

ABSTRACT
The development of chemotherapeutic drugs resistance such as doxorubicin (DOX) and cisplatin (DDP) is the major barrier in gastric cancer therapy. Emerging evidences reveal that microRNAs (miRNAs) contribute to chemosensitivity. In this study, we investigated the role of miR-633, an oncogenic miRNA, in gastric cancer chemoresistance. In gastric cancer tissue and cell lines, miR-633 expression was highly increased and correlated with down regulation of Fas-associated protein with death domain (FADD). Inhibition of miR-633 significantly increased FADD protein level and enhanced DOX/DDP induced apoptosis in vitro. MiR-633 antagomir administration remarkably decreased tumor growth in combination with DOX in vivo, suggesting that miR-633 targets FADD to block gastric cancer cell death. We found that the promoter region of miR-633 contained putative binding sites for forkhead box O 3 (Foxo3a), which can directly repress miR-633 transcription. In addition, we observed that DOX-induced nuclear accumulation of Foxo3a leaded to the suppression of miR-633 transcription. Together, our study revealed that miR-633/FADD axis played a significant role in the chemoresistance and Foxo3a regulated this pathway in gastric cancer. Thus, miR-633 antagomir resensitized gastric cancer cells to chemotherapy drug and had potentially therapeutic implication.
KEYWORDS: miR-633, FADD, Foxo3a, gastric cancer, chemosensitivity
Introduction
Gastric cancer ranks the second leading cause of cancer death worldwide [1], and more than 679100 new cases occurred each year in China [2]. Despite the advances in therapeutic options such as surgery, radiotherapy and chemotherapy, the gastric cancer patients still suffer the pain on poor survival, less than 30% age-standardized 5-year survival rate in most countries [3].
Chemotherapy has been widely used to shrink tumor size, repress tumor growth or metastasis, and improve survival and quality of life in patients with carcinoma. Combinations of chemotherapeutic agents effectively improves survival in gastric cancer [4]. For instance, doxorubicin (DOX), a widely used anthracycline chemotherapy agents in cancer treatments past decades, could bind to topoisomerase-2 (Top2) and DNA to form the ternary top2-doxorubicin-DNA cleavage complex, causing tumor cell death [5]. Cisplatin (DDP) is recommended as first line chemotherapy agent in gastric carcinoma [6], can interfere with DNA replication and induce apoptosis of tumor cells. Although plenty of chemotherapeutic agents were used in cancer treatment, majority of these patients eventually experience tumor recurrence due to drug resistance. The median survival duration is lower than 16 months in metastatic and recurrent gastric cancer patients [7]. The failure of chemotherapy makes urgent that understanding the exact molecular mechanism of chemoresistance and finding new drugs for anticancer treatment.
Fas-associated protein with death domain (FADD) is an adaptor that participates in extrinsic pathway of apoptosis induced by tumor necrosis factor (TNF) receptor superfamily. Death receptors activation induce the FADD recruitment, forming the death-inducing signaling complex (DISC) and caspase-8 cleavage, activating apoptotic executioner caspase-3, eventually leading to cell death [8]. Extensive researches have shown that FADD protein expression decreased in malignant tumors [9]. Yoo et al. have reported the loss of FADD protein in gastric cancer and it was in connection with the stage of gastric cancer [10]. Furthermore, loss of FADD expression might contributed to chemotherapy resistance in cancer cells [11,12]. However, the exact molecular mechanism of FADD expression deficiency in chemotherapy-sensitivity remains to characterized.
MicroRNAs are endogenous 18–22 nt small RNA, which dysregulation has been implicated in a variety of diseases, including malignant tumors. Accumulated evidences lighten that miRNAs serve as oncogenes or tumor suppressors in tumor proliferation, metastasis, invasion and drug resistance. It has previously been observed that miR-633 was differentially expressed in cerebro-spinal fluid between primary progressive multiple sclerosis patients and relapsing-remitting multiple sclerosis patients [13]. Abnormal miR-633 expression in childhood acute lymphoblastic leukemia (ALL) is associated with prednisone response and early relapse [14]. To date, few studies have investigated the association between abnormal miR-633 expression and drug chemoresistance in gastric cancer.
The forkhead box O 3 (Foxo3a) is a member of Foxo transcription factor family, a key player in downstream of insulin growth factor receptors. Served as a tumor suppressor in malignant tumors, Foxo3a is involved in tumor cell proliferation, metabolism, cell cycle and apoptosis processes [15,16]. Phosphorylated by AKT and interacted with 14-3-3, Foxo3a underwent degradation and nuclear exclusion, resulting in repression of its transcriptional activity. Previous study showed that transcriptional activity regulation of Foxo3a have a profound influence on the tumor suppression [17], and was associated with respondence to chemotherapy drugs [18].
In this article, we have demonstrated that miR-633 plays a critical role in mediating gastric carcinoma chemoresistance. In response to chemotherapy, Foxo3a, as a transcription factor which relocated in nuclear, suppressed miR-633 transcriptional activity increasing the expression of the target gene FADD, leading to the activation of the apoptotic pathway. Moreover, our results identified for the first time, to our best knowledge, that miR-633 directly target to FADD and might be potential effective chemoresistance reversal agent.
Results
FADD loss is associated with high expression of miR-633 in gastric carcinoma
FADD loss in malignant tumors has been reported in several researches [12,19,20], including in gastric carcinoma [10]. To identify the molecular mechanism underlying the abnormal FADD expression, we analyzed its potential targets by the bioinformatic program TargetScan. We found that miRNAs were aberrantly expressed in cancer tissues, compared with those in noncancerous tissues. Furtherly, most robustly upregulated miRNAs were verified by qRT-PCR. Among them, miR-633 was significantly upregulated in both GC tissues and cell lines. According to previous reports, no detailed mechanism has been reported about this miR-633, therefore we chose miR-633 for further research. We found human FADD mRNA 3ʹ-untranslated regions (UTRs) contained one binding sites for miR-633 (Figure 1(a)). To determine whether FADD affects gastric tumorigenesis, we first evaluated the FADD protein expression levels in human gastric cancer tissues and matched adjacent noncancerous tissues. Three representative results exhibited that low expression of FADD was more likely detectable in gastric cancer tissues compared to the matched adjacent tissues (Figure 1(b)). Then we measured FADD protein levels in gastric cancer cell lines SGC-7901, BGC-823, MGC-803, NCl-N87 and AGS compared with gastric epithelium cell line GES-1. Endogenous levels of FADD were lower in gastric cancer cells than in GES-1 (Figure 1(c)). Immunohistochemical staining showed FADD loss in gastric cancer tissue, further confirmed this potential clinical relevance (Figure 1(d)).
Figure 1.
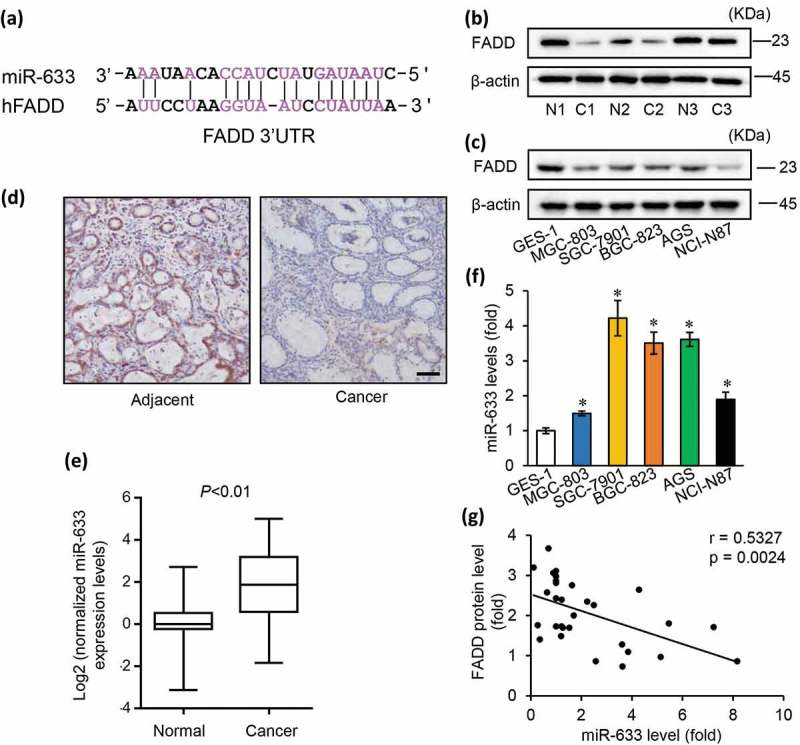
FADD loss is associated with high expressions of miR-633 in gastric carcinoma.
(a) The predicted binding sequences for the miR-633 within the human FADD 3ʹ-UTR. b and c, FADD protein expression levels in gastric carcinoma tissues (b) and gastric cell lines (c), c = cancer tissues; n = matched adjacent tissues. (d) Immunohistochemistry staining of FADD in paraffin sections of patient tissues, scale bar = 50 μm. (e) normalized miR-633 expression levels in normal and cancer tissues from 26 pairs of gastric cancer patients. (f) qRT-PCR data of miR-633 expression in the gastric cell lines. (g) the correlation of miR-633 level and FADD protein level. Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments. *P < 0.05
To verify the potential role of miR-633 in gastric carcinoma, we examined miR-633 expression in 26 pairs of gastric carcinoma and matched normal tissue (Figure 1(e)). The characteristics of the patients were detailed in Supplementary table 1. In corresponded with previous reports [13,21], miR-633 was upregulated in malignant tissue compared with normal tissues. The endogenous levels of miR-633 in gastric cancer cells were relatively higher than in GES-1 cells (Figure 1(f)). The results suggested that miR-633 might play an important role in gastric cancer. The inverse correlation between miR-633 and FADD was confirmed by qRT-PCR analyses of 15 pairs of gastric cancer tissue samples (r = 0.5327, p = 0.0024) (Figure 1(g)). Our data indicate that miR-633 is pathologically and clinically associated with FADD protein expression in gastric carcinoma.
