

ABSTRACT
Ribosome synthesis is an essential cellular process, and perturbation of human ribosome production is linked to cancer and genetic diseases termed ribosomopathies. During their assembly, pre-ribosomal particles undergo numerous structural rearrangements, which establish the architecture present in mature complexes and serve as key checkpoints, ensuring the fidelity of ribosome biogenesis. RNA helicases are essential mediators of such remodelling events and here, we demonstrate that the DEAH-box RNA helicase DHX37 is required for maturation of the small ribosomal subunit in human cells. Our data reveal that the presence of DHX37 in early pre-ribosomal particles is monitored by a quality control pathway and that failure to recruit DHX37 leads to pre-rRNA degradation. Using an in vivo crosslinking approach, we show that DHX37 binds directly to the U3 small nucleolar RNA (snoRNA) and demonstrate that the catalytic activity of the helicase is required for dissociation of the U3 snoRNA from pre-ribosomal complexes. This is an important event during ribosome assembly as it enables formation of the central pseudoknot structure of the small ribosomal subunit. We identify UTP14A as a direct interaction partner of DHX37 and our data suggest that UTP14A can act as a cofactor that stimulates the activity of the helicase in the context of U3 snoRNA release.
KEYWORDS: RNA helicase, ribosome, ribosome biogenesis, snoRNA, protein cofactor, RNA processing
Introduction
The production of eukaryotic ribosomes, which are responsible for the translation of mRNAs into proteins, is a highly complex, dynamic and energy-consuming cellular process involving the maturation of four ribosomal RNAs (rRNAs) and their assembly with approximately 80 ribosomal proteins (RPs) [1]. Numerous ribosome biogenesis factors (RBFs), which associate transiently with pre-ribosomal particles, are responsible for mediating key maturation events and in the yeast Saccharomyces cerevisiae, approximately 200 such factors have been identified (see for example reference [2]). Due to the larger size of human (pre-)ribosomal complexes and the evolution of regulatory networks that coordinate ribosome production with other cellular processes, the inventory of human ribosome biogenesis factors is likely to be much larger than that of yeast. Proteomic analyses of human nucleoli [3,4], together with several recent genome-wide RNAi-based screens [5–8] have uncovered a plethora of factors without yeast homologues that are required for human ribosome assembly. Furthermore, detailed analyses of the human pre-rRNA processing pathway (see for example references [9–11]) and functional characterization of various individual human RBFs have revealed that many human RBFs have different or additional functions compared to their yeast counterparts. A portfolio of genetic diseases, termed ribosomopathies, that are caused by mutations in genes encoding RPs and/or RBFs (reviewed in reference [12]), as well as an increasing body of evidence linking ribosome assembly to cancer (reviewed in reference [13]), emphasize the importance of a detailed knowledge of the human ribosome assembly pathway.
In human cells, ribosome assembly is initiated in the nucleolus by synthesis of a 47S pre-rRNA transcript containing the sequences of three of the four rRNAs (18S, 5.8S and 28S), separated by internal transcribed spacers (ITS) and flanked by external transcribed spacers (ETS). Maturation of the pre-rRNAs involves processing of this transcript by endo- and exo-nucleases and extensive modification of the mature rRNA sequences by small nucleolar RNPs (snoRNPs) and stand-alone modification enzymes [14–16]. A subset of RPs and RBFs are recruited to the nascent pre-rRNA transcript giving rise to an early pre-ribosomal complex termed the small subunit (SSU) processome. The SSU processome is composed of numerous sub-complexes, including the UTP-A, UTP-B and UTP-C complexes, as well as various enzymes that mediate important steps in SSU biogenesis, such as the endonuclease UTP24, the PIN domain protein UTP23, the rRNA methyltransferase EMG1 and the GTPase BMS1 [6,17–19]. A core component of the SSU processome is the U3 snoRNP, which in contrast to most other snoRNPs, does not guide rRNA modification, but instead bridges key interactions within early pre-ribosomes [20]. This is achieved by basepairing interactions between the U3 snoRNA 3ʹ hinge region and the 5ʹ ETS, and the U3 5ʹ region (GAC box, box A and box A’) and the 18S rRNA sequence that forms the central pseudoknot in mature SSU complexes [21–23]. Recent structural analyses of S. cerevisiae SSU processomes [24,25] have revealed that the basepairing interactions formed between the U3 snoRNA and the pre-rRNA tether the SSU processome in a conformation that allows essential pre-rRNA processing events to be co-ordinated. Release of the U3 snoRNP, which in yeast requires the RNA helicase Dhr1 [26], is an essential step in SSU biogenesis as it licences downstream maturation events and allows folding of the 18S rRNA sequence into its mature conformation. A pre-rRNA cleavage step in ITS1 separates the SSU (40S) and LSU (60S) precursor particles, which undergo separate maturation pathways in the nucleolus and nucleoplasm involving further pre-rRNA processing steps, the recruitment and release of various RBFs and significant structural rearrangements. Such remodelling events serve as important checkpoints during the assembly pathway and are often a pre-requisite for establishing export competence. The pre-40S and pre-60S subunits are independently exported to the cytoplasm [27] where final maturation and quality control steps take place before the mature subunits re-join during translation.
RNA helicases, which are typified by a helicase core domain composed of tandem RecA-like domains containing conserved sequence motifs involved in NTP/substrate binding, NTP hydrolysis and its coordination with unwinding activity, are proposed to be important regulators of pre-ribosome remodelling events [28,29]. Structural rearrangements, such as the release of snoRNPs from pre-ribosomes, recruitment or dissociation of RPs and/or RBFs, and altering pre-rRNA basepairing/folding, are proposed functions of RNA helicases during the maturation of pre-ribosomal subunits [26,30–35]. In human cells, the DEAD-box helicases DDX51 and DDX27 are implicated in release of the metazoan-specific snoRNA U8 from its basepairing sites with the 28S rRNA and ITS2 sequences, and maturation of the 3ʹ end of the 28S rRNA respectively [36,37]. Interestingly, the catalytic activity of the DEAH-box RNA helicase DHX15, which is the human homologue of yeast Prp43, is required for a metazoan-specific cleavage event (A’) in the 5ʹ ETS [38]. However, unlike Prp43, DHX15 does not appear to be required for the final steps of 18S rRNA maturation, implying that the functions of this helicase are different in yeast and humans. Furthermore, human ribosome assembly involves RNA helicases not present in yeast; for example, it was recently shown that the action of the metazoan-specific DEAD-box protein DDX21 is required for rDNA transcription and that this enzyme also facilitates the access of late-acting snoRNPs to pre-40S complexes [39,40]. Recent cryo-EM structures of late pre-40S complexes [41,42] provide the first snap-shots of structural rearrangements that take place during the final steps of SSU maturation but the roles of many human RNA helicases during ribosome assembly remain uncharacterized.
Here, we show that the putative RNA helicase DHX37 is an RNA-dependent ATPase that is required for biogenesis of the SSU. Our data show that lack of DHX37 triggers a surveillance pathway that leads to degradation of pre-ribosomal particles. Depletion of DHX37 inhibits the conversion of the 21S pre-rRNA to 18SE and expression of catalytically inactive DHX37 also impairs a pre-rRNA cleavage step in the 5ʹ ETS. Using in vivo protein-RNA crosslinking, we demonstrate that DHX37 binds close to the 3ʹ hinge region of the U3 snoRNA, which basepairs with the 5ʹ ETS in proximity of the A’ cleavage site, and we show that expression of catalytically inactive DHX37 causes accumulation of the U3 snoRNA on pre-ribosomes. Together, our data imply that dissociation of the U3 snoRNA, which requires DHX37, is an important early pre-ribosome remodelling event that licences downstream steps in human ribosome assembly.
Results
The human RNA helicase DHX37 is required for biogenesis of the small ribosomal subunit
Although many components of the ribosome assembly machinery are conserved throughout eukaryotes, the precise functions of many RBFs, especially RNA helicases, differ in yeast and humans. Based on its homology to yeast Dhr1, the putative RNA helicase DHX37 is anticipated to play a role in human ribosome biogenesis, however, a detailed functional characterization of this protein is lacking. To first ascertain the localization of DHX37 in human cells, we performed immunofluorescence in HeLa cells using antibodies against DHX37 and, as a nucleolar marker, the rRNA methyltransferase NSUN5. This revealed that DHX37 predominantly localizes to nucleoli (Figure 1(a)), which is consistent with a role during the early stages of ribosome assembly. Next, to enable the effects of depletion of DHX37 on ribosome assembly to be analyzed, RNAi against DHX37 was established using two alternative siRNAs. HeLa cells were left untreated (wild-type), or were transfected with control siRNAs or siRNAs targeting DHX37, then the levels of the DHX37 protein were examined using western blotting. This demonstrated that the amount of DHX37 in cells treated with either of the two siRNAs against DHX37 was markedly reduced (Figure 1(b)), thereby confirming the effectiveness of both siRNAs. To determine the effect of depletion of DHX37 on ribosomal subunit production, whole cell extracts prepared from cells treated with either non-target siRNAs (siNT) or those targeting DHX37 (siDHX37_1) were separated by sucrose density gradient centrifugation and the relative amounts of small subunits (40S), large subunits (60S) and monosomes (80S) were determined by monitoring the absorbance of each fraction at 260 nm. The obtained profile shows that depletion of DHX37 does not affect production of 60S subunits but leads to a significant decrease in the abundance of 40S particles as well as a decrease in the amount of 80S monosomes (Figure 1(c)), indicating a role for DHX37 in production of the small ribosomal subunit. Consistent with this, pulse-chase labelling of newly synthesized RNAs in siRNA-treated cells as described above confirmed the requirement for DHX37 for production of the 18S rRNA, but not the 28S rRNA (Figure 1(d,e)). To determine at what stage of SSU production DHX37 is required, northern blotting was used to examine the levels of different pre-rRNA species in cells depleted of DHX37. Compared to RNA from wild-type cells or cells treated with non-target siRNAs, depletion of DHX37 caused accumulation of the 21S pre-rRNA and a concomitant decrease in the levels of the 18SE pre-rRNA (Figure 1(d,f)), which is largely produced by exonucleolytic processing of the 21S pre-rRNA following site 2 cleavage [10,11]. Interestingly, several aberrant pre-rRNAs were also detected when DHX37 was depleted; in particular, prominent species that migrated below the 41S and 18SE pre-rRNAs were observed.
