 |
| L. thermotolerans-AR | 0.125 | 2.5E+ 08 | 58.0 | 1.34 | 0.24 |
| Mixed-AR | 0.125 | Sc- 1.1E+ 08 Lt- 2.3E+ 08 |
62.0 | 1.12 |
Fig. 1.

Fermentation kinetics and population dynamics of S. cerevisiae and L. thermotolerans in single and mixed fermentation under anaerobic (left panel) and aerobic (right panel) fermentation conditions. The red line indicates the start of continuous culture, the arrows indicate the sampling point
Global analysis of the transcriptome
The RNA-sequencing was performed for two biological replicates of each fermentation. RNA extractions were performed on samples collected after the two species were in contact for 48 h and had maintained similar cell concentrations (Fig. 1). This was done to allow sufficient metabolic interaction under conditions akin to a steady state. After filtering and trimming, RNAseq samples contained between 11 and 15 Mb total reads (Additional file 1: Table S1). These reads were mapped to the S288CplusLT chimeric genome which was generated after cross-mapping between S. cerevisiae and L. thermotolerans was found to be less than 1%. The data show that the mixed culture transcriptome had low coverage of L. thermotolerans. In particular, the oxygenated fermentation samples generated a total sum of ≈ 2 Mb L. thermotolerans reads between the two biological replicates, which is below the recommended threshold. Nevertheless, the data were considered useful for certain analyses. One of the S. cerevisiae anaerobic monocultures (Sc.AN.2) appeared to be mixed and was therefore not considered in subsequent analyses. Principal Component Analysis (PCA) performed on normalized TPM data showed that the samples clustered together in a yeast and treatment specific manner, with only minor variations between the independent biological replicates (Fig. 2). L. thermotolerans displayed higher levels of transcriptomic change under each condition compared to S. cerevisiae, as evident in the separation of the samples along PC2. The statistical analysis was done using Benjamini-Hochberg on all highly-expressed genes to control the FDR.
Fig. 2.
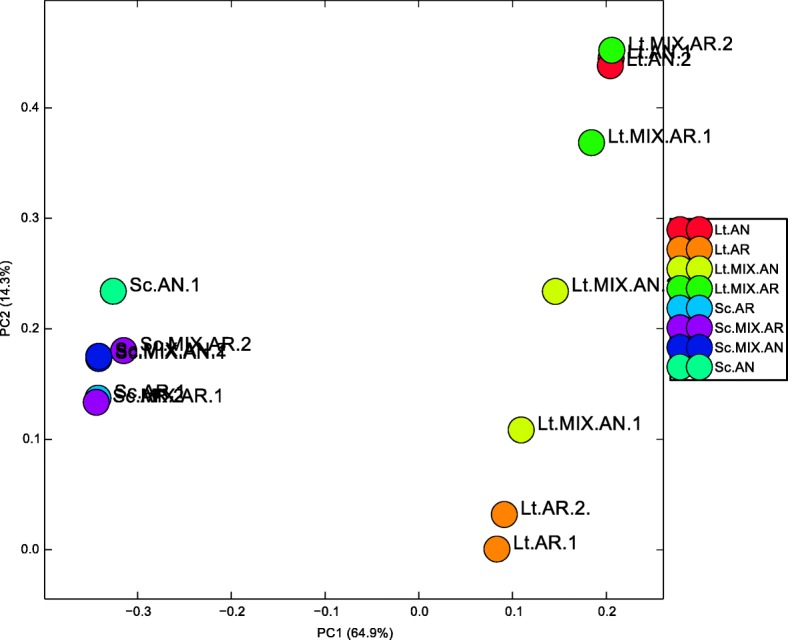
Principal Component Analysis (PCA) plot of the biological replicates of transcripts from Lachancea thermotolerans (Lt) and Saccharomyces cerevisiae (Sc) monocultures and mixed cultures (Lt.MIX and Sc.MIX) under aerobic (AR) and anaerobic (AN) conditions
Overview of transcriptional response in mixed fermentation
The effect of mixing (MIX) on the gene transcription in S. cerevisiae and L. thermotolerans was assessed by comparing the transcriptome of the mixed cultures to the monocultures under both aerobic and anaerobic conditions (i.e Sc-Lt-AN and Sc-Lt-AR compared to Sc-AN, Sc-AR, Lt-AN and Lt-AR). Genes uniquely differentially expressed in the mixed fermentations compared to monocultures were identified (Additional file 2: Table S2). The genes differentially expressed under anaerobic conditions (AN) were determined by comparing the anaerobic cultures to the aerobic cultures (i.e Sc-Lt-AN, Sc-AN and Lt-AN compared to Sc-Lt-AR, Sc-AR and Lt-AR). Furthermore, common genes differentially expressed in both MIX and AN sets were considered to be affected by the interaction (INT) between mixing and anoxia (Additional file 2: Table S2). For the initial global analysis, the differentially expressed genes (DEGs) were visualized by overlaying the data on the Biocyc Omics dashboard. Overall, the interaction between mixing and anoxia (INT) elicited a stronger response in L. thermotolerans than in S. cerevisiae (Fig. 3). In S. cerevisiae, only the degradation of secondary metabolites as well as the biosynthesis of metabolic regulators were significantly up-regulated. These included TSL1, TPS1, TPS2 and TPS3, involved in trehalose biosynthesis (Additional file 3: Table S3). Conversely, L. thermotolerans displayed an up-regulation of several biological processes most notably detoxification, cell death, adhesion as well as response to stimulus (Fig. 3). The highly up-regulated detoxification and adhesion-related genes in L. thermotolerans included ALD2, ALD5, SOD1, SOD2, DLD1, CTT1, and HSP12, FLO9, NRG1, SDS3 and CCW12, respectively (Additional file 3: Table S3).
Fig. 3.

Overall comparison of transcriptional response of Saccharomyces cerevisiae (Sc) and Lachancea thermotolerans (Lt) to mixed-culture environment (Sc-mix and Lt-mix), and to the combined effect of mixing and anoxia (Sc-int and Lt-int)
Response to anoxia in mixed fermentations
The anaerobic and aerobic mixed cultures (Sc-Lt-AN vs Sc-Lt-AR) were compared to assess the transcriptional response of S. cerevisiae and L. thermotolerans to anoxia in a mixed fermentation setting. Considering DEGs with log2FC ≥ 1.5 or ≤ − 1.5, only 8 genes were up-regulated genes and 22 down-regulated genes were common between S. cerevisiae and L. thermotolerans (Fig. 4). Gene ontology (GO) enrichment showed that cation transport, electron transport chain, generation of precursor metabolites and energy, were some of the highly down-regulated biological processes in both yeasts. By contrast, biological processes enriched in up-regulated genes were distinct between the two yeasts. For instance, in S. cerevisiae, sterol transport, cell wall organization and associated processes were enriched while in L. thermotolerans oxidation-reduction process, carbohydrate catabolism as well as cellular lipid biosynthetic processes were enriched (Fig. 4).
