

Abstract
Patient: Male, 6
Final Diagnosis: Chronic cholesteatomatous otitis media
Symptoms: Otalgia and major otorrhea in the right ear
Medication: —
Clinical Procedure: Opened tympanoplasty surgery
Specialty: Otolaryngology
Objective:
Rare disease
Background:
The Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome is characterized by congenital hypothyroidism, facial dysmorphism, postaxial polydactyly, and mental retardation. The SBBYS variant of Ohdo syndrome is extremely rare with only 19 cases previously reported in the literature. A case is presented of chronic otitis media associated with cholesteatoma in a six-year-old boy with the SBBYS variant of Ohdo syndrome.
Case Report:
A 6-year-old boy presented with perforation of the tympanic membrane and a cholesteatoma in the mesotympanic-attic region associated with chronic otitis media. The child had previously been diagnosed with the SBBYS variant of Ohdo syndrome. Following computed tomography (CT) and magnetic resonance imaging (MRI), tympanoplasty was performed with removal of the lesion.
Conclusions:
This is the first case described in the literature of chronic otitis media associated with cholesteatoma in a patient with the SBBYS variant of Ohdo syndrome. This case demonstrates the importance of specialist otolaryngology referral for patient management.
MeSH Keywords: Cholesteatoma; Hospitals, Pediatric; Otorhinolaryngologic Diseases; Rare Diseases
Background
The Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome is characterized by the presence of congenital hypothyroidism, polydactyly, mental retardation, and facial dysmorphism that can include microcephaly, blepharophimosis, a globose nose, thin lips, cleft palate, low-hanging ears, and micrognathia [1]. The incidence of this syndrome variant is less than 1 per 1,000,000 individuals, and fewer than 20 cases have been described in the literature [1]. The SBBYS variant of Ohdo syndrome was first described by Young and Simpson in 1987 [1]. Cryptorchidism is present in males and some patients also present with interventricular cardiomyopathies, hypotonia, and growth retardation [1].
The SBBYS variant of Ohdo syndrome is caused by mutations in the K(lysine) acetyltransferase (KAT6B) gene, which encodes the enzyme, histone acetyltransferase [2]. Histone acetyltransferase modifies histones, which are the structural proteins that bind to DNA and give chromosomes their shape, and deficiency of this enzyme affects the regulation of several genes during early development [2]. However, it is unclear how these changes lead to the specific characteristics of the SBBYS variant of Ohdo syndrome. Almost all reported cases of the SBBYS variant of Ohdo syndrome are the result of new gene mutations and have occurred in individuals with no family history of the condition [3].
This report is the first to describe a case of chronic otitis media associated with cholesteatoma in a child with the SBBYS variant of Ohdo syndrome. After diagnosis by otomicroscopy, computed tomography (CT), and magnetic resonance imaging (MRI), the patient was successfully treated surgically by tympanoplasty.
Case Report
A 6-year-old child presented to the ear, nose, and throat (ENT) department of our hospital in October 2017 with a history of frequent episodes of otalgia and severe otorrhea of the right ear, which had been previously treated, unsuccessfully, with corticosteroids and antibiotic therapy. The patient also had symptoms of chronic nasal obstruction. He had a known diagnosis of the Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome, resulting from de novo mutations of the KAT6B gene encoding histone acetyltransferase. The diagnosis had been made by the Complex Operative Unit of Genetic and Pediatric Immunology of Messina, by a group who have previously diagnosed and reported previous cases of the SBBYS variant of Ohdo syndrome [4]. The child’s parents reported that his past medical history included congenital hypothyroidism, and he had previously undergone bilateral orchidopexy and surgery for trigger finger.