FADD participates in chemotherapy drugs induced apoptosis in gastric tumor cells
We wondered whether abnormal FADD expression was involved in chemotherapy drugs induced apoptosis in gastric cancer. Western blot results showed that SGC-7901 cells (Figure 2(a,b)) and AGS cells (Supplementary Figure S1A, B) exposed to the doxorubicin (2 μM) or cisplatin (30 μM) elevated the expression of FADD, but qRT-PCR results showed that the mRNA expression of FADD remained at the same levels. To determine the role of FADD in gastric cancer, we inhibited FADD expression by transfected FADD siRNA (si-FADD), siRNA control (FADD-sc) as control. Western blot and qRT-PCR were used to confirm that FADD expression can be effectively reduced (Figure 2(c)). Next, we performed apoptotic functional TUNEL assays in SGC-7901 cells. The results showed that inhibition of FADD significantly down-regulated chemotherapy drugs induced apoptosis both in 2 μM DOX (Figure 2(d,e)) or 30 μM cisplatin (Figure 2(f)) treatment for 24 h. The similar results were observed in AGS cells treated with DOX (Supplementary Figure S2C). These data remind us that and FADD might be involved in chemotherapy drugs induced apoptosis.
Figure 2.
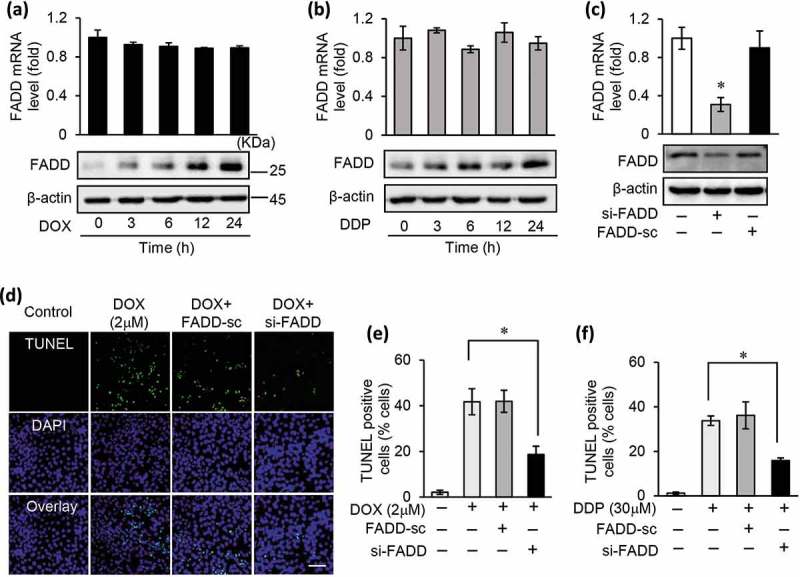
FADD participates in chemotherapy drugs induced apoptosis in gastric tumor cells.
(a and b), the mRNA and protein expression levels of FADD in SGC-7901 cells exposed to 2 μM DOX (a) or 30 μM DDP (b) treatment for 24 h. (c), the mRNA expression and protein expression of FADD in SGC-7901 cells after transfected with FADD siRNA (si-FADD) or scramble siRNA (FADD-sc). (d and e), inhibition of FADD by transfecting si-FADD or FADD-sc prevented apoptosis in SGC-7901 treated with 2 μM DOX for 24 h, apoptotic cells were analyses by TUNEL assay (d) and counted (e). f), inhibition of FADD by transfecting si-FADD or FADD-sc prevented apoptosis in SGC-7901 treated with 30 μM DDP for 24 h.
miR-633 suppresses apoptosis and induced chemoresistance in gastric cancer cells
We hypothesized that dysregulated miR-633 was involved in the development of gastric cancer chemoresistance. To test this hypothesis, we exposed SGC-7901 cells and AGS cells to the doxorubicin (2 μM) or cisplatin (30 μM), and qRT-PCR to assess the expression levels of miR-633 (Figure 3(a), Supplementary Figure S2A, B). The reduction of miR-633 expression induced by doxorubicin or cisplatin suggested that miR-633 might cause apoptosis pathway suppression. In line with this, we synthesized the specific inhibition of miR-633, miR-633 antagomir (Anta-633), antagomir control (Anta-NC) as control. The results confirmed that miR-633 inhibition effectively repressed the miR-633 expression in SGC-7901 cells (Figure 3(b)) and in AGS cells (Supplementary Figure S2C). To examine the possible apoptotic function of miR-633, we performed apoptotic functional TUNEL assays in SGC-7901 cells (Figure 3(c)) and AGS cells (Supplementary Figure S2D). Inhibition of miR-633 alone had slight effect on apoptotic rate. These results suggested that suppression of miR-633 expression could not raise the apoptosis in gastric cancer cells. To further characterize the function of endogenous miR-633, we produced a construct encoding miR-633 to enforce the expression of miR-633 (Supplementary Figure S2E). Next we transfected with miR-633 mimics or mimic control (NC) into SGC-7901 cells treated with 2 μM doxorubicin for 24 h. TUNEL assay showed doxorubicin-induced apoptosis was attenuated by overexpression of miR-633 in SGC-7901 cells (Figure 3(d,e)) and AGS cells (Supplementary Figure S2F). Besides, miR-633 levels were analyzed by qRT-PCR (Figure 2(f) and Supplementary Figure S2G). Western blotting revealed that enforced expression of miR-633 abolished DOX-induced conversion of caspase-3 (Figure 3(g)), suggesting simultaneously declined the cell death ratio in SGC-7901 cells. A similar result was obtained in SGC-7901 cells with the treatment of 30 μM cisplatin (Supplementary Figure S2H). Furthermore, we attempted to investigate the influence of miR-633 on cell susceptibility to chemotherapy. Transfected with miR-633 antagomir into SGC-7901 cells (Figure 2(h–j)) or AGS cells (Supplementary Figure S1I), a limited amount of cells undergoing apoptosis was observed after low-dose doxorubicin (0.2 μM) treatment for 24 h. The data suggested that in response to the same low-dose doxorubicin, inhibition of miR-633 significantly increased the apoptotic cells. The data strongly suggest that miR-633 contributed to chemoresistance in gastric cancer by suppressing the apoptotic pathway.
Figure 3.
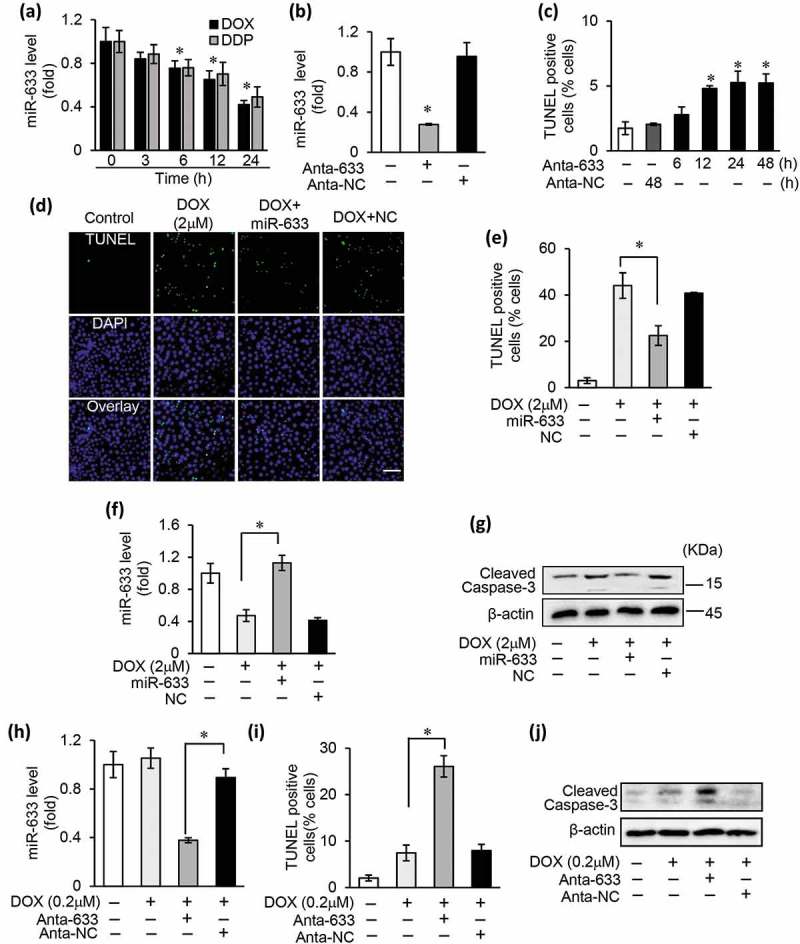
miR-633 suppresses apoptosis and induced chemoresistance in gastric cancer cells.
