

Abstract
Epigenetic mechanisms — including DNA methylation, histone post-translational modifications and changes in nucleosome positioning — regulate gene expression, cellular differentiation and development in almost all tissues, including the brain. In adulthood, changes in the epigenome are crucial for higher cognitive functions such as learning and memory. Striking new evidence implicates the dysregulation of epigenetic mechanisms in neurodegenerative disorders and diseases. Although these disorders differ in their underlying causes and pathophysiologies, many involve the dysregulation of restrictive element 1-silencing transcription factor (REST), which acts via epigenetic mechanisms to regulate gene expression. Although not somatically heritable, epigenetic modifications in neurons are dynamic and reversible, which makes them good targets for therapeutic intervention.
Conrad Waddington coined the term epigenesis in the 1940s to describe his logical deduction that, during embryonic development, a set of mechanisms must exist that resides above (‘epi’) the level of genes such that identical genes can be differentially expressed in different cell types and contexts to determine cell fate1. Epigenetics (as epigenesis is now termed) in its most classical sense encompasses a wide range of heritable changes in gene expression that occur in response to environmental influences and that do not result from alterations in the DNA sequence. These alterations typically arise owing to DNA methylation or hydroxymethylation, histone post-translational modifications and changes in nucleosome positioning; these processes are collectively referred to by the broad term ‘chromatin remodelling’ Recent studies have identified histone variants, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) as additional epigenetic mechanisms2–7 (BOX 1).
Box 1 | Epigenetic modifications
Nucleosomes are the basic unit of chromatin and are composed of a 146 bp stretch of genomic DNA wrapped around an octamer of the core histone proteins H2A, H2B, H3 and H4. Histone octamers are connected to one another by linker DNA, which spans the region between the nucleosomes, and linker histones, which bind to and stabilize the linker DNA97,98. DNA and histone modifications, changes in nucleosome positioning, histone variants, microRNAs and long non-coding RNAs (lncRNAs) constitute the basic mechanisms that underlie chromatin remodelling98.
DNA methylation and hydroxymethylation
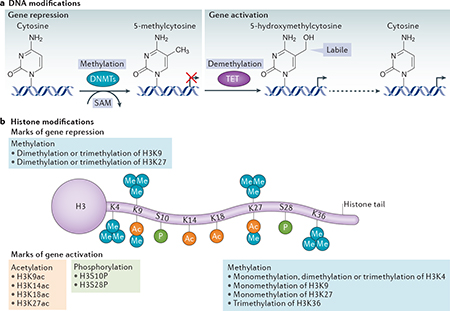
In the brain, DNA methylation and hydroxymethylation denote gene activation or repression, and their effects extend over long chromosomal domains, giving rise to ‘memorized’ states of gene expression99. DNA methyltransferases (DNMTs) — including DNMT1, DNMT3A and DNMT3B — catalyse the transfer of methyl groups from S-adenosyl-L-methionine (SAM) to the 5-position carbon in cytosines within DNA to generate 5-methylcytosine (see the figure, part a). Although this methyl group transfer was originally thought to be irreversible, recent evidence has revealed that DNA can rapidly and reversibly undergo changes in methylation and can undergo demethylation12. A recently discovered family of dioxygenases known as the ten-eleven translocation (TET) proteins (TET1, TET2 and TET3) can catalyse the conversion of 5-methylcytosine to 5-hydroxymethylcytosine16,100. The hydroxymethyl moiety is labile and can rapidly regenerate unmethylated cytosines (indicated by the dashed arrow), which in turn activate genes.
Post-translational histone modifications
Each histone has a central domain and several unstructured amino-terminal tails that contain sites for more than 100 post-translational modifications101, including acetylation (denoted ‘Ac’ in part b of the figure), methylation (denoted ‘Me’), phosphorylation (denoted ‘P’), ubiquitylation, sumoylation, deamination and poly(ADP) ribosylation. Part b of the figure shows the sites at which histone H3 is post-translationally modified by acetylation, methylation and phosphorylation. These marks are added by chromatin-remodelling enzymes known as ‘writers’ and are removed by enzymes known as ‘erasers’. Distinct, site-specific post-translational modifications of core histone proteins act sequentially or combinatorially to create a ‘histone code’ that is recognized by proteins known as ‘readers’. Readers recognize epigenetic marks on core histones through specialized motifs and are thereby recruited to the promoters of target genes98.
Histone variant exchange
The replacement of one histone variant with another is an additional mode of chromatin remodelling that is important in memory consolidation. Unlike canonical histones, histone variants undergo replication-independent expression, and they have pronounced effects on nucleosome stability and positioning102. The variant H2A.Z replaces the canonical histone H2A in the hippocampus, where it blunts memory formation in response to fear conditioning in mice103, which implicates H2A.Z as a negative regulator of memory consolidation104. Moreover, the exposure of mice to an enriched environment promotes the incorporation of the variant H3.3 into the chromatin of active genes in the hippocampus104, whereas blocking the turnover of H3.3 diminishes spine density, miniature excitatory postsynaptic current amplitude at CA1 synapses and hippocampus-based learning104. These findings implicate histone variants as potential therapeutic targets for the impaired cognition that is associated with neurodegeneration.
Part b is adapted with permission from REF. 105, Macmillan Publishers Ltd.
In addition to driving heritable changes in gene expression, epigenetics is also a mechanism through which experience can modify synaptic plasticity, the acquisition and consolidation of memory, the wiring of neural circuits and information flow. Chromatin remodelling can enable alterations in the expression of genes that are associated with synaptic plasticity during development and throughout adult life8, and thus allow genetic predisposition, experience and neuronal activity to act together to modify cellular and behavioural responses. Furthermore, as illustrated by recent striking findings, epigenetic dysregulation is implicated in the impaired cognition and neuronal death that are associated with neurodegenerative diseases9–11. In particular, in several diseases that have different underlying causes and pathophysiologies, impaired cognition and neuronal death have been shown to involve the dysregulation of the restrictive element 1-silencing transcription factor (REST; also known as NRSF), which orchestrates the epigenetic remodelling of genes that are essential for synaptic plasticity and survival.
In this article, we review the mechanisms by which the epigenetic modification of gene expression regulates memory acquisition and consolidation in the healthy brain and describe how epigenetic dysregulation contributes to the impaired cognition and neuronal death that are associated with neurodegenerative diseases. We focus on a subset of neurological disorders (namely, Alzheimer disease, Huntington disease, stroke and global ischaemia) for which the evidence supporting the contribution of epigenetics is most well established. Finally, we highlight the potential power of new therapeutic approaches that target the epigenetic machinery to ameliorate the symptoms associated with these diseases.
The emerging field of neuroepigenetics
As outlined above, epigenetics classically serves to propagate acquired non-DNA-sequence information to progeny cells. Neuroepigenetics (defined as ‘epigenetics as it pertains to neurons’) involves the same chemical modifications of DNA and histones; however, in neurons (which do not divide) these modifications are not propagated to progeny cells. Despite this key difference between classical epigenetics and neuroepigenetics, the same transcription factors and epigenetic mechanisms — including DNA methylation and hydroxymethylation, histone post-translational modifications, histone variants, miRNAs, lncRNAs and changes in nucleosome positioning — function to modify transcriptional output in both settings. As described below, these epigenetic mechanisms are co-opted in neurons for information storage and circuit regulation. As such, the failure of these systems can disrupt basic network-related cognitive function and might contribute to neurodegenerative disease.
DNA methylation and memory
In the brain, DNA methylation — which typically acts to silence gene expression — is an essential mediator of memory acquisition and storage12,13. In addition to their pivotal role in brain development, the DNA meth- yltransferases DNMT1, DNMT3A and DNMT3B also have a key role in the experience-dependent modification of brain function in adulthood10,14. The targeted deletion of Dnmtl and/or Dnmt3a in mice results in the loss of long-term potentiation (LTP) at CA1 synapses in the hippocampus, and deficits in hippocampus-based learning and memory15. DNMTs are upregulated in the adult rat hippocampus in response to contextual fear conditioning, and the inhibition of DNMTs before the stimulus blocks fear conditioning12. Moreover, fear conditioning in rats promotes the rapid DNMT-dependent methylation and transcriptional silencing of the gene encoding the catalytic subunit of the memory suppressor protein phosphatase 1 (PP1), as well as the rapid DNMT- dependent demethylation (resulting in a rapid switching in methylation status) and active transcription of Reln, which encodes a plasticity-associated protein12. Thus, memory formation can be associated with changes in DNA methylation.
Although methylation was originally thought to serve as a stable mark of gene silencing, we now know that changes in the DNA methylation status of synaptic plasticity-associated and memory-associated genes can be both rapid and reversible12. The methylcytosine dioxygenases ten-eleven translocation protein 1 (TET1), TET2 and TET3 catalyse the conversion of 5-methyl- cytosine to 5-hydroxymethylcytosine16. In mice, TET1 acts in an activity-dependent manner to promote the DNA demethylation and activation of genes crucial for memory formation and consolidation17, including those encoding brain-derived neurotrophic factor (BDNF), fibroblast growth factor (FGF), activity-regulated cytoskeletal-associated protein (ARC), early growth response protein 1 (EGR1), the transcription factor FOS, the scaffolding protein HOMER1 and nuclear receptor subfamily 4 group A member 2 (NR4A2). The loss of TET1 activity leads to a deficit in long-term contextual fear memory17. Accordingly, the impaired cognition that is associated with neurodegeneration could potentially involve the dysregulation of both DNA methylation and demethylation, as well as the dysregulation of the rapid switching between the two states (see above)18.
