

Abstract
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause the inherited disorder cystic fibrosis (CF). Lung disease is the major cause of CF morbidity, though CFTR expression levels are substantially lower in the airway epithelium than in pancreatic duct and intestinal epithelia, which also show compromised function in CF. Recently developed small molecule therapeutics for CF are highly successful for one specific CFTR mutation and may have a positive impact on others. However, the low abundance of CFTR transcripts in the airway limits the opportunity for drugs to correct the defective substrate. Elucidation of the transcriptional mechanisms for the CFTR locus has largely focused on intragenic and intergenic tissue-specific enhancers and their activating trans-factors. Here we investigate whether the low CFTR levels in the airway epithelium result from the recruitment of repressive proteins directly to the locus. Using an siRNA screen to deplete ~1500 transcription factors (TFs) and associated regulatory proteins in Calu-3 lung epithelial cells we identified nearly 40 factors that upon depletion elevated CFTR mRNA levels more than two-fold. A subset of these TFs was validated in primary human bronchial epithelial (HBE) cells. Among the strongest repressors of airway expression of CFTR were Krüppel like factor 5 (KLF5) and Ets homologous factor (EHF), both of which have pivotal roles in the airway epithelium. Depletion of these factors, which are both recruited to an airway-selective cis-regulatory element at −35kb from the CFTR promoter, improved CFTR production and function, thus defining novel therapeutic targets for enhancement of CFTR.
Keywords: CFTR, gene regulation, transcription factors, KLF5, EHF, lung epithelium
Introduction
Tissue-specific regulation of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which when mutated causes the common inherited disorder cystic fibrosis (CF), has been a topic of intensive investigation (reviewed in [1], [2], [3]). The results revealed key aspects of locus architecture, intergenic and intronic regulatory elements with cell-specific signatures, and a transcription factor network recruited to these elements reflecting the differentiated functions of the host cell. These features of CFTR are a paradigm for other genes with complex control mechanisms, which involve cis-regulatory elements outside the gene promoter. CFTR lies within a topologically associated domain (TAD) that is limited by regions occupied by the architectural proteins CCCTC–binding factor (CTCF) and cohesin complex [4] [5]. Within the TAD, which extends from −80.1 kb 5’ to the CFTR promoter to +48.9 kb 3’ to the last coding base, cell-selective cis-regulatory elements are recruited to the gene promoter to drive gene expression (Fig. 1). These elements are located both in introns and in intergenic regions and several of them encompass tissue-specific enhancers. In the intestinal epithelium the pioneer transcription factor (TF) Forkhead box A2 (FOXA2) has a pivotal role in driving gene expression through intronic enhancers and in establishing the 3D structure of the active locus [6] [7] [4]. It may also have an important role in the recruitment of other TFs including hepatocyte nuclear factor 1 (HNF1) [8] [9] and Caudal type homeobox 2(CDX2) to the cis-regulatory elements. In the airway epithelium two cis-elements outside the gene at −44 kb and −35 kb upstream of the gene promoter are important in conferring tissue-specific expression [10–12]. Both the sites encompass airway-selective enhancers, which appear to be functionally linked, though they recruit different TFs. The −44 kb site is associated with an antioxidant response element (ARE) that may serve as an environmental/stress sensor. Under normal conditions, this site is occupied by repressor BTB and CNC homology 1, basic leucine zipper transcription factor (Bach1), and v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog K (MafK) heterodimers. After natural antioxidant treatment (sulforaphane, SFN), nuclear factor, erythroid 2-like 2 (Nrf2) translocates into the nucleus and displaces these repressive factors and activates CFTR expression. Site-directed mutagenesis shows that both the ARE and an adjacent NF-κB binding site are required for activation. The −35 kb enhancer is responsive to the immune-mediators interferon regulatory factor 1 and 2 (IRF1 and IRF2), IRF1 activating and IRF2 repressing the enhancer function. However, the functional complexity of this site and the requirement of other factors, including nuclear factor Y (NF-Y), to maintain active histone marks at the site implicate additional mechanisms underlying its function. Our goal was to reveal other key pathways and TFs that contribute to the control of CFTR expression in airway epithelial cells.
Figure 1. Histone modifications across the CFTR locus in airway epithelial cells.

UCSC genome browser graphic showing occupancy of repressive (H3K9me2, H3K9me3 and H3K27me3) and active (H3K4me3 and H3K27ac) histone modifications by ChIP-seq in Calu-3 and primary HBE cells. Blue triangles denote the TAD boundaries. The CFTR promoter is marked by a black arrowhead. Red and grey arrows denote repressive histone marks at cis-regulatory elements. Of note the promoter carries active marks (turquoise arrows) in Calu-3 and HBE cells but also repressive marks (purple arrow) in HBE cells. Cis-regulatory elements/DHS as reviewed in [3].
Of direct relevance to this objective is one aspect of CFTR regulation that is not well understood, specifically how very low CFTR expression is maintained in the airway epithelium in comparison to the much higher levels evident in pancreatic duct, intestinal epithelium and male genital ducts [13], [14], [15], [16], [17]. Moreover, the developmental profile of CFTR suggests that the locus may be subject to repression, since transcript abundance is highest in the early to mid-trimester of human and sheep gestation and then appears to fall gradually to reach the low levels characteristic of the lung epithelium at birth [18]. To test the hypothesis that repressive TFs or other chromatin associated-factors (CAFs) were responsible for this phenomenon we screened a library of siRNAs designed to target about 1500 TFs and CAFs in Calu-3 cells. We identified ~50 siRNAs that reproducibly increased CFTR expression levels more than two-fold in replica screens. Factors with documented global roles in gene activation or repression, together with TFs with very low expression levels in Calu-3 and HBE cells, according to our RNA-seq data (FPKM<1), were not evaluated further. Here we describe 37 TFs, which upon depletion increase CFTR transcript levels and were not previously associated with regulation of CFTR expression. We show that several of these TFs also repress CFTR protein expression and directly regulate CFTR by recruitment to cis-regulatory elements at the locus, most notably the upstream airway-selective enhancers or the promoter. These data generate new targets and potential avenues to augment CFTR transcript and protein expression for therapeutic benefit.
Experimental
Cell culture
Calu-3 cells were obtained from ATCC and cultured by standard methods. Passage 1 (P1) primary bronchial epithelial cells were obtained under protocol #03–1396 approved by the University of North Carolina at Chapel Hill Biomedical Institutional Review Board. Informed consent was obtained from authorized representatives of all organ donors. Cells were grown on collagen-coated plastic in bronchial epithelial growth medium (BEGM) [19]. Passage 2 or 3 primary cells were used for the study.
Transcription Factor siRNA screen and validation.