Fig. 4.

A Venn diagram showing the number of differentially expressed genes (DEGs) commonly regulated in S. cerevisiae and L. thermotolerans in a mixed-culture fermentation in response to anoxia compared to aerobic mixed-culture. The enriched GO terms reflecting the biological processes associated with the DEGs are presented in the associated tables
Response to mixing under anaerobic conditions
Since the mixed aerobic fermentation samples contained a low percentage of L. thermotolerans reads and transcripts, which reduced the power to detect differentially expressed genes, we only considered the mixed culture fermentations under anaerobic conditions for further analyses. The transcriptional response of the two yeasts to mixing was analysed by comparing the mixed culture transcriptome to the monocultures in these conditions (i.e. Sc-Lt-AN vs Sc-AN and Lt-AN). The data revealed that only 62 genes were differentially expressed (31 up-regulated and 31 down-regulated) in S. cerevisiae in response to the presence of L. thermotolerans. In contrast, in L. thermotolerans 687 genes were differentially expressed, amongst them 639 (408 up-regulated and 231 down-regulated) which could be annotated to S. cerevisiae homologs. The DEGs were independently analysed for enriched GO terms at the three hierarchical categories (biological process, molecular function and cellular component). Up-regulated genes in S. cerevisiae were mainly associated with copper and iron ion import, as well as iron ion homeostasis, while the down-regulated genes were associated with cell aggregation and flocculation (Table 2). The genes significantly up-regulated in the copper and iron homeostasis were FRE1 and FRE7 which encode ferric reductases, CTR1 and CTR3 encoding high affinity copper transporters, as well as ENB1 and FIT2 that encode an endosomal ferric enterobactin transporter and a mannoprotein, respectively. In contrast, FLO10, FLO11 and SAG1 required for pseudohyphal and invasive growth, as well as flocculation and agglutination, respectively, were down-regulated (Fig. 5). In L. thermotolerans, biological processes associated with filamentous growth in response to starvation were enriched with up-regulated genes, while processes associated with iron assimilation, iron ion homeostasis as well as siderophore transport were enriched with down-regulated genes (Table 3). The data show that transcriptional factors and repressors (e.g. MIG1, MIG2, OPY2) that are likely involved in the regulation of filamentous growth were up-regulated, while FTR1, FRE3, FET3 and ARN1 encoding a high affinity iron permease, ferric reductase, ferro-O2-oxidoreductase, as well as ARN family transporter for siderophore-iron chelates, respectively, were down-regulated (Fig. 5).
Table 2.
Enriched GO terms (Biological Processes (BP), Biological Function (BF) and Cellular component (CC)) in Saccharomyces cerevisiae in anaerobic mixed fermentations
| Expression | Category | GO Terms | FDR q-value | Description |
|---|---|---|---|---|
| Up regulated | BP | GO:0015677 | 1.70E-04 | Copper ion import |
| GO:0000041 | 9.06E-04 | Transition metal ion transport | ||
| GO:0006825 | 8.15E-04 | Copper ion transport | ||
| GO:0030001 | 8.85E-03 | Metal ion transport | ||
| GO:0006826 | 2.82E-02 | Iron ion transport | ||
| GO:0055072 | 3.28E-02 | Iron ion homeostasis | ||
| GO:0055076 | 1.26E-01 | Transition metal ion homeostasis | ||
| GO:0035434 | 1.50E-01 | Copper ion transmembrane transport | ||
| GO:0055065 | 2.84E-01 | Metal ion homeostasis | ||
| GO:0015688 | 3.12E-01 | Iron chelate transport | ||
| GO:0015891 | 2.83E-01 | Siderophore transport | ||
| GO:0071555 | 3.78E-01 | Cell wall organization | ||
| GO:0045229 | 3.49E-01 | External encapsulating structure organization | ||
| BF | GO:0005199 | 1.70E-02 | Structural constituent of cell wall | |
| GO:0005375 | 1.36E-01 | Copper ion transmembrane transporter activity | ||
| GO:0052851 | 1.90E-01 | Ferric-chelate reductase (NADPH) activity | ||
| GO:0000293 | 1.89E-01 | Ferric-chelate reductase activity | ||
| GO:0016723 | 1.51E-01 | Oxidoreductase activity, oxidizing metal ions, NAD or NADP as acceptor | ||
| GO:0016811 | 2.35E-01 | Hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in linear amides | ||
| GO:0016722 | 3.47E-01 | Oxidoreductase activity, oxidizing metal ions | ||
| CC | GO:0009277 | 4.84E-02 | Fungal-type cell wall | |
| GO:0030312 | 3.04E-02 | External encapsulating structure | ||
| GO:0005618 | 2.30E-02 | Cell wall | ||
| Down-regulated | BP | GO:0098743 | 8.92E-03 | Cell aggregation |
| GO:0098630 | 4.46E-03 | Aggregation of unicellular organisms | ||
| GO:0051704 | 6.15E-03 | Multi-organism process | ||
| GO:0098609 | 8.21E-02 | Cell-cell adhesion | ||
| GO:0000128 | 9.84E-06 | Flocculation | ||
| BF | GO.0035673 | 1.56E-01 | Oligopeptide transmembrane transporter activity | |
| GO:1904680 | 1.00E+ 00 | Peptide transmembrane transporter activity | ||
| CC | GO:0005576 | 2.42E-04 | Extracellular region | |
| GO:0030312 | 7.10E-03 | External encapsulating structure | ||
| GO:0005618 | 4.74E-03 | Cell wall | ||
| GO:0031225 | 6.22E-03 | Anchored component of membrane | ||
| GO:0000322 | 1.08E-02 | Storage vacuole | ||
| GO:0000324 | 8.99E-03 | Fungal-type vacuole |
Fig. 5.

Differentially expressed genes involved in enriched biological processes in S. cerevisiae and L. thermotolerans in response to mixing under anaerobic conditions compared to aerobic conditions
Table 3.