Histone modifications and cognition
Histone acetylation is the modification that is best understood and most tightly associated with memory formation. The addition of acetyl groups to histones is catalysed by histone acetyltransferases (HATs). Histone acetylation relaxes the interactions between histones and DNA, leading to a more open configuration that enables the transcriptional machinery to access gene promoters. One of the earliest studies linking histone modifications to cognition reported that novel taste learning in mice activates extracellular signal-regulated kinase (ERK)- mitogen-activated protein kinase (MAPK) signalling, which in turn activates HATs and thereby drives histone acetylation at gene promoters in the insular cortex, a brain region that is crucial for the formation of novel taste memories19. These findings implicated histone acetylation in single-trial learning and the acquisition of long-term memories. Accordingly, histone deacetylase (HDAC) inhibitors, which increase the acetylation of histone tails, lower the threshold for LTP and increase its magnitude at CA1 synapses, and improve the performance of mice in a contextual fear conditioning par- adigm20. Mice that express a dominant-negative form of cAMP response element-binding protein (CREB)- binding protein (CBP; a HAT and crucial binding partner of CREB) and mice deficient in CBP exhibit deficits in LTP and long-term memory21–23 that are ameliorated by HDAC inhibitors21,22. In humans, mutations in the gene encoding CBP or its homologue p300 result in Rubenstein-Taybi syndrome, which is an inherited, autosomal dominant disease characterized by severe learning disabilities24,25.
These findings established a role for histone acetylation in synaptic plasticity and memory formation, and provided a rationale to explore the role of altered acetylation in the cognitive decline associated with neurodegenerative disease. Indeed, a landmark study that made use of the CK-p25 mouse model of neurodegeneration (in which p25, a protein implicated in neurodegeneration, is conditionally expressed under the control of the promoter of the gene that encodes calcium/calmodulin- dependent protein kinase II) showed that lost memories could be recovered through chromatin modification26. The exposure of the mice to an enriched environment — which increases the acetylation of H3K9 or H3K14, and the methylation of H3K4 (that is, epigenetic marks of active gene transcription) — or the in vivo administration of HDAC inhibitors that boosts histone acetylation could reinstate lost memories even after neuronal loss and brain atrophy had occurred26 by enhancing the performance of the small number of surviving neurons26.
Histone acetylation and neuronal death
In addition to their role in modulating memory function, histone modifications have been linked to the neuronal cell death that drives memory loss in neurodegenerative disease. It has been shown that ischaemic insults activate REST (which silences its target genes through the recruitment of HDAC1 and HDAC2; see below) in selectively vulnerable CA1 neurons before the onset of neuronal death in a clinically relevant model of global ischaemia27,28. Accordingly, in vivo administration of the HDAC inhibitor trichostatin A (TSA) immediately after neuronal insult ameliorated ischaemia-induced neuronal death28.
Transcription factors drive remodelling
Several transcription factors have been identified as master regulators of epigenetic remodelling in the brain. To date, most of our understanding of the role of these factors in coordinating neuroepigenetics comes from work on REST and the Polycomb proteins, which are gene-silencing transcription factors that act through their effects on histone acetylation and methylation to coordinate the expression of genes encoding synaptic vesicle proteins, receptors, channels and adhesion proteins. These transcription factors have been shown to have a role in normal brain development and function, and their dysregulation is thought to contribute to neurological diseases.
REST orchestrates epigenetic remodelling.
REST is widely expressed during embryogenesis29,30. In pluripotent stem cells and neural progenitors, REST actively represses a vast array of coding and non-coding neuron-specific genes, many of which are important for synaptic plasticity and structural remodelling during development. These include genes encoding synaptic vesicle proteins, ion channels, neurotransmitter receptors, and miRNAs that regulate networks of non-neuronal genes31. Bioinformatics analysis predicts that there are more than 2,000 putative REST targets in the mammalian genome32,33. During the late stages of neuronal differentiation, the elimination of REST is crucial for the acquisition of the neuronal phenotype34. In differentiated neurons, REST is normally quiescent, but it can be transiently activated during postnatal development in an experience-dependent manner to promote the acquisition of the mature form of the NMDA receptor (NMDAR)35, and in response to neuronal insults such as seizures36,37, global ischaemia27,28,38 and stroke39,40. As described in more detail below, it has been reported that excessive amounts of REST accumulate in the nuclei of selectively vulnerable striatal cells in Huntington disease41,42 (although a recent study has reported a different result43). In the healthy ageing brain, REST silences genes involved in apoptosis and oxidative stress44, whereas the loss of REST is causally linked to Alzheimer disease44.
A fundamental mechanism by which REST silences target genes in neural progenitors and neurons during development and in differentiated neurons is that of epigenetic remodelling28,34 (BOX 2). Recent evidence suggests that REST not only silences but also activates target genes. For example, REST has been shown to bind to the short form of TET3 (the major TET isoform expressed in neurons) and guide it to the promoters of REST target genes for directed 5-hydroxymethyl- cytosine generation and activation45. In addition, the REST-TET3 complex interacts with histone- lysine N-methyltransferease NSD3 (also known as nuclear SET-domain-containing protein 3) and two other H3K36 methyltransferases to add a trimethylation moiety to the core histone protein H3 at lysine 36 and generate H3K36me3, which is a strong mark of gene activation that promotes the context-specific transcription of REST target genes45. These observations reveal that the mechanisms by which REST influences the expression of neuronal genes are more complex than was initially appreciated.
Box 2 | Restrictive element 1-silencing transcription factor silences target genes by epigenetic remodelling.
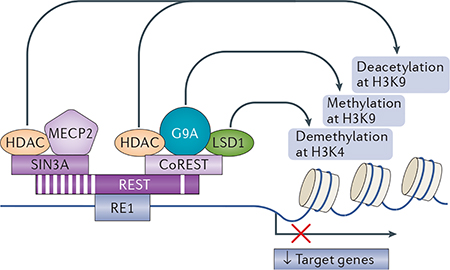
Restrictive element 1-silencing transcription factor (REST) initiates the repression of gene transcription by binding to restrictive element 1 (RE1), a 21–23 bp recognition motif that is contained within the promoters of target genes, and recruiting its co-repressors REST co-repressor (CoREST) and SIN3A106–108. This generates platforms that in turn recruit histone deacetylases (HDACs), including HDAC1 and HDAC2 (see the figure). HDACs deacetylate core histone proteins and mediate gene silencing by tightening the chromatin, which prevents the access of the transcriptional machinery to gene promoters92,93. REST also represses gene expression through its association with G9A, which is a site-specific histone methyltransferase that adds a dimethylation mark to histone 3 at lysine 9 (H3K9me2); the NADH-sensitive co-repressor carboxy-terminal binding protein 1 (CTBP1; not shown)109; and the site-specific histone demethylase LSD1, which removes monomethyl and dimethyl moieties from H3K4, thus promoting gene repression110,111. In addition, REST assembles with methyl-CpG-binding protein 2 (MECP2), which is recruited to epigenetic marks34,112 and promotes epigenetic remodelling in a REST-dependent or REST-independent manner34,112.
It is well known that REST target genes can be repressed even after REST is depleted owing to the continued presence of co-repressors such as CoREST and MECP2 (REF 34). The binding of REST to RE1 sites is stabilized by BRG1 (not shown), a chromatin-remodelling bromodomain- containing enzyme that recognizes histone H4 acetylated at K8 (H4K8ac), which is an epigenetic mark of gene repression that recruits REST to RE1 sites113. Upon binding to DNA, BRG1 promotes nucleosome repositioning, which stabilizes the interaction of REST with RE1 sites and promotes gene repression114. In addition, the REST-co-repressor complex may recruit the DNA methylation machinery, which promotes the methylation of cytosines at the promoters of REST target genes34. These observations link REST not only to histone modifications but also to DNA methylation, and have important implications for drug discovery.
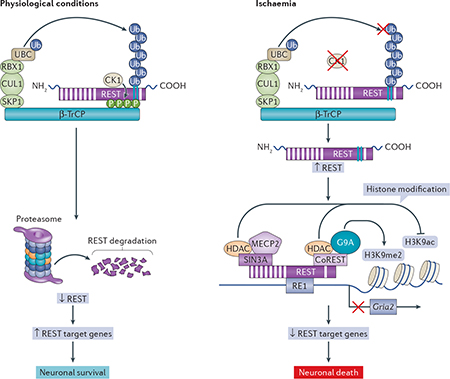
REST abundance is regulated at the level of protein stability. In stem cells and neural progenitors, REST abundance is bidirectionally controlled by ubiquitin-based proteasomal degradation34,46–49 that is dependent on β-TrCP (also known as BTRC and F-box/WD repeat-containing protein 1A) and by deubiquitylation that is dependent on ubiquitin carboxyl-terminal hydrolase 7 (USP7; also known as HAUSP)50, which add a new dimension to the relationship between the ubiquitin-proteasomal pathway and epigenetic regulation. A recent study showed that REST abundance is regulated by β-TrCP-dependent, ubiquitin-based proteasomal degradation in differentiated neurons and identified casein kinase 1 (CK1) as an upstream signal that promotes the interaction of REST with β-TrCP49 (BOX 3).
Box 3 | The regulation of restrictive element 1-silencing transcription factor degradation.
