

Abstract
Hundreds of patients with autosomal recessive, complete IL-12p40 or IL-12Rβ1 deficiency have been diagnosed over the last 20 years. They typically suffer from invasive mycobacteriosis and, occasionally, from mucocutaneous candidiasis. Susceptibility to these infections is thought to be due to impairments of IL-12-dependent IFN-γ immunity, and IL-23-dependent IL-17A/IL-17F immunity, respectively. We report here patients with autosomal recessive, complete IL-12Rβ2 or IL-23R deficiency, lacking responses to IL-12 or IL-23 only, all of whom, surprisingly, display mycobacteriosis without candidiasis. We show that αβ T, γδ T, B, NK, ILC1, and ILC2 cells from healthy donors preferentially produce IFN-γ in response to IL-12, whereas NKT, MAIT, and ILC3 cells preferentially produce IFN-γ in response to IL-23. We also show that the development of IFN-γ- producing CD4+ T cells, including, in particular, mycobacterium-specific Th1* cells (CD45RA-CCR6+), is dependent on both IL-12 and IL-23. Finally, we show that IL12RB1, IL12RB2, and IL23R have similar frequencies of deleterious variants in the general population. The comparative rarity of symptomatic patients with IL-12Rβ2 or IL-23R deficiency, relative to IL-12Rβ1 deficiency, is, therefore, due to lower clinical penetrance. These experiments of Nature show that human IL-12 and IL-23 are both crucial for IFN-γ- dependent immunity to mycobacteria, both individually and much more so cooperatively.
Introduction
Life-threatening infectious diseases during the course of infection in otherwise healthy individuals can result from single-gene inborn errors of immunity (1,2). Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by severe disease upon exposure to weakly virulent mycobacteria, such as Mycobacterium bovis-Bacille Calmette- Guerin (BCG) vaccines and environmental mycobacteria (3, 4). Affected patients are also at risk of severe infections with M. tuberculosis, Salmonella, and, more rarely, other intramacrophagic bacteria, fungi, and parasites (4). All known genetic etiologies of MSMD affect IFN-γ-mediated immunity (4, 5).
The most common etiology is autosomal recessive (AR) IL-12Rβ1 deficiency, which has been reported in over 200 kindreds since 1998 and has probably been diagnosed in many more (4, 6–9). IL-12Rβ1 deficiency displays incomplete clinical penetrance for MSMD, and can also underlie monogenic tuberculosis in families with no history of infection with environmental mycobacteria or BCG (4, 7, 10). IL-12Rβ1 dimerizes with IL-12Rβ2 to form the IL-12 receptor (11), or with IL-23R to form the IL-23 receptor (12). IL12B encodes the p40 subunit common to both IL-12 (with p35) and IL-23 (with p19) (13). Consistently, AR IL12p40 deficiency, which has been reported in over 50 kindreds, is a clinical phenocopy of IL-12Rβ1 deficiency (4, 14).
Both IL-12 immunity and IL-23 immunity are abolished in these two disorders. Unlike other etiologies of MSMD, these two disorders can also underlie chronic mucocutaneous candidiasis (CMC), which is seen in about 25% of patients, due to impaired IL-17A/IL-17F-mediated immunity (6, 15–17). Indeed, all known genetic etiologies of isolated or syndromic CMC affect IL-17A/IL-17F (16, 18–22). Studies of mouse models have suggested that disruption of the IL-12-IFN-γ circuit, particularly in CD4+ helper T cells (Th1), underlies mycobacteriosis in these patients, whereas disruption of the IL-23- IL-17 circuit, particularly in Th17 cells, underlies candidiasis (13). However, the respective contributions of human IL-12 and IL-23 to IFN-γ-dependent MSMD and IL-17A/F- dependent CMC have remained elusive in the absence of inborn errors of IL-12p35, IL- 12RP2, IL-23p19, or IL-23R.