(a), the expression levels of miR-633 were analyzed by qRT-PCR in SGC-7901 cells exposed to 2 μM doxorubicin (DOX) or 30 μM cisplatin (DDP) treatment for 24 h. (b), real-time PCR in SGC-7901 cells to detect the expression of miR-633 after transfected with miR-633 antagomir (Anta-633) or antagomir control (Anta-NC). (c), TUNEL assay in SGC-7901 cells to detect apoptosis after transfected with Anta-633 or Anta-NC as the indicated time. (d, e, f and g), overexpression of miR-633 by transfecting miR-633 mimics or mimics control (NC) prevented apoptosis in SGC-7901 treated with 2 μM DOX for 24 h, representative images show TUNEL-positive cells (d), scale bar = 100 μm. Apoptotic cells were counted (e), miR-633 expression were detected (f) and cleaved caspase-3 was analyzed by western blot (g). (h, i and j), inhibition of endogenous miR-633 transfected with Anta-633 enhanced apoptosis in SGC-7901 after 24 h 0.2 μM DOX treatment. MiR-633 expression were detected (h), the apoptotic cells were counted after DOX treatment (i), and cleaved caspase-3 was analyzed by western blot (j). Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments.*P < 0.05.
miR-633 regulates chemotherapy resistance through FADD in gastric tumor cells
MiRNAs are reported to negatively regulate gene expression [22,23]. Considering the target gene predictions (Figure 1(a)), we investigated whether inhibition of miR-633 promoted the expression of FADD. Validated by qRT-PCR and western blot, the inhibitory effect of miR-633 significantly elevated the FADD protein expression levels but not the mRNA expression levels both in SGC-7901 cells and AGS cells (Figure 4(a) and Supplementary Figure S3A). Different doses of miR-633 and Anta-633 were used to transfect SGC-7901 with or without DOX treatment, with the purpose of effectively affecting FADD expressions (Supplementary Figure S3B, C). The results suggested that 100 nM miR-633 were efficient to reverse DOX-induced increase of FADD expression; meanwhile, 100 nM Anta-633 made a difference in FADD expression decrease. To identify whether FADD participate the miR-633 mediated drug sensitivity, we overexpressed the miR-633 in SGC-7901 cells and treated them with high dilution of DOX/DDP, and then analyzed FADD expression (Figure 4(b) and Supplementary Figure S3D). Western blot results showed that miR-633 overexpression reversed the promoting effect of FADD expression. In contrast, administration of miR-633 antagomir improved the elevation of FADD protein levels upon low dilution of doxorubicin (Figure 4(c)) or cisplatin (Supplementary Figure S3E) treatment. The data suggested that miR-633 might modulate FADD expression at the post-transcriptional level. To determine the role of FADD in the miR-633 pathway, we also performed functional rescue studies by modulating FADD expression levels and functions. We found that inhibition of FADD expression reversed miR-633 antagomir induced cell death (Figure 4(d)). Based on the results above, it seems that miR-633 modulates FADD expression at the post transcriptional level. Wherefore, we cloned FADD 3ʹ-UTR containing one miR-633 binding sites downstream of the luciferase reporter gene (FADD 3ʹ-UTR-Wt) to testify whether miR-633 directly targets to FADD, mutated luciferase constructs (FADD 3ʹ-UTR-Mut) was used as control (Supplementary Figure S3F). After transfected synthesized miR-633 mimics or miRNA mimic control into HEK-293 cells (Figure 4(e)), the luciferase reporter assays demonstrated that miR-633 repressed the luciferase activity in cells simultaneously transfected with wild-type FADD reporter plasmids, but not with the mutant ones. Furthermore, biotinylated miR-633 (Bio-633), miR-633 mutant (Bio-633mut), or RNA control (Bio-nc) was separately transfected into SGC-7901 cells for FADD 3ʹ-UTR pull-down assays. As shown in Figure 4(f), FADD 3ʹ-UTR could only be enriched significantly in cells transfected by Bio-633. These results indicate that miR-633 could directly bind to FADD 3ʹ-UTR and plays a negative regulatory role for FADD expression. Thus, our data suggest that FADD is a direct target of miR-633, besides miR-633 regulates chemotherapy resistance through FADD in gastric tumor cells.
Figure 4.
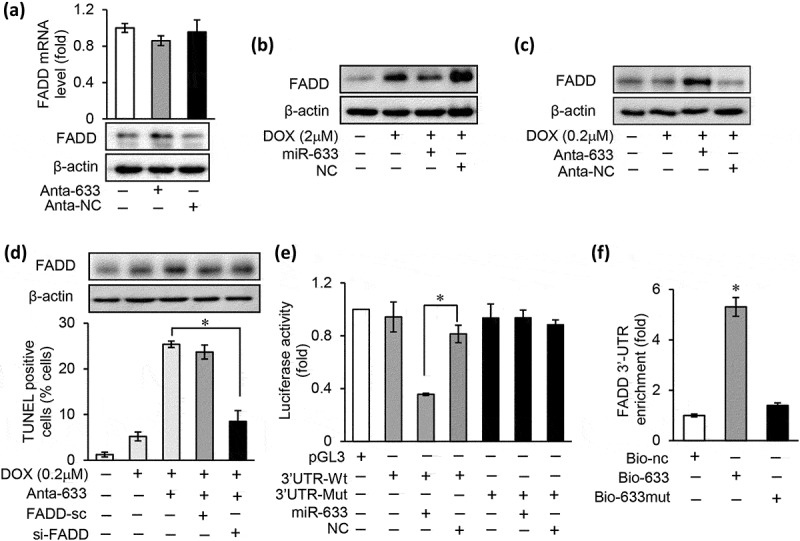
miR-633 regulates chemotherapy resistance through FADD in gastric tumor cells.
(a), the mRNA and protein expression levels of FADD were analyzed by qRT-PCR and western blot in SGC-7901 cells after miR-633 inhibition. (b), overexpression of miR-633 prevented the FADD protein expression increase in SGC-7901 treated with 2 μM DOX for 24 h. (c), inhibition of miR-633 promoted the FADD protein expression in SGC-7901 treated with 0.2 μM DOX 24 h. (d), inhibition of miR-633 enhanced cell death in SGC-7901 in response to 0.2 μM DOX treatment for 36 h, which was reversed by FADD siRNA. (e), luciferase activity detected in HEK-293 transfected with synthesized miR-633 mimics or mimic control, along with human FADD 3ʹUTR luciferase reporter constructs as indicated. (f), detection of FADD 3ʹUTR by qRT-PCR in SGC-7901 cells transfected with Bio-633, Bio-633mut, or Bio-nc by RNA pull-down assay and results were normalized to human GAPDH (n = 3). Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments.*P < 0.05. Mut, mutated; Wt, wild type.
miR-633 is a transcriptional target of Foxo3a
The aberrant miRNAs expression regulated by transcription factors occurred via primary transcription regulation mechanism [24,25], and is associated with tumorigenesis [26,27]. To understand the regulatory mechanism involved in miR-633 induced chemoresistance, we analyzed the promoter region of miR-633. Two putative biding sites for Foxo3a were identified, located at 1259 and 905 nt upstream from the transcription starting site (Figure 5(a)). The chromatin immunoprecipitation (CHIP) analysis was performed to identify whether Foxo3a participated in the regulation of miR-633 expression. The results showed that Foxo3a directly bounded to the binding site 1 (BS1) region but not the binding site 2 (BS2) region in SGC-7901 cells treated with DOX or DDP (Figure 5(b)). Next, we analyzed whether Foxo3a was involved in doxorubicin induced apoptotic program. As a transcriptional factor, Foxo3a was activated by AKT phosphorylation and translocated from nuclear to cytoplasm [28]. In line with this, we analyzed the subcellular distributions of Foxo3a after 2 μM doxorubicin for 24 h. Western blot results showed that total Foxo3a increased in nuclear meanwhile Foxo3a reduced in cytoplasm (Figure 5(c)). The phosphorylated Foxo3a in cytoplasm display a time-dependent decrease, which is in agreement with our previous study [29]. The immunofluorescence of SGC-7901 cells revealed that Foxo3a mainly located in cytoplasm without drug treatment, while after DOX treatment, a large proportion of Foxo3a relocated in nuclear. These results suggested that doxorubicin mediated nuclear relocalization of Foxo3a (Figure 5(d)). The foregoing data led us to consider whether Foxo3a participated in the regulation of miR-633 expression. We next made the assumption that Foxo3a transcriptional repressed miR-633 expression. MiR-633 promotor upstream region was cloned to luciferase reporter gene which contained transcriptional factor Foxo3a binding site 1 (BS1) and binding site 2 (BS2) as previous depicted (Figure 5(a)). Overexpression of Foxo3a induced a significant repression of miR-633 promoter luciferase activity after transfected with Foxo3a wild type promoter 1 (Wt1), and this loss was reversed by introducing mutation of BS1 (m-BS1) (Figure 5(e)). There was no significant difference in luciferase activity between wild type promoter only contained BS2 (Wt2) and BS2 mutation promoter (m-BS2). To further confirm the influence of chemotherapy drug on miR-633 transcription, we transfected si-Foxo3a and Wt1 promotor, and performed luciferase reporter assays (Figure 5(f)). The results presented that DOX/DDP induced luciferase activity repression was rescued by inhibiting Foxo3a expression. These data suggested that Foxo3a directly bound to miR-633 upstream binding site 1 and negatively regulate miR-633 transcriptional activity. Enforced expression of Foxo3a, infected by adenovirus Foxo3a, led to an expression decrease of miR-633 (Figure 5(g)). Thus, our data suggest that Foxo3a had a transcriptional repressive effect on miR-633.
Figure 5.