Figure 1.
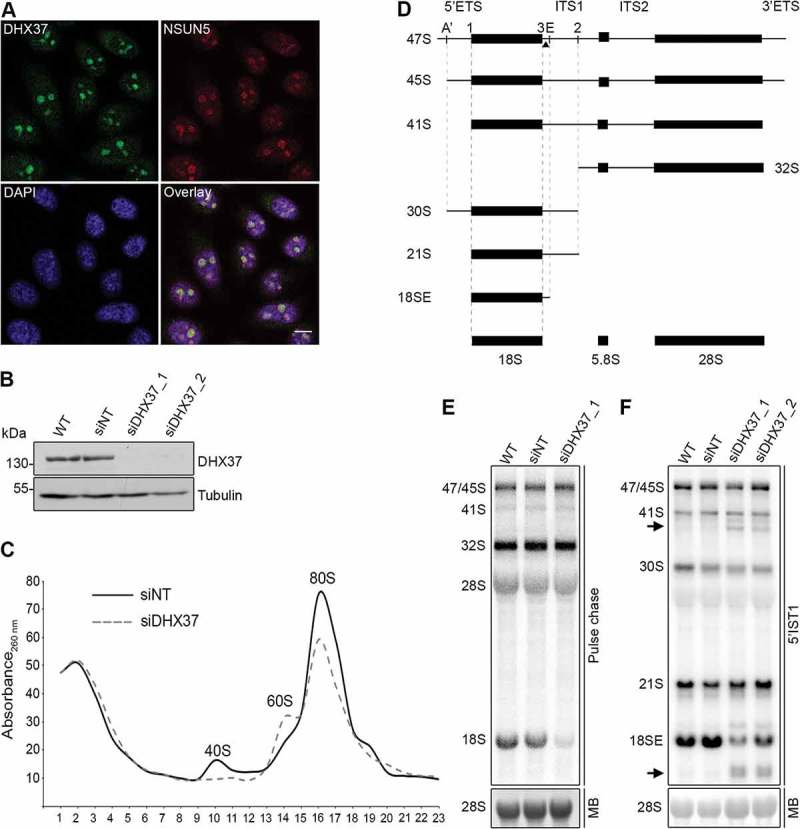
Depletion of the nucleolar RNA helicase DHX37 affects early and late stages of 18S rRNA maturation. (a) HeLa cells were fixed and the localization of DHX37 was determined by immunofluorescence using an antibody against endogenous DHX37 (green). Immunofluorescence against NSUN5 (red) served as a nucleolar marker and nuclear material was visualized by DAPI staining (blue). An overlay of the images is provided and a scale bar representing 10 μm is depicted. (b) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting DHX37 (siDHX37_1 and siDHX37_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (c) Whole cell extracts from cells treated with non-target siRNAs (siNT) or a siRNA targeting DHX37 (siDHX37_1) were separated by sucrose density gradient centrifugation. The absorbance of each fraction at 260 nm was used to generated a profile on which the positions of the ribosomal and pre-ribosomal complexes are indicated. (d) Schematic view of the major pre-rRNA intermediates detected in human cells. Mature rRNA regions are shown as black rectangles, and internal (ITS) and external transcribed spacer (ETS) regions are represented by black lines. Cleavage sites relevant for maturation of the 18S rRNA are named above the 47S rRNA transcript and the hybridization position of the 5ʹ ITS1 probe used for northern blotting is indicated by the triangle. (e) Untransfected HeLa cells, or HeLa cells that had been treated with non-target siRNAs (siNT) or those targeting DHX37 (siDHX37_1) for 72 h were starved of phosphate then grown in media supplemented with [32P]-orthophosphate for 1 h before growth in normal DMEM for 3 h. Total RNA was extracted, separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and nascent RNAs were detected using a phosphorimager. The mature 28S rRNA was visualized using methylene blue staining (MB). (f) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (b). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. Aberrant pre-rRNA species detected upon depletion of DHX37 are indicated by arrows. Experiments were performed at least in triplicate and representative data are shown.
The detection of these aberrant pre-rRNA species upon depletion of DHX37 raised the question of how they are produced. The pre-rRNA regions present in each of the prominent intermediates were therefore determined by northern blotting using a series of probes hybridizing within the 5ʹ ETS and ITS1 (Figure 2(a)). The 16S* species was detected with probes hybridizing to ITS1 upstream, but not downstream, of site 2 (Figure 2(b)), implying that the 3ʹ end of this pre-rRNA is generated by site 2 cleavage. The finding that a probe hybridizing within the 5ʹ ETS, immediately upstream of site 1 was not able to detect 16S*, together with the estimated size of this intermediate, indicates that its 5ʹ end lies within the mature 18S rRNA sequence. While the 38S* intermediate could be visualized using probes spanning ITS1, it was not detected with probes hybridizing in the 5ʹ ETS (Figure 2(a,(b)), implying that, similarly, the 5ʹ end of this species lies within the mature 18S rRNA sequence. A series of northern blot probes spanning the 18S rRNA sequence (Figure 2(a)) were therefore utilized to map the 5ʹ end of the 38S* intermediate. While 38S* was readily observed using probes 18S-1285, 18S-1377 and 18S-1670, it was only very weakly detected by probe 18S-1230 and was not visualized using probes 18S-1 or 18S-1004 (Figure 2(c)). This suggests that the 5ʹ end of the 38S*, and by analogy, the 16S* species, is somewhat heterogeneous but primarily lies between nucleotides 1253 and 1285 (helix 32) of the 18S rRNA (Supplementary Figure S1A). The finding that the 38S* and 16S* intermediates are not part of the normal pre-rRNA processing pathway, but instead represent turnover intermediates derived from the 41S and 30S/21S pre-rRNAs respectively, suggests the existence of a surveillance pathway that targets pre-ribosomal complexes lacking DHX37 for degradation. Relatively little is known about how incorrectly assembled pre-ribosomes are turned over in human cells, however, an enzyme that has been implicated in the degradation of aberrant pre-rRNAs is the 5ʹ-3ʹ exonuclease XRN2 [43]. To determine if the 5ʹ ends of the 38S* and 16S* intermediates are generated by the stalling of XRN2 during exonucleolytic processing, pre-rRNA intermediates were examined in HeLa cells depleted of DHX37, XRN2 or both DHX37 and XRN2. As previously reported, depletion of XRN2 impaired A’ cleavage, leading to accumulation of the 30SL5ʹ intermediate [10,43] and the previously observed defect in conversion of the 21S pre-rRNA to 18SE was detectable when the levels of DHX37 were reduced both in the absence and presence of XRN2 (Figure 2(c)). Upon co-depletion of DHX37 and XRN2, the 38S* and 16S* fragments were clearly observed, implying that this surveillance pathway does not rely on XRN2 and that other endo-/exonuclease(s) mediate the degradation of pre-rRNAs within aberrant pre-ribosomes lacking DHX37.
Figure 2.
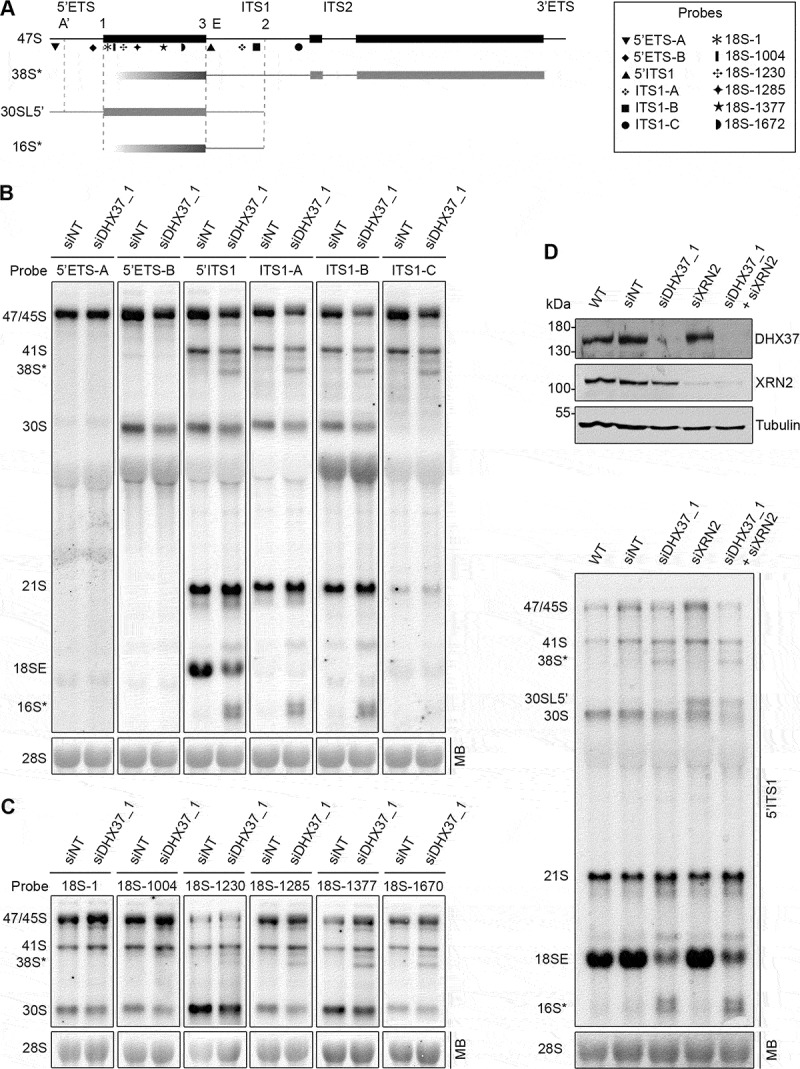
Mapping of the aberrant 16S* and 38S* pre-rRNA intermediates that accumulate upon depletion of DHX37. (a) Schematic view of the 47S pre-rRNAs (black) and the aberrant 38S*, 16S* and 30SL5ʹ pre-rRNA species not normally detected in human cells (grey). Mature rRNA regions are shown as rectangles, and internal (ITS) and external transcribed spacer (ETS) regions are represented by lines. Cleavage sites are named above the 47S pre-rRNA and the hybridization position of probes used for northern blotting are indicated. (b,c) RNA extracted from HeLa cells transfected with non-target siRNAs (siNT) or siRNAs targeting DHX37 (siDHX37_1) was separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was detected by methylene blue staining (MB). Northern blotting was performed using probes hybridizing to different positions within the 5ʹ ETS and ITS1 (b) or the 18S rRNA (c) and pre-rRNAs were visualized using a phosphorimager. (d) RNA and proteins were extracted from wildtype (WT) HeLa cells or cells that had been transfected with non-target siRNAs (siNT), or siRNAs targeting DHX37 (siDHX37_1) or XRN2 (siXRN2). Proteins were analyzed by western blotting using the antibodies indicated to the right (upper panel) and pre-RNAs were detected by northern blotting using a probe hybridizing to the 5' end of ITS1.