Results
Homozygous IL12RB2 and IL23R variants in two MSMD kindreds
We searched for rare, biallelic, non-synonymous and splice variants of IL12A, IL12RB2, IL23A, and IL23R, by whole-exome sequencing (WES) in patients with unexplained MSMD only (n=555), CMC only (n=214), or both MSMD and CMC (n=27). We identified two MSMD kindreds with mutations of IL12RB2 or IL23R. Kindred A (Fig 1A) is a consanguineous Turkish family, the proband of which (P1; 7.6% homozygosity; Fig 1A) had MSMD (Case Reports, SOM, Fig. S1A). Kindred B (Fig. 1B) is a consanguineous Iranian family with two MSMD-affected children (P4 and P5; 3.5% of homozygosity for P5, Fig. 1B, Fig. S1B-D). These two kindreds were analyzed independently, by a combination of WES and genome-wide linkage (GWL) analysis (Fig. S1E, F). In kindred A, the c.412C>T mutation of IL12RB2 results in a premature stop codon (Q138X) upstream from the segment encoding the transmembrane domain of IL-12Rβ2 (Fig. 1C). In kindred B, the c.344G>A mutation of IL23R results in the C115Y substitution, at a key residue in the extracellular domain of IL-23R (23) (Fig. 1D). The IL12RB2 and IL23R loci are located in tandem on chromosome 1, and within the largest MSMD-linked chromosomal regions in kindreds A and B, respectively (Fig. S1E-G). The three MSMD patients are homozygous for the mutant alleles (Fig. 1A-B, Fig. S1H-I). Incomplete clinical penetrance for MSMD was observed in kindred A, as IL12RB2-homozygous and BCG- vaccinated individual II.3 (P2; 9.1% of homozygosity) had tuberculosis but not MSMD, and II.4 (P3; 10.8% of homozygosity) had neither (Case Reports, SOM). None of the five homozygous mutant individuals from kindreds A (P1-P3) and B (P4-P5) had ever displayed signs of CMC. Principal component analysis (PCA) based on WES data confirmed the ancestries of these patients (Fig S1J) (24). The IL12RB2 Q138X mutation was reported in the heterozygous state in two of 123,098 individuals in the gnomAD database (http://gnomad.broadinstitute.org), whereas the IL23R C115Y mutation was not reported in any of the 122,998 individuals sequenced at this site. These mutations were not found in a cohort of 3,869 individuals of Middle Eastern descent with neurological diseases (data not shown). Finally, both variants have CADD scores well above their respective MSCs (25, 26). These data suggested that MSMD patients from kindreds A and B had AR deficiencies of IL-12Rβ2 and IL-23R, respectively, the former disorder displaying incomplete penetrance for MSMD.
Figure 1: Identification of homozygous complete loss-of-function IL-12Rβ2 Q138X and IL-23R C115Y mutations in families with MSMD.
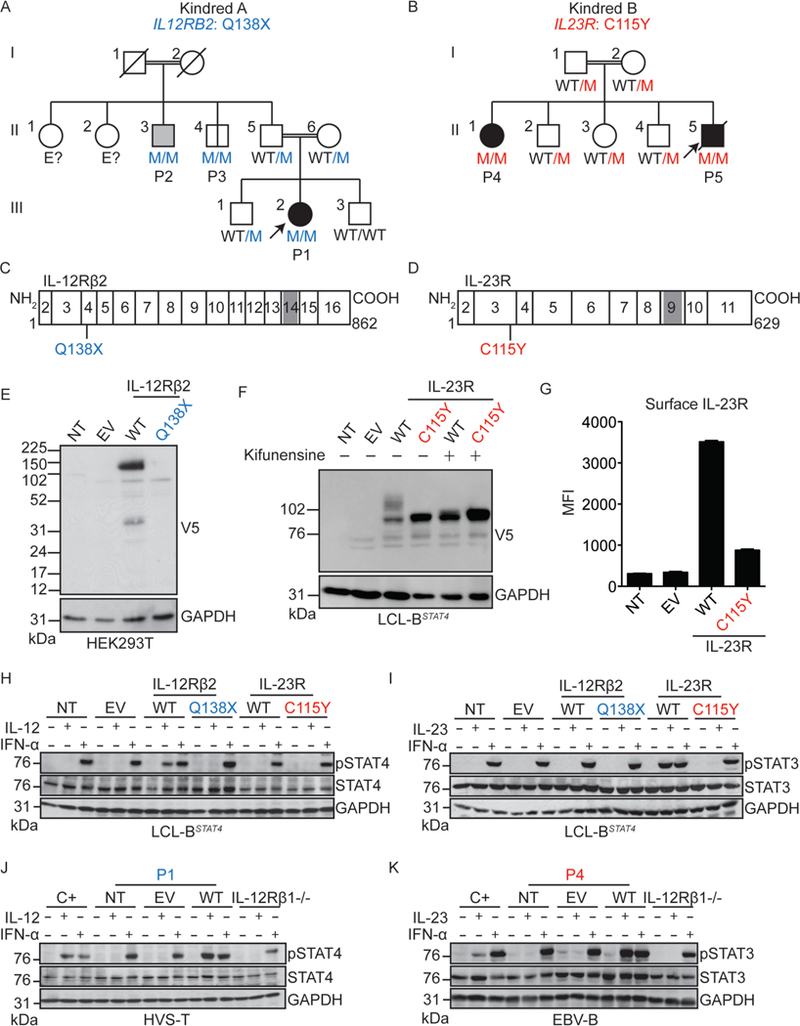