The Dharmacon siGENOME SMARTpool siRNA Library- Human Transcription Factor (G-005805 Lot Number 13107) was used (GE Healthcare). This library comprises twenty 96-well plates with each well containing a pool of four siRNAs that target a single human transcription factor. In total 1528 TFs were targeted. Either 6 pMols of an siRNA pool or non-targeting negative control (D-001210–02-05) were reverse transfected with Lipofectamine RNAiMax (ThermoFisher) into 25,000 Calu-3 cells in a 96-well plate in duplicate. After 72h Calu-3 cells were lysed for RNA extraction, DNase I - treated and cDNA synthesized using the Cell-to-Ct Kit (Ambion). Untransfected Calu-3 cells were used as controls to ensure similar CFTR expression levels across all plates used in the screen. CFTR and β2 microglobulin (β2M) transcript levels were determined in duplicate using 4uL of cDNA in a multiplexed Taqman assay (CFTR probe 5’FAM; β2M probe 5’JOE) on a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). CFTR transcript values were normalized to β2M and compared to the non-targeting negative controls to determine fold changes in CFTR expression. Select factors that showed an increase of CFTR/β2M >2.0 fold were repeated. For validation experiments, CFTR mRNA expression was then measured relative to the geometric mean of 3 normalizers β2M, phosphoglycerate kinase (PGK1) and β actin (ACTB) Taqman primer/probe sets (Suppl. Table 1A) in optimized multiplex RT qPCR reactions. All factors were validated with Dharmacon siRNA reagents and for the factors of most immediate interest KLF5, EHF and BRD8, siRNAs from Ambion (LT) were also used (Fig.2A and Fig 4A). SYBR green RT-qPCR assays were used to confirm siRNA- mediated depletion of each factor (Suppl. Figure 1, Suppl. Table 1C). The negative control siRNA value is taken as 1 and specific siRNA levels for each factor compared to that.
Figure 2. Depletion of multiple transcription factors enhances CFTR transcript levels in Calu-3 cells.
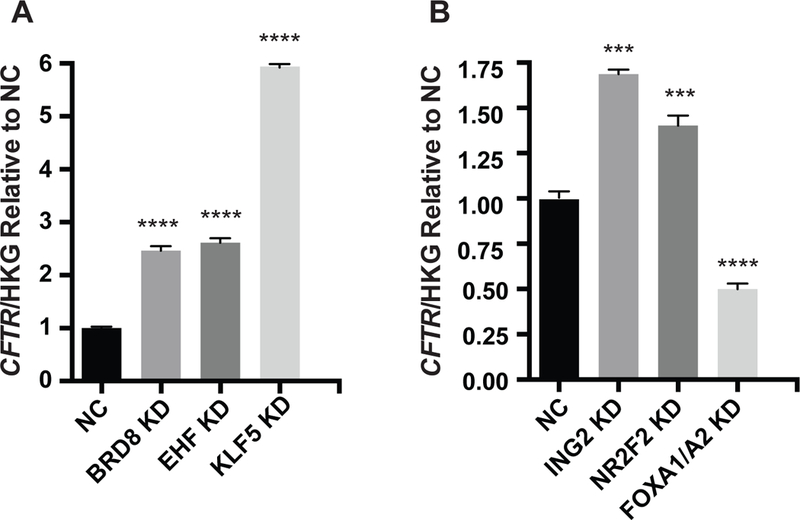
siRNA-mediated depletion (KD) of A) BRD8, EHF, KLF5 and B) ING2, NR2F2, enhances CFTR mRNA abundance. RT-qPCR measurements of CFTR normalized to the geometric mean of three house-keeping gene controls, β2 microglobulin (β2M), phosphoglycerate kinase (PGK) and β actin (ACTB) and shown relative to negative control (NC). Bar shows average of 3 experiments with standard error of the mean. Key: *** P<0.001, **** P<0.0001 by an unpaired two-tailed Student’s t test.
Figure 4. Depletion of KLF5, BRD8 and ING2 enhance CFTR transcript levels in HBE cells.
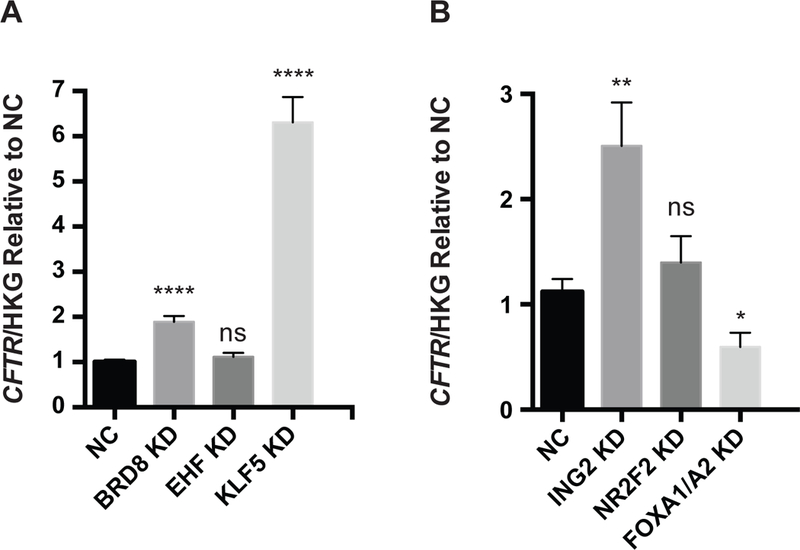
siRNA-mediated depletion (KD) of A) BRD8, EHF, KLF5 and B) ING2, NR2F2, enhances CFTR mRNA abundance. RT-qPCR measurements of CFTR normalized to the geometric mean of three house-keeping gene controls, β2 microglobulin (β2M), phosphoglycerate kinase (PGK) and β actin (ACTB) and shown relative to negative control (NC). Bar shows standard error of the mean. Key: not significant, ns. P>0.05, * P< 0.05, ** P<0.01, **** P<0.0001 by an unpaired two-tailed Student’s t test.
CFTR protein detection by western blot
To assay the effect of siRNA depletion TFs on CFTR protein expression, transfections were done as above and at 72h post-transfection whole cell lysates were collected using NET buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1 mM Na2EDTA) supplemented with Triton-X 100 and protease inhibitors (Sigma). Protein concentration was determined by Bradford assay. SDS- sample loading buffer with β-mercaptoethanol was added and samples were heated at 55°C for 15 minutes. These samples were resolved by SDS-PAGE, transferred to Immobilon membrane and western blots probed with anti-CFTR 596 (Cystic Fibrosis Foundation) and β-tubulin (T4026, Sigma-Aldrich) with ECL detection. Each western blot was only probed once. Films were scanned by densitometry and the intensities of CFTR and β-tubulin compared using Photoshop CS6 Extended and/or ImageJ.
ChIP-seq and ChIP-qPCR
Antibodies used in ChIP-seq and ChIP-qPCR were against Ets homologous factor (EHF) (Clone 5A.5) [20] and Krüppel-like factor 5 (KLF5) (Active motif 61100; Santa Cruz sc-398470) ChIP-seq was performed as described previously [21], [22]. For primers see Suppl. Table 1B. EHF ChIP-seq data are deposited at GEO (GSE:85403).