Interestingly, the 38S* and 16S* pre-rRNA intermediates are also detected upon depletion of the rRNA methyltransferase DIMT1L, which installs two evolutionarily conserved N6-N6-dimethylations at positions A1850 and A1851 of the 18S rRNA sequence that forms the decoding centre in the mature SSU, and TSR1, which acts during the late stages of SSU biogenesis where it is proposed to prevent premature association of the small and large ribosomal subunits (Supplementary Figure S1B,C) [6,44–46]. This suggests that the presence of DHX37, DIMT1L and TSR1 in early pre-ribosomal complexes may all be monitored by a common surveillance pathway.
DHX37 is an RNA-dependent ATPase and its catalytic activity is required for the conversion of the 21S pre-rRNA to the 18SE pre-rRNA
Several enzymes involved in ribosome biogenesis have been shown to have both catalytic and non-catalytic functions in the pathway (see for example references [47–49]). Given the extensive pre-rRNA processing defects detected upon depletion of DHX37 and the fact that DHX37 is a putative DEAH-box RNA helicase, we next aimed to demonstrate that DHX37 is a catalytically active enzyme and determine if this activity is required for its function(s) in biogenesis of the small ribosomal subunit. A characteristic feature of RNA helicases is their capability to hydrolyze nucleotide triphosphates (NTPs) in an RNA-dependent manner [28,29] and we therefore expressed recombinant His6-tagged DHX37 in Escherichia coli and purified the protein for use in in vitro ATPase assays(Figure 3(a)). To confirm the specificity of any ATPase activity detected, DHX37 carrying a threonine to alanine substitution (T282A) in the evolutionarily conserved ‘GKT’ sequence of motif I that is implicated in NTP binding [26,28], was also expressed and purified (Figure 3(a)). NADH-coupled ATPase assays were performed using DHX37 or DHX37T282A in the absence and presence of RNA. Compared to a control reaction containing neither protein nor RNA, DHX37 showed mild ATPase activity in the absence of RNA and the addition of RNA stimulated ATP hydrolysis by DHX37 almost four-fold (Figure 3(b)), demonstrating that DHX37 is an RNA-dependent ATPase. While DHX37T282A. also weakly hydrolyzed ATP in the absence of RNA, the effect of RNA addition on the activity of DHX37T282A was minimal, indicating that this amino acid substitution within the GKT motif impairs the catalytic activity of the helicase. To enable the requirement for the catalytic activity of DHX37 for small subunit biogenesis to be examined in human cells, HEK293 cell lines were generated in which endogenous DHX37 could be depleted using an established siRNA (Figure 1(b)) and a C-terminally His6-PreScission protease-2x Flag (Flag) tagged version of DHX37 or DHX37T282A could be expressed from a genomic locus via a tetracycline regulatable promoter. HEK293 cells were transfected with non-target siRNAs (siNT) or siRNAs targeting DHX37 (siDHX37_1) and expression of the Flag tag, DHX37-Flag or DHX37T282A-Flag was induced (Figure 3(c)). Western blot analysis confirmed specific expression of the Flag-tagged proteins, albeit at slightly higher than endogenous levels. Examination of pre-rRNA levels in these cells by northern blotting using a probe hybridizing to the 5ʹ end of ITS1 demonstrated that depletion of DHX37 from HEK293 cells leads to pre-rRNA processing defects similar to those observed in HeLa cells (Figure 1(f)) and that expression of wild-type DHX37 from a transgene can rescue these defects (Figure 3(d)). Interestingly, the aberrant 38S* and 16S* pre-rRNA fragments did not accumulate upon expression of DHX37T282A (Figure 3(d)), implying the presence of the protein rather than its ATPase activity is sensed by the quality control pathway that monitors DHX37 recruitment to pre-ribosomal complexes. In contrast, the accumulation of the 21S pre-rRNA and reduced level of the 18SE pre-rRNA observed upon depletion of DHX37 were also detected in cells expressing DHX37T282A (Figure 3(d)); left panel), demonstrating that lack of the catalytic activity of DHX37 is responsible for these defects. Expression of DHX37T282A also lead to accumulation of a pre-rRNA longer than 30S that was not detected in cells lacking DHX37. Northern blotting with a probe hybridizing close the 5ʹ end of the 47S pre-rRNA transcript confirmed this species to be the 5ʹ extended version of 30S, 30SL5ʹ (Figure 3(d)); right panel), which accumulates when A’ processing is impaired. These data suggest that while cleavage at the A’ site in the 5ʹ ETS can take place when DHX37 is lacking, the presence of inactive DHX37 in pre-ribosomal particles reduces the efficiency of this cleavage event or promotes bypassing of this step.
Figure 3.
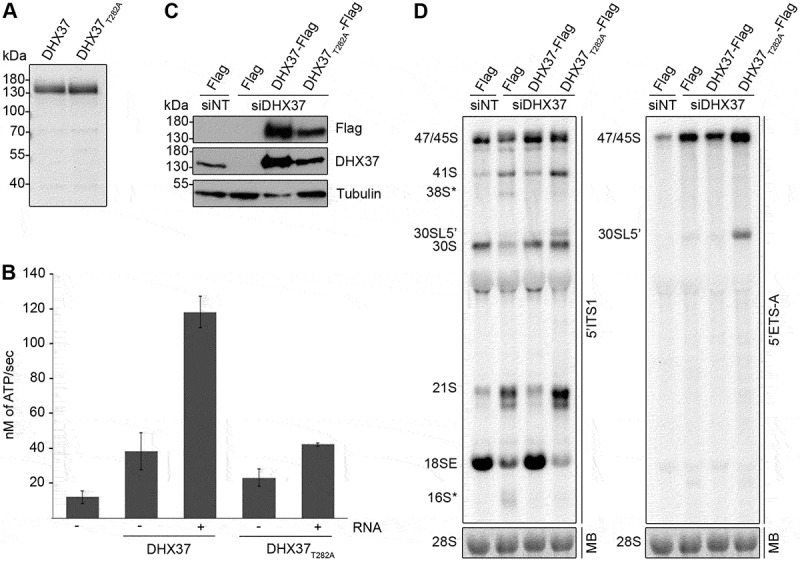
Pre-rRNAs are degraded upon depletion of DHX37 and expression of catalytically inactive DHX37 leads to defects in A’ cleavage and the conversion of 21S to 18SE. (a) C-terminally His6-tagged DHX37 or DHX37 carrying a threonine to alanine substitution at amino acid 282 within the evolutionarily conserved ‘GKT’ motif (DHX37T282A) was recombinantly expressed in E. coli and purified. Purified proteins were separated by SDS-PAGE and visualized by Coomassie staining. (b) The amount of ATP hydrolyzed by recombinant DHX37 or DHX37T282A in the presence (+) or absence (-) of RNA was determined using an in vitro NADH-coupled ATPase assay. Experiments were performed in triplicate and error bars represent mean ± standard deviation. (c) HEK293 cell lines were transfected with either non-target siRNAs (siNT) or siRNAs targeting DHX37 (siDHX37_1) and expression of the Flag tag, or C-terminally Flag-tagged DHX37 or DHX37T282A was induced by addition of tetracycline 24 h before harvesting. Proteins were extracted and analyzed by western blotting using antibodies against DHX37, tubulin and the Flag tag. (d) Total RNA extracted form HEK293 cell lines treated as described in (c) was separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and analyzed by northern blotting using probes hybridizing at the 5ʹ end of ITS1 (left panel) or within the 5ʹ ETS (right panel). Pre-rRNAs were detected using a phosphorimager and the mature 28S rRNA was visualized by methylene blue staining (MB).