On the most recent hospital admission, general physical examination of the child showed a delay in growth, with hypotonia, blepharophimosis (bilateral ptosis with reduced eyelid size), and a globose nose. Otomicroscopy of the right ear showed perforation of the upper posterior quadrant of the tympanic membrane, with spontaneous atticotomy and the presence of white scales in the mesotympanic and attic region. On the left side, tympanic membrane was intact with a clean posterior retraction pocket. Pure tone audiometry (PTA) testing demonstrated moderate bilateral conductive hearing loss (Figure 1). Nasal endoscopy showed patent bilateral nasal passages and a mild degree of enlargement of the adenoidal tissue in the nasopharynx.
Figure 1.

Pure tone audiometry shows mild to moderate bilateral transmissive hearing loss.
Computed tomography (CT) imaging was performed three weeks later, which showed the presence of amorphous tissue in the right ear, which appeared to invade the mastoid cells, the antrum, the aditus of the mastoid antrum and the tegmen tympani, and involved the ossicles and the hypotympanum, the mesotympanum, and the protympanum. Magnetic resonance imaging (MRI) was performed while the child was sedated, which supported a diagnosis of cholesteatoma that showed hypointensity in the T1-weighted image and hyperintensity in the T2-weighted image (Figure 2). MRI was able to define the size and composition of the lesion and to identify any intratemporal and intracranial complications.
Figure 2.
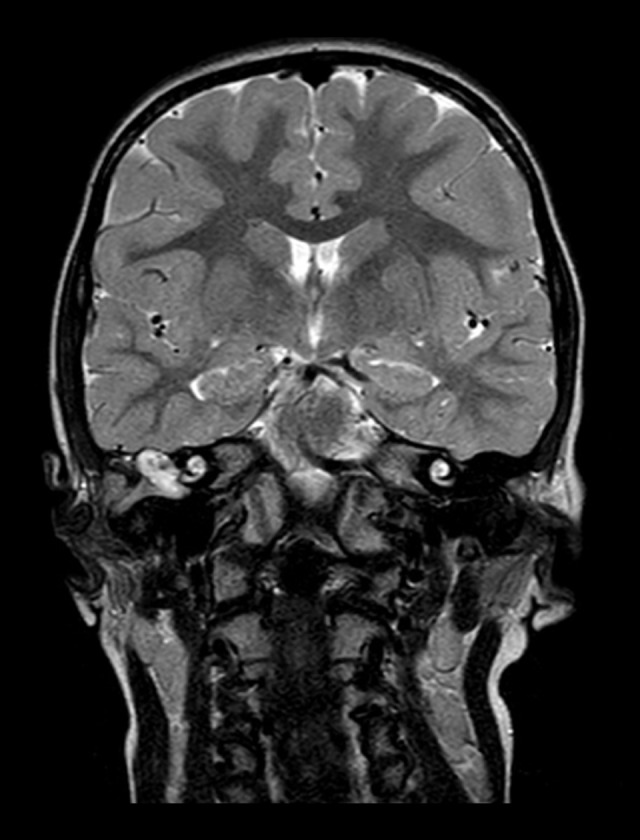
T2-weighted magnetic resonance imaging (MRI) of the brain shows the cholesteatoma of the right middle ear.
The use of imaging using diffusion-weighted imaging (DWI) MRI sequences supported a diagnosis of cholesteatoma of the middle ear. The high content of keratin in the lesion resulted in a hyperintense signal in the DWI sequences. The imaging findings did not support a diagnosis of cholesterol granuloma of the petrous apex, which typically shows hyperintense signals in both T1-weighted and T2-weighted MRI sequences, which were not present in this case [5]. Following the imaging findings, surgery was planned. Preoperative cardiologic evaluation and electrocardiographic (ECG) examination were performed and showed that the child was fit for surgery. Hematologic and laboratory investigations were normal.