Chloride conductance assay
Assays were performed as described previously [23] and calibration of intracellular MQAE was performed as in [24]. Briefly, once confluent, Calu-3 cells growing on 96-well tissue culture plates were loaded with 1mM MQAE (N-(Ethoxycarbonylmethyl)- 6-Methoxyquinolinium Bromide) in DMEM low glucose culture medium with 10% FCS in 5% CO2, overnight at 37°C. Cells were washed and then equilibrated in a high chloride, loading buffer (137mM NaCl, 2.7mM KCl, 0.7mM CaCl2, 1.5mM KH2PO4, 8.1mM Na2HPO4, pH 7.4) for 1h. This buffer was then replaced with a high nitrate, efflux buffer (137mM NaNO3, 2.7mM KNO3, 0.7mM Ca(NO3)2, 1.5mM KH2PO4, 8.1mM Na2HPO4, pH 7.4), with or without 5μM the CFTR-activator Forskolin. MQAE fluorescence readings at 360nm excitation /460nm emission were immediately recorded every minute for 10 mins in a Spectramax M5 fluorescence plate reader (Molecular Devices).
Results
Repressive histone marks are evident at cis-regulatory elements in the CFTR locus in airway epithelial cells.
To test the hypothesis that the low abundance of CFTR transcripts in the airway epithelium was a consequence of the recruitment of transcriptional repressors we first sought evidence for repressive histone modifications at the locus. We inspected our genome-wide ChIP-seq data for active (H3K27ac and H3K4me3) and repressive (H3K9me2, H3K9me3and H3K27me3) histone marks in Calu-3 lung adenocarcinoma cells and primary human bronchial epithelial (HBE) cells [21], [22], [25]. These data revealed repressive marks at a number of known cis-regulatory elements in addition to other sites within the CFTR locus (red and grey arrows in Fig. 1).
siRNA screen for factors that repress CFTR expression.
Next, to identify factors that might recruit negative histone marks to the locus we performed a siRNA screen for repressive transcription factors in Calu-3 cells. The Dharmacon siRNA Library targeting human transcription factors includes TFs, some CAFs and certain other factors associated with transcriptional regulation. Here we consider all factors together and do not subdivide them according to specific functions (activating or repressing) since these are often cell-context dependent. In the first replicated screen of siRNAs against 1528 TFs we applied a filter to the data analysis to remove factors with low expression levels (FPKM <1) in Calu-3 cells, which reduced the total factors analyzed further to 1058. CFTR expression after specific factor depletion was normalized to β2 microglobulin transcript levels and compared to replicated (n=4) negative control (NC) siRNA values. Fifty-four factors were identified that upon siRNA-mediated depletion increased the normalized (CFTR/β2M compared to NC2) ratio of CFTR expression more than 2 fold. For each siRNA plate positive controls for transfection efficiency were provided by siRNAs specific for FOXA1 and FOXA2, which we showed previously to be activating transcription factors at the CFTR locus [6], [7]. An additional replica experiment was performed on the 54 factors with 2 independent transfections to generate n=4 assays on each factor. The ratio of CFTR to β2 microglobulin compared to NC was then averaged across experiments and siRNAs against TFs that no longer caused a CFTR change of >2 fold were excluded from further analysis, reducing the number to 50. Thirty-seven of these are shown in Table 1. Also shown in Table 1 are the expression levels (FPKM) of each factor in RNA-seq data (average from n=3) from Calu-3 and HBE cells. In all further experiments on a smaller number of candidate factors that repressed CFTR expression, siRNA-mediated depletion of each factor was confirmed by RT-qPCR (Suppl. Fig 1).
Table 1:
Transcription factors that when depleted increase CFTR mRNA levels ≥ 2 fold in Calu-3 cells.
| GENE | CFTR/β2M Change* |
Calu-3 FPKM** |
HBE FPKM** |
|---|---|---|---|
| ING2 | 3.00 | 7.38 | 10.10 |
| ANKFY1 | 2.83 | 8.01 | 11.52 |
| NR2F2 | 2.71 | 44.65 | 10.21 |
| BRD8 | 2.58 | 37.76 | 9.47 |
| KLF5 | 2.58 | 79.54 | 122.97 |
| ZNF148 | 2.52 | 13.44 | 6.93 |
| TCF7L2 | 2.45 | 22.52 | 4.15 |
| TGIF1 | 2.41 | 47.48 | 38.61 |
| VGLL1 | 2.39 | 80.18 | 9.71 |
| TLE1 | 2.36 | 30.09 | 10.32 |
| ANKRD54 | 2.34 | 16.48 | 24.21 |
| EHF | 2.32 | 72.53 | 189.09 |
| SMARCC2 | 2.31 | 28.88 | 17.51 |
| MDM2 | 2.30 | 14.84 | 35.66 |
| BRCA1 | 2.29 | 8.00 | 2.89 |
| ANKZF1 | 2.29 | 22.62 | 23.95 |
| GTF2IRD1 | 2.28 | 11.93 | 24.06 |
| ATF5 | 2.25 | 34.38 | 59.74 |
| ECSIT | 2.25 | 20.23 | 13.86 |
| SATB2 | 2.24 | 1.146 | 2.69 |
| SREBF2 | 2.23 | 131.37 | 67.47 |
| HDAC1 | 2.19 | 206.77 | 42.60 |
| SMARCE1 | 2.17 | 315.31 | 39.85 |
| BTG1 | 2.16 | 52.28 | 34.37 |
| PLRG1 | 2.14 | 36.62 | 21.04 |
| IRF2BP1 | 2.13 | 15.08 | 7.32 |
| RBAK | 2.13 | 4.05 | 8.48 |
| CASKIN1 | 2.13 | 5.51 | 5.59 |
| FOXE1 | 2.11 | 1.21 | 5.26 |
| GATAD2A | 2.10 | 40.31 | 14.27 |
| ZNF232 | 2.07 | 3.63 | 4.23 |
| CDKN2B | 2.07 | 63.80 | 16.03 |
| TARBP1 | 2.05 | 9.47 | 11.10 |
| TEAD4 | 2.05 | 21.13 | 1.91 |
| NR1D1 | 2.04 | 23.90 | 22.81 |
| ZNF33A | 2.02 | 6.19 | 5.81 |
| TAF4B | 2.00 | 3.82 | 1.62 |
Denotes average of duplicate or
triplicate assays.
Highlighted factors were studied further.
Validation of TFs that repress CFTR expression.