DHX37 associates directly with the U3 snoRNA in vivo
Elucidating the precise functions of RNA helicases in ribosome biogenesis necessitates identification of the precise targets of their remodelling activity. To gain insight into the in vivo RNA substrates of DHX37, the crosslinking and analysis of cDNA (CRAC) approach was applied [50,51]. HEK293 cells expressing either the Flag tag or Flag-tagged DHX37 were grown in the presence of 4-thiouridine before in vivo crosslinking using light at 365 nm. Cell extracts were used for tandem affinity purification under native and denaturing conditions, and a partial RNase digestion was performed to ensure that only RNA sequences directly contacted by DHX37 were recovered. RNA fragments co-purified with DHX37-Flag or the Flag tag were ligated to sequencing adaptors and labelled with [32P]. RNA-protein complexes were separated by denaturing polyacrylamide gel electrophoresis (PAGE), transferred to a nitrocellulose membrane and then visualized by autoradiography (Figure 4(a)). A signal migrating at the molecular weight of DHX37 was detected in the sample derived from cells expressing DHX37-Flag but not the Flag tag, confirming the direct interaction of DHX37 with RNA in vivo. This area of the membrane, and a corresponding region for the control sample (Figure 4(a)), were excised and RNA fragments were isolated. After reverse transcription, the cDNA library was amplified by PCR and subjected to Illumina sequencing. The obtained sequence reads were mapped to the human genome using a filter to allow only mapping of reads containing a specific T-C mutation introduced during the reverse transcription step at positions where an amino acid remained crosslinked to a 4-thiouridine. Relatively few sequence reads mapped to the rDNA and no specific crosslinking site(s) could be identified. This suggests that, similar to yeast Dhr1 [26], DHX37 interacts only transiently or unstably with pre-rRNA/pre-ribosomal complexes. Consistent with this, sucrose density gradient analysis of the distribution of DHX37 between pre-ribosomal and non-ribosomal complexes showed that under normal conditions, only a small proportion of the helicase co-migrates with pre-ribosomal particles (Supplementary Figure 2). A higher proportion of sequence reads mapping to snoRNA genes was, however, observed in the DHX37-Flag CRAC dataset compared to the control, suggesting the association of DHX37 with snoRNA(s) (Figure 4(b)). Analysis of the relative distribution of sequence reads mapping to snoRNA genes in the two datasets highlighted an enrichment of reads corresponding to the U3 snoRNA (Figure 4(c); Supplementary Table 1). To confirm the association of DHX37 with the U3 snoRNA, extracts from cells expressing Flag-DHX37 or the Flag tag were used in immunoprecipitation experiments and the amounts of the U3, and as a control the U8, snoRNAs present in the inputs and eluates were examined by northern blotting. While the U8 snoRNA was not retrieved in either eluate, the U3 snoRNA was specifically co-purified with Flag-DHX37 but not the Flag tag, further supporting the interaction of DHX37 with the U3 snoRNA in vivo (Figure 4(d)). We next examined the distribution of CRAC sequencing reads mapping to each nucleotide of the U3 snoRNA sequence. Almost no sequence reads were observed to be derived from the very 5ʹ end of the snoRNA containing the U3-specific GAC and A/A’ boxes, which basepairs with an 18S rRNA sequence that forms part of the central pseudoknot in the mature SSU particle. Instead, the majority of sequence reads in the DHX37-Flag dataset were mapped towards the 3ʹ end of the snoRNA containing the box C/D elements and close to the 3ʹ hinge region of the U3 snoRNA that basepairs with pre-rRNA sequences present in the 5ʹ ETS proximal to the A’ cleavage site (Figure 4(e); upper panel). The binding of DHX37 to these sequences is supported by the detection of T-C mutations in the sequencing reads as these are specifically introduced at nucleotides crosslinked to amino acids (Figure 4(e); lower panel).
Figure 4.
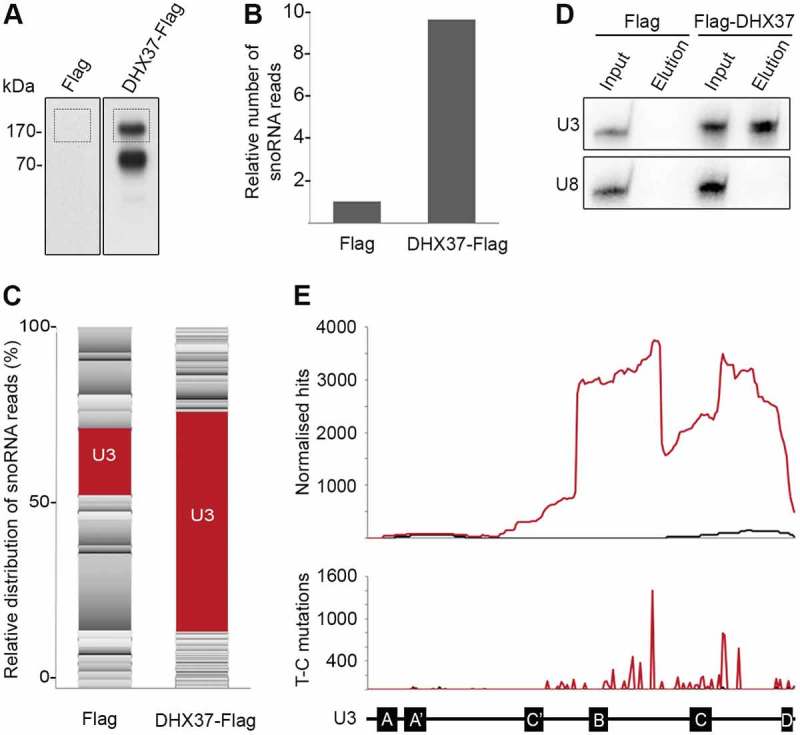
DHX37 associates with the 3ʹ region of the U3 snoRNA that contains the box C/D motifs. (a) HEK293 cells expressing DHX37-Flag or the Flag tag were grown in the presence of 4-thiouridine before crosslinking in vivo using light at 365 nm. RNA-protein complexes were tandem affinity purified under native and denaturing conditions, then RNAs co-purified with DHX37 were ligated to sequencing adaptors and labelled using [32P]. Complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and labelled RNAs were detected by autoradiography. The regions of the membrane excised for subsequent analysis are indicated by boxes. (b) The region of the membrane containing DHX37-Flag-RNA complexes as indicated in (a), and a corresponding region of the lane containing the Flag sample, were excised. RNAs were isolated and subjected to reverse transcription and PCR amplification to generate a cDNA library upon which Illumina deep sequencing was performed. The obtained sequence reads were mapped to the human genome and, after normalization, the relative number of reads derived from snoRNAs in the Flag and DHX37-Flag samples was determined. (c) The relative distribution of sequence reads derived from each box C/D snoRNA in the Flag and DHX37-Flag datasets is shown. The proportion of sequence reads mapping to the U3 snoRNA is highlighted in red while all other snoRNAs are shown in shades of grey. (d) Cell extracts prepared from HEK293 cells expressing Flag-DHX37 or the Flag tag were incubated with anti-Flag beads. After thorough washing steps, complexes were eluted and RNA was extracted. RNAs extracted from inputs (1%) and eluates were separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using probes hybridizing to the U3 and U8 snoRNAs. RNAs were detected using a phosphorimager. (e) The normalized number of reads mapping to each nucleotide of the gene encoding the U3 snoRNA in the Flag (black) and DHX37-Flag (red) datasets is shown graphically (upper panel). The normalized number of mutations mapping to each position are indicated in the lower panel. A schematic representation of the U3 snoRNA is shown below with the relative positions of key features indicated.
The U3 snoRNA accumulates on pre-ribosomes when the catalytic activity of DHX37 is impaired
The direct interaction of DHX37 with the U3 snoRNA suggests that, like its yeast counterpart Dhr1, DHX37 may regulate the interactions of this snoRNA with pre-ribosomal complexes. The amounts of U3 present on pre-ribosomal complexes in cells expressing either wild-type DHX37 or catalytically inactive DHX37T282A was examined. Endogenous DHX37 was depleted and expression of DHX37-Flag or DHX37T282-Flag was induced to equal levels (Figure 5(a)). Extracts from these cells were then subjected to sucrose density gradient centrifugation followed by northern blotting for the U3 snoRNA (Figure 5(b)). In cells expressing wild-type DHX37, the U3 snoRNA was present in fractions containing non-ribosomal complexes as well as those containing larger pre-ribosomal complexes. Expression of DHX37T282A lead to a notable increase in the proportion of the U3 snoRNA associated with pre-ribosomal complexes (Figure 5(b)), implying that the catalytic activity of DHX37 is required for release of this snoRNA from pre-ribosomal particles.
Figure 5.
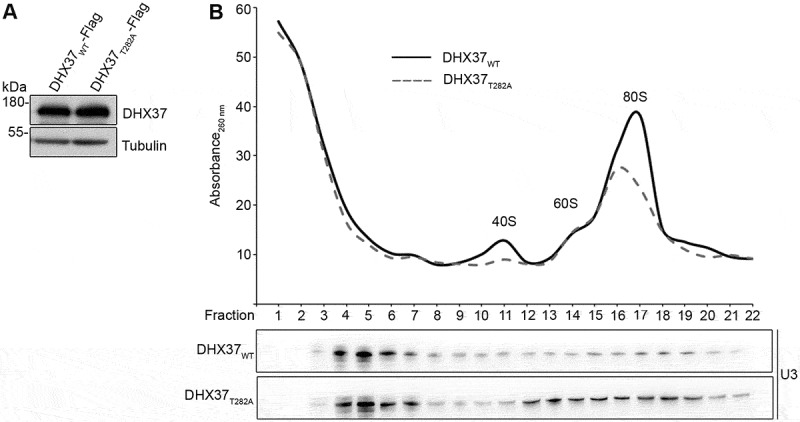
The U3 snoRNA accumulates on pre-ribosomes upon expression of catalytically inactive DHX37. (a) HEK293 cells capable of expression of DHX37-Flag or DHX37T282A-Flag were transfected with siRNAs against DHX37 and 24 h prior to harvesting, expression of the tagged proteins was induced by addition of tetracycline. To confirm equal expression levels, proteins were analyzed by western blotting using antibodies against DHX37 and tubulin. (b) Whole cell extracts prepared from HEK293 cells treated as in (a) were separated by sucrose density gradient centrifugation. The optical density of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal and pre-ribosomal complexes are indicated. RNA extracted from the gradient fractions described in (b) was separated by denaturing PAGE and transferred to a nylon membrane. Northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. The experiments shown in this figure were performed in triplicate and representative data are shown.