The patient underwent tympanoplasty of the right ear in January 2018. The surgery included externalization of the atticus, adidus, and antrum, with the removal of the posterior wall of the meatus and lowering of the facial ridge. At surgery, the cholesteatoma was found to invade the atticus, the mesotympanum, the tympanic sinus, the region of the oval and round windows, part of the protympanum, and the hypotympanum. The cholesteatoma had destroyed the incus and stapedius. The cholesteatoma was removed with surgical clearance and irrigation of the cavity. The remaining anterior tympanic membrane was reversed to create a new anterior tympanic box. The patient was discharged from the hospital four days after surgery and had a good postoperative course. Follow-up at three months included otomicroscopy, which showed good outcome with the absence of residual cholesteatoma.
Discussion
There have been few previously reported cases of the Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome. Previously reported otological associations with the SBBYS variant of Ohdo syndrome include low attachment of the ears, the presence of a prominent antihelix, and malformations of the earlobes and helix [1]. Because the previously reported associations affect the outer ear, there have no previous reports of cases of hearing loss. To our knowledge, this is the first case of the SBBYS variant of Ohdo syndrome presenting with chronic otitis media associated with cholesteatoma.
In cases of pediatric cholesteatoma, it is important to determine whether a congenital or acquired cholesteatoma is present. Congenital cholesteatoma originates from residual embryonic epithelium that remains trapped in the tympanic cavity during embryogenesis. Congenital cholesteatoma presents as a pale mass that may be visible through an intact and transparent tympanic membrane, and patients have a negative history of otitis media [6]. In this case, there was perforation of the tympanic membrane and the patient had a history of chronic otitis media, previously treated with corticosteroids and antibiotics, but with poor results. Given the clinical history, the diagnosis was that of otitis media due to acquired cholesteatoma. It was assumed that the cholesteatoma might have arisen as a consequence of altered drainage of the middle ear due to Eustachian tube dysfunction.
In this case, Eustachian tube dysfunction might have been present for two reasons. First, the patient had general hypotonia, which is a feature of the SBBYS variant of Ohdo syndrome and which could cause dysfunction of the main muscles that regulate the physiological opening and closing of the Eustachian tube, including the tensor veli palatini, the levator veli palatini, the salpingopharyngeus, and the tensor tympani [7]. Second, the craniofacial malformations associated with the SBBYS variant of Ohdo syndrome can alter the physiology and the anatomy of the Eustachian tube. The major functions of the Eustachian tube are drainage, pressure equalization, and protection. Cleft palate may affect all these functions, with drainage probably being the most important.
There has been some speculation regarding the relationship between the cranial base and the Eustachian tube and the development of otitis media. When the cranial base is relatively flat, and the Eustachian tube is horizontal, the risk of developing otitis media increases. Also, there may be dysfunction of the Eustachian tube dysfunction caused by abnormal cartilage and abnormal musculature of the Eustachian tube or because the cranial base is abnormal and the musculature is altered. Children with craniostenosis have a small nasopharynx, which also may result in dysfunction of the Eustachian tube [8,9]. In the patient presented in this report, there was a conductive hearing loss in the left ear, and a tympanic membrane with posterior retraction pocket, which were two findings that would require careful follow-up, due to the association with cholesteatoma.
During growth, it would be important to follow-up patients with the SBBYS variant of Ohdo syndrome in specialist otolaryngology departments, because craniofacial malformations can worsen nasal breathing and function of the Eustachian tube, causing obstructive sleep apnea syndrome and recurrent otitis media [10,11]. It would also be appropriate to perform tests for atopy because the presence of allergic rhinitis would be additionally disabling in patients with the SBBYS variant of Ohdo syndrome [12,13].
Conclusions
Although rare, chronic otitis media associated with cholesteatoma can lead to severe complications such as meningitis, brain abscess, and sinus thrombosis with otogenic hydrocephalus. These complications place the patient’s life at serious risk. Pediatricians should recommend an early evaluation by an experienced otolaryngologist of patients with the Say-Barber-Biesecker-Young-Simpson (SBBYS) variant of Ohdo syndrome to prevent significant complications from the malformations associated with this condition.
Footnotes
Conflict of interest
None.