Enriched GO terms (Biological Processes (BP), Biological Function (BF) and Cellular component (CC)) in Lachancea thermotolerans under anaerobic mixed fermentations
| Expression | Category | GO Terms | FDR q-value | Description |
|---|---|---|---|---|
| Up regulated | BP | GO:0005975 | 9.65E-03 | Carbohydrate metabolic process |
| GO:0044281 | 8.15E-03 | Small molecule metabolic process | ||
| GO:0035434 | 4.67E-02 | Copper ion transmembrane transport | ||
| GO:0044282 | 4.19E02 | Small molecule catabolic process | ||
| GO:0043436 | 4.79E02 | Oxoacid metabolic process | ||
| GO:0006082 | 4.34E-02 | Organic acid metabolic process | ||
| GO:1900434 | 3.91E-02 | Regulation of filamentous growth of a population of unicellular organisms in response to starvation | ||
| GO:0019752 | 3.79E-02 | Carboxylic acid metabolic process | ||
| BF | GO:0001078 | 1.96E-03 | Transcriptional repressor activity, RNA polymerase II proximal promoter sequence-specific DNA binding | |
| GO:0001227 | 1.47E-03 | Transcriptional repressor activity, RNA polymerase II transcription regulatory region sequence-specific DNA binding | ||
| GO:0000982 | 5.40E-02 | Transcription factor activity, RNA polymerase II proximal promoter sequence-specific DNA binding | ||
| CC | GO:0005576 | 1.39E-03 | Extracellular region | |
| GO:0005618 | 6.98E-02 | Cell wall | ||
| Down-regulated | BP | GO:0055072 | 2.97E-05 | Iron ion homeostasis |
| GO:0055076 | 4.61E-04 | Transition metal ion homeostasis | ||
| GO:0006879 | 6.37E-04 | Cellular iron ion homeostasis | ||
| GO:0048878 | 1.03E-03 | Chemical homeostasis | ||
| GO:0055065 | 1.98E-03 | Metal ion homeostasis | ||
| GO:0015688 | 2.89E-03 | Iron chelate transport | ||
| GO:0015891 | 2.48E-03 | Siderophore transport | ||
| GO:0046916 | 5.32E-03 | Cellular transition metal ion homeostasis | ||
| GO:0051321 | 5.84E-03 | Meiotic cell cycle | ||
| GO:0042592 | 7.78E-03 | Homeostatic process | ||
| GO:1901678 | 8.35E-03 | Iron coordination entity transport | ||
| GO:0098771 | 8.65E-03 | Inorganic ion homeostasis | ||
| GO:0055080 | 1.04E-02 | Cation homeostasis | ||
| GO:0006826 | 1.09E-02 | Iron ion transport | ||
| GO:0050801 | 1.97E-02 | Ion homeostasis | ||
| BF | GO:0016491 | 5.40E-01 | Oxidoreductase activity | |
| GO:0016722 | 4.45E-01 | Oxidoreductase activity, oxidizing metal ions | ||
| GO:0050662 | 6.57E-01 | Coenzyme binding | ||
| CC | GO:1990351 | 2.83E-01 | Transporter complex | |
| GO:0097249 | 1.66E-01 | Mitochondrial respiratory chain supercomplex | ||
| GO:0033573 | 2.19E-01 | High-affinity iron permease complex | ||
| GO:19–05862 | 1.64E-01 | Ferroxidase complex |
Within the category of Cellular component, GO enrichment revealed that the extracellular region and the cell wall were the most enriched cellular component GO terms in S. cerevisiae with both up- and down-regulated genes (Table 2). The enrichment is mainly associated with the up-regulation of mannoprotein encoding genes (FIT2 and FIT3) as well as the seripauperin protein encoding genes (PAU5, PAU12, PAU17 and PAU24) and down-regulation of FLO10, FLO11, SPO19, SAG1 and PHO5 (Fig. 7). Conversely, in L. thermotolerans the cell wall was only enriched with up-regulated genes. The genes associated with this enrichment included KRE9, BGL2, DSE4, CWP1, PIR1, HSP150 (a paralog of PIR3), UTH1, and CRH1 (Fig. 6) all required for cell wall biogenesis and stability, particularly β-glucan formation.
Fig. 7.
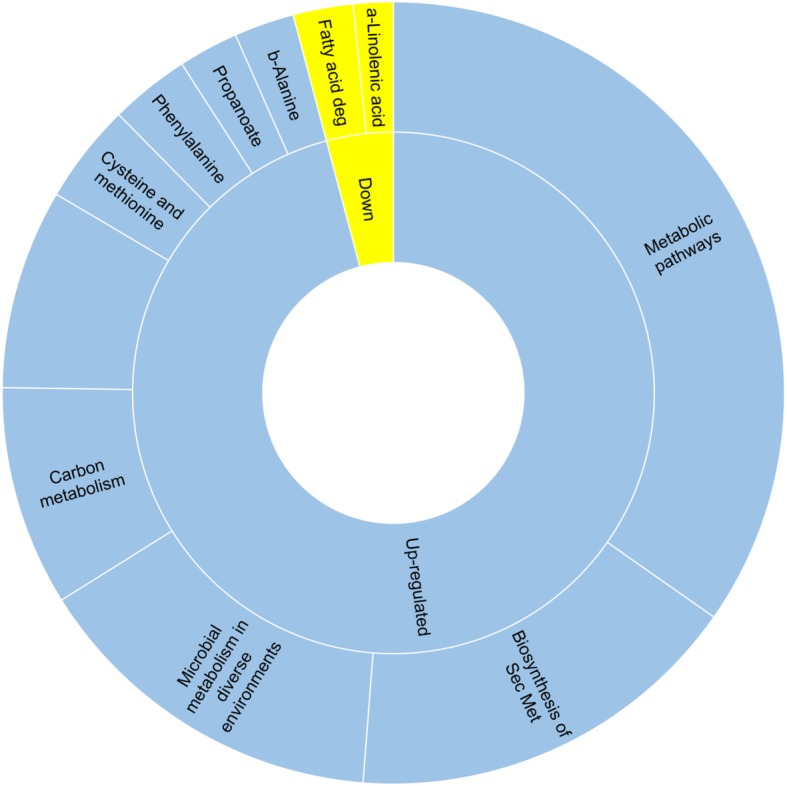
KEGG pathways enriched in L. thermotolerans in mixed culture fermentation under anaerobic conditions compared to aerobic conditions
Fig. 6.