Restrictive element 1-silencing transcription factor (REST) is widely expressed throughout embryogenesis31, and its elimination at the end of neuronal differentiation is crucial for the elaboration of the neuronal phenotype. In pluripotent stem cells, neural progenitors and cancer cells, REST abundance is regulated by ubiquitin-based proteasomal degradation. In differentiated neurons under physiological conditions, casein kinase 1 (CK1) is activated and ensures that REST abundance remains at low basal levels (see the figure, left panels). CK1 phosphorylates REST at serine residues within its two degron motifs, thus enabling the recognition and binding of the E3 ligase β-TrCP (phosphorylation is denoted ‘P’ in the figure). β-TrCP is a member of the F-box family of E3 ligases that binds to several target proteins — including the transcription factors REST, β-catenin115 and nuclear factor-kB2 (NF-kB2; also known as NF-kB p100 subunit)116 — and initiates their ubiquitylation (denoted ‘Ub’ in the figure). Upon binding, β-TrCP initiates the ubiquitylation of REST (the substrate) and primes it for ubiquitin-based proteasomal degradation. A recent study identified a proline-rich motif upstream of the phospho-degron motifs that, when phosphorylated by extracellular signal-regulated kinase 1 (ERK1) or ERK2, promotes REST degradation in neural progenitor cells, a mechanism that is implicated in the timing of REST elimination in the end stages of neural differentiation117. Neuronal insults, such as ischaemia28,61, reduce the levels of CK1 and β-TrCP, allowing the levels of REST to rise in a spatially restricted set of neurons (see the figure, right panels). Upon induction, REST binds to the restrictive element 1 (RE1) elements in target genes and assembles into a large co-repressor complex that includes SIN3A, REST co-repressor (CoREST), histone deacetylase 1 (HDAC1), HDAC2, G9A, carboxy-terminal binding protein 1 (CTBP1; not shown)109, LSD1 (not shown)110,111 and methyl-CpG-binding protein 2 (MECP2). The co-repressor complex orchestrates epigenetic modifications and gene silencing in a cell-specific and context-specific manner (BOX 4).
REST can also be degraded through the autophagy pathway, as occurs, for example, in Alzheimer disease44 and possibly in other neurodegenerative disorders. The activation of autophagy by serum deprivation in SH-SY5Y cells results in the translocation of REST from the nucleus to the cytoplasm, where it colocalizes to punctate structures that also contain autophagosome markers44. Moreover, REST colocalizes with amyloid-β in a subset of autophagosomes44. This might occur because the ubiquitin-proteasome system becomes overloaded or impaired in neurodegenerative disorders such as Alzheimer disease. A number of proteasome substrates have been shown to also be degraded by autophagy118.
CUL1, cullin 1; Gria2, gene encoding mouse glutamate receptor 2; H3K9me2, histone H3 lysine 9 dimethylation; H3K9ac, H3K9 acetylation; SKP1, suppressor of kinetochore protein 1; UBC, polyubiqutin-C. Republished with permission of Society for Neuroscience, from Casein kinase 1 suppresses activation of REST in insulted hippocampal neurons and halts ischemia-induced neuronal death, Kaneko N., Hwang J. Y, Gertner M., Pontarelli F, Zukin R. S., J. Neurosci. 34, 6030–6039, 2014; permission conveyed through Copyright Clearance Center, Inc.
Polycomb proteins coordinate epigenetic remodelling.
First discovered for their role in specifying the variegation and patterning of the wings and legs of Drosophila melanogaster51, Polycomb proteins are transcriptional repressors that are activated in differentiated neurons by ischaemic preconditioning52. Bioinformatics analyses predict that there are more than 500 putative Polycomb protein targets. Polycomb proteins form large multimeric complexes known as Polycomb repressive complex 1 (PRC1) and PRC2, which are recruited to the genome where they enforce global gene silencing by altering local, higher-order chromatin structure53. The histone-lysine N-methyltransferases enhancer of zeste homologue 1 (EZH1) and EZH2, which are the catalytically active components of PRC2, add a H3K27me3 mark, which is a strong mark of gene repression. The recruitment of PRC1 to the genome also initiates repression through the deposition of trimethylation marks. Subsequently, PRC1 is recruited to and stabilized at regions of the genome marked by H3K27me3. Two recent studies demonstrate that EZH2 also recruits DNMTs, thus providing a direct link between two key epigenetic silencing mechanisms: the Polycomb proteins and DNA methylation54,55.
Polycomb proteins mediate the protection of hippocampal neurons in response to ischaemic preconditioning52, as demonstrated by a proteomics profiling study that revealed an abundance of proteins involved in chromatin remodelling — including histone H2A and H2B variants and the Polycomb proteins BMI1 and SCMH1 — in the ischaemia-tolerant brain52. Polycomb proteins act through chromatin remodelling to repress the expression of proteins that mediate neuronal death and to promote cellular arrest, which is a state that is reminiscent of hibernation and enables neurons to survive the hypoxia that is associated with ischaemic stroke (FIG. 1). In addition, Polycomb proteins are implicated in Huntington disease (see below).
Figure 1 |. Polycomb proteins in epigenetic remodelling and neuroprotection.

Ischaemic preconditioning is a well-known phenomenon in which a brief, sublethal ischaemic insult confers hippocampal CA1 neurons with robust protection against a subsequent severe ischaemic challenge123–125. Ischaemic preconditioning promotes the expression of the Polycomb proteins SCMH1, BMI1 and RING2, and the core histone protein H2A, as well as the formation of Polycomb protein complex 1 (PRC1) and PRC2. PRC2 associates with the promoters of target genes. The catalytically active subunits of PRC2, enhancer of zeste homologue 1 (EZH1) and EZH2, catalyse the trimethylation of histone H3 at lysine 27 (H3K27me3), a hallmark of EZH2-mediated gene repression. Chromodomain- containing protein (CBX; also known as chromobox protein homologue 1), a component of PRC1, recognizes and binds to H3K27me3, and thereby recruits PRC1 to the promoter. SCMH1 is a component of PRC1 (REF 126) and is crucial for the neuroprotection induced by ischaemic preconditioning52. The PRC1 protein BMI1 is a Polycomb ring-finger protein that protects against stress-induced cell death127 and p53-mediated cell death128. RING2 catalyses the mono-ubiquitylation of histone H2A at lysine 119 (H2AK119ub1), a post-translational modification that represses transcript elongation by RNA polymerase. It has been shown that ischaemic preconditioning is associated with the appearance of these epigenetic marks in the promoters of the genes Kcna5 and Kcnab2 (which encode voltage-gated potassium channel subfamily A member 5 and voltage-gated potassium channel subunit β2, respectively). The presence of these marks represses Kcna5 and Kcnab2 expression, and thereby attenuates the activity of voltage-gated potassium channels in the ischaemia-tolerant brain52, resulting in neuroprotection. PHC1, polyhomeotic-like protein 1; RPAP, RNA polymerase-associated protein.
Non-coding RNAs and synaptic plasticity
Accumulating evidence implicates miRNAs and lncRNAs in the epigenetic regulation of gene expression. miRNAs are small non-coding RNA molecules (each containing ~22 nucleotides) that function in RNA silencing and the post-transcriptional regulation of gene expression. In early brain development, miRNAs are crucial for synapse formation and maturation, and for dendritogenesis56–58. A powerful feature of miRNAs is their ability to control entire networks of genes in neurons in response to neural activity56–58. Moreover, neuron-specific miRNAs can act in dendrites to regulate the local protein synthesis that is crucial for synaptic plasticity and cell survival56–58.
Recent findings suggest that there is an interaction between REST and miRNAs in the transition of pluripotent stem cells into neurons. A subset of miRNAs, including miR-9 and miR-9*, are functionally validated targets of REST. In addition, a reciprocal double-negative feedback loop exists in which miR-9 targets REST and miR-9* targets REST co-repressor (CoREST; also known as RCOR1): thus, the suppression of miR-9 and miR-9* expression results in increased REST and CoREST expression59. This is important because miR-9 and miR-9* are misregulated in Huntington disease, suggesting that REST activity may also be altered in this disease59. The loss of another miRNA that is a target of REST, miR-132, is causally related to the pathophysiology of focal and global ischaemia, and is linked to neuronal death60,61.
lncRNAs are >200 nucleotides in length and serve as molecular scaffolding proteins that tether two or more transcription factors to target genes. Long intergenic non-coding RNAs (lincRNAs) are located and transcribed within intergenic stretches of DNA where they serve to influence gene expression. A prominent example of a lincRNA implicated in REST-mediated epigenetic remodelling is HOX transcript antisense RNA (HOTAIR). EZH2 is recruited by HOTAIR to REST at restrictive element 1 (RE1; also known as NRSE) sites within the genome, where REST and EZH2 act synergistically to silence target genes62.
Epigenetics and neurodegeneration
Many of the epigenetic mechanisms discussed above have been shown to be altered in specific neurodegenerative diseases. The following section summarizes the role of DNA methylation and histone modifications in neurodegenerative diseases. We focus on the neurological disorders for which the supporting data are strongest: namely, Alzheimer disease, Huntington disease, stroke and global ischaemia.
Alzheimer disease
Alzheimer disease is the most common late-onset neurodegenerative disorder and is characterized by progressive cognitive decline and neuronal death. As most cases of Alzheimer disease are sporadic and develop over time, we believe that environmental factors result in alterations in gene expression that are not ‘hardwired’ into the DNA sequence and that may contribute to disruptions in synaptic signalling and neuronal survival. Recent findings implicate the dysregulation of REST and REST-dependent epigenetic remodelling in this process.