DHX37 interacts directly with the SSU biogenesis factor UTP14A
The finding that the catalytic activity of DHX37 is required for release of the U3 snoRNA from pre-ribosomal particles raises the question of how this activity is regulated in the cellular environment. The promiscuity of many RNA helicases is limited by their inherently low catalytic activity, which is stimulated by dedicated cofactors upon binding to their cognate RNA substrates. Furthermore, protein cofactors can bestow specificity to RNA helicases by directing their recruitment to particular target RNAs in vivo (reviewed in [52]). In yeast, Dhr1 physically interacts with the 18S rRNA m7G methyltransferase Bud23 and the SSU biogenesis factor Utp14 to facilitate its access to pre-ribosomal particles, and it has been suggested that the unwinding activity of Dhr1 is stimulated by a direct interaction with Utp14 [26,53,54]. To determine if DHX37 interacts with the human orthologues of Bud23 (WBSCR22) and/or Utp14 (UTP14A) immunoprecipitation assays were performed. Extracts from cells expressing DHX37-Flag or the Flag tag were treated with RNase A or left untreated to determine if any interactions detected are bridged by RNA, and inputs (1%) and eluates were analyzed by western blotting. This revealed that neither tubulin nor WBSCR22 were recovered with DHX37-Flag in the presence or absence of RNase treatment (Figure 6(a)). In yeast, Bud23 interacts with amino acids in the N-terminal region of Dhr1 [54,55] and the lack of a direct interaction between DHX37 and WBSCR22 likely reflects the poor sequence conservation of this region of the helicase between yeast and humans (Supplementary Figure 3). Furthermore, in contrast to yeast Bud23, which is recruited to early pre-ribosomal complexes, WBSCR22 is reported to be recruited to late pre-40S complexes [56], consistent with the lack of an interaction with DHX37, which is likely to only be present in early complexes. UTP14A was, however, efficiently co-precipitated with DHX37-Flag, but not the Flag tag alone, implying the existence of pre-ribosomal complexes containing both these proteins (Figure 6(a)). The observed interaction between DHX37-Flag and UTP14A was not affected by RNase treatment, suggesting that this interaction is mediated by protein-protein contacts (Figure 6(a)). As depletion of DIMT1L or TSR1 appears to trigger the same quality control pathway as depletion of DHX37 (indicated by the accumulation of the aberrant 38S* and 16S* pre-rRNA species; Supplementary Figure 1), we speculated that these proteins might interact in human cells. Analysis of the IP inputs and eluates revealed that while both DIMT1L and TSR1 could be co-precipitated with DHX37-Flag from cell extracts, these interactions were dependent on RNA (Supplementary Figure 4A and B), implying that DHX37, DIMT1L and TSR1 are present in common pre-ribosomal complexes but that they do not form direct interactions.
Figure 6.
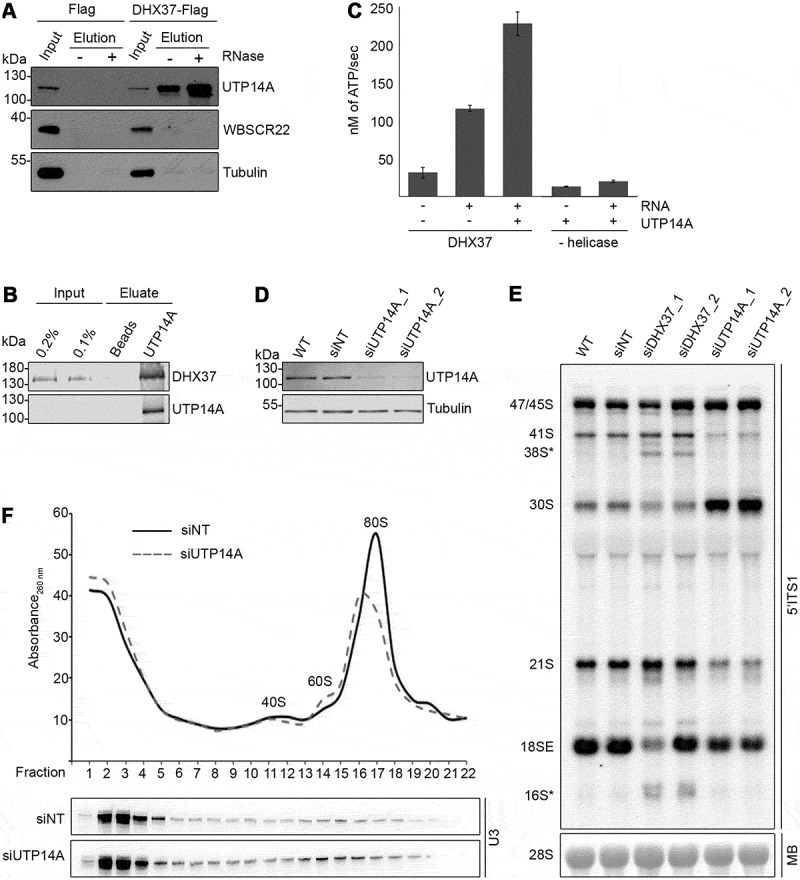
UTP14A interacts with DHX37 and stimulates its ATPase activity in vitro. (a) Cell extracts prepared from HEK293 cells expressing either DHX37-Flag or the Flag tag were used for immunoprecipitation experiments in the presence (+) or absence (-) of RNase. Inputs (1%) and eluates were separated by SDS-PAGE and analyzed by western blotting using antibodies against DHX37, UTP14A, WBSCR22 and tubulin. (b) Recombinant DHX37-His6 was incubated with either IgG sepharose (beads) or with IgG sepharose on which ZZ-UTP14A-His10 had been immobilized. After washing steps, proteins were eluted, and inputs and eluates were separated by SDS-PAGE then analyzed by western blotting using antibodies against DHX37 and UTP14A. (c) The amount of ATP hydrolyzed by recombinant DHX37 in the presence (+) or absence (-) of RNA and the presence (+) or absence (-) of UTP14A was determined using an in vitro NADH-coupled ATPase assay. Experiments were performed in triplicate and error bars represent mean ± standard deviation. (d) HeLa cells were left untransfected (WT), or were transfected with non-target siRNA (siNT) or siRNAs targeting UTP14A (siUTP14A_1 and siUTP14A_2). After 72 h, cells were harvested, and proteins were analyzed by western blotting using antibodies against UTP14A (upper panel) and tubulin (lower panel). (e) Total RNA was extracted from wild-type (WT) HeLa cells or HeLa cells transfected with siRNAs as in (D). RNAs were separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane and the mature 28S rRNA was visualized by methylene blue staining (MB). Northern blotting with a probe hybridizing to the 5ʹ end of ITS1 (5ʹ ITS1) was used to detect precursors of the 18S RNA, which were visualized using a phosphorimager. (f) Whole cell extracts prepared from HEK293 cells treated with control siRNAs (siNT) or siRNAs targeting UTP14A (siUTP14A_1) were separated by sucrose density gradient centrifugation. The absorbance of each faction at 260 nm was determined and used to generate a profile on which the peaks corresponding to ribosomal subunits (40S and 60S) and 80S monosomes are indicated. RNA extracted from the gradient fractions was separated by denaturing PAGE, transferred to a nylon membrane and northern blotting was performed using a [32P]-labelled probe hybridizing to the U3 snoRNA. Replica experiments have been performed and representative data is shown in this figure.
To test the hypothesis that UTP14A represents a direct interaction partner of DHX37, an in vitro binding assay was performed using recombinant ZZ-UTP14A-His10 and DHX37-His6. DHX37-His6 was incubated with either IgG sepharose or IgG sepharose on which ZZ-UTP14A-His10 had been immobilized. After thorough washing steps, bound proteins were eluted and analyzed by western blotting alongside input samples. DHX37-His6 was co-precipitated with the ZZ-UTP14-His10-bound beads, but not with the empty IgG sepharose (Figure 6(b)), confirming that these proteins can interact directly. We next explored the influence of UTP14A on the catalytic activity of DHX37 using in vitro ATPase assays performed in the presence and absence of UTP14A. As previously shown (Figure 3(b)), DHX37 displayed RNA-dependent ATPase activity (Figure 6(c)) whereas the amount of ATP hydrolyzed by UTP14A in the presence or absence of RNA was minimal. However, addition of UTP14A stimulated the RNA-dependent ATPase activity of DHX37 more than two-fold. To verify the specificity of UTP14A in stimulating the ATPase activity of DHX37, we also examined the influence of UTP14A on the ATPase activity of another DEAH-box RNA helicase, DHX15. While recombinant MBP-DHX15-His10 displayed RNA-dependent ATPase activity, the rate of ATP hydrolysis was not stimulated by the addition of UTP14A (Supplementary Figure 5). To determine if UTP14A acts as a cofactor of DHX37 during SSU biogenesis, we first established RNAi against UTP14A (Figure 6(d)) and compared the pre-rRNA processing defects observed upon depletion of the helicase and its cofactor. Depletion of UTP14A caused accumulation of the 30S pre-rRNA and a corresponding decrease in the levels of the 21S and 18SE pre-rRNAs (Figure 6(e)). The aberrant 38S* and 16S* pre-rRNA fragments observed upon depletion of DHX37 did not accumulate when the level of UTP14A was reduced, suggesting that the presence of the helicase in pre-ribosomal particles, but not its cofactor, is monitored. The finding that depletion of UTP14A impedes a pre-rRNA processing step (conversion of 30S to 21S) upstream of the processing event for which the catalytic activity of DHX37 is required (conversion of 21S to 18SE) could suggest that UTP14A has an additional role in SSU biogenesis that is not linked to its stimulation of DHX37. To investigate if, like DHX37, UTP14A is required for the release of the U3 snoRNA from pre-ribosomes, extracts from cells treated with control siRNAs (siNT) or siRNAs targeting UTP14A were separated by sucrose density gradient centrifugation and the amounts of U3 co-migrating with non-ribosomal and (pre-)ribosomal complexes were determined by northern blotting. Similar to expression of catalytically inactive DHX37 (Figure 5(b)), depletion of UTP14A lead to increased amounts of the U3 snoRNA present in fractions containing pre-ribosomal complexes (Figure 6(f)), suggesting that stimulation of DHX37 by UTP14A may be linked to the requirement for the helicase for the release of U3 from pre-ribosomes.