A,B) Pedigrees of the two kindreds studied in this report. The gene and mutation are indicated under the kindred name. Solid black symbols indicate patients with MSMD and solid gray symbols indicate cases of primary tuberculosis during childhood. Symbols linked with a double line indicate consanguinity. The genotype is indicated under each symbol, with M corresponding to the mutation found in each kindred. Arrows indicate the index case in each family. C, D) Schematic representation of IL-12Rβ2 (C) and IL-23R (D). Rectangles represent individual exons of the gene, with the exon numbers indicated within the rectangle. In each case, the N terminal portion of the protein is the extracellular domain, and gray shaded areas represent the transmembrane domain. The mutations studied here are indicated below each protein, in the corresponding exons. E) HEK293T cells were either non-transfected (NT) or transfected with an empty vector (EV), a vector containing the C-terminal V5-tagged WT or Q138X mutant versions IL12RB2. Western blotting was performed with antibodies against the V5 tag, or GAPDH as a loading control. F) LCL-BSTAT4 cells were either non-transduced (NT), or transduced with retroviruses generated with an empty vector (EV) or with vectors containing the WT or C115Y mutant versions of IL23R. Cells were either left untreated, or treated with kifunensine to inhibit N- glycosylation. Western blotting was performed with antibodies against the V5 tag, or GAPDH as a loading control. G) LCL-BSTAT4 cells from (F) were stained with an anti-IL- 23R biotinylated primary antibody, then with avidin-PE, and the mean fluorescence intensity (MFI) for the PE signal was quantified by flow cytometry. H) LCL-BSTAT4 cells were either left non-transduced (NT), or were transduced with retroviruses generated with an empty vector (EV) or vectors containing V5-tagged WT or Q138X mutant versions of IL12RB2, or WT or C115Y versions of IL23R. The cells were left unstimulated, or were stimulated with IL-12 or IFN-α. Cell lysates were prepared, and western blotting was performed with antibodies against pSTAT4, total STAT4, and GAPDH. I) LCL-BSTAT4 cells from (H) were left unstimulated, or were stimulated with IL-23 or IFN-α as a positive control. Cell lysates were prepared, and western blotting was performed with antibodies against pSTAT3, total STAT3, and GAPDH. J) HVS-T cells from a healthy control (C+), an IL-12Rβ−/− patient or P1 (IL-12Rβ2 Q138X) were not transduced (NT), or were transduced with a retrovirus generated with an empty vector (EV) or a WT allele of IL12RB2 and then stimulated with IL-12 or IFN-α. Western blotting was performed as described in (H). K) EBV-B cells from a healthy control (C+), an IL-12Rβ1−/− patient or P4 (IL-23R C115Y) were not transduced (NT), or were transduced with a retrovirus generated with an empty vector (EV) or a WT allele of IL23R and then stimulated with IL-23 or IFN- α. Western blotting was performed as described in (I).