References:
- 1.Masuno M, Imaizumi K, Okada T, et al. Young-Simpson syndrome: Further delineation of a distinct syndrome with congenital hypothyroidism, congenital heart defects, facial dysmorphism, and mental retardation. Am J Med Genet. 84(1):8–11. doi: 10.1002/(sici)1096-8628(19990507)84:1<8::aid-ajmg2>3.0.co;2-2. 199. [DOI] [PubMed] [Google Scholar]
- 2.Clayton-Smith J, O’Sullivan J, Daly S, et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am J Hum Genet. 2011;89(5):675–81. doi: 10.1016/j.ajhg.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li G, Li N, Li J, et al. De dovo mutation of KAT6B gene causing atypical Say-Barber-Biesecker-Young-Simpson syndrome or genitopatellar syndrome. Fetal Pediatr Pathol. 2017;36(2):130–38. doi: 10.1080/15513815.2017.1281364. [DOI] [PubMed] [Google Scholar]
- 4.Szakszon K, Salpietro C, Kakar N, et al. De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber/Biesecker/Young-Simpson syndrome. Am J Med Genet A. 2013;161A(4):884–88. doi: 10.1002/ajmg.a.35848. [DOI] [PubMed] [Google Scholar]
- 5.Grinblat G, Vashishth A, Galetti F, et al. Petrous apex cholesterol granulomas: Outcomes, complications, and hearing results from surgical and wait-and-scan management. Otol Neurotol. 2017;38(10):e476–85. doi: 10.1097/MAO.0000000000001578. [DOI] [PubMed] [Google Scholar]
- 6.Mostafa BE, El Fiky L. Congenital cholesteatoma: The silent pathology. ORL J Otorhinolaryngol Relat Spec. 2018;80(2):108–16. doi: 10.1159/000490255. [DOI] [PubMed] [Google Scholar]
- 7.Verloes A, Bremond-Gignac D, Isidor B, et al. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006;140(12):1285–96. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- 8.Sando I, Shibahara Y, Wood RP. Congenital anomalies of the external and middle ear. In: Bluestone CD, Stool SE, editors. Pediatric Otolaryngology. 2nd edn. Vol. 1. Philadelphia: WB Saunders; 1990. pp. 271–304. [Google Scholar]
- 9.Fria TJ, Paradise JL, Sabo DL, Elster BA. Conductive hearing loss in infants and young children with cleft palate. J Pediatr. 1987;3:84–87. doi: 10.1016/s0022-3476(87)80348-7. [DOI] [PubMed] [Google Scholar]
- 10.Ottaviano G, Maculan P, Borghetto G, et al. Nasal function before and after rapid maxillary expansion in children: A randomized, prospective, controlled study. Int J Pediatr Otorhinolaryngol. 2018;115:133–38. doi: 10.1016/j.ijporl.2018.09.029. [DOI] [PubMed] [Google Scholar]
- 11.Cammaroto G, Montevecchi F, D’Agostino G, et al. Tongue reduction for OSAHS: TORSs vs coblations, technologies vs techniques, apples vs oranges. Eur Arch Otorhinolaryngol. 2017;274(2):637–45. doi: 10.1007/s00405-016-4112-4. [DOI] [PubMed] [Google Scholar]
- 12.Colavita L, Catalano N, Sposito G, et al. Local allergic rhinitis in pediatric patients: Is IgE dosage in nasal lavage fluid a useful diagnostic method in children? Int J Mol Cell Med. 2017;6(3):174–82. doi: 10.22088/acadpub.BUMS.6.3.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galletti B, Santoro R, Mannella VK, et al. Olfactory event-related potentials: A new approach for the evaluation of olfaction in nasopharyngeal carcinoma patients treated with chemoradiotherapy. J Laryngol Otol. 2016;130(5):453–61. doi: 10.1017/S0022215116000761. [DOI] [PubMed] [Google Scholar]