Among the factors with the most repressive effect on CFTR transcript levels (Table 1) were several of particular interest: firstly, Inhibitor of growth family member 2 (ING2), which associates with both histone acetyltransferases (HATs) and histone deacetylases (HDACs) to modulate gene expression. It is a component of the Sin3A deacetylase (repressive) complex, which interacts with HDAC1 and HDAC2 [26], and may repress CFTR transcription by several mechanisms [10, 27]. Next, Nuclear receptor subfamily 2 group F, member 2 (NR2F2) is a member of steroid hormone superfamily of nuclear receptors, which were implicated in CFTR regulation previously [28, 29]. Bromodomain containing protein 8 (BRD8), also a strong repressor of CFTR transcription in this study, interacts with the thyroid hormone receptor, likely as a co-activator and amplifies thyroid hormone-dependent activation of genes [30], [31]. Krüppel like factor 5 (KLF5) is involved in lung development and function [32] and is a candidate CFTR transcriptional regulator since we showed previously that its motif is overrepresented in open chromatin in human tracheal epithelial (HTE) cells [33]. Forkhead box E1 (FOXE1) is involved in thyroid morphogenesis/function and CFTR is expressed in the thyroid epithelium. Both TGFβ-Induced Factor Homeobox 1 (TGIF1) and the co-factor Transducin-like Enhancer of Split 1 (TLE1) are logical candidates for a role in CFTR expression, which may be regulated by immune and inflammatory response in the airway [2, 10]. Moreover, the important role of Ets-homologous factor (EHF) in the human lung epithelium was reported in our recent work [21] [22]. Validation of the significant repressive effect of BRD8, EHF, KLF5, ING2 and NR2F2 on CFTR mRNA expression in Calu-3 cells was provided by 3 independent siRNA transfection experiments (Fig. 2). CFTR expression was normalized to the geometric means of 3 housekeeping gene (HKG) control transcripts, β2M, PGK1 and ACTB, to ensure that results were not influenced by siRNAs also altering the expression of the normalizer. As before siRNAs specific for FOXA1/FOXA2 provided the transfection control, reducing CFTR mRNA expression by more than 50%. Of note for BRD8, EHF and KLF5, siRNA from a different supplier (Ambion) with the relevant negative control was used to validate the earlier results obtained using the Dharmacon siRNAs (Fig.2A). The most repressive factors were KLF5 (6 fold), EHF and BRD8 (both ~ 3-fold).
Impact of TF depletion on CFTR protein abundance in Calu-3 cells.
Little is known about the correlation between CFTR mRNA levels and the abundance of CFTR protein, so we could not assume that an increase in CFTR mRNA abundance would be therapeutically useful. To determine the impact on CFTR protein abundance of depletion of five factors that repress CFTR mRNA levels (BRD8, EHF, KLF5, ING2 and NR2F2), western blots were performed. Protein was extracted from Calu-3 cells 72hr after TF siRNA delivery and CFTR was detected with antibody 596 (CFF) and normalized to β tubulin levels on the same membrane. Figure 3A shows representative western blots, where depletion of BRD8, EHF, KLF5, ING2 and NR2F2 augments CFTR protein expression in three replicates of Calu-3 cells in comparison to non-targeting control siRNAs. Again the impact on CFTR protein of repressing KLF5 was the greatest effect. Quantification of data from ≥ 3 western blots from independent experiments is shown in Figure 3B, where CFTR abundance relative to β tubulin is compared to the relevant negative control (Ambion or Dharmacon) for each siRNA, though only the Ambion NC is shown.
Figure 3. Depletion of KLF5, EHF, ING2, NR2F2 and BRD8 enhances CFTR protein abundance in Calu-3 cells.
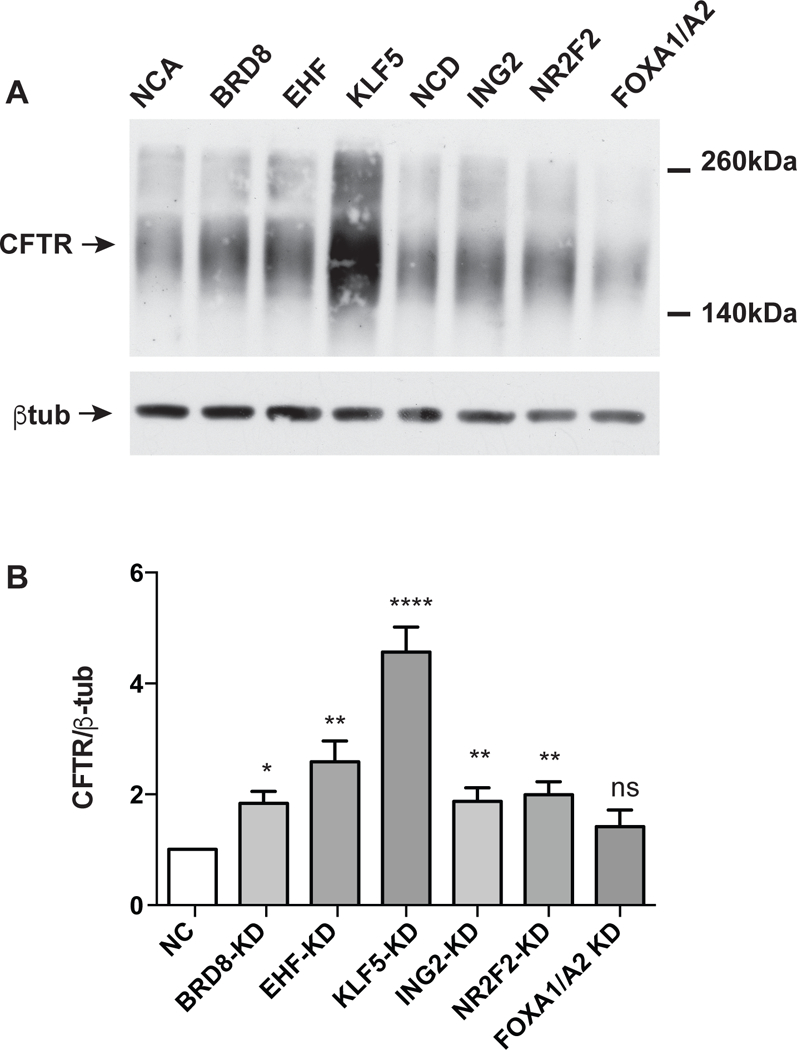
siRNA-mediated depletion of BRD8, EHF, KLF5, compared to NCA and ING2, NR2F2, FOXA1/A2 compared to NCD. A) Western blot probed with CFTR antibody 596 (CFF) or β tubulin as a loading control. FOXA1/A2 are known activators of CFTR expression. (NCA = Ambion, NCD =Dharmacon negative controls). B) Quantification of densitometry from ≥3 western blots shows pooled data on the impact of depletion of each factor on CFTR protein abundance. Bar shows average with standard error of the mean. Key: not significant, ns. P>0.05, * P< 0.05, ** P<0.01, **** P<0.0001 by an unpaired two-tailed Student’s t test.
Impact of TF depletion in primary HBE cells.