The IL12RB2 and IL23R mutant alleles are loss-of-function
We assessed the expression and function of these mutant alleles, by using retrovirus-mediated gene transfer to express C-terminal V5-tagged wild-type (WT) or mutant IL-12Rβ2 or IL-23R proteins in a B-lymphocyte cell line (LCL) that expressed STAT3 and IL-12Rβ1 and was engineered to express STAT4IRS-GFP in a stable manner (referred to here as LCL-BSTAT4 cells) (27). The IL12RB2 Q138X mutation resulted in abundant IL-12Rβ2 mRNA production (Fig. S1K), but the corresponding protein was undetectable (Fig. 1E, Fig. S1L). The IL23R C115Y mutation did not alter the amount of mRNA (Fig. S1K) or protein detected (Fig. 1F, Fig. S1L). However, the mutant IL-23R protein did not undergo normal N-glycosylation (Fig. 1F) and was much less abundant on the cell surface than the exogenous WT protein (Fig. 1G, Fig. S1M). Next, we stimulated the LCL-BSTAT4 cells expressing either the WT or mutant IL-12Rβ2 and IL-23R proteins with IL-12, IL-23, or IFN-α as a positive control. Human IL-12-dependent signaling via IL-12Rβ1/IL-12Rβ2 results in STAT4 activation, whereas IL-23-dependent signaling via IL-12Rβ1/IL-23R preferentially results in STAT3 activation, although STAT4 may also be activated (13). LCL-BSTAT4 cells expressing WT, but not mutant IL-12Rβ2, displayed STAT4 phosphorylation in response to IL-12 (Fig. 1H). Similarly, the expression of WT IL-23R, but not mutant IL-23R, conferred responsiveness to IL-23, as assessed by the level of phosphorylation of STAT3 (Fig. 1I) and STAT4 (Fig. S2A) and their upstream Janus kinases TYK2 and JAK2 (Fig. S2B). Cells expressing WT, but not mutant IL-12Rβ2 or IL-23R, also produced CXCL10 in response to IL-12 or IL-23 stimulation, respectively (Fig. S2C). Pretreatment of the cells with kifunensine, an inhibitor of glycosylation that can rescue mild misfolding and gain-of-glycosylation mutants (28), did not restore the function of IL-23R C115Y, suggesting that the mutation is severely deleterious (Fig. S2D). Thus, the Q138X IL12RB2 and C115Y IL23R mutant alleles are completely loss-of- function (LOF) for the IL-12 and IL-23 signaling pathways, respectively, at least in these experimental conditions. These experiments demonstrated the role of each mutant allele cDNA over-expressed in isolation. Our next set of experiments used patient-derived cells, which capture the effects of the patients’ full genotypes at these loci in the context of their own genome.
Autosomal recessive IL-12Rβ2 and IL-23R complete deficiencies
We assessed the expression and function of IL-12Rβ2 and IL-23R in patient- derived cell lines. Herpesvirus saimiri-transformed T (HVS-T) cells from a healthy control, P1 (IL-12Rβ2 Q138X), and an IL-12Rβ1-deficient patient (4) were left unstimulated, or stimulated with IL-12 or IFN-α. The healthy control cell line, but not cells from P1 or the IL-12Rβ1-deficient patient, displayed STAT4 phosphorylation in response to IL-12 (Fig. 1J). This defect was confirmed in HVS-T cells from P2 and P3 (Fig. S2E). Furthermore, the retroviral transduction of P1 cells with WT IL12RB2 restored STAT4 phosphorylation in response to IL-12, whereas retrovirus-mediated transduction with an empty vector did not (Fig. 1J). In parallel, EBV-B cells from a healthy control, P4 (IL-23R C115Y), and an IL-12Rβ1-deficient patient were left unstimulated, or were stimulated with IL-23 or IFN-α. Control EBV-B cells responded to IL-23 by phosphorylating STAT3, whereas cells from P4 and the IL-12Rβ1-deficient patient did not (Fig. 1K). The transduction of EBV-B cells from P4 with WT IL23R rescued both the weak IL-23R expression on the surface of P4 EBV-B cells (Fig. S2F-G), and the defective IL-23 response (Fig. 1K, Fig S2H), whereas retrovirus-mediated transduction with an empty vector did not. IL-12Rβ1- and TYK2- deficient cells were used as negative controls for the IL-23- and IFN-α-dependent phosphorylation of STAT3, respectively (Fig. S2H). The IL-23 response was not restored by the prior treatment of EBV-B cells from P4 with kifunensine (Fig. S2I-J). Thus, the patients from kindreds A and B had complete forms of AR IL-12Rβ2 deficiency and AR IL-23R deficiency, respectively.