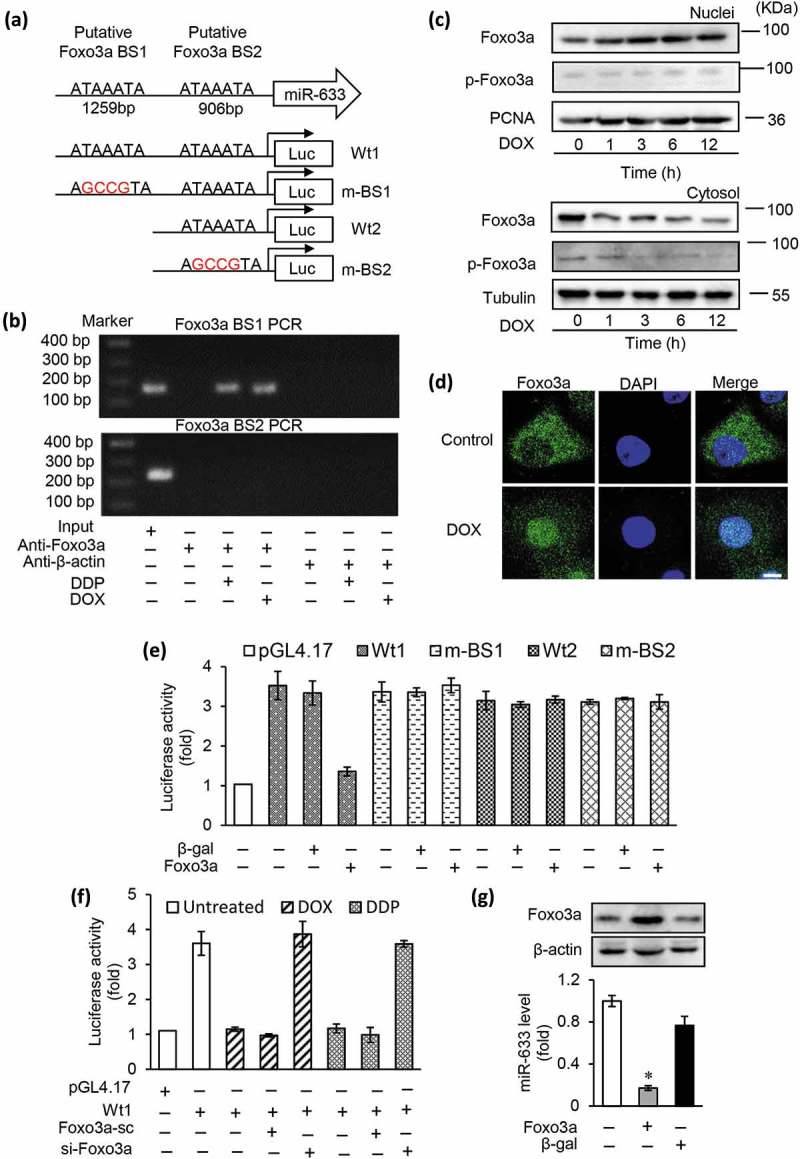
miR-633 is a transcriptional target of Foxo3a.
(a), the promoter region of human miR-633 contains two optimal Foxo3a binding sites (BS1 and BS2). The wild type miR-633 promotors (Wt1 and Wt2) linked to luciferase report gene. The mutations were introduced into the potential binding sites (m-BS1 and m-BS2) as indicated. (b), CHIP analysis was performed in SGC-7901 cells to identify the binding site of Foxo3a in miR-633 promoter treated with 2 μM DOX or 30 μM DDP for 12 h. (c), immunoblotting was performed to analyze the phosphorylated Foxo3a (p-Foxo3a) and total Foxo3a in the cellular fractions of nuclei or cytosol. Proliferating cell nuclear antigen (PCNA) is a nucleic marker. Tubulin is a cytosolic marker. (d), 2 μM DOX induced Foxo3a nuclear relocation from cytoplasm in SGC-7901 cells with 2 μM DOX treatment as the indicated time. Subcellular location was definitude by immunofluorescent staining, cell nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI), scale bar = 10 μm. (e and f), Luciferase activity of reporter constructs harboring wild-type or mutated miR-633 promoter in HEK-293 cells infected with adenoviral Foxo3a as indicated. Foxo3a inhibits miR-633 promotor activity (e), and DOX or DDP induced miR-633 promotor activity reduction was rescued by inhibition of Foxo3a siRNA (f). (g), the miR-633 level and Foxo3a protein expression were analyzed by qRT-PCR and western blot in SGC-7901 cells after infected with adenoviral Foxo3a. Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments.*P < 0.05. Mut, mutated; Wt, wild type, β-gal, β-galactosidase.
Foxo3a regulated chemoresistance dependent on miR-633 and FADD in gastric cancer cells
He et al. reported that Foxo3a contributed to drug resistance in gastric cancer, and low Foxo3a expression levels were detected in cisplatin-resistance phenotype [30]. We identified the Foxo3a expression differences between SGC-7901 cells and cisplatin-resistant gastric cancer cells SGC-7901/DDP (SGC-7901*), and the results indicated Foxo3a protein level was significantly decreased in SGC-7901/DDP cells (Supplementary Figure S4A), which was similar to the previous report. These data remained us that Foxo3a may participate in the miR-633 induced chemoresistance process. To testify this hypothesis, we enforced expressed Foxo3a in SGC-7901 cells. TUNEL staining results showed an increase in the apoptotic cells by Foxo3a overexpression (Figure 6(a)). To examine whether the Foxo3a is involved in the miR-633 pathway, two Foxo3a siRNAs (si-Foxo3a) were selected to lower the expression of Foxo3a (Supplementary Figure S4B). Si-Foxo3a and Foxo3a siRNAs control (Foxo3a-sc) were transfected separately into the SGC-7901 cells with high DOX (Figure 6(b,c), and Supplementary Figure S4C) or DDP (Supplementary Figure S4E, F) dilution treatment. The results showed that the suppression of Foxo3a could restore the loss of miR-633 treated by anticancer drugs. 2 μM DOX-induced cell death abolished by si-Foxo3a transfection, and there were coincide results under 30 μM DDP treatment. On the contrary, enforced expression of Foxo3a significant decreased miR-633 levels, and significantly promoted the apoptosis with the treatment of 0.2 μM DOX (Figure 5(d,e), Supplementary Figure S4F). These results suggested that Foxo3a negatively regulate the expression of miR-633.
Figure 6.
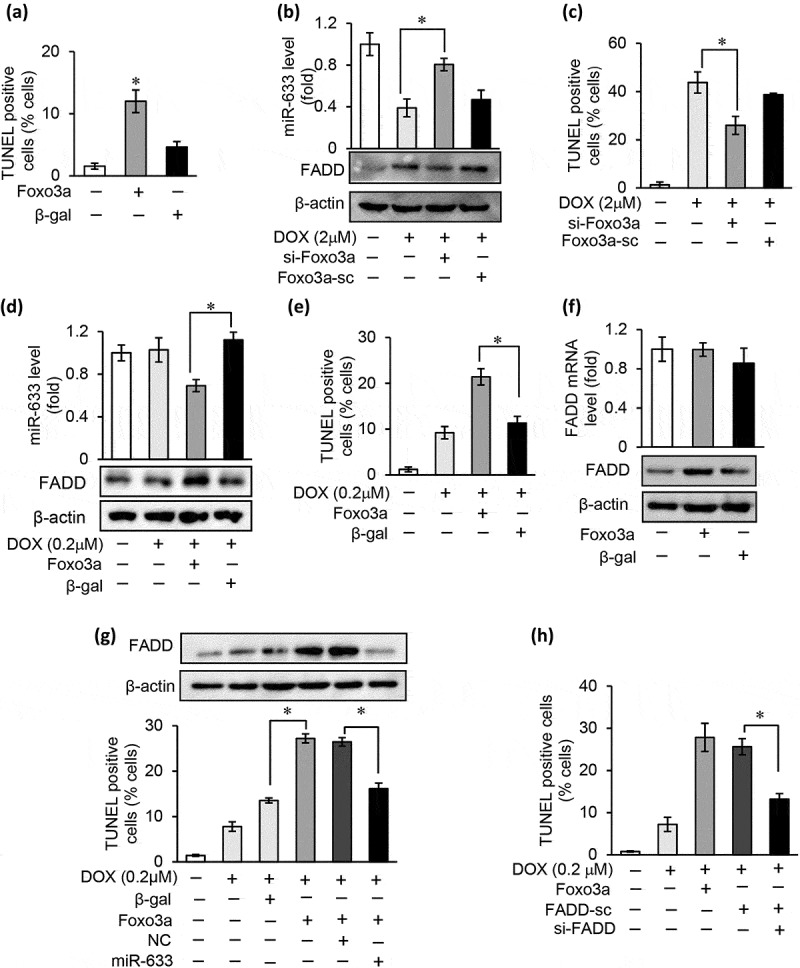
Foxo3a regulates miR-633-induced chemoresistance dependenting on FADD in gastric cancer cells.
(a), TUNEL assay in SGC-7901 cells to detect apoptosis after infected with adenoviral Foxo3a. (b and c), inhibition of Foxo3a by transfect with Foxo3a siRNA (si-Foxo3a) or scramble siRNA (Foxo3a-sc) rescued 2 μM DOX induced cell death in SGC-7901. Real-time PCR to detecte miR-633 level, FADD protein expression were detected by western blot (b) and TUNEL assay to detect cell apoptosis in SGC-7901 cells (c). (d and e), overexpression of Foxo3a treated with 0.2 μM DOX for 24 h in SGC-7901 cells, miR-633 level, FADD protein expression (d) and TUNEL assay (e) were detected. (f), the mRNA and protein expression levels of FADD analyzed by qRT-PCR and western blot in SGC-7901 cells after infected with adenoviral Foxo3a. (g), overexpression of Foxo3a elevated FADD protein level in SGC-7901 cells in response to 0.2 μM DOX treatment for 36 h, which reversed by miR-633 mimics. (h), overexpression of Foxo3a increased cell death in SGC-7901 in response to 0.2 μM DOX treatment for 36 h, which was reversed by FADD siRNA. FADD protein level analyzed by western blot. Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments.*P < 0.05.