A recent study44 revealed that REST expression and function — as assessed by measuring the binding of REST to canonical RE1 sites in its target genes — are ubiquitous features of normal ageing, and that the loss of REST is associated with mild or severe cognitive impairment and Alzheimer disease (FIG. 2a). In this study, immunofluorescence revealed a striking increase in REST abundance in the nuclei of neurons in the prefrontal cortex and in hippocampal CA1 and CA3 of normal ageing subjects that is lost in subjects with mild or severe cognitive impairment or Alzheimer disease. The authors showed that, during normal ageing, REST is activated at least in part by the WNT signalling pathway63. Subsequent chromatin immunoprecipitation followed by sequencing (ChIP-seq) experiments in a neural cell line44 revealed the enrichment of REST at targets involved in neuronal death (namely, the genes encoding p38 MAPK, FAS (also known as TNFRSF6), FAS-associated death domain protein (FADD), tumour necrosis factor receptor type 1-associated death domain protein (TRADD), BAX, BH3-interacting domain death agonist (BID), BCL-2-binding component 3 (BBC3; also known as PUMA), mitochondrial permeability transition pore proteins and cytochrome c) and those involved in Alzheimer disease pathology (namely, the genes encoding γ-secretase, presenilin 2, presenilin enhancer protein 2 and cyclin-dependent kinase 5 activator 1). Thus, REST regulates the expression of genes that are involved in oxidative stress and β-amyloid toxicity, which is consistent with the concept that the sustained maintenance of the REST-driven programme of gene repression confers neuroprotection. REST was also enriched at the promoters of genes involved in neurotransmission in normal ageing, but its binding to these promoters was markedly reduced in both early-stage and late-stage Alzheimer disease44. mRNA expression of these neurotransmission-related genes is indeed derepressed (that is, their expression is increased relative to controls) in early-stage Alzheimer disease; however, this is not the case in late-stage Alzheimer disease44, which might reflect either the loss of neurons or other regulatory changes. For example, during neuronal development, genes can still be repressed after REST is depleted, owing to the continued presence of co-repressors such as CoREST and methyl-CpG-binding protein 2 (MECP2)34. In addition to repressing genes that promote neuronal death, REST promoted the expression of anti-apoptotic genes and genes that mediate resistance to oxidative stress. Consistent with this, mice deficient in REST exhibited increased vulnerability to oxidative stress.
Figure 2 |. Restrictive element 1-silencing transcription factor in neurodegenerativo disease.

a | In -the healthy aged brain, stress increases WNT-β-catenin signalling (specifically that mediated by WNT3A and WNT7A) in the hippocampus and the frontal cortex. This promotes the nuclear translocation of restrictive element 1-silencing transcription factor (REST) and increased binding of REST to the restrictive element 1 (RE1) sites of REST target genes. This induction of REST nuclear abundance promotes neuroprotection by repressing the transcription of genes that encode proteins involved in neuronal death (such as BCL-2-binding component 3 (BBC3), BAX, death domain-associated protein 6 (DAXX), tumour necrosis factor receptor type 1-associated death domain protein (TRADD) and BCL-2-like protein 11 (BCL2L11)) and genes that encode proteins involved in Alzheimer disease (AD) pathology (such as members of the γ-secretase complex, which is implicated in amyloid-β (Aβ) generation). In AD-affected brains, oxidative stress activates autophagy129. The autophagosomes that are generated engulf REST, together with misfolded proteins such as Aβ and tau, which reduces REST abundance in the nucleus. The loss of REST in the nucleus causes an increase in expression of genes that are involved in oxidative stress and neuronal death. b | In the striatal neurons of wild-type (WT) mice, huntingtin (HTT) binds to and sequesters REST in the cytoplasm away from target genes that are important for neuronal activity and survival, such as brain-derived neurotrophic factor (BDNF)71,130,131. In the striatal neurons of mice carrying Huntington disease (HD)-related mutations, mutant HTT (mHTT) is unable to bind to and sequester REST, enabling REST to translocate to the nucleus where it silences target genes, thus promoting neuronal death41,42. A recent study suggests that this mechanism may not apply to neurons, but rather to glial cells, in the striata of humans with HD43. CoREST, REST co-repressor; H3K9ac, histone H3 lysine 9 acetylation; HDAC, histone deacetylase.
This study was important, as it demonstrated a neuroprotective role for REST in the ageing brain that transcends its role in Alzheimer disease. REST was also significantly depleted in neurons from subjects with frontotemporal dementia or dementia with Lewy bodies44. Moreover, recent findings have revealed that REST is protective in an animal model of Parkinson disease. Substantia nigra neurons from mice deficient in REST are more vulnerable than wild-type neurons to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) toxicity64. Consistent with this, MPTP promotes an increase in REST expression but at the same time drives the translocation of REST from the nucleus to the cytoplasm, eliciting the derepression of REST target genes and neuronal death.
Two recent independent epigenome-wide association studies involving cohorts of subjects with Alzheimer disease and healthy subjects identified increased methylation at four loci — namely, those encoding ankyrin 1 (ANK1), ribosomal protein L13 (RPL13), rhomboid 5 homologue 2 (RHBDF2) and cadherin-related protein 23 (CDH23) — in the Alzheimer disease cohort65,66. Hypermethylation of ANK1 occurred predominantly at CpG sites65,66. Moreover, ANK1 expression is altered in AD65,66. ANK1 localizes to the plasma membrane, where it binds to the actin cytoskeleton. ANK1, RPL13 and RHBDF2 are biologically linked to protein tyrosine kinase 2β (PTK2β), which is encoded by a known Alzheimer disease-associated gene65. These studies were the first replicable, robust epigenome-wide association studies to demonstrate increased DNA methylation in post-mortem tissues from cohorts of patients with Alzheimer disease and healthy controls65,66. Moreover, an independent study involving individuals with late-onset Alzheimer disease and sex-matched, age-matched healthy controls found single-nucleotide polymorphisms (SNPs) adjacent to the hypermethylated CpG sites in ANK1 in the Alzheimer disease samples67, which suggests that SNPs might be involved in the methylation of the ANK1 CpG sites. Indeed, one of these SNPs (the A allele of the rs515071 SNP in ANK1) was strongly associated with late-onset Alzheimer disease. This study implicates ANK1 polymorphisms as novel genetic risk factors for late-onset Alzheimer disease67. The relevance of the final gene shown to be hypermethylated in Alzheimer disease in this study, CDH23, remains unknown.
Despite the compelling evidence from each of these well-designed studies, there are important limitations. Notably, the microarray used in these studies covered only a small proportion (~2%) of the human genome68. In addition, the experiments were performed on heterogeneous brain tissue samples and, as such, could not distinguish altered DNA methylation in neurons from that in non-neuronal cells such as glia68. This problem is compounded in Alzheimer disease, in which there is substantial neuronal loss accompanied by glial proliferation69. Moreover, this approach cannot distinguish between methylation and hydroxymethylation, which have opposing effects on gene expression.
Huntington disease
Huntington disease is an autosomal dominant, late- onset neurodegenerative disease that is characterized by progressive motor and cognitive decline, and psychiatric disturbances that may result in premature death. Huntington disease is caused by a polyglutamine (CAG) repeat in the 5’-coding region of the gene that encodes huntingtin. Neuropathological hallmarks of the disease include the loss of selectively vulnerable aspiny striatal neurons and the presence of aggregates of mutant huntingtin. A key feature of the effects of Huntington disease on the synapses onto aspiny striatal neurons is a marked reduction in the abundance of components of the NMDAR signalling complex, which is a crucial player in synaptic plasticity and cognition. The debilitating chorea exhibited by patients with Huntington disease has been attributed to the degeneration of the caudate putamen, which is caused, at least in part, by the loss of the supply of BDNF from the cortex42,43.
Transcriptional silencing (independent of the CAG expansion) is also central to the pathophysiology of Huntington disease and is presumed to occur, at least in part, as result of the interaction of mutant huntingtin with the transcriptional machinery. In addition, miRNA-mediated gene silencing results from the interaction of mutant huntingtin with the Argonaute protein family member AGO2 — which is the endonuclease involved in RNA-induced silencing complexes (RISCs) — and from the localization of huntingtin within processing bodies70. Accordingly, a focus of research has been the identification of the mechanisms that underlie this gene silencing.
A major advance occurred when it was shown that the dysregulation of REST and REST-dependent epigenetic remodelling has a central role in the gene silencing and progressive neurodegeneration that are associated with Huntington disease41,42,71. In wild-type mice, it was shown that huntingtin tightly binds to REST in the cytoplasm of vulnerable cells within the striatum, thus sequestering it away from target genes. In Huntington disease, the trinucleotide-repeat expansion in hunting- tin disrupts the binding of huntingtin to REST, enabling REST to translocate to the nucleus, where it silences genes that are crucial for neuronal survival41 (FIG. 2b). Accordingly, the depletion of endogenous huntingtin in cells in culture and in mice is associated with the reduced transcription of genes that have RE1 sites42. The delivery of a dominant-negative REST mutant directly into the motor cortex of symptomatic mouse models of Huntington disease reduced REST occupancy and rescued the transcription of a subset of REST target genes in the brain72. These included the genes encoding the prosurvival peptide BDNF, proenkephalin and muscarinic acetylcholine receptor M4. Although the delivery of dominant-negative REST rescued transcription, it did not correct motor deficits, which suggests that there is another mechanism — in addition to REST-dependent gene silencing — that is required to induce the neuronal death that is associated with Huntington disease.
It is important to note that a more recent paper reported that REST abundance is increased in the nuclei of glia but not in those of neurons in autopsy tissue from humans with Huntington disease43. The discrepancy between this finding and that of earlier papers may have arisen because the previous studies that reported an enrichment of REST at the promoters of target genes by means of ChIP-on-chip analysis were performed on samples of parietal cortex tissue from patients with Huntington disease and, as such, could not distinguish neuronal REST from glial REST41,42,71. It is also possible that, despite a decrease in REST abundance in selectively vulnerable striatal neurons in humans with Huntington disease, the recruitment of REST to target genes such as BDNF might be increased43.
Polycomb proteins have also been implicated in transcriptional dysregulation in Huntington disease. A recent study revealed that PRC2 silences genes that are involved in neuronal death and that the loss of PRC2 in medium striatal neurons induces progressive and fatal neurodegeneration in mice that resembles the neurodegeneration seen in humans with Huntington disease73. Moreover, loss of PRC2 in forebrain neurons induces the upregulation of genes involved in Huntington disease73.