Among critical cell types that will be a target of new CF therapeutics are the epithelial cells lining the human lung. Hence, we next investigated whether the TFs that were the most effective repressors of CFTR mRNA and protein expression in Calu-3 cells had similar properties in human bronchial epithelial (HBE) cells. siRNAs specific for BRD8, EHF, KLF5, ING2, NR2F2 and relevant NCs were transfected into p2 primary HBE cells and RNA extracted after 96 hours. Consistent with the results in Calu-3 cells, depletion of BRD8, KLF5, and ING2 caused significant enhancement of the CFTR to normalizer ratio (geometric mean of 3 control transcripts, β2M, PGK1 and ACTB) in HBE cells (n= 3 donors) (Fig 4). Depletion of EHF and NR2F2 increased CFTR expression somewhat though values did not reach statistical significance. CFTR expression levels in HBE cells are too low to reliably assay the effect of siRNA depletion on CFTR protein abundance by western blot.
EHF and KLF5 regulate CFTR expression through an upstream cis-regulatory element
Though it is likely that many of the factors that activate or repress CFTR expression do so indirectly, through complex networks of transcription factors, we were particularly interested in those that targeted the CFTR locus through direct occupancy of its cis-regulatory elements. We recently revealed the role of EHF in modulating pathways that contribute to lung disease severity in CF patients [21, 22] and in that context we generated genome-wide occupancy data for EHF by ChIP-seq. Inspection of these data suggests that EHF may directly regulate CFTR in HBE cells. Peaks of EHF occupancy are seen at previously-characterized regulatory elements for CFTR, in DNase I hypersensitive sites −35 kb from the transcription start site [10], within intron 23 (legacy nomenclature) [16],[12], and at 48.9kb 3’ to the locus [4] (Fig. 5A). Though the impact of EHF depletion on CFTR expression was not significant in HBE cells, this may be due to the substantial donor-to-donor variation in EHF levels that was observed in the primary cells ([22], data not shown). Since, CFTR protein is at very low abundance in HBE cells grown on plastic, below the levels that can be reliably detected by western blot, the effect of EHF on its expression was again investigated in Calu-3 cells. EHF was depleted by siRNA in Calu-3 cells, and CFTR mRNA and protein expression were measured by RT-qPCR and western blot, respectively (Fig. 5B-C). Confirming our initial screening data that showed EHF as a repressor of the CFTR gene, EHF depletion significantly increased CFTR mRNA abundance, and this correlated with increased CFTR protein.
Figure 5. Repression of CFTR expression by EHF.
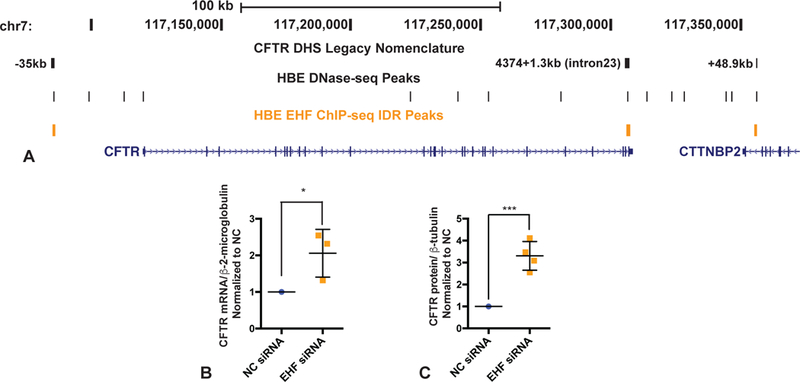
A) UCSC genome browser graphic showing EHF ChIP-seq peaks (yellow) at the CFTR locus. Legacy nomenclature for CFTR DHS is shown. HBE DHS track from [4]. B) CFTR mRNA expression relative to β2M control in NC and EHF depleted (siRNA) Calu-3 cells. C) CFTR protein (antibody 596) relative to β−, assayed by scanning western blots of NC and EHF depleted (siRNA) Calu-3 cells. B, C) Bars show average of experiments (B, n=3; C, n=4) with SEM. * p<0.05; *** p<0.001 by an unpaired two-tailed Student’s t test.
Next we examined ChIP-seq data for KLF5 in Calu-3 cells and also observed a peak of occupancy for this factor at the −35 kb DHS in replicate experiments (Fig. 6A), though not at the intron 23 and +48.9 kb cis-elements which bound EHF. These data suggest the −35 kb site may encompass an element with a key role in fine-tuning airway expression of CFTR.
Figure 6. Depletion of KLF5 enhances CFTR chloride channel activity in Calu-3 cells.
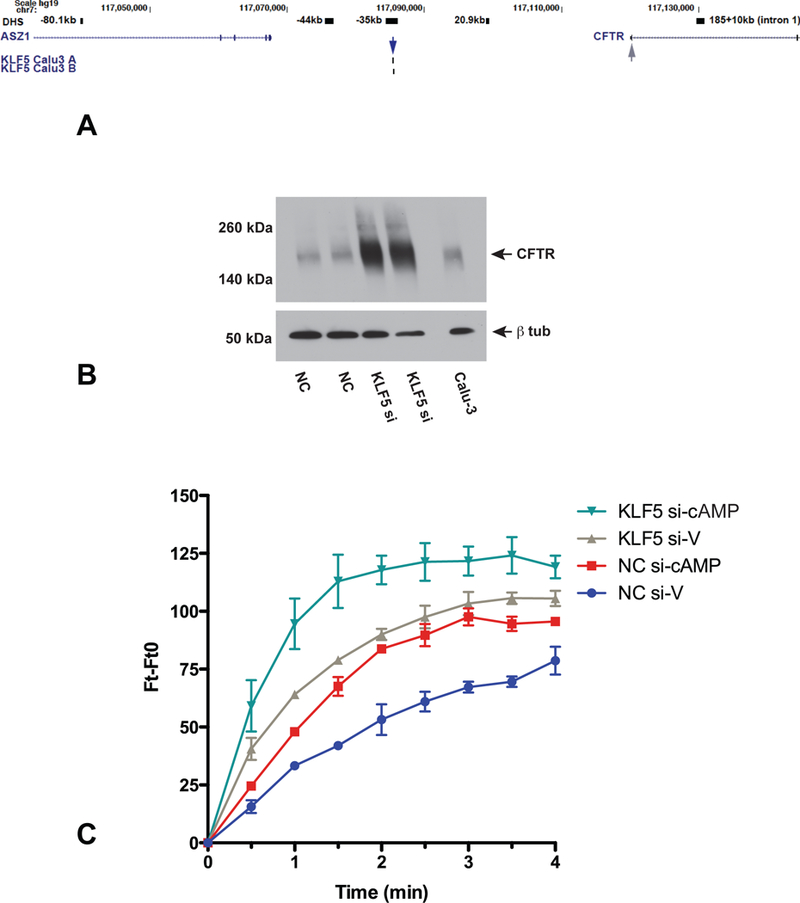
A) UCSC genome browser graphic showing KL5 ChIP-seq peak at the −35kb airway-selective DHS at the CFTR locus (blue arrow), with the CFTR gene promoter highlighted (grey arrow). B) Western blot probed with CFTR antibody 596 and β tubulin as a loading control shows the impact of siRNA-mediated depletion of KLF5 compared to NC on CFTR abundance in representative samples used for MQAE assays shown in C). C) MQAE assay shows change in fluorescence with time, with (cAMP) or without (V) the cAMP agonist forskolin. NC siRNA (V) blue, (cAMP) red; KLF5 siRNA (V) gray, (cAMP) teal.