IL12RB1, IL12RB2, and IL23R have not evolved under purifying selection
The much smaller number of biallelic mutations responsible for MSMD found in IL12RB2 and IL23R than in IL12RB1 suggested that IL12RB2 and IL23R might have evolved under stronger selective constraints, preventing the accumulation of monoallelic deleterious variants. We tested this hypothesis by estimating the levels of purifying selection acting on all human genes with a generalized linear mixed model approach, comparing protein-coding polymorphisms and divergence data (29). Exome data from the 1,000 Genomes project (30) were aligned with the chimpanzee reference genome, yielding exploitable data for a total of 18,969 protein-coding genes (Methods, SOM). The proportion of non-deleterious amino-acid variants (f) was estimated at 46.7% for IL12RB1 [95% confidence interval: 30.4%−71.7%], 61.7% for IL12RB2 [95%CI: 42.1%−90.3%], and 49.0% for IL23R [95%CI: 31.1%−77.4%] (Table S1). These three genes were among the 50% least constrained genes, and their high levels of amino acid-altering polymorphisms, relative to divergence, were consistent with the action of weak negative selection (as shown by the negative estimates for the population selection coefficient y), as observed for most human protein-coding genes. Moreover, the neutrality index (NI) and gene damage index (GDI, (31)) of IL12RB1, IL12RB2, and IL23R were intermediate (Fig. 2A-B). The results were similar when looking at alternative parameters of selection such as RVIS (32) and f (33) (data not shown). Consistently, numerous missense and predicted LOF variants of IL12RB1, IL12RB2, and IL23R are present, in the heterozygous state, in gnomAD (Fig. S3A-C). There are also fewer homozygous missense variants for these three genes (Fig. 2C-D, Fig. S3D). Overall, this computational study suggested that neither IL12RB2 nor IL23R had evolved under purifying selection, or even under strong negative selection, and that they were no less mutated than IL12RB1 in the general population. These findings support the alternative hypothesis that the paucity of MSMD patients with IL- 12Rβ2 or IL-23R deficiency, relative to the number of patients identified with IL-12Rβ1 deficiency, results from lower clinical penetrance for MSMD.
Figure 2: IL12RB2 and IL23R are evolving under weak purifying selection, and homozygous complete loss-of-function mutations of these genes occur rarely in the general population.
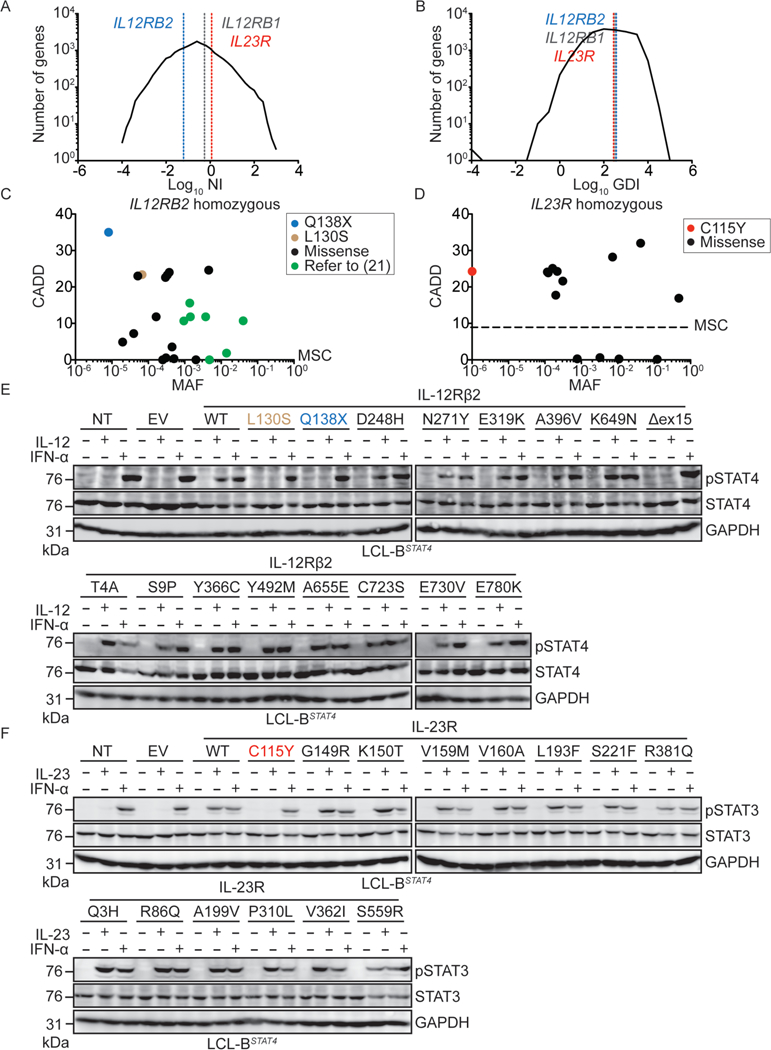
A,B) Graphic representation of all genes of the human genome according to the log10 of their (A) neutrality index (NI) or (B) gene damage index (GDI, (31)). IL12RB2 (blue), IL23R (red), and IL12RB1 (grey) are each indicated by a dashed vertical line. C, D) All homozygous variations found in gnomAD for IL12RB2 (C) and IL23R (D) are plotted according to their CADD score (y-axis) and minor allele frequency (MAF, x-axis). The dashed line indicates the mutation significance cutoff (MSC, (26)) for each gene. E) LCL- BSTAT4 