DEGs associated with the enrichment of the cellular component GO terms: cell wall and cell exterior
The DEGs were further mapped to terms in the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/) database. No pathways were enriched in S. cerevisiae probably due to the limited number of genes (62) differentially expressed in response to mixing. Conversely, in L. thermotolerans out of the 639 annotated DEGs,116 up-regulated and 5 down-regulated genes revealed enriched KEGG pathways. As illustrated in Fig. 7, metabolic pathways, biosynthesis of secondary metabolites as well as microbial metabolism in diverse environments were the largest categories in the up-regulated genes. Furthermore, the data show that carbon metabolism as well as biosynthesis of amino acids were highly enriched. Moreover, the metabolism of β-alanine, phenylalanine, cysteine and methionine was enriched. However, not all the genes involved in the metabolic processes were significantly upregulated. For instance, in the β-alanine metabolism only PAN6 and GAD1 were significantly up-regulated, while ARO8 and AAT2, and CYS3, ARO8, MCY1 and AAT2, were significantly up-regulated in the phenylalanine and the cysteine and methionine metabolism, respectively (Additional file 4: Table S4). Amongst the down-regulated genes, fatty acid degradation as well as α-linoleic acid metabolism were the main enriched pathways.
Discussion
The current study aimed to unravel transcriptional responses of S. cerevisiae and L. thermotolerans in mixed culture fermentation. By manipulating dilution rates in a continuous culture, the two yeasts were maintained at approximately equal concentrations for 24 h. L. thermotolerans transcripts accounted for 24% of the sequences reads in anaerobic mixed cultures and ≈ 8% in aerobic mixed cultures. However, there was sufficient depth in the anaerobic mixed fermentation transcriptome to extract key genes that are influenced by mixing and interaction between S. cerevisiae and L. thermotolerans. Related studies have employed strategies, which only allowed for analysis of early transcriptional responses to mixed culture or analyses of S. cerevisiae transcriptional response only. For instance, Tronchoni et al. [25] first pre-cultured S. cerevisiae and the T. delbrueckii in separate bioreactors and then mixed the two at equal volumes, followed by withdrawal of samples for RNAseq before the cells started proliferating and in the early exponential phase to avoid over-representation of only one species. In contrast, Barbosa et al. [17] conducted mixed-culture fermentations of S. cerevisiae with Hanseniaspora guilliermondii in a typical wine fermentation batch set-up, extracted RNA at different fermentation stages and only focused on the transcriptional response of S. cerevisiae. The specific conditions also aimed at maintaining relatively stable environmental conditions and cell densities during the co-culture of the two species by using a set-up similar to a continuous fermentation. This strategy was intended to limit the transcriptomic signature to the interaction-relevant responses by reducing genetic responses that would be due to the continuous dynamic adaptation to changing conditions that are prevalent in batch cultures [17, 25]. The relatively limited number of S. cerevisiae genes that are significantly affected by the mixed culture conditions in our study when compared to other such data sets suggests that this strategy has succeeded, and that the highlighted genes are indeed the core of a perpetual interaction response.
Overall response to the interaction between mixing and anoxia
The transcriptomic data revealed divergent responses of the two yeasts to the interaction between mixing and anoxia. These conditions elicited a stronger response in L. thermotolerans. Indeed, on average L. thermotolerans displayed a marked up-regulation of genes associated with cell aggregation, cell death and response to osmotic stress. Furthermore, several genes encoding catalase, superoxide dismutases, and peroxidases were significantly up-regulated, suggesting that L. thermotolerans cells are experiencing some level of oxidative stress. This is not surprising since other studies have demonstrated that hypoxia and anoxia induce transient oxidative stress in other yeasts such as S. cerevisiae and Kluyveromyces lactis [26]. Our data also suggest that L. thermotolerans redirects its metabolic flux from glycolysis to the PPP similar to K. lactis and Pichia pastoris [26, 27].
Under anoxic conditions, mitochondrial and cytochrome c activities were down-regulated in both yeasts. However, differences were observed in the up-regulated processes, where seemingly S. cerevisiae increased its sterol uptake while L. thermotolerans increased the expression of genes required for ergosterol biosynthesis including those encoding enzymes that catalyse oxygen-dependent reactions e.g. ERG3, ERG11 and ERG25. This induction of oxygen-dependent genes has been observed in other yeasts including S. cerevisiae and P. pastoris albeit transiently, and has been attributed to cells adjusting to anaerobic conditions following cultivations in oxygenated environments [27].
Transcriptional responses to mixed culture fermentation – Iron and copper acquisition
In the current study, it was evident that in mixed-culture fermentation, S. cerevisiae increased the expression of iron and copper acquisition systems. Indeed, the data show that FRE1, FRE7, ENB1, FIT2, CTR1 and CTR3 were significantly upregulated. Fre1p, Enb1p and Fit2p form a cluster of proteins required for the uptake of siderophores. Fre1p has broad substrate specificity and can catalyse the reduction of iron-sidephore chelates such as ferrichrome, ferrioxamine B and enterobactin. Enb1p exclusively recognizes and transports enterobactin, while Fit2p contributes to the retention of siderophore-iron chelates in the cell wall [28]. Regarding copper uptake, Ctr1p and Ctr3p together with Fre1p and Fre7p are important in high affinity uptake of copper ions. Their expression is a cellular response to inadequate intracellular copper levels [29]. Conversely, L. thermotolerans shown a down-regulation of genes encoding nonreductive (copper independent) siderophore-iron transport (ARN1, FIT2, FIT3), as well as those required for reductive iron uptake (FRE3) and copper-dependent iron import (FTR1 and FET3). Fet3p is multicopper oxidase that also exhibit ferrous oxidase activity and forms a high-affinity iron transport complex with Ftr1p as well as Fet5p and Fth1p [30], which were not differentially expressed in the current study. Hodgins-Davis et al. [31] showed that FTR1 and FET3 were uniformly downregulated under copper deprivation in different S. cerevisiae strains. Since high-affinity copper and iron acquisition systems are homeostatically regulated, we can infer from the current data that both yeasts experience the growth conditions to be limited in bioavailable iron and copper, and therefore activate different systems to allow them to thrive in such environments. In S. cerevisiae, high expression of CTR1 gene is reported to be induced when copper levels are below 10 μM [30], while high concentrations of copper result in down-regulation of both CTR1 and CTR3 [29]. The synthetic grape juice medium used in the current study contains 0.11 μM copper. Copper is required for iron homeostasis in yeast and the link between copper and iron metabolism in S. cerevisiae is well recognized [29]. Evidently, in mixed-culture fermentation, S. cerevisiae competes with L. thermotolerans by activating a full set of genes to acquire different forms of iron from the environment and to store it in the cells in bound-form (e.g. ferrichrome).