KLF5 depletion enhances CFTR channel activity.
To be of therapeutic use, the inhibition of a repressive TF that increased CFTR mRNA and protein abundance would also need to enhance activity of the CFTR chloride channel activity in relevant epithelial cells. Since KLF5 was apparently the TF that most robustly repressed CFTR levels we tested the effect of its depletion on CFTR channel activity by fluorescent MQAE assays in Calu-3 cells. Enhanced CFTR protein levels after siRNA-mediated depletion of KLF5 (KLF5si) are shown by western blot (Fig. 6B) in one of the samples used in the MQAE assays shown in Fig. 6C. In replicate experiments we observed that not only did siRNA-mediated depletion of KLF5 increased basal chloride channel activity in Calu-3 cells (grey compared to blue traces), it also substantially elevated forskolin-activated CFTR conductance (teal compared to red) (Fig. 6C). The specificity of the forskolin response was confirmed by CFTR inhibitor inh 172 (Suppl. Fig. 2).
Discussion
The CFTR locus is among the best-studied single gene loci in the human genome, in part due to its association with a common genetic disease (reviewed in [1], [2], [3]). Its control mechanisms provide many insights into how cis-regulatory elements in non-coding regions of the genome organize chromatin structure and coordinate tissue-specific expression. Most studies to date focused on pathways that activate CFTR gene expression and the associated TF repertoire. Here we identify a network of TFs that repress CFTR transcription in airway epithelial cells, which maintain low levels of expression in comparison to epithelial cells in the digestive tract. These repressive TFs function both directly through cis-elements at the locus and indirectly by other mechanism including through other TFs and possibly microRNAs. The data reveal an important component of the complex fine-tuning of CFTR expression, which is directly relevant to the development of novel therapeutics for CF.
Repression of about 100 factors increased CFTR levels between 1.6 and 2 fold, and of ~ 50 factors > 2 fold in the initial replicated screen. However, for practical reasons we chose the arbitrary > 2 fold cut off for further analysis, though it is possible that factors with an important role in CFTR regulation are in the 1.6 – 2 fold group. Among the 37 factors that were validated to repress CFTR transcription more than 2 fold, a smaller group is of particular interest due either to their relevance to CF biology and pathology or their status as therapeutic targets in other contexts. For example, NR2F2 is an orphan (no endogenous ligand identified) nuclear receptor in the steroid thyroid hormone superfamily. Several members of this superfamily are already targets for pharmacological intervention due to their potential role in cancer [34]. Another CFTR repressor is BRD8, which interacts with the thryroid hormone receptor and may function as a co-activator of hormone-activated nuclear receptors. BRD8 contains two bromodomains, protein modules that specifically recognize acetylated lysine residues on histone tails and other CAFs. The distinct hydrophobic cleft within bromodomains makes them attractive targets for synthetic small molecule drugs [35–37]. Indeed several clinical studies using small molecule inhibitors of bromodomains are currently underway, and another orphan bromodomain protein BRD9 was recently effectively targeted by this approach [38].
Human KLF5, originally cloned from placenta as BTEB2 (basic transcription element binding protein 2 [39]) is a zinc finger (GC-box) transcription factor with key roles in multiple tissues. It is a member of the Krüppel-like family of transcription factors and its mouse homologue was initially named IKlf5 due to its abundance in the digestive tract. It showed developmental regulation, being enriched in early endoderm [40], though its expression was restricted to the intestinal epithelium with highest levels in intestinal crypt cells [41]. Later studies also found Klf5 to be developmentally regulated in the mouse bronchi and trachea [42] and to be involved in beta-catenin-independent Wnt-1 signaling [43]. Most relevant to our observations on the role of KLF5 in repression of CFTR in lung epithelial cells are data showing that Klf5 is required for morphogenesis and function of the perinatal mouse lung [32]. Conditional loss of Klf5 from the respiratory epithelium caused mice to die immediately after birth due to respiratory distress, in part caused by immaturity of the respiratory epithelium and decreased surfactant levels. Loss of KLF5 from bronchial epithelial cells was associated with reduced levels of the CAAT/enhancer binding protein alpha (CEBPα) and secretoglobulin family 1A Member 1 (SCGB1A1/CCSP) transcription factors, though likely through an indirect mechanism [32]. The impact of loss on Klf5 on other activating transcription factors was not investigated in detail. MatInspector analysis of the airway-selective −35kb cis-element in the CFTR locus shows 3 predicted KLF-binding motifs (EKLF or GKLF) though none specifically for KLF5, despite the observed peak of occupancy of this factor in Calu-3 ChIP-seq data. This raises the possibility that KLF5 is recruited to this site by another DNA-binding protein, through which it exerts its repressive action. Most studies on KLF5 focus on its role as a transcriptional activator and it is known to be a core regulator of intestinal oncogenesis [44]. KLF5 is down-regulated during epithelial- mesenchymal transition (EMT) and its loss induces EMT in absence of EGF/TGFβ activation of EMT. In this context, KLF5 maintains epithelial characteristics by transcriptionally activating miR-200 family member in epithelial cells [45]. Interestingly miR-200b was recently shown to down-regulate CFTR expression during hypoxia [46], suggesting an additional pathway for repression of CFTR by KLF5.
EHF was originally cloned from mouse pituitary somatotroph tumors [47]. In parallel with the observations of KLF5 as a repressive factor acting through miRNAs, EHF represses miR-424 in normal prostate epithelial cells. However, miR-424 is upregulated in prostate tumors and is associated with the aggressive features of the tumor, since it impairs the E3 ubiquitin ligase COP1 and in turn activates STAT3 [48]. Prostate tumors show an inverse correlation between EHF levels and miR-424. Though EHF is quite well studied in epithelial cancers, its role in normal epithelial cells has received less attention. It is involved in the wound response in the mouse cornea [49], in differentiation of the skin [50] and it regulates important functions of the airway epithelium including wound repair, inflammation and barrier function [21, 22]. We also showed by ChIP-seq for EHF and RNA-seq after EHF depletion in primary human bronchial epithelial cells that EHF directly regulates a number of other transcription factors with key role in the airway epithelium [22]. Among genes with nearby peaks of EHF occupancy in replicated EHF ChIP-seq datasets and/or differentially expressed genes (DEG) after EHF depletion in primary HBE cells were GATA Binding Protein 6 (GATA6), HOP Homeobox (HOPX), KLF5, SAM Pointed Domain Containing ETS Transcription Factor (SPDEF), Retinoic Acid Receptor Beta (RARB), FOXA1 and FOXA2. HOPX, KLF5 and RARB abundance significantly increased after EHF depletion and SPDEF levels significantly decreased. EHF occupancy at intergenic sites 5’ of the HOPX, RARB and FOXA1 genes and at the promoter of SPDEF was confirmed by ChIP-qPCR in primary HBE cells. We also showed that FOXA1 co-regulates gene expression with EHF [22].