cells were left non-transduced (NT), or were transduced with retroviruses generated with an empty vector (EV) or vectors containing V5-tagged WT or mutant versions of IL12RB2, including the 16 mutations indicated. These IL12RB2 mutations are present in at least one individual in the gnomAD database. Cells were left unstimulated, or were stimulated with IL-12 or IFN-α. Cell lysates were prepared, and western blotting was performed with antibodies against pSTAT4, total STAT4, and GAPDH. F) LCL-BSTAT4 cells were left non-transduced (NT), or were transduced with retroviruses generated with an empty vector (EV) or vectors containing V5-tagged WT or mutant versions of IL23R, for the 14 mutations indicated. With the exception of the C115Y mutation found in kindred B, these IL23R mutations are each present in at least one individual in the gnomAD database. Cells were left unstimulated, or were stimulated with IL-23 or IFN-α. Cell lysates were prepared, and western blotting was performed with antibodies against pSTAT3, total STAT3, and GAPDH.
Null mutations in IL12RB1, IL12RB2 and IL23R are rare
We tested these computational predictions through experimental determinations of the actual frequency of IL12RB2 and IL23R LOF mutations in the general population, by using the same retroviral system to express all previously untested variants of both genes present in the homozygous state in gnomAD (Fig. 2C-D, Fig. S3E-F), in LCL-BSTAT4 cells. All homozygous IL12RB1 variants present in the general population that have been tested (6 of 16) have been shown to be neutral (34). We stimulated the LCL-BSTAT4 cells expressing either the WT or homozygous gnomAD variants of IL-12Rβ2 (15 variants) and IL-23R (13 variants) proteins with IL-12, IL-23, or IFN-α as a positive control. The IL12RB2 mutation L130S encoded a protein that was expressed (Fig. S3G) but not functional (Fig. 2E). The L130S mutation of IL12RB2 was found to be present in the homozygous state in 1 of 123,092 individuals in the gnomAD database. The IL12RB2 K649N mutation affects a splice site, and may, therefore, cause a missense or splicing mutation, resulting in the deletion of exon 15 (Δex15) (Fig S3H-I). The IL12RB2 K649N mutation was found to encode a protein that was both expressed (Fig. S3G) and functional (Fig. 2E), like the WT protein, whereas the IL12RB2 Δex15 mutation encoded a protein with a lower molecular weight (Fig. S3G) that was non-functional (Fig. 2E). The IL12RB2 K649N mutation was identified in 1 of 138,539 individuals in the gnomAD database. None of the IL23R mutations tested resulted in a complete loss of protein expression or function (Fig. S3J, Fig. 2F). These and previous data (34) suggest that homozygous complete LOF mutations of IL12RB2, IL23R and IL12RB1 occur in the general population at a frequency of <1 in 100,000 individuals. This frequency is similar to that of autosomal recessive IL- 12Rβ1 deficiency, the penetrance of which for MSMD reaches a plateau of about 80% in adults (6, 7).