Discussion
Here, we have analyzed the putative RNA helicase DHX37, which is the homologue of the yeast DEAH-box protein Dhr1. In yeast, Dhr1 is required during the early steps of SSU maturation where it is proposed to trigger dissociation of the U3 snoRNA from its pre-rRNA basepairing site(s) allowing formation of the central pseudoknot, a key structural feature of the 18S rRNA within the small ribosomal subunit[26]. Our data demonstrate that in human cells, DHX37 is also required for pre-40S maturation and that lack of DHX37 impairs various steps in the maturation of the 18S rRNA leading to reduced levels of both the mature 18S rRNA and 40S subunits. We further show that DHX37 crosslinks to the U3 snoRNA and that expression of catalytically inactive DHX37 leads to increased levels of U3 on pre-ribosomal complexes, suggesting that the function of this helicase in facilitating U3 snoRNA release may be conserved from yeast to humans. In yeast, changes in the solvent accessibility of a residue within the 18S sequence that basepairs with the 5ʹ region of the U3 snoRNA have been observed upon Dhr1 depletion, and Dhr1 has been reported to crosslink to the 5ʹ region of the U3 sequence containing the GAC and A/A’ boxes [26]. However, in vitro Dhr1 has also been shown to unwind a duplex formed between the U3 3ʹ hinge region and the corresponding 5ʹ ETS sequence [26]. It therefore remains unclear whether the remodelling activity of Dhr1 is directed at both of these U3-pre-rRNA duplexes or if disruption of one interaction is sufficient to trigger spontaneous dissociation of the U3 snoRNA from the other site leading to release of the snoRNA from early pre-ribosomal complexes. This open question is likely best addressed by structural analyses of Dhr1-containing pre-ribosomal complexes; however, Dhr1 is not present in any of the currently available high resolution structures of SSU processomes, as these particles still contain the U3 snoRNA and evidence suggests that Dhr1 does not interact stably with pre-ribosomes [24–26]. Interestingly, in vivo crosslinking in human cells revealed direct interactions between DHX37 and U3 sequences that lie close to the 3ʹ hinge region, which is predicted to basepair with the 5ʹ ETS, implying that DHX37 may influence U3-5ʹ ETS interactions. Consistent with this, we observed that expression of catalytically inactive DHX37 impairs processing at the A’ cleavage site in the 5ʹ ETS, a pre-rRNA cleavage event that has previously been shown to be sensitive to depletion of the U3-associated protein U3-55K (RRP9) [57]. Our complementation assays show that the catalytic activity of DHX37 is required for the conversion of the 21S pre-rRNA to 18SE, indicating that removal of the 5ʹ ETS and site 2 cleavage in ITS1 can take place but that subsequent processing of ITS1 is inhibited. The finding that removal of the 5ʹ ETS and site 2 processing can take place although A’ is impaired is consistent with previous data showing that A’ cleavage can be bypassed [9]. The pre-rRNA processing defects observed upon depletion of DHX37 could suggest that failure to release the U3 snoRNA during earlier stages of pre-40S maturation may not inhibit pre-rRNA processing until later. However, it is not yet known at precisely which stage of ribosome biogenesis the U3 snoRNA is released from human pre-ribosomes. Although production of the 21S pre-rRNA involves cleavages in 5ʹ ETS, which would be anticipated to remove U3 basepairing sites, it remains to be determined how U3 release is co-ordinated with pre-rRNA processing events. An alternative possibility is that the requirement for DHX37 for the conversion of 21S to 18SE represents an additional function for the helicase in SSU biogenesis, beyond its proposed role in facilitating U3 dissociation.
Similar to yeast Dhr1, DHX37 does not appear to associate stably with pre-ribosomal complexes in sucrose density gradient experiments. Nevertheless, the binding of DHX37 to the U3 snoRNA observed by immunoprecipitation and CRAC is anticipated to take place in the context of pre-ribosomal particles. Consistent with this, our complementation assays suggest that the presence of DHX37 in early pre-ribosomal complexes is monitored and that in the absence of the helicase, pre-ribosomes are targeted for degradation. Relatively little is known about the quality control pathways that are responsible for turnover of aberrant pre-ribosomes, especially in human cells, but it has been suggested that exonucleases involved in pre-rRNA processing steps could be utilized as degradation machines if particular ribosome biogenesis steps have not taken place correctly. Mapping of the aberrant 38S* and 16S* pre-rRNAs detected upon depletion of DHX37 demonstrates that they are 5ʹ truncated derivatives of the 41S and 21S pre-rRNA, but depletion of the 5ʹ-3ʹ exonuclease XRN2 did not influence production of these species. This implies that other nucleases are involved in the turnover of pre-ribosomes lacking DHX37. Stalling of exonucleases at highly structured pre-rRNA sequences, or pre-rRNA-bound ribosome biogenesis factors or ribosomal proteins may lead to formation of discrete pre-rRNA intermediates, and the diffuse nature of the 16S* intermediate, which often appears as two bands, suggests that this species may be heterogenous as would be expected if it is produced by exonucleolytic trimming. It is possible however, that during surveillance, the 18S rRNA sequence is first targeted by an endonuclease, as was recently reported in yeast [57]. In yeast, Utp24 cleaves the 18S rRNA sequence at nucleotide 618 (Q1 site) between helices 19 and 20, whereas the 5ʹ end of 38S* maps to helix 32 of the human 18S rRNA. While both these rRNA sequences lie in close proximity to the Utp24 binding site identified in yeast, as UTP24 is a core component of the SSU processome, expression of a catalytically inactive form of UTP24 would be required to investigate its potential role in pre-ribosome quality control in human cells.
The finding that pre-ribosomes lacking DHX37 are targeted for degradation further highlights the importance of this protein for ribosome assembly. Interestingly, the surveillance pathway also appears to be triggered when the pre-40S biogenesis factors DIMT1L or TSR1 are lacking. While our data indicate that DHX37, DIMT1L and TSR1 can be present in the same pre-ribosomal particles, their association in vivo is dependent on RNA, implying that they do not form a discrete complex whose presence is monitored by this surveillance system. It is possible that the quality control pathway instead monitors maturation or folding of the central domain of the 18S rRNA as human TSR1 and yeast Dim1 have been demonstrated to bind this region, and DHX37 is implicated, either directly or indirectly, in regulating assembly of the central pseudoknot structure via U3 release. Alternatively, as the evolutionarily conserved rRNA modifications installed by DIMT1L are implicated in maintaining the fidelity of translation, and TSR1 plays an important role in preventing premature subunit joining, the requirement for a quality control pathway to confirm the pre-ribosomal recruitment of these three factors may reflect the critical roles they individually play during ribosome assembly.
In yeast, Dhr1 interacts with the rRNA methyltransferase Bud23 and with the pre-40S biogenesis factor Utp14 [53–55]. While Dhr1 has been shown to be required for the recruitment of Bud23 to pre-ribosomes, Bud23 and Utp14 are also necessary for the association of Dhr1 [54,55]. As these proteins also form direct interactions, it has been proposed that they are recruited to pre-ribosomes as a complex. Our data reveal that although the interaction between Dhr1/DHX37 and Utp14/UTP14A is conserved between yeast and humans, DHX37 does not co-precipitate the Bud23 orthologue WBSCR22 from human cells. On a molecular level this difference can readily be explained as structural analysis of the Dhr1-Bud23-Trm112 complex revealed that amino acids in the N-terminal region of Dhr1 contact Bud23 [54] and this region of the protein is largely absent from human DHX37. In yeast, Bud23 is recruited to early pre-ribosomal complexes but is reported to install the 18S-m7G1575 modification at a later stage [54,58]. In contrast, and consistent with the lack of interaction with DHX37, WBSCR22 is only present in later pre-40S complexes [38,56], implying that pre-ribosomal recruitment of these two proteins is independent in human cells. However, as in yeast, we discovered a direct, RNA-independent interaction between DHX37 and UTP14A in human cells. The interaction surface between yeast Dhr1 and Utp14 has been identified and amino acid substitutions within this region have been shown to impair the association of these proteins [55]. Surprisingly, introduction of the equivalent amino acid substitutions in DHX37 did not block its interaction with UTP14A (data not shown), suggesting that the precise contacts between these proteins are different in humans than yeast. Yeast Utp14 has been reported to stimulate the unwinding, but not ATPase, activity of the Dhr1 in vitro and is proposed to act as a cofactor for the helicase [26]. The precise mechanism by which Utp14 regulates Dhr1 therefore remains unclear. We observe a mild increase in the rate of ATP hydrolysis by DHX37 in the presence of UTP14A compared to the helicase alone, suggesting that human UTP14A can also function as a helicase cofactor. Depletion of either yeast Utp14 [55] or human UTP14A causes accumulation of the U3 snoRNA on pre-ribosomes. As lack of UTP14A causes defects in early pre-rRNA processing steps, we cannot rule out the possibility that the observed accumulation of U3 on pre-ribosomes is an indirect effect cause by impaired ribosome assembly. However, taken together with the stimulation of DHX37 by UTP14A and data from a yeast complementation system showing that disruption of the interaction between these proteins causes a similar phenotype, it is likely that, on a cellular level, UTP14A acts together with DHX37 to promote U3 release.