Our observation that EHF represses CFTR expression may also be pertinent to understanding the mechanisms underlying the association of modifier genes with CF lung disease severity [51], [52]. In these genome-wide association studies (GWAS) among the highest P-value single nucleotide polymorphisms (SNPs) correlating with CF lung phenotype were several within a non-coding (intergenic) region of chromosome 11p13. Though the relationship of these SNPs to expression of nearby loci has yet to be determined, they appear to lie within the same TAD or sub-TAD as the EHF locus [25], suggesting a connection with cis-regulatory elements for this gene.
This remarkable complexity of interactions in the TF network in airway epithelial cells makes direct correlation between the role of a single TF in the regulation of expression of CFTR, or indeed any locus, a major challenge. However, since a major therapeutic goal is to identify TFs that repress CFTR expression, in order to compromise their activity, whether they do so directly or indirectly or both, may not be important. Of more concern is the involvement of KLF5 and EHF, together with several other CFTR repressive TFs identified in this study, in epithelial cancers. This will necessitate a highly focused approach to inhibiting recruitment of the repressive factors to specific regulatory elements at the CFTR locus. For example, targeting the airway cis-regulatory element for CFTR at −35 kb with specific guide RNAs, Cas9 and a transcriptional activator (reviewed in [53], [54]) could be used to nudge this element towards enhancer function and prevent the recruitment of repressive factors.
Supplementary Material
Acknowledgements
The authors thank Drs S. Ghosh and A. Hoffman for generating ChIP-seq data for negative histone marks in Calu-3 and HBE cells; Dr J.L. Kerschner for helpful discussions; also Dr S. Randell and colleagues for HBE cells, which were provided with support from NIH [P30DK065988]; Cystic Fibrosis Foundation [BOUCHE15R0] and for contributions.
Funding
This work was funded by the Cystic Fibrosis Foundation (Harris14G0, Harris 16G0 and 15XX0); National Institutes of Health: R01HL094585, R01HD068901 and R01HL117843 (PI:A.H.); F30HL124925 (S.F).
Footnotes
Declaration of interest
The authors have nothing to declare.
References
- 1.Ott CJ, Blackledge NP, Leir SH and Harris A (2009) Novel regulatory mechanisms for the CFTR gene. Biochem Soc Trans. 37, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillen AE and Harris A (2012) Transcriptional regulation of CFTR gene expression. Front Biosci (Elite Ed). 4, 587–592 [DOI] [PubMed] [Google Scholar]
- 3.Gosalia N and Harris A (2015) Chromatin Dynamics in the Regulation of CFTR Expression. Genes (Basel). 6, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang R, Kerschner JL, Gosalia N, Neems D, Gorsic LK, Safi A, Crawford GE, Kosak ST, Leir SH and Harris A (2016) Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 44, 3082–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith EM, Lajoie BR, Jain G and Dekker J (2016) Invariant TAD Boundaries Constrain Cell-Type-Specific Looping Interactions between Promoters and Distal Elements around the CFTR Locus. Am J Hum Genet. 98, 185–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerschner JL and Harris A (2012) Transcriptional networks driving enhancer function in the CFTR gene. Biochem J. 446, 203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerschner JL, Gosalia N, Leir SH and Harris A (2014) Chromatin remodeling mediated by the FOXA1/A2 transcription factors activates CFTR expression in intestinal epithelial cells. Epigenetics : official journal of the DNA Methylation Society. 9, 557–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mouchel N, Henstra SA, McCarthy VA, Williams SH, Phylactides M and Harris A (2004) HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem J. 378, 909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ott CJ, Suszko M, Blackledge NP, Wright JE, Crawford GE and Harris A (2009) A complex intronic enhancer regulates expression of the CFTR gene by direct interaction with the promoter. J Cell Mol Med. 13, 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Leir SH and Harris A (2013) Immune Mediators Regulate CFTR Expression through a Bifunctional Airway-Selective Enhancer. Molecular and cellular biology. 33, 2843–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Leir SH and Harris A (2014) Oxidative Stress Regulates CFTR Gene Expression in Human Airway Epithelial Cells Through a Distal Antioxidant Response Element. Am J Respir Cell Mol Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Ott CJ, Lewandowska MA, Leir SH and Harris A (2012) Molecular mechanisms controlling CFTR gene expression in the airway. Journal of cellular and molecular medicine. 16, 1321–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris A, Chalkley G, Goodman S and Coleman L (1991) Expression of the cystic fibrosis gene in human development. Development. 113, 305–310 [DOI] [PubMed] [Google Scholar]
- 14.Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, Gatter KC, Harris A and Higgins CF (1991) Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci U S A. 88, 9262–9266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyde K, Reid CJ, Tebbutt SJ, Weide L, Hollingsworth MA and Harris A (1997) The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology. 113, 914–919 [DOI] [PubMed] [Google Scholar]
- 16.Ott CJ, Blackledge NP, Kerschner JL, Leir SH, Crawford GE, Cotton CU and Harris A (2009) Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc Natl Acad Sci U S A. 106, 19934–19939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leir SH, Browne JA, Eggener SE and Harris A (2015) Characterization of primary cultures of adult human epididymis epithelial cells. Fertil Steril. 103, 647–654 e641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broackes-Carter FC, Mouchel N, Gill D, Hyde S, Bassett J and Harris A (2002) Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum Mol Genet. 11, 125–131 [DOI] [PubMed] [Google Scholar]
- 19.Fulcher ML, Gabriel S, Burns KA, Yankaskas JR and Randell SH (2005) Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 107, 183–206 [DOI] [PubMed] [Google Scholar]
- 20.Tugores A, Le J, Sorokina I, Snijders AJ, Duyao M, Reddy PS, Carlee L, Ronshaugen M, Mushegian A, Watanaskul T, Chu S, Buckler A, Emtage S and McCormick MK (2001) The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem. 276, 20397–20406 [DOI] [PubMed] [Google Scholar]
- 21.Fossum SL, Mutolo MJ, Yang R, Dang H, O’Neal WK, Knowles MR, Leir SH and Harris A (2014) Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 42, 13588–13598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fossum SL, Mutolo MJ, Tugores A, Ghosh S, Randell SH, Jones LC, Leir SH and Harris A (2017) Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J Biol Chem. 292, 10938–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.West MR and Molloy CR (1996) A microplate assay measuring chloride ion channel activity. Analytical biochemistry. 241, 51–58 [DOI] [PubMed] [Google Scholar]
- 24.Amaral MD and Kunzelmann K (2011) Cystic Fibrosis: Diagnosis and Protocols Springer [Google Scholar]