Genome-wide association studies have been performed in humans with Huntington disease, and in mouse and D. melanogaster models of the disease. Of note, a deficiency in 5-hydroxymethylcytosine, an epigenetic mark on DNA that enables rapid switching between ‘on’ and ‘off’ states of transcription (see above), was found in a mouse model of Huntington disease74. Ingenuity Pathway Analysis revealed the aberrant methylation and silencing of genes that are important in neuronal development and differentiation in the striatum and cortex of a mouse model of Huntington disease during postnatal development74. Well-known pathways that were shown to be silenced in the Huntington disease model include the WNT-β-catenin-SOX signalling that is implicated in axonal guidance; the NMDAR-calcium- CREB signalling that is important in neuronal function and survival; GABA type A receptor signalling; and dopamine-PP1 regulatory subunit 1B (PP1R1B; also known as DARPP32) signalling74. These findings link a deficiency of 5-hydroxymethylcytosine to the impaired neurogenesis, aberrant neuronal function and neuronal death that are associated with Huntington disease.
Stroke
Stroke is the leading cause of disability in adults in the United States and is the second leading cause of death worldwide75. Symptoms result from an interruption of blood flow caused by thrombosis or embolism or, less frequently, haemorrhage or cardiac arrest. Although a vast number of pathways contribute to the neuronal injury that is associated with stroke, a considerable amount of evidence supports a role for DNA methylation in this process. Mutant mice that express reduced levels of DNMT1 exhibit protection from mild ischaemic damage, which suggests that DNA methylation exacerbates neuronal injury76,77. Moreover, mice in which Dnmtl was conditionally knocked out in one allele exhibited markedly reduced infarct volume in the transient middle cerebral artery occlusion (MCAO) model, which is a clinically relevant model of stroke76,77. Accordingly, the demethylating agent 5-aza-2’-deoxycytidine ameliorated ischaemia-induced neuronal death in wild-type mice77.
Recent studies implicate REST in the pathophysiology of cerebral stroke. Rats subjected to transient MCAO exhibited marked expression of REST in the deep cortical layers and REST-induced silencing of sodium/calcium exchanger 1 (NCX1) in layer V of the temporoparietal cortex39,40. This is notable because NCX1 enables the rapid transport of Ca2+ ions and is thought to have an important role in promoting the return of Ca2+ homeostasis following neuronal insults such as stroke78,79. The acute knockdown of REST or site-directed mutagenesis of the RE1 site in the Ncxl promoter rescued NCX1 expression, establishing a causal relationship between REST and the suppression of NCX1 expression in post-ischaemia tissue. Administration of entinostat (also known as MS-275) — an HDAC inhibitor that is currently in clinical trials for the treatment of cancer — in combination with the sirtuin 1 (SIRT1) activator resveratrol elicited neuroprotection in mice subjected to transient MCAO80. By blocking HDAC activity, entinostat promotes the acetylation of both transcription factor p65 (also known as RELA) and histone H3 (REF 80). Entinostat and other HDAC inhibitors act, at least in part, by preventing the binding of a dimer comprising the nuclear factor-kB (NF-kB) p50 subunit and transcription factor p65 to the promoter of the gene encoding BCL-2-like protein 11 (BCL2L11; also known as BIM), which has a pro-apoptotic function, thereby preventing the expression of BCL2L11 and the onset of apoptosis80.
Endogenous brain repair stimulated in response to stroke involves a set of highly interactive processes — including angiogenesis, neurogenesis, synaptogenesis, oligodendrogenesis and axonal outgrowth — which together promote neurological recovery81. Emerging evidence implicates miRNAs in this process. Ischaemia substantially alters the expression of miRNAs that are involved in neurogenesis and brain repair82. Of note, stroke downregulates miR-124a, which normally acts via Notch and sonic hedgehog signalling to suppress the proliferation of neural progenitor cells in the sub-ventricular zone of adult animals83. Ischaemia-induced downregulation of miR-124a derepresses the expression of the Notch ligand Jagged 1, leading to increased cellular proliferation and neurogenesis.
Global ischaemia
Transient forebrain or global ischaemia is a neurological disorder that occurs in association with cardiac arrest, open-heart surgery, profuse bleeding or carbon monoxide poisoning84–86. A prominent neurological sequela is cognitive impairment. Although all forebrain areas experience oxygen and glucose deprivation, neuronal death arising from a brief ischaemic event is restricted primarily to hippocampal CA1 and is not detected until 2–3 days after the insult. By 1 week after ischaemia, the CA1 pyramidal cell layer is ablated86.
Although the mechanisms that underlie neuronal death in global ischaemia are complex, the considerable delay between the insult and the occurrence of cell death is consistent with a role for changes in gene expression. Within hours after an ischaemic episode, the expression of CK1 decreases, and REST is activated in the nuclei of CA1 pyramidal neurons27,28,38. Ischaemia also triggers the activation of the co-repressors of REST28. Upon ischaemia-mediated activation, REST assembles with CoREST, SIN3A, HDAC1, HDAC2, G9A (also known as EHMT2), LSD1 (also known as KMT1A) and MECP2 (to form the REST-co-repressor complex) at the promoters of target genes and orchestrates epigenetic remodelling (BOX 2). ChIP-on-chip profiling and bioinformatics analysis have revealed that the REST-co-repressor complex orchestrates the silencing of an ensemble of ‘transcriptionally responsive’ target genes (including those encoding the AMPA receptor subunit glutamate receptor 2 (GluA2), the NMDAR subunit glutamate receptor ionotropic NMDA 1, neuronal acetylcholine receptor subunit β2, muscarinic acetylcholine receptor M4, neurofilament heavy polypeptide, NF-kB subunit 2, transient receptor potential cation channel subfamily V member 1 and synaptotagmin 6), of which the gene encoding GluA2 was a top hit. Genes with enriched REST binding exhibited decreased mRNA-level and protein-level expression in CA1 after ischaemia.
In addition to these transcriptionally responsive coding genes, REST also promotes the epigenetic remodelling and silencing of miRNAs such as miR-132 in the neurons of animals subjected to global ischaemia in vivo61. miR-132 is crucial for activity-dependent synaptic plasticity, the sculpting of dendritic arborization56,87 and neuronal survival88,89, and it suppresses the expression of the genes encoding phosphatase and tensin homologue, forkhead box protein O3 and p300, which are known to promote apoptosis88,89. Accordingly, the overexpression of miR-132 reduces ischaemia-induced cell death61. These findings indicate a protective role for miR-132 in the context of global ischaemia.
Targeting the epigenetic machinery
An effective treatment for the cognitive and motor deficits that are associated with most neurodegenerative diseases remains an unmet medical need. Components of the epigenetic machinery — such as DNMTs, the TET family of DNA demethylases, HDACs, HATs, methyltransferases, miRNAs and lncRNAs — all represent potential targets for the development of epigenetic drugs. Modifications of histones and DNA are reversible, which makes them good targets for therapeutic intervention. Moreover, the transcriptional repressor REST assembles with other chromatin-remodelling enzymes that together orchestrate changes in not only histone modifications but also DNA methylation. In this section, we highlight the potential power of new therapeutic approaches that target the epigenetic machinery.
The past decade has witnessed the development of new chromatin-modifying drugs and their advance to clinical trials for use in patients with brain disorders. The most promising drugs to date are DNA-demethylating agents and HDAC inhibitors (TABLE 1). Members of these two classes are approved by the US Food and Drug Administration for haematological malignancies and have been in use for the treatment of cancer for nearly two decades. The recent approval of two drugs that target DNA methylation (azactidine (also known as azacytidine; marketed as Vidaza by Celgene) and decitabine (also known as 5-aza-2’-deoxycytidine; marketed as Dacogen by Eisai)) and two drugs that target HDACs (suberoylanilide hydroxamic acid (SAHA; also known as vorinostat; marketed as Zolinza by Merck) and romidepsin (also known as depsipeptide or FK228)) represents a major advance for epigenetic-based therapies90.
Table 1 |.
Drugs that target the epigenetic machinery
| Inhibitor | Drug target | Stage of development | Animal models tested | Refs |
|---|---|---|---|---|
| DNMT inhibitors | ||||
| Decitabine (also known as 5-aza-2- deoxycytidine; marketed as Dacogen by Eisai) | DNMTs | Approved in 2006 | Stroke | 76,77 |
| Zebularine | DNMTs | Preclinical | Stroke | 132 |
| HDAC inhibitors | ||||
| Valproic acid | Class I and II HDACs | Approved in 1986 | AD, HD, stroke and global ischaemia | 133–139 |
| Phenylbutyrate | Class I and II HDACs | Approved in1996 | AD and HD | 140–143 |
| Suberoylanilide hydroxamic acid (also known as vorinostat; marketed as Zolinza by Merck) | Class I and II HDACs | Approved in 2006 | AD, HD and stroke | 133, 144–147 |
| Nicotinamide (sirtuin inhibitor) | Class III HDACs | Phase III | AD and HD | 94,95 |
| Entinostat (also known as MS-275) | Class I HDACs | Phase II | AD and stroke | 80,148 |
| Trichostatin A | Class I and II HDACs | Preclinical | HD, stroke and global ischaemia | 28,149,150 |
| AGK2 (sirtuin 2 inhibitor) | Class III HDACs | Preclinical | HD and PD | 151,152 |
| Sodium butyrate | Class I and II HDACs | Preclinical | AD, HD, stroke, brain atrophy and the bi-transgenic CK-p25 mouse model of neurodegeneration | 26,133,140, 153–156 |
AD, Alzheimer disease; CK-p25, mouse model in which p25 is conditionally expressed under the control of the promoter of the gene that encodes calcium/calmodulin-dependent protein kinase II; DNMT, DNA methyltransferase; HD, Huntington disease; HDAC, histone deacetylase; PD, Parkinson disease.