Transcriptional responses to mixed culture fermentation – Cell wall integrity and adhesion
Our data show a strong response to cell wall integrity in both yeasts in mixed fermentation when compared to single species fermentation. Indeed, both yeast species up-regulate genes that are involved in cell wall integrity under stress conditions although different genes and processes are induced. Indeed, under anaerobic mechanisms in mixed fermentations. In particular, S. cerevisiae shows a significant up-regulation of 5 PAU genes (PAU5, PAU9, PAU12, PAU17, PAU24). These genes have been reported to play an important role in promoting fitness under anaerobic and fermentative condition as well as in interactions between natural strains of S. cerevisiae [32, 33]. In particular, PAU5 was shown to play a key role in competition by providing protection against killer toxins [33]. The abundance of the PAU genes in the S. cerevisiae transcriptome in the presence of L. thermotolerans could therefore suggest that they also play a role in interactions with other yeast species which are phylogenetically closely related to S. cerevisiae. Indeed, in another study, Tronchoni et al. [25] reported an induction of 20 of the 24 PAU genes in the primary response of S. cerevisiae to Torulaspora delbrueckii. Our data suggest that these genes are indeed specifically associated with yeast-yeast interactions. Interestingly, in the mixed fermentation under anaerobic conditions, L. thermotolerans increases the expression of endoglucanases (e.g. BGL2), while simultaneously reinforcing and stabilizing its cell wall, as evident in the up-regulation of genes involved in the biogenesis, assembly and maintenance of glucan and chitin. The overexpression of BGL2 in S. cerevisiae has been shown to retard growth, which could suggest that L. thermotolerans compensates for this possible impact by strengthening the cell wall. In contrast, S. cerevisiae protects itself through the upregulation of PAU genes, which are amongst the genes that encode cell wall proteins thought to be important in cell wall remodelling and maintenance of cell wall integrity during stress [32]. In the current study, the recovery of L. thermotolerans reads in mixed culture fermentations was consitently lower than those of S. cerevisiae (Additional file 1: Table S1). We may deduce from our data that this alludes to poor RNA extraction which could partly be due to cell wall thickness and rigidity, that rendered the L. thermotolerans cells less sensitive to mechanical disruption. However, this will require further evaluation.
Transcriptional responses to mixed culture fermentation – Amino acid metabolism
Our data suggest that in mixed culture fermentation under anaerobic conditions, L. thermotolerans specifically increases the expression of genes involved in the metabolism of four amino acids viz. cysteine, methionine, phenylalanine and β-alanine. The up-regulation of cystein and methionine-related metabolic activities may suggest that sulphur-related processes are directly affected by co-culture conditions. Furthermore, all genes involved in the conversion of phenylalanine to phenylethanol were up-regulated. These data support our previous findings which showed that L. thermotolerans produces high levels of phenylethanol in monoculture, and to enhance phenylethanol production in mixed fermentations with S. cerevisiae [5]. This trait seems common in various L. thermotolerans since other studies have reported increased levels of phenylethanol in combinations of different strains of L. thermotolerans and S. cerevisiae [10, 11].
Conclusions
Overall, our study reveals divergent molecular signatures underlying the performance of S. cerevisiae and L. thermotolerans in mixed culture fermentation. The data shows that S. cerevisiae is better able to deal with the fermentation environment possibly due to its efficient competitive uptake of sterols, copper and iron, accompanied by cell wall remodelling to accommodate additional mannoproteins and PAU proteins. These strategies allow the yeast to regulate membrane fluidity and cell wall porosity, and withstand an anaerobic, high ethanol environment. Conversely, the fermentation environment seems highly toxic to L. thermotolerans, which mainly features a molecular signature that is characterized by detoxification, cell aggregation and cell death associated genes. The strong cell wall-related responses in both species suggest the importance of this organelle in the cellular response to other species. In particular, the data support that the regulation of adhesion properties may play a central role in modulating the physical and ecological interactions between species [16].
The data are also a confirmation of many studies that have reported a rapid decline of L. thermotolerans in wine fermentation especially in mixed cultures with S. cerevisiae. The study also underlines the usefulness of a global approach to the study of yeast-yeast interactions to shed light on the molecular basis of yeast dynamics during wine fermentation. Besides a general contribution to a better understanding of yeast ecological interactions, the data will be useful for the rational development of mixed-starter cultures in the winemaking industry.
Methods
Yeast strains and media
S. cerevisiae (Cross evolution-285) was obtained from Lallemand SAS (Blagnac, France), while L. thermotolerans (IWBT-Y1240) was obtained from the culture collection of the Institute for Wine Biotechnology (Stellenbosch University). Yeast strains were maintained cryogenically (− 80 °C) and were reactivated by streaking out on YPD agar plates containing (per litre) 10 g yeast extract, 20 g peptone and 20 g glucose and 20 g bacteriological agar. Cultures were stored at 4 °C for short-term use.
Batch fermentation
Batch fermentations were performed in synthetic grape juice medium containing (per litre) 100 g glucose, 100 g fructose (Merck), 1 g yeast extract (Oxoid), 0.3 g citric acid, 5 g l-malic acid, 5 g l-tartaric acid, 0.4 g MgSO4, 5 g KH2PO4, 0.2 g NaCl, 0.05 g MnSO4 (Sigma-Aldrich) and anaerobic factors (ergosterol 10 mg (Sigma-Aldrich), tween 80 0.5 mL (Merck)) [34, 35]. Fermentations were conducted in 1.3 L BioFlo 110 bench top bioreactors (New Brunswick, NJ, USA) using 900 mL of final working volume, a temperature of 25 °C and an agitation speed of 200 rpm. Fermentations were performed under two conditions: anaerobic and aerobic at 5% (0.41 mg L− 1) dissolved oxygen (DO). The anaerobic conditions were created by initially sparging N2 to bring down the DO level to 0%, and then to minimize diffusion of atmospheric oxygen into the cultures, the entire fermentation set-up was equipped with Norprene tubing. The 5% DO level was maintained through the supplementary addition of 4 gasses (CO2, N2, O2 and compressed air) whenever required, using an automated gas flow controller. The DO levels in the cultures were monitored with an oxygen electrode.
Fermentation conditions
In order to maintain similar environmental conditions in mixed and single-culture fermentations, a system similar to continuous fermentation using continuous in-flow and out-flow of the medium was optimised for single and mixed fermentations. Samples for RNAseq analysis were withdrawn at 48 h when total viable cell count was similar between the mixed and single culture fermentation. The feeding medium contained glucose and fructose, each at 50 g L− 1. The working volume was maintained at 0.7 L using a peristaltic effluent pump. All fermentations were conducted in duplicate.
Analysis of population dynamics
Serial dilutions of the cell suspensions were performed with 0.9% (w/v) NaCl. One hundred microliter samples were spread on YPD agar and incubated at 30 °C for 2–3 days. For yeast enumeration in mixed culture fermentations, both species were distinguished based on colony morphology. Colony counts were performed on plates with 30–300 colonies.