- 25.Stolzenburg LR, Yang R, Kerschner JL, Fossum S, Xu M, Hoffmann A, Lamar KM, Ghosh S, Wachtel S, Leir SH and Harris A (2017) Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res. 45, 8773–8784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E and Eisenman RN (1997) Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 89, 349–356 [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran S, Karp PH, Jiang P, Ostedgaard LS, Walz AE, Fisher JT, Keshavjee S, Lennox KA, Jacobi AM, Rose SD, Behlke MA, Welsh MJ, Xing Y and McCray PB Jr. (2012) A microRNA network regulates expression and biosynthesis of wild-type and {Delta}F508 mutant cystic fibrosis transmembrane conductance regulator. Proceedings of the National Academy of Sciences of the United States of America. 109, 13362–13367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yigit E, Bischof JM, Zhang Z, Ott CJ, Kerschner JL, Leir SH, Buitrago-Delgado E, Zhang Q, Wang JP, Widom J and Harris A (2013) Nucleosome mapping across the CFTR locus identifies novel regulatory factors. Nucleic acids research. 41, 2857–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blackledge NP, Ott CJ, Gillen AE and Harris A (2009) An insulator element 3’ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res. 37, 1086–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monden T, Wondisford FE and Hollenberg AN (1997) Isolation and characterization of a novel ligand-dependent thyroid hormone receptor-coactivating protein. J Biol Chem. 272, 29834–29841 [DOI] [PubMed] [Google Scholar]
- 31.Hollenberg AN, Monden T, Madura JP, Lee K and Wondisford FE (1996) Function of nuclear co-repressor protein on thyroid hormone response elements is regulated by the receptor A/B domain. J Biol Chem. 271, 28516–28520 [DOI] [PubMed] [Google Scholar]
- 32.Wan H, Luo F, Wert SE, Zhang L, Xu Y, Ikegami M, Maeda Y, Bell SM and Whitsett JA (2008) Kruppel-like factor 5 is required for perinatal lung morphogenesis and function. Development. 135, 2563–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bischof JM, Ott CJ, Leir SH, Gosalia N, Song L, London D, Furey TS, Cotton CU, Crawford GE and Harris A (2012) A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 67, 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Safe S, Jin UH, Hedrick E, Reeder A and Lee SO (2014) Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Mol Endocrinol. 28, 157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filippakopoulos P and Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews. Drug discovery. 13, 337–356 [DOI] [PubMed] [Google Scholar]
- 36.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH and Knapp S (2012) Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 149, 214–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature. 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, McConnell D, Pearson M, Gerstberger T, Gottschamel T, Thompson D, Suzuki Y, Koegl M and Vakoc CR (2016) Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol. 12, 672–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sogawa K, Imataka H, Yamasaki Y, Kusume H, Abe H and Fujii-Kuriyama Y (1993) cDNA cloning and transcriptional properties of a novel GC box-binding protein, BTEB2. Nucleic Acids Res. 21, 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B and Melton DA (2004) Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development. 131, 165–179 [DOI] [PubMed] [Google Scholar]
- 41.Conkright MD, Wani MA, Anderson KP and Lingrel JB (1999) A gene encoding an intestinal-enriched member of the Kruppel-like factor family expressed in intestinal epithelial cells. Nucleic Acids Res. 27, 1263–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohnishi S, Laub F, Matsumoto N, Asaka M, Ramirez F, Yoshida T and Terada M (2000) Developmental expression of the mouse gene coding for the Kruppel-like transcription factor KLF5. Developmental dynamics : an official publication of the American Association of Anatomists. 217, 421–429 [DOI] [PubMed] [Google Scholar]
- 43.Ziemer LT, Pennica D and Levine AJ (2001) Identification of a mouse homolog of the human BTEB2 transcription factor as a beta-catenin-independent Wnt-1-responsive gene. Mol Cell Biol. 21, 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakaya T, Ogawa S, Manabe I, Tanaka M, Sanada M, Sato T, Taketo MM, Nakao K, Clevers H, Fukayama M, Kuroda M and Nagai R (2014) KLF5 regulates the integrity and oncogenicity of intestinal stem cells. Cancer Res. 74, 2882–2891 [DOI] [PubMed] [Google Scholar]
- 45.Zhang B, Zhang Z, Xia S, Xing C, Ci X, Li X, Zhao R, Tian S, Ma G, Zhu Z, Fu L and Dong JT (2013) KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol Cell Biol. 33, 4919–4935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartoszewska S, Kamysz W, Jakiela B, Sanak M, Kroliczewski J, Bebok Z, Bartoszewski R and Collawn JF (2017) miR-200b downregulates CFTR during hypoxia in human lung epithelial cells. Cell Mol Biol Lett. 22, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bochert MA, Kleinbaum LA, Sun LY and Burton FH (1998) Molecular cloning and expression of Ehf, a new member of the ets transcription factor/oncoprotein gene family. Biochem Biophys Res Commun. 246, 176–181 [DOI] [PubMed] [Google Scholar]
- 48.Dallavalle C, Albino D, Civenni G, Merulla J, Ostano P, Mello-Grand M, Rossi S, Losa M, D’Ambrosio G, Sessa F, Thalmann GN, Garcia-Escudero R, Zitella A, Chiorino G, Catapano CV and Carbone GM (2016) MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J Clin Invest. 126, 4585–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephens DN, Klein RH, Salmans ML, Gordon W, Ho H and Andersen B (2013) The Ets transcription factor EHF as a regulator of cornea epithelial cell identity. J Biol Chem. 288, 34304–34324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, Kim DS, Boxer LD, Cairns J, Spivakov M, Wingett SW, Shi M, Zhao Z, Greenleaf WJ, Kundaje A, Snyder M, Chang HY, Fraser P and Khavari PA (2017) Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet. 49, 1522–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L, Berthiaume Y, Cutler D, Cojocaru A, Collaco JM, Corey M, Dorfman R, Goddard K, Green D, Kent JW Jr., Lange EM, Lee S, Li W, Luo J, Mayhew GM, Naughton KM, Pace RG, Pare P, Rommens JM, Sandford A, Stonebraker JR, Sun W, Taylor C, Vanscoy LL, Zou F, Blangero J, Zielenski J, O’Neal WK, Drumm ML, Durie PR, Knowles MR and Cutting GR (2011) Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 43, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, Accurso FJ, Clement A, Collaco JM, Dang H, Dang AT, Franca A, Gong J, Guillot L, Keenan K, Li W, Lin F, Patrone MV, Raraigh KS, Sun L, Zhou YH, O’Neal WK, Sontag MK, Levy H, Durie PR, Rommens JM, Drumm ML, Wright FA, Strug LJ, Cutting GR and Knowles MR (2015) Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nature communications. 6, 8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.La Russa MF and Qi LS (2015) The New State of the Art: Cas9 for Gene Activation and Repression. Mol Cell Biol. 35, 3800–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vora S, Tuttle M, Cheng J and Church G (2016) Next stop for the CRISPR revolution: RNA-guided epigenetic regulators. Febs J. 283, 3181–3193 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.