HDAC inhibitors have a long history of use in psychiatry and neurology as mood stabilizers and anti-epileptics. More recently, they have been investigated as potential therapeutic agents for the treatment of neurodegenerative diseases primarily on the basis of their actions as anti-inflammatory agents91. Preclinical studies suggest that HDAC inhibitors may have therapeutic potential in neurodegenerative diseases including acute brain injury, stroke, Parkinson disease, Huntington disease and Alzheimer disease91 (TABLE 1). HDAC inhibitors can be classified into four main chemical families: namely, hydroxamic acids (TSA and SAHA), epoxyketones (such as trapoxin), short-chain fatty acids (sodium butyrate, phenylbutyrate and valproic acid) and benzamides92,93. Of these, butyrates are known for their ability to penetrate the blood-brain barrier, which makes them potentially suitable for the treatment of brain disorders. Intense interest has focused on SIRT1, a Class III HDAC that is implicated in the neurodegeneration that is associated with Alzheimer disease and Huntington disease94,95. The development of new drugs that block the binding of nicotinamide to SIRT1 and other NAD-dependent HDACs could enable the development of a new generation of drugs that target the epigenetic machinery with increased specificity. However, the application of these drugs for the treatment of neurodegenerative disorders is in its infancy.
The major challenge for the future is the development of drugs that can penetrate the blood-brain barrier, and ameliorate neurodegeneration and cognitive deficits with increased specificity and minimal toxicity. To date, most drugs that target the epigenetic machinery are nonspecific. Despite off-target effects and a lack of specificity, the HDAC inhibitor sodium butyrate restored learning and synaptogenesis in the CK-p25 model of neurodegeneration26, and the HDAC inhibitor TSA ameliorated ischaemia-induced neuronal death28. Thus, nonspecific treatments that target the epigenetic machinery can ameliorate impaired cognition and neuronal death in animal models. Another approach is to develop inhibitors that are specific for a single HDAC isoform. However, even an isoform-specific inhibitor can have multiple, even contradictory, actions. For example, HDAC6 localizes to the cytoplasm where it has a role in diverse and even opposing functions such as the degradation of proteins by autophagy and the deacetylation of tubulin, thereby disrupting axonal transport96. Thus, a class II inhibitor that blocks HDAC6 may have contradictory effects on neuronal function. Another problem is that drugs that target elements of the epigenetic machinery — such as HDACs and DMNTs — are not selective for a given brain region, cell type or gene. A possible strategy to overcome this lack of specificity would be to target a specific epigenetic modification rather than affect global modifications; this would increase clinical efficiency and, at the same time, reduce toxicity and side effects.
Concluding remarks
The epigenetics of neurodegenerative diseases and disorders is an emerging topic that has a great potential to inform the design of novel therapeutic strategies for ameliorating the cognitive deficits and neurodegeneration that are associated with neurological disorders and diseases. In the past decade, remarkable progress has been made in documenting a role for epigenetics in neurodegenerative diseases. New evidence implicates REST dysregulation in many of these disorders. A case in point is the discovery that the expression of REST in the prefrontal cortex is a universal feature of normal ageing and that the loss of REST in the prefrontal cortex of aged humans is associated not only with Alzheimer disease but also with frontotemporal dementia and dementia with Lewy bodies44. By contrast, REST expression in differentiated neurons is low at early ages, and the activation of REST in response to neuronal insults is causally linked to ischaemia-induced neuronal death. Collectively, these studies reveal that the impact of REST on its target genes varies in a context- specific and cell-specific manner (BOX 4).
Box 4 | The specificity of restrictive element 1-silencing transcription factor-dependent gene silencing.
It is important to note that the subset of target genes that are responsive to restrictive element 1-silencing transcription factor (REST) — which are known as the ‘transcriptionally responsive’ genes — at a given developmental stage or in a particular disease state varies in a cell type-dependent and context-dependent manner. For example, the ensemble of REST target genes that exhibit altered expression after global ischaemia28 differs from the ensemble of genes that show altered expression in the hippocampi of mice with kainite-induced seizures (an animal model of epilepsy)119, in the prefrontal cortex of humans with Huntington disease42 and in the prefrontal cortex of healthy aged humans44. Moreover, the subset of transcriptionally responsive genes identified in unbiased, genome-wide studies that used REST chromatin immunoprecipitation followed by sequencing (ChIP-seq) in a neuroblastoma cell line44 differs from that identified by large-scale ChIP-seq analysis in Jurkat cells120 and from that identified by ChIP-on-chip analysis in mouse neural stem cells121. Thus, in different cell types, at different ages and in different disease states, REST regulates the expression of different subsets of target genes.
In silico analyses predict that there are approximately 2,000 putative REST target genes within the mammalian genome32,122. The presence of noncanonical restrictive element 1 (RE1) motifs in the genome120 accounts for an even broader repertoire of dynamically regulated, but lower affinity, REST targets. These observations raise the question of what factors determine the specificity of interaction between REST and its targets. An attractive possibility is that the epigenetic landscape — which varies with developmental stage, cell type, brain region and disease state — influences the affinity of REST for a subset of target genes. For example, the chromatin-remodelling protein BRG1 recruits REST to a specific ensemble of target genes114. It is therefore possible that other transcription factors may influence the enrichment of REST at the promoters of different sets of target genes. A case in point are the Polycomb group proteins, which serve as “global enforcers of epigenetically repressed states” in various cells types including neurons51. The Polycomb repressive complex 2 (PRC2) is recruited to RE1 sites within REST target genes by the long non-coding RNA (lncRNA) HOX transcript antisense RNA (HOTAIR)62. HOTAIR binds via its 3’ domain to the LSD1-REST co-repressor (CoREST)-REST complex, which is consistent with the concept that lncRNAs serve as scaffolds by providing binding platforms for the assembly of unique ensembles of chromatin-remodelling enzymes and thereby modify the expression of target genes63.
Despite recent progress in our understanding of the role of epigenetics in learning and memory, and its dysregulation in neurodegenerative disorders, many questions remain unanswered. Is the dysregulation of REST and REST-dependent epigenetic remodelling causally related to the impaired cognition that is associated with neurodegenerativa disorders? What are the mechanisms that underlie the ‘disconnect’ between the nuclear abundance and binding affinity of transcription factors such as REST and Polycomb proteins at the promoters of target genes? Is a change in the epigenetic landscape sufficient to substantially alter the recruitment of transcription factors to their target genes? Does chromatin remodelling promote an increased vulnerability to neuronal insults, and/or do neuronal insults promote chromatin modifications that drive neuronal survival or cell death? Is it possible to identify a signalling pathway that is associated with a specific component of the epigenetic machinery (such as CK1, which negatively regulates REST) and that could serve as a novel target for the treatment of neurodegenerative disorders or for slowing their progression?
In summary, the past decade has witnessed the accumulation of a huge amount of evidence that collectively points to a role for the dysregulation of REST, a master transcriptional regulator of neuronal genes, in neurodegenerative disorders including Alzheimer disease, Huntington disease, stroke and global ischaemia. In each case, REST assembles to form a large macromolecular complex that orchestrates chromatin remodelling34. These observations link REST to epigenetic modifications of histones and DNA methylation, and are consistent with the possibility that drugs that target the epigenetic machinery will also target REST and thereby ameliorate the impaired cognition and neuronal death that are associated with these disorders.
Histone post-translational modifications.
Covalent modifications of histone proteins, including methylation, phosphorylation, acetylation, ubiquitylation and sumoylation.
Nucleosome.
The basic building unit of chromatin. It comprises 147 bp of DNA wrapped around a histone octamer that contains two molecules of each of the four histones H2A, H2B, H3 and H4.
Long-term potentiation.
(LTP). A long-lasting (hours or days) increase in the response of neurons to the stimulation of their afferents following a brief patterned stimulus (for example, a 100 Hz stimulus).
Contextual fear conditioning.
A form of conditioning in which animals associate the conditioning context (the ‘neutral’ conditioned stimulus) with an aversive stimulus (for example, a footshock).
Oxidative stress.
A disturbance in the pro-oxidant–antioxidant balance in favour of the former, which can lead to cellular damage. Indicators of oxidative stress include damaged DNA bases, protein oxidation products and lipid peroxidation products.
Ubiquitin-based proteasomal degradation.
A process that is initiated by a protein complex and is based on ubiquitin, a 76‑amino acid protein that forms a covalent link with (and thereby marks) proteins destined for degradation. Proteins tagged by a poly-ubiquitin chain are targeted to the proteasome, which is a large, multimeric, barrel-like complex that degrades proteins by proteolysis.
Single-nucleotide polymorphisms.
(SNPs). A type of genetic variation within a DNA sequence that occurs when a single nucleotide (for example, thymine) replaces one of the other three nucleotides (for example, cytosine).
RNA-induced silencing complexes.
(RISCs). A complex of proteins that is involved in silencing target mRNAs.
Ingenuity Pathway Analysis.
A web-based software application that enables the analysis, integration and understanding of data from gene expression, microRNA, and single-nucleotide polymorphism microarrays; and metabolomics, proteomics and RNA sequencing experiments.
Acknowledgements
The authors acknowledge all the authors whose valuable work they could not include owing to the limited number of citations allowed. This work was supported by US National Institutes of Health grants NS046742, MH092877and HD083828; a generous grant from the F. M. Kirby Foundation and a National Alliance for Research on Schizophrenia and Depression (NARSAD) Distinguished Investigator Grant to R.S.Z.; and American Heart Association Scientist Development Grant 16SDG31500001 and NARSAD Young Investigator Grant 25369 to J.-Y.H. R.S.Z. is the F. M. Kirby Chair in Neural Repair and Protection.