To exam whether FADD is needed in miR-633 induced chemoresistance regulated by Foxo3a, we infected SGC-7901 cells with adenovirus to overexpress Foxo3a. The results presented that FADD protein expression level elevated in responded to the Foxo3a overexpression, but not on the mRNA level (Figure 6(f)). To confirm that FADD is a direct downstream of Foxo3a/miR-633 dependent cell death resistance, we infected SGC-7901 cells with adenovirus overexpressing Foxo3a in the presence or absence of miR-633 mimics. Foxo3a overexpression significantly increased FADD protein expression with 0.2 μM DOX treatment, which was suppressed in the presence of miR-633 mimics (Figure 6(g), Supplementary Figure S4G).The data indicated that Foxo3a participated in chemoresistance through regulating miR-633 and its target FADD. To confirm the results, we silenced FADD expression in Foxo3a overexpressing SGC-7901 cells treated with 0.2 μM DOX. As expected, inhibition of FADD expression blocked Foxo3a-induced cell death (Figure 6(h)). Together, these data indicated that Foxo3a-dependent chemoresistance to DOX or DDP develops through suppression of miR-633 and upregulation of FADD in gastric cancer cells.
Inhibition of miR-633 promoted chemotherapeutic effect in vivo
To validate the role of miR-633 as a regulator of gastric cancer chemotherapy resistance in vivo, we examined the miR-633 antagomir function in xenograft models. A total of 1 × 107 SGC-7901 cells were injected subcutaneously into nude mice. Xenograft tumor formation was monitored over a 36-day time course. When xenograft tumors reached 0.25–0.3 cm3, nude mice were randomly distributed into 5 groups. To verify whether miR-633 antagomir alone could inhibit gastric cancer tumor growth, we did intratumor injection of saline (Control), antagomir control (Anta-NC) and miR-633 antagomir (Anta-633) separately. Compared with controls, mice injected miR-633 antagomir resulted in a smaller tumor size and weight (Figure 7(a)), which indicated that inhibition of miR-633 restricted gastric tumor growth in vivo. To identify the drug resistance reversal of miR-633 antagomir, a dose of doxorubicin, 2 mg/kg, were intraperitoneal injected every other day. During 4 weeks of therapy, we monitored the tumor growth (Figure 7(b)) and body weight (not shown) of the mice. In comparison with the drug treatment alone, miR-633 antagomir injection combined with doxorubicin exhibited obvious tumor suppression effect after the 4-week course. The tumors were weighted. The tumor weight results showed significant decrease both in miR-633 antagomir group and antagomir combined with DOX group (Figure 7(a), bottom), in accord with the tumor volume (Figure 7(b)). The expression of miR-633 levels was confirmed by qRT-PCR and FADD expression by western blot (Figure 7(c)). MiR-633 levels remarkably reduced and FADD protein increased, which might contributed to the restriction of tumor growth. TUNEL assay in xenograft exhibit significant elevation of apoptosis in combination therapy (Figure 7(d)). These in vivo results presented that inhibition of miR-633 effective restricted tumor growth, were consistent with results in vitro.
Figure 7.
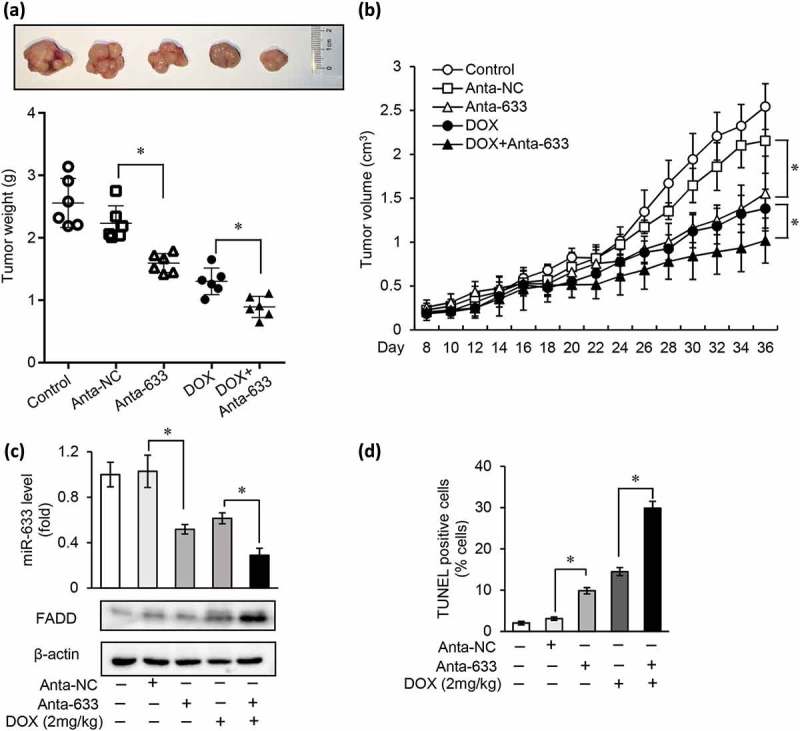
Inhibition of miR-633 promotes chemotherapeutic effect in vivo.
BALB/c nude mice subcutaneously transplanted with a total of 1 × 107 SGC-7901 cells. When tumors reached 250–300 mm3, intratumorally injected with miR-633 antagomir or antagomir control and intraperitoneally treated with DOX (DOX; 2 mg/kg) or PBS (Control) was given every other day. (a and b), Representative images of xenografts (a, upper), tumor weight (a, bottom) and tumor volume (b) are shown. (c), immunoblot of FADD protein (upper) and miR-633 expression levels (bottom) in generated xenograft tumors were analyzed. (d), TUNEL assays were performed to detect tumor cell apoptosis in xenograft tumor tissues. n = 6 each group. Error bars represent S.D. Data are expressed as the mean ± SD of 3 independent experiments.*P < 0.05 compared with DOX (1 mg/kg) alone.
Altogether, we reasoned that accumulation of miR-633 in gastric cancer caused a selective loss of FADD expression and apoptosis suppression, subsequently resulted in chemoresistance, and this miR-633 accumulation transcriptional regulated by Foxo3a nuclear relocalization.
Discussion
Chemotherapy in combination with surgical resection is the predominant therapy strategy in gastric carcinomas. Cisplatin is recommended with routinely used chemotherapy in ECF regimen as first-line setting for patients, which showed a significant survival benefit in patients with gastric cancer [6]. Doxorubicin is a common used anticancer drug both in hematogenous and solid cancers including in gastric cancer [31]. Although most gastric cancer patients responded to multi-agent chemotherapy effectively, over 50% or greater, chemoresistance occurred [32]. Since the novel immune checkpoint inhibitor has not been available in gastric cancer clinical application, understanding the mechanisms of chemoresistance in gastric cancer is essential to therapy regimen optimization.
Cancer chemoresistance results from complex mechanisms such as evasion of apoptosis, DNA repair and insensitivity to drug-induced apoptosis [33]. As tumor suppressors or oncogenes, miRNAs participate in chemoresistance through several ways, including cell proliferation and apoptosis [34,35]. MiR-214 was proved to induce cell survival and cisplatin resistance by targeting to phosphatase and tensin homolog (PTEN), a tumor suppressor that regulates cell proliferation and cell cycle by its phosphatase activity, modulating PTEN/PI3K/Akt pathway in ovarian cancer cells [18]. In Shang’s study, miR-508-5p reversed multidrug resistance through drug transporters ABCB1 regulation. The restoration of miR-508-3p expression led to markedly decreased after chemotherapy in gastric tumor size [36]. Previous investigations implied that miR-633 possibly related to drug resistance in tumor therapy. In endometrial carcinoma cells, miR-633 was down-regulated with the progesterone treatment, a tumor suppressor therapeutically applied to endometrial carcinoma [21]. However, there was few reports about the potential function of miR-633 in cancer. In our research, we identified elevation of miR-633 expression both in gastric cancer tissues and cell lines (Figure 1(e,f)), and the enhancement of chemotherapy sensitivity of gastric cancer cells in low dilution drug treatments both in vitro (Figure 3(h-j)) and in vivo (Figure 7(c,d)). Inhibition of miR-633 effectively restricted the tumor growth both on tumor size and weight (Figure 7(a,b)). These data suggested miR-633 act as an oncogene and could reverse the chemosensitivity in gastric cancer. This chemosensitivity reversal is dependent on death reporter apoptosis, which was further confirmed by later experiments.
FADD is a pivotal member of the extrinsic death signaling apoptosis and participated death-inducing signaling complex formation [37]. In this study, we observed the loss of FADD both in gastric carcinoma tissue and cells (Figure 1(b,c)), which is accord with previous studies [10]. The death receptor function deficiency, including Fas, TNFR1, DR3, DR4, DR5 and FADD loss, contributed to cell death resistance in tumor cells, and anthracyclines such as doxorubicin induced Fas-mediated cell death was suppressed in vitro [8]. In Yan’s report, H19/miR-675 axis regulated cell proliferation and apoptosis, and overexpression of miR-675 or H19 inhibited FADD expression; subsequently suppressed caspase-8 and caspase-3 cleavage cascades [11]. Another possible explanation is post-translational modification in cancer cells, which might contribute to non-apoptosis functions. For example, FADD is post-translationally regulated by Makorin Ring Finger Protein 1(MKRN1) E3 ligase-mediated ubiquitination and proteasmal degradation. Knockdown MKRN1 promoted the stabilization of FADD and sensitivity to extrinsic apoptosis in breast cancer cells, and tumor growth suppression was reversed by both MKRN1 and FADD depletion in a xenograft model [38]. In the present study, we verified that FADD was a direct target of miR-633 in gastric cancer (Figure 4(e,f)). Moreover, in our study expression of miR-633 was inversely correlated with FADD in gastric tissues (Figure 1(g)), suggesting the negatively regulation of miR-633 on FADD expression. In addition, inhibition of miR-633 induced apoptosis was rescued by FADD siRNAs (Figure 4(d)), which demonstrated this apoptotic progress rely on FADD exist.