Analytical methods
Supernatants were obtained by centrifuging cell suspensions at 5000×g for 5 min. The concentrations of fructose, glucose, acetaldehyde and acetic acid were measured using specific enzymatic kits, Enytec™ Fluid d-fructose, glucose, acetic acid (Thermo Fisher Scientific Oy, Finland), Boehringer Mannheim / R-Biopharm-acetaldehyde (R-Biopharm AG, Darmstadt) and analyzed using Arena 20XT photometric analyzer (Thermo Electron Oy, Helsinki, Finland). Ethanol was analysed by high performance liquid chromatography (HPLC) on an AMINEX HPX-87H ion exchange column using 5 mM H2SO4 (Sigma-Aldrich) as the mobile phase as described by Rossouw et al. [24]. The major volatiles were extracted with diethyl ether and analysed by Gas Chromatography with Flame Ionization Detection (GC-FID) as described in previously [36].
Sampling, RNA-extraction and RNA-sequencing
Cell samples for RNA-sequencing were obtained from both single and mixed culture fermentations (anaerobic and aerobic, respectively) at 48 h when population and sugar levels were approximately same in all fermentations. Total RNA extractions were performed according to the hot phenol method [37]. Concentration and purity of RNA were determined by spectrophotometry and integrity was confirmed using an Agilent 2100 Bioanalyzer with an RNA 6000 Nano Assay (Agilent Technologies, Palo Alto, CA, USA). The RNA samples with RNA integrity number (RIN) more than 8, and 280:260 ratios more than 2 were further used for the RNA-sequencing purpose. Library preparation and sequencing was performed by VIB Nucleomics core (KU, Leuven (Belgium). Complementary DNA (cDNA) library was generated using TruSeq® Library Prep Kit v2. Paired-end reads were sequenced on the Illumina NextSeq platform.
RNAseq data processing
Low quality reads (< Q20), polyA-reads as well as ambiguous reads (containing N) were removed using FastX 0.0.13 [38]. Furthermore, reads shorter than 35 bp were removed with ShortRead 1.16.3 [39] and adapters on the remaining reads were trimmed with cutadapt 1.7.1 [40].
RNAseq data analysis
Annotation of genomic features was performed using the reference genomes of S. cerevisiae S288c and L. thermotolerans CBS6340. In the case of L. thermotolerans unknown genes were identified by the homology with the S. cerevisiae S288c genome. Reads from L. thermotolerans and S. cerevisiae monoculture fermentation samples were aligned to the reference genomes of the two yeasts with TopHat v2.0.13 [41]; and reads that were non-primary mapping or had a mapping quality ≤20, were removed. Subsequently, cross-mapping between S. cerevisiae S288c and L. thermotolerans was evaluated to determine the impact of merging the genomes. Cross-mapping between the two yeasts was found to be less than 1%; consequently, pre-processed reads of all fermentations were aligned to the reference genome of S288cplusLT. The obtained bam files were further converted in to gff files to analyse the data further. The number of reads in the alignments that overlap with gene features were counted using htseq-count 0.6.1p1 [42]. Genes for which all samples had less than 1 count-per-million were removed and full quantile normalization using the EDASeq package from Bioconductor was applied to correct for sample-specific variation typically introduced by differences in library size and RNA composition. Transcript abundance was measured in Fragments Per Kilobase of exon per Million mapped reads (FPKM).
Identification and statistical analysis of differentially expressed genes
For the selection of differentially expressed genes statistical modelling was used to design the following experiments:
For each gene the coefficients β were estimated with the edgeR 3.8.6 package of Bioconductor [43], by fitting a negative binomial generalized linear model (GLM) [44]. Offsets were used to estimate the models. Subsequently, the model estimates were used to compute contrasts of primary interest which were (i) MIX vs PURE (MIX effect) (ii) AN vs AR (AN effect), (iii) the interaction between MIX and AN effect (INT), (iv) MIX vs PURE, only in AR samples, (v) MIX vs PURE, only in AN samples and (vi) AN vs AR, only in MIX samples. The differential expression was tested with a GLM likelihood ratio test, also implemented in the edgeR 3.8.6 package. The resulting p-values were corrected for multiple testing with Benjamini-Hochberg to control the false discovery rate (FDR) [45]. Genes with an absolute log2-ratio larger than 1 and an adjusted p-value < 0.05 were considered differentially expressed.
Additional files
Table S1. The table provides details of number of reads per sample sequenced and after number of reads used to analyse the data after removing bad sequences and reads less than 35 bp (DOCX 27 kb)
Table S2. A list of genes differentially expressed in Saccharomyces cerevisiae and Lachancea thermotolerans in mixed fermentations compared to monocultures. (XLSX 1539 kb)
Table S3. A summary of genes differentially expressed in different metabolic processes in Saccharomyces cerevisiae and Lachancea thermotolerans due to mixing and the interaction between mixing and anoxia (DOCX 49 kb)
Table S4. Up-regulated genes involved in the metabolism of cysteine, methionine, phenylalanine and β-alanine in Lachancea thermotolerans (DOCX 28 kb)
Acknowledgments
We would like to thank Arrie Arends, a Senior technical officer in Department of Biochemistry for helping with Bioreactors; VIB Nucleomics core in Leuven, Belgium for performing RNA-sequencing, analysing data and their support and assistance at several points; International office, Stellenbosch University, for providing the travel grant to Kirti Shekhawat for a visit to VIB Genomics.
Funding
This research for the present study was funded by the National Research Foundation (NRF) Grant Unique Number 83471 and the National Research Foundation-Technology and Human Resources for Industry Programme (grant TP14070874355). The opinions expressed and conclusions arrived at, are those of the authors and are not necessarily to be attributed to the funding agencies.
Availability of data and materials
The data set supporting the results of this article is included within the article and its additional files. The raw data from the RNAseq samples were submitted to the National Centre for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database, under project GSE112581, accession numbers GSM3073202 - GSM3073211. The data are publicly available and accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112581
Abbreviations
- ADP
Adenosine di-phosphate
- AN
Anaerobic
- AR
Aerobic
- ATP
Adenosine tri-phosphate
- BF
Biological function
- BP
Biological processes
- CC
Cellular component
- cDNA
Complementary DNA
- CFU
Colony forming units
- DEG
Differentially expressed genes
- DNA
Deoxyribonucleic acid
- DO
Dissolved oxygen
- FC
Fold change
- FDR
False discovery rate
- FPKM
Fragments per kilobase of exon per million mapped reads
- GC-FID
Gas chromatography with flame ionization detection
- GEO
Gene expression omnibus
- GLM
Generalized linear model
- GO
Gene ontology
- HPLC
High performance liquid chromatography
- IWBT
Institute for wine biotechnology
- KEGG
Kyoto encyclopedia of genes and genomes
- Lt
Lachancea thermotolerans
- Mb
Million base
- NADP
Nicotinamide adenine dinucleotide phosphate
- NCBI
National Centre for Biotechnology Information
- NRF
National Research Foundation
- PCA
Principal component analysis
- RIN
RNA integrity number
- RNA
Ribonucleic acid
- Sc
Saccharomyces cerevisiae
- TPM
Transcripts per million
- YPD
Yeast potato dextrose
Authors’ contributions
KS, FFB and MES conceptualized and designed the experiments, KS performed all the experiments, analysed data and wrote the draft manuscript; FFB and MES supervised the project and edited the manuscript. HP provided bioinformatics support and guidance. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kirti Shekhawat, Email: kirtishekhawat9@gmail.com.