Taken together, while our analysis of DHX37 suggests that the requirement for this helicase for efficient dissociation of the U3 snoRNP from pre-ribosomes is conserved in eukaryotes, we also highlight differences in the precise RNA and protein interactions formed by this helicase in yeast and human cells. To gain further mechanistic insights into Dhr1/DHX37 regulation by Utp14/UTP14A and to understand precisely how Dhr1/DHX37 contributes to U3 release, structural analyses of the helicase alone and within the context of pre-ribosomes will likely be required.
Materials and methods
Molecular cloning
The coding sequence of DHX37 (NM_032656.3) was cloned into a pcDNA5-based vector for the expression of proteins with a C-terminal His6-PreScission protease cleavage site-2xFlag (Flag) tag from a defined genomic locus via a tetracycline-regulatable promoter in human cells. To express RNAi-resistant DHX37, site-directed mutagenesis was used to introduce five silent mutations within the binding site of siDHX37_1 (Supplementary Table 2). The coding sequences of DHX37 and UTP14A (NM_006649.3) were also cloned into pQE80-derivatived vectors (A42 and A15) for expression of proteins with a C-terminal His6 tag or an N-terminal ZZ tag and C-terminal His10 tag from IPTG-regulatable promoters in E. coli. Site-directed mutagenesis was performed on both the DHX37 constructs to introduce point mutations that lead to a threonine 282 to alanine substitution (T282A) in the expressed protein.
Human cell culture, generation of cell lines and RNAi
Hela CCL2 and HEK293 cells were cultured according to standard protocols at 37 °C with 5% CO2 in 1x Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% foetal calf serum. To generate cell lines for expression of RNAi-resistant, wild-type or mutant DHX37, or the Flag tag, HEK293 Flp-In-T-Rex (Thermo Scientific) cells were transfected with the appropriate pcDNA5 constructs according to the manufacturer’s instructions. Selection of stably transfected cells was achieved using hygromycin and blasticidin. Protein expression was induced by addition of 0.5–1000 ng/mL tetracycline for 24 h before harvesting. For RNAi, cells were transfected with 20–50 nmol of siRNA (Supplementary Table 2) using lipofectamine RNAiMAX (Thermo Scientific) according to the manufacturer’s instructions. Cells were harvested 72 h after transfection unless otherwise stated.
Immunofluorescence
Immunofluorescence was essentially performed as previously described in [19]. HeLa cells were grown on coverslips before fixing using 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were first permeabilised using 0.1% Triton-X-100 in PBS for 20 min then blocked with 10% FCS and 0.1% Triton-X-100 in PBS for 1 h at room temperature. Cells were incubated with anti-DHX37 and anti-NSUN5 antibodies (Supplementary Table 3) diluted in 10% FCS in PBS for 2 h at room temperature. Coverslips were washed and cells were incubated with Alexa Fluor 488-conjugated and Alexa Fluor 657-conjugated secondary antibodies for 1 h at room temperature. Coverslips were washed again and then mounted using Vectashield (Vector Laboratories) containing DAPI. Cells were analysed by confocal microscopy using a ConfoCor2 microscope (Carl Zeiss).
RNA extraction, northern blotting and pulse-chase metabolic labelling
Total RNA was extracted from human cells using TRI reagent (Sigma Aldrich) according to the manufacturer’s instructions. Detection of pre-rRNAs by northern blotting was performed as previously described in [9]. In brief, 5 μg of total RNA were separated on a 1.2% agarose-glyoxal gel and transferred to a nylon membrane by vacuum blotting or alternatively, RNAs were separated on a 10% denaturing (7 M urea) polyacrylamide gel and transferred to nylon membrane by wet electroblotting. Membranes were pre-hybridized in SES1 (0.25 M sodium phosphate pH 7.0, 7 % SDS (w/v), 1 mM EDTA) buffer before incubation with 5ʹ [32P]-labelled DNA oligonucleotides (Supplementary Table 4) in SES1 buffer at 37°C overnight. After washing, signals were visualized using a Typhoon FLA9500 phosphorimager. Pulse-chase labelling of nascent RNAs was essentially performed as previously described in [10]. siRNA-treated cells were grown in phosphate-free DMEM for 1 h before exchanging the media for phosphate-free DMEM supplemented with 15 μCi/mL [32P]-labelled orthophosphate and growth for a further 1 h. The labelled media was then removed, cells were washed and grown in unlabelled DMEM for an additional 3 h before harvesting. Total RNA was extracted, separated by denaturing agarose gel electrophoresis, transferred to a nylon membrane by capillary blotting and abundant, labelled RNA species were visualized using a phosphorimager.
Immunoprecipitation of complexes from human cells
Expression of DHX37-Flag or the Flag tag was induced in HEK293 cells for 24 h and cells were harvested. Cells were lyzed by sonication in a buffer containing 50 mM Tris- HCl pH 7.8, 150 mM NaCl, 0.5 mM ethylenediaminetetraacetic acid (EDTA), 5 mM β-mercaptoethanol and extracts were centrifuged at 20,000 x g for 10 min to remove insoluble material. The cleared lysate was supplemented with 1.5 mM MgCl2 and was either treated with RNase A (2.5 U) or was left untreated. The extracts were then incubated with anti-Flag magnetic beads (Sigma Aldrich) for 2 h at 4 °C, and bound proteins and RNAs were eluted with 250 μg/mL Flag peptide. Proteins were precipitated with 20% trichloroacetic acid (TCA) and analyzed by western blotting using antibodies listed in Supplementary Table 3. Alternatively, RNAs were extracted using phenol:chloroform:isoamylalcohol (PCI; 25:24:1), ethanol precipitated and analysed by northern blotting using probes listed in Supplementary Table 4.
Sucrose density gradient centrifugation
HeLa cells, or HEK293 cell lines expressing DHX37-Flag or DHX37T282-Flag were lysed by sonication in a buffer containing 50mM Tris pH 7.5, 100mM NaCl, 5mM MgCl2 and 1mM DTT before centrifugation at 20,000 x g for 10 min at 4°C to remove insoluble material. The cleared extracts were separated on 10–45% sucrose density gradients in an SW-40Ti rotor for 16 h at 23,500 rpm. 530 μL fractions were collected and the absorbance of each fraction at 260 nm was determined using a nanodrop (Peqlab). The proteins present in each fraction were precipitated using 20% TCA, separated by SDS-PAGE and analyzed by western blotting using antibodies listed in Supplementary Table 3. RNAs present in each fraction were extracted using PCI, ethanol precipitated and analyzed by northern blotting using probes listed in Supplementary Table 4.
Crosslinking and analysis of cDNA (CRAC)
CRAC was performed as previously described in references 38,40,51,52. Briefly, expression of DHX37-Flag or the Flag tag was induced in HEK293 cell lines for 24 h before growth in media supplemented with 100 μM 4-thiouridine for 6 h. In vivo crosslinking was then performed using 2 cycles of 180 mJ/cm2 light at 365 nm in a Stratalinker (Agilent Technologies). Cells were harvested and RNA-protein complexes were enriched by tandem affinity purification on anti-Flag M2 magnetic beads and NiNTA. After a limited RNase digestion, co-precipitated RNAs were 5ʹ end labelled with [32P] and sequencing adaptors were ligated. RNA-protein complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and labelled RNAs were visualized by autoradiography. Regions of the membrane were excised and RNAs were eluted by Proteinase K digestion then extracted using PCI. RNA fragments were reverse transcribed, amplified by PCR and the resulting library was subjected to Illumina sequencing. The obtained sequence reads were mapped to the human genome (GRCh37.p13).
Purification of recombinant proteins, and in vitro binding and ATPase assays
Expression of DHX37-His6 or ZZ-UTP14A-His10 was induced in BL21 codon plus cells by addition of 1 mM IPTG for 16 h at 18°C. Cells were pelleted, resuspended in Lysis buffer (50 mM Tris-HCl pH 7.0, 500 mM NaCl, 1 mM MgCl2, 10% glycerol and 5 mM imidazole) and disrupted by sonication. The cell lysate was cleared by centrifugation at 50,000 x g for 30 min and the soluble fraction was incubated with cOmplete His-tag purification resin (Roche) for 2 h at 4°C. After thorough washing steps with low and high salt buffers (50 mM Tris-HCl pH 7.0, 500 mM/1000 mM NaCl, 1 mM MgCl2, 10% glycerol and 25 mM imidazole), bound proteins were eluted with lysis buffer supplemented with 500 mM imidazole. The eluate was dialyzed against a buffer containing 50 mM Tris-HCl pH 7.0, 150 mM NaCl and 20% glycerol. MBP-DHX15-His10 was prepared as previously described in reference 38.
For in vitro binding assays, 200 pmol of recombinant ZZ-UTP14A-His10 was immobilized on IgG sepharose (GE Healthcare) that had been pre-equilibrated in 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1.5 mM MgCl2, 0.2 % Triton X-100 and 1 mM DTT. After washing steps to remove unbound protein, IgG sepharose and ZZ-UTP14A-His10-IgG sepharose was incubated with 200 pmol DHX37-His6 for 1 h rotating at 4 °C. The beads were then washed and elution was performed by treatment with 2 μM TEV protease overnight at 4 °C in 50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1.5 mM MgCl2, 0.2 % Triton X-100 and 1 mM DTT. Inputs and eluates were separated by SDS-PAGE then analyzed by western blotting using antibodies listed in Supplementary Table 3.
Hydrolysis of ATP was monitored in an NADH-coupled assay [59]. Reactions were set up containing 45 mM Tris-HCl pH 7.4, 25 mM NaCl, 2 mM MgCl2, 1 mM phosphoenolpyruvate, 300 μM NADH, 20 U/ml pyruvate kinase and 4 mM ATP and were supplemented with 500 nM recombinant DHX37/DHX37T282/DHX15, 500 nM recombinant UTP14A and/or 1.5 μM RNA (5ʹ-GUAAUGUAAGUGAACGUAAAACAAAACAAAAC-3ʹ) as indicated. The decrease in absorbance at 340 nm was monitored using a BioTEK Synergy plate reader and this was used to calculate the rate of ATP hydrolysis.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [BO3442/1-2];Deutsche Forschungsgemeinschaft [SFB1190].