Foxo3a plays a vital role in gene regulation in tumorigenesis. Resent research report that as transcription factor, Foxo3a inhibited proliferation and induced cell cycle arrest by controlling cyclin-dependent kinase inhibitor p27kip1 [39], contributing to cancer cell apoptosis by regulation of proapoptotic member [40]. Previous reports have showed Foxo3a were overexpressed in less aggressive types of GC [41,42]. Foxo3a aberrantly downregulation in chemo-resistant gastric cancer cells was related to chemoresistance [30], which were verified in our research (Supplementary Figure S4A). Hence, it is of great crucial to explore regulatory mechanisms of expressed Foxo3a in gastric cancer. Foxo3a is known to be a target of Akt pathway, and Akt pathway is activated in many malignant tumors, such as gastric cancer and non-small cell lung cancer [43,44]. The phosphorylated Foxo3a might change the Foxo3a protein formation, loose the combination between Foxo3a and DNA [45], eventually Foxo3a combined 14-3-3 protein to translocate. As the downstream of PI3K-Akt pathway, phosphorylated Foxo3a sequestrate in the cytoplasm in cancer cells, while Foxo3a phosphorylation reduced and translocated into nuclear under stress condition. In this work, we investigated the Foxo3a location in gastric cancer cells, and found out that Foxo3a translocated in nuclear under the DOX treatment and the phosphorylation level was decreased (Figure 5(c)). The phosphorylation site Ser253 locates in nuclear localization signal, and might start nuclear relocation process [46]. Our present study also unveiled that Foxo3a has the transcriptional suppression on miR-633 expression. In our present study, the cytoplasm to nuclear translocation might restrain Foxo3a transcriptional suppression to miR-633, and eventually miR-633 accumulated in gastric carcinoma. It was reported that Foxo3a transactivated miR-34b/c to modulate WNT signaling, resulting in the suppression of epithelial-to-mesenchymal transition in prostate cancer cells [47]. In our research, the subsequent rescue experiments (Figure 6(g,h)) demonstrated that miR-633-induced chemoresistance might be dependent on FADD exist. Our present data revealed that Foxo3a participated in the chemoresistance through transcription suppress miR-633.
As regulators of tumorigenesis and disease occurrence, miRNAs exhibit the potential function of therapeutic intervention. miR-122 has been proved to upregulate hepatitis C virus (HCV) RNA genome, which causes chronic infection [48]. Using miR-122-targeted locked nucleic acids (LNAs) to prevent HCV infection showed promising activity [49], and currently as a therapy in phase Ⅱ clinical trials. MiR-34 has been reported to significantly inhibit tumor growth in mouse models [40]. Therefore, miR-34 mimics, encapsulated in nanoparticles are currently being tested in a phase Ⅰ clinical trial in several solid malignancies. miR-15/16 family were reported to show a higher therapeutic efficiency in reducing tumor weight by combination with cisplatin, suggesting the improvement of the therapeutic response [50]. In our study, the inhibitor of miR-633 alone exhibited restriction of gastric cancer tumor growth. What’s more, the combination of miR-633 inhibitor and chemotherapy agent significantly promoted apoptosis of gastric cancer cells and shrinked the tumor size (Figure7(a,d)). Thus, it is feasible to establish novel chemotherapy strategy with the consideration of potential anticancer miRNAs.
In conclusion, our study presented that miR-633 lowered the chemotherapy sensitivity in gastric cancer. Inhibition of miR-633 exerted tumor-suppressing function in vitro and in vivo through negatively regulating FADD. In addition, this study highlighted the function of Foxo3a at transcriptional level, uncovered the miR-633 regulation axis in response to doxorubicin treatment in gastric cancer cells. Our data suggested that miR-633 may advance our understanding of the chemoresistance of gastric cancer and could be a potential therapeutic strategy for the treatment of chemoresistance in gastric cancer in the future.
Material and methods
Clinical gastric cancer samples and cell lines
We studied patients with gastric cancer samples between 2015–2016 from PLA Army General Hospital (Beijing, China) and Chinese people’s Liberation Army No. 401 (Qingdao, China). Pairs of gastric cancer tissues and matched noncancerous gastric tissues from 26 patients (21 males and 5 females; mean age, 60 years) were collected. These patients were randomly selected from the patient pool of the hospital’s gastrointestinal clinic; none of the patients received chemotherapy or radiotherapy. Human tissues were collected during gastroscopy or surgery. Fresh tissues were immediately frozen in liquid nitrogen. The study was approved by the ethics committee of PLA Army General Hospital and Chinese people’s Liberation Army No. 401. Informed consent was obtained from all study subjects.
Human gastric cancer cell line SGC7901 and cisplatin-resistant human gastric adenocarcinoma SGC7901/DDP cell lines were obtained from Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Human gastric epithelium cell, GES-1, and human gastric cancer cells, MGC-803, BGC-823, AGS, NCI-N87, were obtained from Beijing Institute for Cancer Research (Beijing, China). The cells were grown in Dulbecco’s modified Eagle’s medium and RPMI 1640 medium (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. The treatment with doxorubicin (Sigma) and cisplatin (Sigma) was performed as previous described [51].
RNA extraction and qRT-PCR
Total RNA was extracted from the gastric cancer tissues or cell lines using Trizol reagent (Invitrogen, Carlsbad, CA), and 1 mg of total RNA was reverse transcribed using the PrimeScript RT Reagent Kit (Perfect Real Time; Takara, Japan) to detect relative mRNAs. For the mature miRNAs, 1 mg of total RNA was reverse transcribed using PrimeScript RT Reagent Kit with gDNA Eraser (Perfect Real Time; Takara, Japan) according to manufacturer’s instructions in a total reaction volume of 25 ml. Stem-loop RT-PCR was carried out as described [52] on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The Ct values obtained from different samples were compared using the 2−ΔΔCt method. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 served as internal reference genes, respectively. MiR-633 levels were measured using SYBR Green Real-Time PCR Master Mix (Takara) according to the manufacturer’s instructions. The sequences of miR-633 primers were: forward, 5ʹ-CGCCGCTAATAGTATCTACCAC −3ʹ; reverse, 5ʹ-GTGCAGGGTCCGAGGT-3ʹ. The sequences of U6 primers were: forward, 5ʹ-CTCGCTTCGGCAGCACA-3ʹ; reverse, 5ʹ-AACGCTTCACGAATTTGCGT-3ʹ. Quantitative detection of FADD was performed using the same strategy. The primers used for FADD mRNA were: forward, 5ʹ-CGAGTCCGAAGATTCCTGA-3ʹ; reverse, 5ʹ-GACCCTCCGGAGTTTATTCA-3ʹ. The mRNAs levels were normalized to GAPDH. The sequences of GAPDH primers were: forward, 5ʹ-GTCGGAGTCAACGGATTTG-3ʹ; reverse, 5ʹ-TGGGTGGAATCATATTGGAA-3ʹ.
Cell transfection with miRNA duplexes or miRNA inhibitors
Overexpression of miR-633 was achieved by transfecting gastric cancer cells with miRNA mimics (synthetic RNA oligonucleotides mimicking precursors of miR-633). Chemically modified single-stranded antisense oligonucleotides designed to specifically sequester mature miR-633. Synthetic miR-633 mimics, miR-633 antagomir and scrambled negative control RNAs (mimic control and antagomir control) were purchased from GenePharma (Shanghai, China). The miR-633 mimic sequence was 5′-CUAAUAGUAUCUACCACAAUAAA-3′. The mimic control sequence was 5′- UUCUCCGAACGUGUCACGUTT −3′. The miR-633 antagomir sequence was 5′-UUUAUUGUGGUAGAUACUAUUAG-3′. The antagomir control sequence was 5′- CAGUACUUUUGUGUAGUACAA −3′. Cells were transfected with miRNA duplexes (50 nM) or antagomirs (50 nM) using Lipofectamine 2000 (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions
RNA interference (RNAi)
The FADD and Foxo3a siRNA sequences were designed on website Invitrogen Block-iT RNAi Designer (http://rnaidesigner.thermofisher.com/rnaiexpress/) following the guide instructions and synthesized by GenePharma Co. Ltd. The FADD-siRNA sequence was 5′-GGTCCTGCCAGATGAACCT-3′. The FADD scramble (FADD-sc) sequence was 5′-CAGUACUUUUGUGUAGUACAA-3′. The Foxo3a-siRNA sequence si-Foxo3a-1: 5′-GAGCTCTTGGTGGATCATC-3′; si-Foxo3a-2: 5′-GCUGUCUCCAUGGACAAUATT-3′. The Foxo3a scramble (Foxo3a-sc) sequence was 5′-CAGUACUUUUGUGUAGUACAA-3′. The gastric cancer cells or HEK-293 cells were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) using Opti-MEM Reduced Serum Medium (Gibco, Carlsbad, CA, USA). The specificity of the oligonucleotides was confirmed through comparing with all other sequences in Genbank using Nucleotide BLAST.
Apoptosis assays
Apoptosis was determined by TUNEL assay using a kit from Roche Applied Science (Hamburg, Germany). The procedures were followed as per the instructions in the kit. The samples were imaged using a laser scanning confocal microscope (Zeiss LSM 510 META, Carl Zeiss, Jena, Germany).