Hugh Patterton, Email: hpatterton@sun.ac.za.
Florian F. Bauer, Email: fb2@sun.ac.za
Mathabatha E. Setati, Email: setati@sun.ac.za
References
- 1.Moenne MI, Saa P, Felipe Laurie V, Ricardo Pérez-Correa J, Agosin E. Oxygen incorporation and dissolution during industrial-scale red wine fermentations. Food Bioprocess Technol. 2014;9:2627–36. doi: 10.1007/s11947-014-1257-2. [DOI] [Google Scholar]
- 2.Boulton RB, Singleton VL, Bisson LF, Kunkee RE. Principles and practices of winemaking. New York: Springer-Verlag Inc.; 1996. [Google Scholar]
- 3.Hansen EH, Nissen P, Sommer P, Nielsen JC, Arneborg N. The effect of oxygen on the survival of non-Saccharomyces yeasts during mixed culture fermentations of grape juice with Saccharomyces cerevisiae. J Appl Microbiol. 2001;91:541–547. doi: 10.1046/j.1365-2672.2001.01426.x. [DOI] [PubMed] [Google Scholar]
- 4.Quirós M, Rojas V, Gonzalez R, Morales P. Selection of non-Saccharomyces yeast strains for reducing alcohol levels in wine by sugar respiration. Int J Food Microbiol. 2014;181:85–91. [DOI] [PubMed]
- 5.Shekhawat K, Bauer FF, Setati ME. Impact of oxygenation on the performance of three non-Saccharomyces yeasts in co-fermentation with Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2017;101:2479–2491. doi: 10.1007/s00253-016-8001-y. [DOI] [PubMed] [Google Scholar]
- 6.Ciani M, Beco L, Comitini F. Fermentation behavior and metabolic interactions of multistarter wine yeast fermentations. Int J Food Microbiol. 2006;108:239–245. doi: 10.1016/j.ijfoodmicro.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Ciani M, Capece A, Comitini F, Canonico L, Siesto G, Romano P. Yeast interactions in inoculated wine fermentation. Front Microbiol. 2016;7:1–7. doi: 10.3389/fmicb.2016.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciani M, Comitini F, Mannazzu I, Domizio P. Controlled mixed culture fermentation: a new perspective on the use of non-Saccharomyces yeasts in winemaking. FEMS Yeast Res. 2010;10:123–133. doi: 10.1111/j.1567-1364.2009.00579.x. [DOI] [PubMed] [Google Scholar]
- 9.Comitini F, Gobbi M, Domizio P, Romani C, Lencioni L, Mannazzu I, Ciani M. Selected non-Saccharomyces wine yeasts in controlled multistarter fermentations with Saccharomyces cerevisiae. Food Microbiol. 2011;28:873–882. doi: 10.1016/j.fm.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Gobbi M, Comitini F, Domizio P, Romani C, Lencioni L, Mannazzu I, Ciani M. Lachancea thermotolerans and Saccharomyces cerevisiae in simultaneous and sequential co-fermentation: a strategy to enhance acidity and improve the overall quality of wine. Food Microbiol. 2013;33:271–281. doi: 10.1016/j.fm.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Sadoudi M, Tourdot-Marechal R, Rousseaux S, Steyer D, Gallardo-Chacon JJ, Ballester J, Vichi S, Guerin-Schneider R, Caixach J, Alexandre H. Yeast-yeast interactions revealed by aromatic profile analysis of sauvignon blanc wine fermented by single or co-culture of non-Saccharomyces and Saccharomyces yeasts. Food Microbiol. 2012;32:243–253. doi: 10.1016/j.fm.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Soden A, Francis IL, Oakey H, Henschke PA. Effect of co-fermentation with Candida stellata and Saccharomyces cerevisiae on the aroma and composition of chardonnay wine. Aust J Grape Wine Res. 2000;6:21–30. doi: 10.1111/j.1755-0238.2000.tb00158.x. [DOI] [Google Scholar]
- 13.Nissen P, Nielsen D, Arneborg N. Viable Saccharomyces cerevisiae cells at high concentrations cause early growth arrest of non-Saccharomyces yeasts in mixed cultures by a cell-cell contact- mediated mechanism. Yeast. 2003;20:331–341. doi: 10.1002/yea.965. [DOI] [PubMed] [Google Scholar]
- 14.Pérez-Nevado F, Albergaria H, Hogg T, Girio F. Cellular death of two non-Saccharomyces wine-related yeasts during mixed fermentations with Saccharomyces cerevisiae. Int J Food Microbiol. 2006;108:336–345. doi: 10.1016/j.ijfoodmicro.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Albergaria H, Francisco D, Gori K, Arneborg N, Girio F. Saccharomyces cerevisiae CCMI 885 secretes peptides that inhibit the growth of some non-Saccharomyces wine-related strains. Appl Microbiol Biotechnol. 2010;86:965–972. doi: 10.1007/s00253-009-2409-6. [DOI] [PubMed] [Google Scholar]
- 16.Rossouw D, Meiring S, Bauer FF. Modifying Saccharomyces cerevisiae adhesion properties regulates yeast ecosystem dynamics. mSphere. 2018;3:e00383–18. [DOI] [PMC free article] [PubMed]
- 17.Barbosa C, Mendes-Faia A, Lage P, Mira NP, Mendes-Ferreira A. Genomic expression program of Saccharomyces cerevisiae along a mixed-culture wine fermentation with Hanseniaspora guilliermondii. Microb Cell Factories. 2015;14:124. doi: 10.1186/s12934-015-0318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bron PA, Wels M, Bongers RS, van Bokhorst-van V, Wiersma A, Overmars L, Marco ML, Kleerebezem M. Transcriptomes reveal genetic signatures underlying physiological variations imposed by different fermentation conditions in Lactobacillus plantarum. PLoS One. 2012;7:e38720. [DOI] [PMC free article] [PubMed]
- 19.de Groot MJL, Daran-Lapujade P, van Breukelen B, Knijnenburg TH, pronk JT, Slijper M, Heck AJR. Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post-transcriptional regulation of key cellular processes. Microbiology. 2007;153:3864–3878. doi: 10.1099/mic.0.2007/009969-0. [DOI] [PubMed] [Google Scholar]
- 20.Gasch AP, Spellma PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koskenniemi K, Laakso K, Koponen J, Kankainen M, Greco D, Auvinen P, Savijoki K, Nyman TA, Surakka A, Salusjarvi T, de Vos WM, Tynkkynen S, Kalkkinen N, Varmanen P. Proteomics and transcriptomics characterization of bile stress response in probiotic Lactobacillus rhamnosus GG. Mol Cell Proteomics. 2011;10:M110.002741. [DOI] [PMC free article] [PubMed]