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Waddington CH The epigenotype. 1942. Int.J. Epidemiol 41, 10–13 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Sweatt JD The emerging field of neuroepigenetics. Neuron 80, 624–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maze I, Noh KM, Soshnev AA & Allis CD Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet 15, 259–271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piletic K & Kunej T MicroRNA epigenetic signatures in human disease. Arch. Toxicol 90, 2405–2419 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Saeidimehr S, Ebrahimi A, Saki N, Goodarzi P & Rahim F MicroRNA-based linkage between aging and cancer: from epigenetics view point. Cell J. 18, 117–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leone S & Santoro R Challenges in the analysis of long noncoding RNA functionality. FEBS Lett. 590, 2342–2353 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Roberts TC, Morris KV & Wood MJ The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Phil. Trans. R. Soc. B 369, 20130507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fagiolini M, Jensen CL & Champagne FA Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol 19, 207–212 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang JY, Aromolaran KA & Zukin RS Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology 38, 167–182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day JJ, Kennedy AJ & Sweatt JD DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu. Rev. Pharmacol. Toxicol 55, 591–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landgrave-Gomez J, Mercado-Gomez O & Guevara- Guzman R Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci 9, 58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller CA & Sweatt JD Covalent modification of DNA regulates memory formation. Neuron 53, 857–869 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Day JJ & Sweatt JD DNA methylation and memory formation. Nat. Neurosci 13, 1319–1323 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graff J, Kim D, Dobbin MM & Tsai LH Epigenetic regulation of gene expression in physiological and pathological brain processes. >Physiol. Rev 91,603–649 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Feng J et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430 (2010).This paper is the first to show that DNMT1 and DNMT3A are required for synaptic plasticity, and learning and memory, and that they act via DNA methylation and regulate the expression of neuronal genes.
- 16.Tahiliani M et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaas GA et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79, 1086–1093 (2013).This paper is the first to show that TET1 demethylates DNA and that its expression, independently of its catalytic activity, regulates memory formation.
- 18.Levenson JM et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem 281, 15763–15773 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Swank MW & Sweatt JD Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J. Neurosci 21,3383–3391 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levenson JM et al. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem 279, 40545–40559 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Alarcon JM et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42, 947–959 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Korzus E, Rosenfeld MG & Mayford M CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourtchouladze R et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl Acad. Sci. USA 100, 10518–10522 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrij F et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP Nature 376, 348–351 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Blough RI et al. Variation in microdeletions of the cyclic AMP-responsive element-binding protein gene at chromosome band 16p13.3 in the Rubinstein- Taybi syndrome. Am. J. Med. Genet 90, 29–34 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Fischer A, Sananbenesi F, Wang X, Dobbin M & Tsai LH Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182 (2007).This paper shows that environmental enrichment reinstates memory and learning after substantial neuronal loss and atrophy in a mouse model of neurodegeneration.
- 27.Calderone A et al. Ischaemic insults de-repress the gene silencer rest in neurons destined to die. J. Neurosci 23, 2112–2121 (2003).This is the first paper to show that REST expression is altered in a disease state and that REST-mediated gene repression is pivotal to a cellular response.
- 28.Noh KM et al. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc. Natl Acad. Sci. USA 109, E962–E971 (2012).This is the first paper to show that REST assembles with co-repressors at the promoters of transcriptionally responsive target genes and orchestrates epigenetic remodelling.
- 29.Ballas N & Mandel G The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol 15, 500–506 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Ooi L & Wood IC Chromatin crosstalk in development and disease: lessons from REST. Nat. Rev. Genet 8, 544–554 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Baldelli P & Meldolesi J The transcription repressor REST in adult neurons: physiology, pathology, and diseases. eNeuro 10.1523/ENEURO.0010-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruce AW et al. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron- restrictive silencing factor (REST/NRSF) target genes. Proc. Natl Acad. Sci. USA 101, 10458–10463 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conaco C, Otto S, Han JJ & Mandel G Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl Acad. Sci. USA 103, 2422–2427 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballas N, Grunseich C, Lu DD, Speh JC & Mandel G REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657 (2005).This is the first paper to show that REST assembles with co-repressors at the promoters of target genes and orchestrates epigenetic remodelling in neural progenitors.
- 35.Rodenas-Ruano A, Chavez AE, Cossio MJ, Castillo PE & Zukin RS REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci 15, 1382–1390 (2012).This is the first paper to show that REST assembles with co-repressors at the promoter of the gene encoding a synaptic protein (ionotropic glutamate receptor NMDA 2B (GluN2B)) and fine-tunes the expression of genes involved in synaptic plasticity.
- 36.Palm K, Metsis M & Timmusk T Neuron-specific splicing of zinc finger transcription factor REST/NRSF/ XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res. Mol. Brain Res 72, 30–39 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, Doherty JJ & Dingledine R Altered histone acetylation at glutamate receptor 2 and brain- derived neurotrophic factor genes is an early event triggered by status epilepticus. J. Neurosci 22, 8422–8428 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Formisano L et al. Ischemic insults promote epigenetic reprogramming of μ opioid receptor expression in hippocampal neurons. Proc. Natl Acad. Sci. USA 104, 4170–4175 (2007).This is the first paper to show the dysregulation of REST and REST-dependent epigenetic remodelling in a clinically relevant model of ischaemic stroke.
- 39.Formisano L et al. NCX1 is a new rest target gene: role in cerebral ischemia. Neurobiol. Dis 50, 76–85 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Formisano L et al. Sp3/REST/HDAC1/HDAC2 complex represses and Sp1/HIF-1/p300 complex activates ncx1 gene transcription, in brain ischemia and in ischemic brain preconditioning, by epigenetic mechanism. J. Neurosci 35, 7332–7348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuccato C et al. Huntingtin interacts with REST/ NRSF to modulate the transcription of NRSE- controlled neuronal genes. Nat. Genet 35, 76–83 (2003).This is the first paper to show that REST expression is altered in the striatal tissue of humans and mice with Huntington disease.
- 42.Zuccato C et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron- restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci 27, 6972–6983 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiffer D et al. Repressor element-1 silencing transcription factor (REST) is present in human control and Huntington’s disease neurones. Neuropathol. Appl. Neurobiol 40, 899–910 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Lu T et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014).This is the first paper to show that REST is upregulated in normal ageing and that its decline is associated with the oxidative stress observed in Alzheimer disease and with the onset of Alzheimer disease.
- 45.Perera A et al. TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell Rep. 11, 283–294 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Guardavaccaro D et al. Control of chromosome stability by the ß-TrCP-REST-Mad2 axis. Nature 452, 365–369 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westbrook TF et al. SCFβ-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 452, 370–374 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh A et al. Retinoic acid induces REST degradation and neuronal differentiation by modulating the expression of SCF(β-TRCP) in neuroblastoma cells. Cancer 117, 5189–5202 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Kaneko N, Hwang JY, Gertner M, Pontarelli F & Zukin RS Casein kinase 1 suppresses activation of REST in insulted hippocampal neurons and halts ischemia-induced neuronal death. J. Neurosci 34, 6030–6039 (2014).This is the first paper to show that CK1 is the upstream signal that negatively regulates REST in neurons.
- 50.Huang Z et al. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat. Cell Biol 13, 142–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zukin RS Eradicating the mediators of neuronal death with a fine-tooth comb. Sci. Signal 3, e20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stapels M et al. Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci. Signal 3, ra15 (2010).This is the first paper to show that Polycomb proteins are activated in postmitotic vertebrate neurons and are crucial for ischaemic tolerance.
- 53.Blackledge NP, Rose NR & Klose RJ Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat. Rev. Mol. Cell Biol 16, 643–649 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vire E et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439, 871–874 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Schlesinger Y et al. Polycomb-mediated methylation on lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet 39, 232–236 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Schratt G MicroRNAs at the synapse. Nat. Rev. Neurosci 10, 842–849 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Aksoy-Aksel A, Zampa F & Schratt G MicroRNAs and synaptic plasticity — a mutual relationship. Phil. Trans. R. Soc. B 369, 20130515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woldemichael BT & Mansuy IM Micro-RNAs in cognition and cognitive disorders: potential for novel biomarkers and therapeutics. Biochem. Pharmacol 104, 1–7 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Packer AN, Xing Y, Harper SQ, Jones L & Davidson BL The bifunctional microRNA miR-9/ miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci 28, 14341–14346 (2008).This paper documents a reciprocal relationship between the regulatory factors REST and CoREST and the miRNAs miR-9 and miR-9* in the brains of mice with Huntington disease.
- 60.Lusardi TA et al. Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J. Cereb. Blood Flow Metab 30, 744–756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang JY, Kaneko N, Noh KM, Pontarelli F & Zukin RS The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J. Mol. Biol 426, 3454–3466 (2014).This paper is the first to show that REST-dependent silencing of a miRNA is crucial for global ischaemia-induced neuronal death.