Adenovirus
Adenovirus Foxo3a and adenovirus β-galactosidase (β-gal) were as we described previously [53]. All adenoviruses were amplified in HEK-293 cells. Adenoviral infection of cancer cells or HEK-293 was performed as we described previously.
Immunohistochemistry
Immunohistochemical staining was carried out as follow: Tumor samples were fixed in 4% paraformaldehyde overnight, dehydrated and embedded in paraffin. The paraffin matrix were cut into 6 μm thick section for staining. After deparafinized and rehydrated, tissue sections were heated by microwave for antigen retrieval in 0.001 mol/L sodium citrate buffer. 3% H2O2 treatment 10 min, washed by PBS and 5% BSA in PBS for blocking. Incubation with primary antibody and horseradish peroxidase-conjugated secondary antibody, the slides were stained with 3,3ʹ-diaminobenzidine (DAB) substrate. Hematoxylin was used for counterstaining
Immunoblot and immunofluorescence
Immunoblotting was carried out as follow: briefly, cells were lysed for 1 h at 4°C in a lysis buffer (20 mM Tris, pH 7.5, 2 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol (DTT), 250 mM sucrose, 0.1 mM phenylmethylsulfonyl fluoride, 1% Triton X-100) containing a protease inhibitor cocktail. Samples were subjected to 12% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes pretreated with methanol. Equal protein loading was controlled by Ponceau Red staining of membranes. Western blots were probed using the primary antibodies. The anti-Foxo3a and the anti-phospho Foxo3a antibody (Ser253) were from Cell Signaling. The anti-FADD antibody was from Abcam. The anti-PCNA antibody, anti-tubulin antibody and the anti-actin antibody were from Santa Cruz Biotechnology. After four washes with PBS-Tween 20, the horseradish peroxidase-conjugated secondary antibodies were added. Antigen-antibody complexes were visualized by enhanced chemiluminescence. For immunofluorescence, samples were fixed in 4% paraformaldehyde overnight and washed by PBS. After dehydrated, pre-frozen samples embedded in OCT and frozen in −80°C to form hardened blocks, tumor tissues were cut into 4 μm thick section. Fixed with precooled acetone and permeablization by 0.025% TritonX-100 in PBS, the samples were blocked by 10% goat serum with 1% BSA in PBS. Incubation with primary antibody and anti-mouse IgG (Alexa Fluor 488) (Abcam, Cambridge, MA) secondary antibody, the slides were imaged using a laser scanning confocal microscope Zeiss LSM 510 META.
Pull-down assay with biotinylated miRNAs
Briefly, we synthesized miR-633 and miR-633mut which were 5ʹ-biotin-labeled. SGC-7901 cells were transfected with biotinylated miRNA, harvested 24 h after transfection. Pulldown assay was performed as formerly described [54]. The miR-633mut sequence was 5ʹ-CACCACCUAUCUACCACAAUAAA-3ʹ. The cells were washed with PBS followed by brief vortex, and incubated in a lysis buffer on ice for 30 min. The lysates were precleared by centrifugation, and 100 μl of the samples were aliquoted for input. The remaining lysates were incubated with streptavidin magnetic beads (Thermo Scientific). After coated with RNase-free bovine serum albumin and yeast tRNA (both from Sigma), the beads were incubated at 4°C for 3 h, washed twice with ice-cold lysis buffer, three times with the low salt buffer and once with the high salt buffer. The bound RNAs were purified using TRIzol and then for FADD 3ʹ-UTR analysis by qRT-PCR.
Chromatin immunoprecipitation (ChIP) analysis
Cells were washed two times with phosphate-buffered saline (PBS), and then were incubated for 10 min with 4% formaldehyde at room temperature. After washing two times with PBS, cells were lysed in lysis buffer for 1 h at 4°C. The cell lysates were sonicated into chromatin fragments (500–800 bp length). The samples were precleared with Protein A-agarose (Roche Applied Science) at 4°C. The anti-Foxo3a antibody or anti-actin antibody was added and rocked overnight. Immunoprecipitates were captured with 10% (v/v) Protein A-agarose. To analyze Foxo3a binding to the promoter of miR-633, PCRs were carried out using primers that encompass Foxo3a BS1 or BS2 of the miR-633 promoter. The primers were: BS1 (forward, 5ʹ-CACTTGGGATGTGTTTATTAT-3ʹ; reverse, 5ʹ-TCCTCCACCTCTCTCTAGCTT-3ʹ); BS2 (forward, 5ʹ-GCTGACATTTGGGAAATTAGAT-3ʹ; reverse, 5ʹ-AAAAGTTTTGAGAAGAACCTTG-3ʹ).
Establishment of tumor xenografts in mice
For the xenograft experiments in Figure 6, male BALB/c nude mice (4–5 weeks old) was selected for each group. Gastric cancer cells (approximately 1 × 107 SGC-7901 cells) were injected subcutaneously into the right axilla to establish the gastric cancer xenograft model. Eight days after subcutaneous inoculation, mice were randomly divided into 5 groups (6 mice per group) for experiments. According to the experimental design, the miR-633 antagomir and antagomir control were given at a dose of 15 μg in a small volume (0.2 ml per injection) by multipoint intratumoral injection, twice per week for four weeks. Chemotherapeutic agents (doxorubicin, 2 mg/kg) were administered by intraperitoneal injection, three times per week for four weeks. The tumor size and body weight was monitored every other day. The tumor volume was calculated using the formula (A × B2)/2, where A and B are the long and short dimensions, respectively. After four weeks, all mice were sacrificed and subcutaneous tumors were separated and weighed. All experiments were performed according to the protocols approved by the Institute Animal Care Committee.
Statistical analysis
All statistical analyses were performed using the SPSS 19.0 statistical software package. The results are expressed as means ± S.D. of at least three independent experiments. The differences among experimental groups were evaluated by one-way analysis of variance (ANOVA) to determine whether the means were significantly different. The differences were considered significant when the p value was <0.05. All statistical analyses were performed using GraphPad Prism Software.
Funding Statement
This work was supported by the Shandong Provincial Natural Science outstanding youth fund (grant numbers JQ201815), National Natural Science Foundation of China (grant numbers 81602341) and Qingdao Postdoctoral Application Research Project (grant number 2016066);Natural Science Foundation of Shandong Province (CN) [JQ201815].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementry material
Supplemental data for this article can be accessed here.
References
- [1].Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet. 2016. November 26;388(10060):2654–2664. PubMed PMID: 27156933; eng. [DOI] [PubMed] [Google Scholar]
- [2].Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016. Mar-Apr;66(2):115–132. PubMed PMID: 26808342; eng. [DOI] [PubMed] [Google Scholar]
- [3].Allemani C, Matsuda T, Di Carlo V, et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet. 2018. March 17;391(10125):1023–1075. PubMed PMID: 29395269; PubMed Central PMCID: PMCPmc5879496. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wagner AD, Syn NL, Moehler M, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2017. August 29;8:Cd004064 PubMed PMID: 28850174; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tewey KM, Rowe TC, Yang L, et al. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science (New York, NY). 1984. October 26;226(4673):466–468. PubMed PMID: 6093249; eng. [DOI] [PubMed] [Google Scholar]
- [6].Ajani JA, D’Amico TA, Almhanna K, et al. Gastric cancer, version 3.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016. October;14(10):1286–1312. PubMed PMID: 27697982; eng. [DOI] [PubMed] [Google Scholar]
- [7].Yoshida K, Yamaguchi K, Okumura N, et al. Is conversion therapy possible in stage IV gastric cancer: the proposal of new biological categories of classification. Gastric Cancer. 2016. April;19(2):329–338. PubMed PMID: 26643880; PubMed Central PMCID: PMCPmc4824831. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang L, Yang JK, Kabaleeswaran V, et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010. November;17(11):1324–1329. PubMed PMID: 20935634; PubMed Central PMCID: PMCPmc2988912. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cimino Y, Costes A, Damotte D, et al. FADD protein release mirrors the development and aggressiveness of human non-small cell lung cancer. Br J Cancer. 2012. June 5;106(12):1989–1996. PubMed PMID: 22669160; PubMed Central PMCID: PMCPmc3388563. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yoo NJ, Lee SH, Jeong EG, et al. Expression of nuclear and cytoplasmic phosphorylated FADD in gastric cancers. Pathol Res Pract. 2007;203(2):73–78. PubMed PMID: 17207586; eng. [DOI] [PubMed] [Google Scholar]
- [11].Yan J, Zhang Y, She Q, et al. Long noncoding RNA H19/miR-675 axis promotes gastric cancer via FADD/Caspase 8/Caspase 3 signaling pathway. Cell Physiol Biochem. 2017;42(6):2364–2376. PubMed PMID: 28848149; eng. [DOI] [PubMed] [Google Scholar]
- [12].Tourneur L, Delluc S, Levy V, et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004. November 1;64(21):8101–8108. PubMed PMID: 15520222; eng. [DOI] [PubMed] [Google Scholar]
- [13].Haghikia A, Haghikia A, Hellwig K, et al. Regulated microRNAs in the CSF of patients with multiple sclerosis: a case-control study. Neurology. 2012. November 27;79(22):2166–2170. PubMed PMID: 23077021; eng. [DOI] [PubMed] [Google Scholar]
- [14].Xu L, Liang YN, Luo XQ, et al. Association of miRNAs expression profiles with prognosis and relapse in childhood acute lymphoblastic leukemia. Zhonghua Xue Ye Xue Za Zhi. 2011. March;32(3):178–181. PubMed PMID: 21535956; chi. [PubMed] [Google Scholar]