Acknowledgments
We would like to thank Jens Kretschmer for help with bioinformatics analyzes and cloning, and Claudia Schneider for helpful discussions. This work was funded by the Deutsche Forschungsgemeinschaft (SFB1190 and BO3442/1-2) and the University Medical Centre Göttingen.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest.
Supplementary material
Supplemental Material footnote should be located in the CFN. The statement to be included: Supplemental data for this article can be accessed on the publisher’s website.
References
- [1].Anger AM, Armache J-P, Berninghausen O, et al. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. [DOI] [PubMed] [Google Scholar]
- [2].Woolford JLJ, Baserga SJ.. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Andersen JS, Lyon CE, Fox AH, et al. Directed proteomic analysis of the human nucleolus. Curr Biol. 2002;12:1–11. [DOI] [PubMed] [Google Scholar]
- [4].Scherl A, Coute Y, Deon C, et al. Functional proteomic analysis of human nucleolus. Mol Biol Cell. 2002;13:4100–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wild T, Horvath P, Wyler E, et al. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010;8:e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tafforeau L, Zorbas C, Langhendries J-L, et al. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol Cell. 2013;51:539–551. [DOI] [PubMed] [Google Scholar]
- [7].Badertscher L, Wild T, Montellese C, et al. Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep. 2015;13:2879–2891. [DOI] [PubMed] [Google Scholar]
- [8].Farley-Barnes KI, McCann KL, Ogawa LM, et al. Diverse regulators of human ribosome biogenesis discovered by changes in nucleolar number. Cell Rep. 2018;22:1923–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sloan KE, Bohnsack MT, Schneider C, et al. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. Rna. 2014;20:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sloan KE, Mattijssen S, Lebaron S, et al. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J Cell Biol. 2013;200:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Preti M, O’Donohue M-F, Montel-Lehry N, et al. Gradual processing of the ITS1 from the nucleolus to the cytoplasm during synthesis of the human 18S rRNA. Nucleic Acids Res. 2013;41:4709–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mills EW, Green R. Ribosomopathies: there’s strength in numbers. Science. 2017;358:eaan2755. [DOI] [PubMed] [Google Scholar]
- [13].Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18:51–63. [DOI] [PubMed] [Google Scholar]
- [14].Henras AK, Plisson-Chastang C, O’Donohue M-F, et al. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6:225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. [DOI] [PubMed] [Google Scholar]
- [16].Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wells GR, Weichmann F, Colvin D, et al. The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 2016;44:5399–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wells GR, Weichmann F, Sloan KE, et al. The ribosome biogenesis factor yUtp23/hUTP23 coordinates key interactions in the yeast and human pre-40S particle and hUTP23 contains an essential PIN domain. Nucleic Acids Res. 2017;45:4796–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Warda AS, Freytag B, Haag S, et al. Effects of the Bowen-Conradi syndrome mutation in EMG1 on its nuclear import, stability and nucleolar recruitment. Hum Mol Genet. 2016;25:5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kass S, Tyc K, Steitz JA, et al. The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell. 1990;60:897–908. [DOI] [PubMed] [Google Scholar]
- [21].Sharma K, Venema J, Tollervey D. The 5ʹ end of the 18S rRNA can be positioned from within the mature rRNA. Rna. 1999;5:678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hughes JM. Functional base-pairing interaction between highly conserved elements of U3 small nucleolar RNA and the small ribosomal subunit RNA. J Mol Biol. 1996;259:645–654. [DOI] [PubMed] [Google Scholar]
- [23].Borovjagin AV, Gerbi SA. The spacing between functional Cis-elements of U3 snoRNA is critical for rRNA processing. J Mol Biol. 2000;300:57–74. [DOI] [PubMed] [Google Scholar]
- [24].Barandun J, Chaker-Margot M, Hunziker M, et al. The complete structure of the small-subunit processome. Nat Struct Mol Biol. 2017;24:944–953. [DOI] [PubMed] [Google Scholar]
- [25].Cheng J, Kellner N, Berninghausen O, et al. 3.2-A-resolution structure of the 90S preribosome before A1 pre-rRNA cleavage. Nat Struct Mol Biol. 2017;24:954–964. [DOI] [PubMed] [Google Scholar]
- [26].Sardana R, Liu X, Granneman S, et al. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol. 2015;13:e1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sloan KE, Gleizes P-E, Bohnsack MT. Nucleocytoplasmic Transport of RNAs and RNA-Protein Complexes. J Mol Biol. 2016;428:2040–2059. [DOI] [PubMed] [Google Scholar]
- [28].Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–516. [DOI] [PubMed] [Google Scholar]
- [29].Jarmoskaite I, Russell R. RNA helicase proteins as chaperones and remodelers. Annu Rev Biochem. 2014;83:697–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liang X-H, Fournier MJ. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol Cell Biol. 2006;26:7437–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bohnsack MT, Kos M, Tollervey D. Quantitative analysis of snoRNA association with pre-ribosomes and release of snR30 by Rok1 helicase. EMBO Rep. 2008;9:1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bohnsack MT, Martin R, Granneman S, et al. Prp43 bound at different sites on the pre-rRNA performs distinct functions in ribosome synthesis. Mol Cell. 2009;36:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dembowski JA, Kuo B, Woolford JL. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic Acids Res. 2013;41:7889–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pertschy B, Schneider C, Gnadig M, et al. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J Biol Chem. 2009;284:35079–35091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Martin R, Straub AU, Doebele C, et al. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013;10:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Srivastava L, Lapik YR, Wang M, et al. Mammalian DEAD box protein Ddx51 acts in 3ʹ end maturation of 28S rRNA by promoting the release of U8 snoRNA. Mol Cell Biol. 2010;30:2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kellner M, Rohrmoser M, Forne I, et al. DEAD-box helicase DDX27 regulates 3ʹ end formation of ribosomal 47S RNA and stably associates with the PeBoW-complex. Exp Cell Res. 2015;334:146–159. [DOI] [PubMed] [Google Scholar]
- [38].Memet I, Doebele C, Sloan KE, et al. The G-patch protein NF-kappaB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids Res. 2017;45:5359–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Calo E, Flynn RA, Martin L, et al. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;518:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sloan KE, Leisegang MS, Doebele C, et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015;43:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Larburu N, Montellese C, O’Donohue M-F, et al. Structure of a human pre-40S particle points to a role for RACK1 in the final steps of 18S rRNA processing. Nucleic Acids Res. 2016;44:8465–8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ameismeier M, Cheng J, Berninghausen O, et al. Visualizing late states of human 40S ribosomal subunit maturation. Nature. 2018;558:249–253. [DOI] [PubMed] [Google Scholar]
- [43].Wang M, Pestov DG. 5ʹ-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res. 2011;39:1811–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zorbas C, Nicolas E, Wacheul L, et al. The human 18S rRNA base methyltransferases DIMT1L and WBSCR22-TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol Biol Cell. 2015;26:2080–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Carron C, O’Donohue MF, Choesmel V, et al. Analysis of two human pre-ribosomal factors, bystin and hTsr1, highlights differences in evolution of ribosome biogenesis between yeast and mammals. Nucleic Acids Res. 2011;39:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].McCaughan UM, Jayachandran U, Shchepachev V, et al. Pre-40S ribosome biogenesis factor Tsr1 is an inactive structural mimic of translational GTPases. Nat Commun. 2016;7:11789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Leulliot N, Bohnsack MT, Graille M, et al. The yeast ribosome synthesis factor Emg1 is a novel member of the superfamily of alpha/beta knot fold methyltransferases. Nucleic Acids Res. 2008;36:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meyer B, Wurm JP, Kotter P, et al. The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Psi1191 in yeast 18S rRNA. Nucleic Acids Res. 2011;39:1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Haag S, Kretschmer J, Bohnsack MT. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. Rna. 2015;21:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bohnsack MT, Tollervey D, Granneman S. Identification of RNA helicase target sites by UV cross-linking and analysis of cDNA [Internet]. 1st ed. Elsevier Inc; 2012. DOI: 10.1016/B978-0-12-396546-2.00013-9. [DOI] [PubMed] [Google Scholar]
- [51].Haag S, Kretschmer J, Sloan KE, et al. Crosslinking methods to identify RNA methyltransferase targets in vivo. Methods Mol Biol. 2017;1562:269–281. [DOI] [PubMed] [Google Scholar]
- [52].Sloan KE, Bohnsack MT. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem Sci. 2018;43:237–250. [DOI] [PubMed] [Google Scholar]
- [53].Sardana R, Zhu J, Gill M, et al. Physical and functional interaction between the methyltransferase Bud23 and the essential DEAH-box RNA helicase Ecm16. Mol Cell Biol. 2014;34:2208–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Létoquart J, Huvelle E, Wacheul L, et al. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. Proc Natl Acad Sci USA. 2014;111:E5518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhu J, Liu X, Anjos M, et al. Utp14 recruits and activates the RNA helicase Dhr1 to undock U3 snoRNA from the preribosome. Mol Cell Biol. 2016;36:965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ounap K, Kasper L, Kurg A, et al. The human WBSCR22 protein is involved in the biogenesis of the 40S ribosomal subunits in mammalian cells. PLoS One. 2013;8:e75686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Choque E, Schneider C, Gadal O, et al. Turnover of aberrant pre-40S pre-ribosomal particles is initiated by a novel endonucleolytic decay pathway. Nucleic Acids Res. 2018;46:4699–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].White J, Li Z, Sardana R, et al. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol Cell Biol. 2008;28:3151–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Heininger AU, Hackert P, Andreou AZ, et al. Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol. 2016;13:320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.