- 62.Tsai MC et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willert J, Epping M, Pollack JR, Brown PO. & Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC. Dev. Biol 2, 8 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu M et al. NRSF/REST neuronal deficient mice are more vulnerable to the neurotoxin MPTP. Neurobiol. Aging 34, 916–927 (2013). [DOI] [PubMed] [Google Scholar]
- 65.De Jager PL et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANKJ1, BIN1, RHBDF2 and other loci. Nat. Neurosci 17, 1156–1163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lunnon K et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 17, 1164–1170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chi S et al. Association of single-nucleotide polymorphism in ANK1 with late-onset Alzheimer’s disease in Han Chinese. Mol. Neurobiol 53, 6476–6481 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Birney E, Smith GD & Greally JM Epigenome- wide association studies and the interpretation of disease -omics. PLoS. Genet 12, e1006105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mattson MP Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Savas JN et al. Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc. Natl Acad. Sci. USA 105, 10820–10825 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buckley NJ, Johnson R, Zuccato C, Bithell A & Cattaneo E The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis 39, 28–39 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Conforti P et al. In vivo delivery of DN:REST improves transcriptional changes of REST-regulated genes in HD mice. Gene Ther. 20, 678–685 (2013). [DOI] [PubMed] [Google Scholar]
- 73.von SM et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci 19, 1321–1330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang F et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet 22, 3641–3653 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Mozaffarian D et al. Heart Disease and Stroke Statistics—2016 Update: a report from the American Heart Association. Circulation 133, e38–e60 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Endres M, Fan G, Meisel A, Dirnagl U & Jaenisch R Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport 12, 3763–3766 (2001). [DOI] [PubMed] [Google Scholar]
- 77.Endres M et al. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 20, 3175–3181 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Annunziato L, Pignataro G & Di Renzo GF Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol. Rev 56, 633–654 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Boscia F et al. Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J. Cereb. Blood Flow Metab 26, 502–517 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Lanzillotta A et al. Targeted acetylation of NF-kB/ RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis 49, 177–189 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Zhang ZG & Chopp M Promoting brain remodeling to aid in stroke recovery. Trends Mol. Med 21, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu XS, Chopp M, Zhang RL & Zhang ZG MicroRNAs in cerebral ischemia-induced neurogenesis. J. Neuropathol. Exp. Neurol 72, 718–722 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu XS et al. MicroRNA profiling in subventricular zone after stroke: miR- 124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS ONE 6, e23461 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liou AK, Clark RS, Henshall DC, Yin XM & Chen J To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog. Neurobiol 69, 103–142 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Moskowitz MA, Lo EH & Iadecola C The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ofengeim D, Miyawaki T & Zukin RS in Stroke: Pathophysiology, Diagnosis and Management 5th edn Ch. 6 (eds Mohr JP et al. ) 75–106 (Saunders, 2011). [Google Scholar]
- 87.Siegel G, Saba R & Schratt G MicroRNAs in neurons: manifold regulatory roles at the synapse. Curr. Opin. Genet. Dev 21,491–497 (2011). [DOI] [PubMed] [Google Scholar]
- 88.Wong HK et al. De-repression of FOXO3a death axis by microRNA-132 and - 212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet 22, 3077–3092 (2013). [DOI] [PubMed] [Google Scholar]
- 89.Lagos D et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol 12, 513–519 (2010). [DOI] [PubMed] [Google Scholar]
- 90.DeWoskin VA & Million RP The epigenetics pipeline. Nat. Rev. DrugDiscov 12, 661–662 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Chuang DM, Leng Y, Marinova Z, Kim HJ & Chiu CT Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Falkenberg KJ et al. A genome scale RNAi screen identifies GLIJ as a novel gene regulating vorinostat sensitivity. Cell Death Differ 23, 1209–1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kazantsev AG & Thompson LM Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov 7, 854–868 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Green KN et al. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci 28, 11500–11510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hathorn T, Snyder-Keller A & Messer A Nicotinamide improves motor deficits and upregulates PGC-Jα and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol. Dis 41, 43–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.d’Ydewalle C, Bogaert E & Van Den Bosch L HDAC6 at the intersection of neuroprotection and neurodegeneration. Traffic 13, 771–779 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 98.Gardner KE, Allis CD & Strahl BD Operating on chromatin, a colorful language where context matters. J. Mol. Biol 409, 36–46 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bird A DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Kriaucionis S & Heintz N The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tan M et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jakovcevski M & Akbarian S Epigenetic mechanisms in neurological disease. Nat. Med 18, 1194–1204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM & Sweatt JD Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 515, 582–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maze I et al. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsankova N, Renthal W, Kumar A & Nestler EJ Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci 8, 355–367 (2007). [DOI] [PubMed] [Google Scholar]
- 106.Grimes JA et al. The co-repressor mSin3A is a functional component of the REST-CoREST repressor complex. J. Biol. Chem 275, 9461–9467 (2000). [DOI] [PubMed] [Google Scholar]
- 107.Naruse Y, Aoki T, Kojima T & Mori N Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc. Natl Acad. Sci. USA 96, 13691–13696 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang Y, Myers SJ & Dingledine R Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci 2, 867–872 (1999). [DOI] [PubMed] [Google Scholar]
- 109.Zhang Q, Piston DW & Goodman RH Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Lee MG, Wynder C, Cooch N & Shiekhattar R An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435 (2005). [DOI] [PubMed] [Google Scholar]
- 111.Shi YJ Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864 (2005). [DOI] [PubMed] [Google Scholar]
- 112.Lunyak VV et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 298, 1747–1752 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Battaglioli E et al. REST repression of neuronal genes requires components of the hSWI.SNF complex. J. Biol. Chem 277, 41038–41045 (2002). [DOI] [PubMed] [Google Scholar]
- 114.Ooi L, Belyaev ND, Miyake K, Wood IC & Buckley NJ BRG1 chromatin remodeling activity is required for efficient chromatin binding by repressor element 1 -silencing transcription factor (ResT) and facilitates REST-mediated repression. J. Biol. Chem 281, 38974–38980 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheong JK & Virshup DM Casein kinase 1: complexity in the family. Int. J. Biochem. Cell Biol 43, 465–469 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Chen H et al. DNA damage regulates UHRF1 stability via the SCF(β-TrCP) E3 ligase. Mol. Cell. Biol 33, 1139–1148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nesti E, Corson GM, McCleskey M, Oyer JA & Mandel G C-Terminal domain small phosphatase 1 and MAP kinase reciprocally control REST stability and neuronal differentiation. Proc. Natl Acad. Sci. USA 111, E3929–E3936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pandey UB et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007). [DOI] [PubMed] [Google Scholar]
- 119.McClelland S et al. The transcription factor NRSF contributes to epileptogenesis by selective repression of a subset of target genes. eLife 3, e01267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Abrajano JJ et al. Corepressor for element-1-silencing transcription factor preferentially mediates gene networks underlying neural stem cell fate decisions. Proc. Natl Acad. Sci. USA 107, 16685–16690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mortazavi A, Thompson EC, Garcia ST, Myers RM & Wold B Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res. 16, 1208–1221 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu C et al. A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J. Neurosci. Methods 107, 101–106 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Tanaka H et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J. Neurosci 24, 2750–2759 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Miyawaki T et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc. Natl Acad. Sci. USA 105, 4892–4897 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takada Y et al. Mammalian Polycomb Scmh1 mediates exclusion of Polycomb complexes from the XY body in the pachytene spermatocytes. Development 134, 579–590 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Lee K et al. Expression of Bmi-1 in epidermis enhances cell survival by altering cell cycle regulatory protein expression and inhibiting apoptosis. J. Invest. Dermatol 128, 9–17 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chatoo W et al. The Polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J. Neurosci 29, 529–542 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lipinski MM et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 14164–14169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zuccato C & Cattaneo E Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol 5, 311–322 (2009). [DOI] [PubMed] [Google Scholar]
- 131.Shimojo M Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150Glued. J. Biol. Chem 283, 34880–34886 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dock H, Theodorsson A & Theodorsson E DNA methylation inhibitor zebularine confers stroke protection in ischemic rats. Transl Stroke Res. 6, 296–300 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Kilgore M et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35, 870–880 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qing H et al. Valproic acid inhibits Aß production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med 205, 2781–2789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chiu CT, Liu G, Leeds P & Chuang DM Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 36, 2406–2421 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zadori D, Geisz A, Vamos E, Vecsei L & Klivenyi P Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol. Biochem. Behav 94, 148–153 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Wang Z, Leng Y, Tsai LK, Leeds P & Chuang DM Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab 31, 52–57 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu XS et al. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 220, 313–321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xuan A et al. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 90, 463–468 (2012). [DOI] [PubMed] [Google Scholar]
- 140.Sadri-Vakili G et al. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet 16, 1293–1306 (2007). [DOI] [PubMed] [Google Scholar]
- 141.Ricobaraza A et al. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 34, 1721–1732 (2009). [DOI] [PubMed] [Google Scholar]
- 142.Ricobaraza A, Cuadrado-Tejedor M, Marco S, Perez-Otano I & Garcia-Osta A Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Cuadrado-Tejedor M, Ricobaraza AL, Torrijo R, Franco R & Garcia-Osta A Phenylbutyrate is a multifaceted drug that exerts neuroprotective effects and reverses the Alzheimer’s disease-like phenotype of a commonly used mouse model. Curr. Pharm. Des 19, 5076–5084 (2013). [DOI] [PubMed] [Google Scholar]
- 144.Giorgini F et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J. Biol. Chem 283, 7390–7400 (2008). [DOI] [PubMed] [Google Scholar]
- 145.Hockly E et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl Acad. Sci. USA 100, 2041–2046 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Faraco G et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol 70, 1876–1884 (2006). [DOI] [PubMed] [Google Scholar]
- 147.Baltan S, Bachleda A, Morrison RS & Murphy SP Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl Stroke Res. 2, 411–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang ZY & Schluesener HJ Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol 72, 178–185 (2013). [DOI] [PubMed] [Google Scholar]
- 149.Dompierre JP et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci 27, 3571–3583 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yildirim F et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp. Neurol 210, 531–542 (2008). [DOI] [PubMed] [Google Scholar]
- 151.Outeiro TF et al. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519 (2007). [DOI] [PubMed] [Google Scholar]
- 152.Luthi-Carter R et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl Acad. Sci. USA 107, 7927–7932 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F & Fischer A Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J. Alzheimers Dis 26, 187–197 (2011). [DOI] [PubMed] [Google Scholar]
- 154.Ferrante RJ et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci 23, 9418–9427 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim HJ, Leeds P & Chuang DM The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem 110, 1226–1240 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cruz JC & Tsai LHA Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr. Opin. Neurobiol 14, 390–394 (2004). [DOI] [PubMed] [Google Scholar]