- [15].Qian Z, Ren L, Wu D, et al. Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis. Int J Cancer. 2017. June 15;140(12):2792–2804. PubMed PMID: 28295288. [DOI] [PubMed] [Google Scholar]
- [16].Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010. February 4;463(7281):676–680. PubMed PMID: 20130650; eng. [DOI] [PubMed] [Google Scholar]
- [17].Haque I, Banerjee S, De A, et al. CCN5/WISP-2 promotes growth arrest of triple-negative breast cancer cells through accumulation and trafficking of p27(Kip1) via Skp2 and FOXO3a regulation. Oncogene. 2015. June 11;34(24):3152–3163. PubMed PMID: 25132260; eng. [DOI] [PubMed] [Google Scholar]
- [18].Yang H, Kong W, He L, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008. January 15;68(2):425–433. PubMed PMID: 18199536; eng. [DOI] [PubMed] [Google Scholar]
- [19].Tourneur L, Mistou S, Michiels FM, et al. Loss of FADD protein expression results in a biased Fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene. 2003. May 8;22(18):2795–2804. PubMed PMID: 12743602; eng. [DOI] [PubMed] [Google Scholar]
- [20].Mishima K, Nariai Y, Yoshimura Y.. Carboplatin induces Fas (APO-1/CD95)-dependent apoptosis of human tongue carcinoma cells: sensitization for apoptosis by upregulation of FADD expression. Int J Cancer. 2003. July 10;105(5):593–600. PubMed PMID: 12740905; eng. [DOI] [PubMed] [Google Scholar]
- [21].Bae J, Won M, Kim DY, et al. Identification of differentially expressed microRNAs in endometrial cancer cells after progesterone treatment. Int J Gynecological Cancer. 2012. May;22(4):561–565. PubMed PMID: 22543862; eng. [DOI] [PubMed] [Google Scholar]
- [22].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009. January 23;136(2):215–233. PubMed PMID: 19167326; PubMed Central PMCID: PMCPmc3794896. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005. May;37(5):495–500. PubMed PMID: 15806104; eng. [DOI] [PubMed] [Google Scholar]
- [24].Michlewski G, Guil S, Semple CA, et al. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008. November 7;32(3):383–393. PubMed PMID: 18995836; PubMed Central PMCID: PMCPmc2631628. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gregory RI, Yan KP, Amuthan G, et al. The microprocessor complex mediates the genesis of microRNAs. Nature. 2004. November 11;432(7014):235–240. PubMed PMID: 15531877; eng. [DOI] [PubMed] [Google Scholar]
- [26].Lv C, Li F, Li X, et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat Commun. 2017. October 19;8(1):1036 PubMed PMID: 29051494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guo SL, Ye H, Teng Y, et al. Akt-p53-miR-365-cyclin D1/cdc25A axis contributes to gastric tumorigenesis induced by PTEN deficiency. Nat Commun. 2013;4:2544 PubMed PMID: 24149576; PubMed Central PMCID: PMCPmc3826643. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999. March 19;96(6):857–868. PubMed PMID: 10102273; eng. [DOI] [PubMed] [Google Scholar]
- [29].Wang K, Li PF. Foxo3a regulates apoptosis by negatively targeting miR-21. J Biol Chem. 2010. May 28;285(22):16958–16966. PubMed PMID: 20371612; PubMed Central PMCID: PMCPmc2878079. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].He J, Qi H, Chen F, et al. MicroRNA-25 contributes to cisplatin resistance in gastric cancer cells by inhibiting forkhead box O3a. Oncol Lett. 2017. November;14(5):6097–6102. PubMed PMID: 29113252; PubMed Central PMCID: PMCPmc5661442. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cascinu S, Galizia E, Labianca R, et al. Pegylated liposomal doxorubicin, 5-fluorouracil and cisplatin versus mitomycin-C, 5-fluorouracil and cisplatin for advanced gastric cancer: a randomized phase II trial. Cancer Chemother Pharmacol. 2011. July;68(1):37–43. PubMed PMID: 20821330; eng. [DOI] [PubMed] [Google Scholar]
- [32].Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008. January 3;358(1):36–46. PubMed PMID: 18172173; eng. [DOI] [PubMed] [Google Scholar]
- [33].Somasagara RR, Spencer SM, Tripathi K, et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene. 2017. November 30;36(48):6680–6690. PubMed PMID: 28806395; PubMed Central PMCID: PMCPmc5709226. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ye FG, Song CG, Cao ZG, et al. Cytidine deaminase axis modulated by miR-484 differentially regulates cell proliferation and chemoresistance in breast cancer. Cancer Res. 2015. April 1;75(7):1504–1515. PubMed PMID: 25643696; eng. [DOI] [PubMed] [Google Scholar]
- [35].Zarogoulidis P, Petanidis S, Kioseoglou E, et al. MiR-205 and miR-218 expression is associated with carboplatin chemoresistance and regulation of apoptosis via Mcl-1 and survivin in lung cancer cells. Cell Signal. 2015. August;27(8):1576–1588. PubMed PMID: 25917317; eng. [DOI] [PubMed] [Google Scholar]
- [36].Shang Y, Zhang Z, Liu Z, et al. miR-508-5p regulates multidrug resistance of gastric cancer by targeting ABCB1 and ZNRD1. Oncogene. 2014. June 19;33(25):3267–3276. PubMed PMID: 23893241; eng. [DOI] [PubMed] [Google Scholar]
- [37].Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J. 1995. November 15;14(22):5579–5588. PubMed PMID: 8521815; PubMed Central PMCID: PMCPmc394672. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee EW, Kim JH, Ahn YH, et al. Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat Commun. 2012;3:978 PubMed PMID: 22864571; eng. [DOI] [PubMed] [Google Scholar]
- [39].Tang YL, Huang LB, Lin WH, et al. Butein inhibits cell proliferation and induces cell cycle arrest in acute lymphoblastic leukemia via FOXO3a/p27kip1 pathway. Oncotarget. 2016. April 5;7(14):18651–18664. PubMed PMID: 26919107; PubMed Central PMCID: PMCPmc4951317. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wiggins JF, Ruffino L, Kelnar K, et al. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010. July 15;70(14):5923–5930. PubMed PMID: 20570894; PubMed Central PMCID: PMCPmc2913706. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Park SH, Jang KY, Kim MJ, et al. Tumor suppressive effect of PARP1 and FOXO3A in gastric cancers and its clinical implications. Oncotarget. 2015. December 29;6(42):44819–44831. PubMed PMID: 26540566; PubMed Central PMCID: PMCPmc4792594. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yu S, Yu Y, Sun Y, et al. Activation of FOXO3a suggests good prognosis of patients with radically resected gastric cancer. Int J Clin Exp Pathol. 2015;8(3):2963–2970. PubMed PMID: 26045805; PubMed Central PMCID: PMCPmc4440114. eng. [PMC free article] [PubMed] [Google Scholar]
- [43].Xie X, Tang B, Zhou J, et al. Inhibition of the PI3K/Akt pathway increases the chemosensitivity of gastric cancer to vincristine. Oncol Rep. 2013. August;30(2):773–782. PubMed PMID: 23743572; eng. [DOI] [PubMed] [Google Scholar]
- [44].Cumberbatch M, Tang X, Beran G, et al. Identification of a subset of human non-small cell lung cancer patients with high PI3Kbeta and low PTEN expression, more prevalent in squamous cell carcinoma. Clin Cancer Res. 2014. February 1;20(3):595–603. PubMed PMID: 24284056; PubMed Central PMCID: PMCPmc4503252. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tsai KL, Sun YJ, Huang CY, et al. Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007;35(20):6984–6994. PubMed PMID: 17940099; PubMed Central PMCID: PMCPmc2175300. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Singh A, Ye M, Bucur O, et al. Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Mol Biol Cell. 2010. March 15;21(6):1140–1152. PubMed PMID: 20110348; PubMed Central PMCID: PMCPmc2836964. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu H, Yin J, Wang H, et al. FOXO3a modulates WNT/beta-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells. Cell Signal. 2015. March;27(3):510–518. PubMed PMID: 25578861; eng. [DOI] [PubMed] [Google Scholar]
- [48].Jopling CL, Yi M, Lancaster AM, et al. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science (New York, NY). 2005. September 2;309(5740):1577–1581. PubMed PMID: 16141076; eng. [DOI] [PubMed] [Google Scholar]
- [49].Elmen J, Lindow M, Silahtaroglu A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008. March;36(4):1153–1162. PubMed PMID: 18158304; PubMed Central PMCID: PMCPmc2275095. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bandi N, Vassella E. miR-34a and miR-15a/16 are co-regulated in non-small cell lung cancer and control cell cycle progression in a synergistic and Rb-dependent manner. Mol Cancer. 2011. May 16;10:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang JX, Li Q, Li PF. Apoptosis repressor with caspase recruitment domain contributes to chemotherapy resistance by abolishing mitochondrial fission mediated by dynamin-related protein-1. Cancer Res. 2009. January 15;69(2):492–500. PubMed PMID: 19147562; eng. [DOI] [PubMed] [Google Scholar]
- [52].Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005. November 27;33(20):e179 PubMed PMID: 16314309; PubMed Central PMCID: PMCPmc1292995. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang K, Lin ZQ, Long B, et al. Cardiac hypertrophy is positively regulated by microRNA miR-23a. J Biol Chem. 2012. January 2;287(1):589–599. PubMed PMID: 22084234; PubMed Central PMCID: PMCPmc3249113. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang JX, Zhang XJ, Li Q, et al. MicroRNA-103/107 regulate programmed necrosis and myocardial Ischemia/Reperfusion injury through targeting FADD. Circ Res. 2015. July 31;117(4):352–363. PubMed PMID: 26038570; eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.