- 22.Lee JY, Pajarillo EA, Kim MJ, Chae JP, Kang DK. Proteomic and transcriptional analysis of Lactobacillus johnsonii PF01 during bile salt exposure by iTRAQ shotgun proteomics and quantitative RT-PCR. J Proteome Res. 2013;12:432–443. doi: 10.1021/pr300794y. [DOI] [PubMed] [Google Scholar]
- 23.Mostert TT, Divol B. Investigating the proteins released by yeasts in synthetic wine fermentations. Int J Food Microbiol. 2014;171:108–118. doi: 10.1016/j.ijfoodmicro.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Rossouw D, Du Toit M, Bauer FF. The impact of co-inoculation with Oenococcus oeni on the transcriptome of Saccharomyces cerevisiae and on the flavour-active metabolite profiles during fermentation in synthetic must. Food Microbiol. 2012;29:121–131. doi: 10.1016/j.fm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Tronchoni J, Curiel JA, Morales P, Torres-Pérez R, Gonzalez R. Early transcriptional response to biotic stress in mixed starter fermentations involving Saccharomyces cerevisiae and Torulaspora delbrueckii. Int J Food Microbiol. 2017;241:60–68. doi: 10.1016/j.ijfoodmicro.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 26.González Siso MI, Cerdán ME. Kluyveromyces lactis: a suitable yeast model to study cellular defense mechanisms against hypoxia-induced oxidative stress. Oxidative Med Cell Longev. 2012;2012:634674. [DOI] [PMC free article] [PubMed]
- 27.Baumann K, Dato L, Graf AAB, Frascotti G, Dragositis M, Porro D, Mattanovich D, Ferrer P, Branduardi P. The impact of oxygen on the transcriptome of recombinant S. cerevisiae and P. pastoris – a comparative analysis. BMC Genomics. 2011;12:218. doi: 10.1186/1471-2164-12-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Philpott CC. Iron uptake in fungi: a system for every source. Biochim Biophys Acta. 2006;1763:636–645. doi: 10.1016/j.bbamcr.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Gross C, Kelleher M, Iyer V, Brown P, Winge D. Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. J Biol Chem. 2000;275:32310–32316. doi: 10.1074/jbc.M005946200. [DOI] [PubMed] [Google Scholar]
- 30.van Bakel H, Strengman E, Wijmenga C, Holstege F. Gene expression profiling and phenotype analyses of S. cerevisiae in response to changing copper reveals six genes with new roles in copper and iron metabolism. Physiol Genomics. 2005;22:356–367. doi: 10.1152/physiolgenomics.00055.2005. [DOI] [PubMed] [Google Scholar]
- 31.Hodgins-Davis A, Adomas AB, Warringer J, Townsend JP. Abundant gene-by-environment interactions in gene expression reaction norms to copper within Saccharomyces cerevisiae. Genome Biol Evol. 2012;11:1061–1079. doi: 10.1093/gbe/evs084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Z, van Vuuren HJJ. Functional analyses of PAU genes in Saccharomyces cerevisiae. Microbiology. 2009;155:4036–4049. doi: 10.1099/mic.0.030726-0. [DOI] [PubMed] [Google Scholar]
- 33.Rivero D, Berná L, Stefanini I, Baruffini E, Bergerat A, Csikász-Nagy A, Filippo C, Cavalieri D. Hsp12p and PAU genes are involved in ecological interactions between natural yeast strains. Environ Microbiol. 2015;17:3069–3081. doi: 10.1111/1462-2920.12950. [DOI] [PubMed] [Google Scholar]
- 34.Henschke P, Jiranek V. Yeasts-metabolism of nitrogen compounds. In: Fleet GH, editor. Wine microbiology and biotechnology. Chur: Harwood Academic; 1993. pp. 77–164. [Google Scholar]
- 35.Ough CS, Davenport M, Joseph K. Effect of certain vitamins on growth and fermentation rate of several commercial active dry wine yeasts. Am J Enol Vitic. 1989;40:208–213. [Google Scholar]
- 36.Louw L, Tredoux AGJ, Van Rensburg P, Kidd M, Naes T, Nieuwoudt HH. Fermentation-derived aroma compounds in varietal young wines from South Africa. S Afr J Enol Vitic. 2010;31:213–225. [Google Scholar]
- 37.Schmitt ME, Brown TA, Trumpower BL. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 1990;18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.HannonLab. Fastx-toolkit. 2010. http://hannonlab.cshl.edu/fastx_toolkit/index.html. Accessed May 2016.
- 39.Morgan M, Anders S, Lawrence M, Aboyoun P, Pagès H, Gentleman R. ShortRead: a Bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics. 2009;25:2607–2608. doi: 10.1093/bioinformatics/btp450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:1. [Google Scholar]
- 41.Trapnell C, Pachter I, Salzberg SL. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The table provides details of number of reads per sample sequenced and after number of reads used to analyse the data after removing bad sequences and reads less than 35 bp (DOCX 27 kb)
Table S2. A list of genes differentially expressed in Saccharomyces cerevisiae and Lachancea thermotolerans in mixed fermentations compared to monocultures. (XLSX 1539 kb)
Table S3. A summary of genes differentially expressed in different metabolic processes in Saccharomyces cerevisiae and Lachancea thermotolerans due to mixing and the interaction between mixing and anoxia (DOCX 49 kb)
Table S4. Up-regulated genes involved in the metabolism of cysteine, methionine, phenylalanine and β-alanine in Lachancea thermotolerans (DOCX 28 kb)
Data Availability Statement
The data set supporting the results of this article is included within the article and its additional files. The raw data from the RNAseq samples were submitted to the National Centre for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database, under project GSE112581, accession numbers GSM3073202 - GSM3073211. The data are publicly available and accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112581