In vivo Th development in IL-12Rβ2 and IL-23R deficiency
As a first approach to testing the hypothesis that IL-12Rβ2 and IL-23R deficiencies have a lower clinical penetrance for MSMD than IL-12Rβ1 deficiency, we set out to decipher the cellular basis of MSMD in patients with these disorders. In humans and mice, IL-12 and IL-23 are thought to promote mutually exclusive CD4+ T-helper cell fates (13). Before testing for IL-12- and IL-23-dependent effects on the differentiation of human Th cells, we compared the frequencies of leukocyte subsets in healthy controls, an IL12RB2 Q138X patient, an IL23R C115Y patient, and at least one IL-12Rβ1-deficient patient. The frequencies of innate lymphoid cells (ILCs), B, NK, γδ T, total T (CD3+), CD4+ αβ T, CD8+ αβ T, Treg, and TFH cells were normal in patients of all genotypes (Fig. S4A). Patients with IL-12Rβ2-, IL-23R-, or IL-12Rβ1-deficiency had a low frequency of MAIT cells (Fig. S4A), consistent with previous findings for IL-12Rβ1- and STAT3-deficient patients (35). The frequency of naive cells in the CD4+ and CD8+ T-cell populations was higher, and that of memory cells was lower in IL-12Rβ2-, IL-23R-, and IL-12Rβ1-deficient patients than in healthy controls (Fig. 3A, left panel, Fig. S4B-C). IL-12Rβ2-, IL-23R-, and IL-12Rβ1- deficient patients had lower frequencies of memory Th1 (CXCR3+CCR6-) and Th1* (CXCR3+CCR6+) cells than healthy controls, IL-12Rβ1-deficient patients also had lower frequencies of Th17 (CCR4+CCR6+) cells, and the frequency of Th2 (CCR4+CCR6-) cells was similar in patients of all genotypes (Fig. 3A, right panel). However, within the CD4+ memory compartment, the percentages of Th1* and Th17 cells were low in some IL- 12Rβ1-deficient patients, but not in IL-12Rβ2- or IL-23R-deficient patients, whereas Th1 and Th2 percentages were normal in patients of all genotypes (Fig. S4D). These surprising findings suggested that isolated deficiencies of IL-12 or IL-23 responses had similar and slightly milder consequences, in terms of Th1, Th1*, and perhaps Th17 development, than a combined deficiency.
Figure 3: Frequencies and responses of CD4+ T cells from IL-12Rβ1-, IL-12Rβ2- and IL-23R-deficient individuals to polyclonal stimuli or Th-polarizing cytokines.
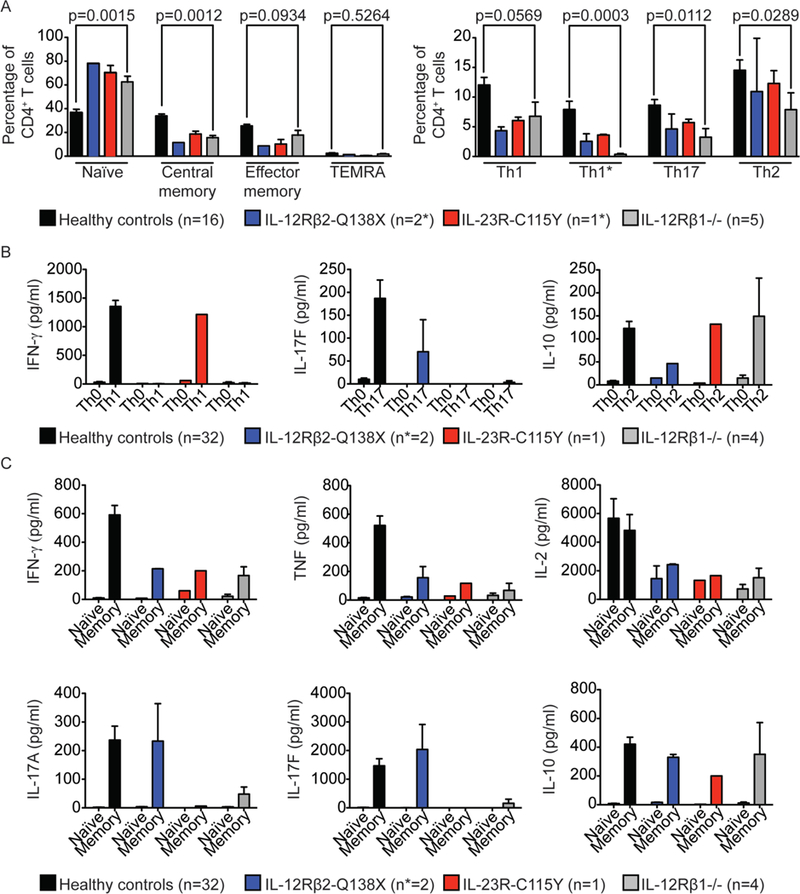
A) On the left, frequencies of naive (CCR7+CD45RA+), central memory (CCR7+CD45RA−), effector memory (CCR7−CD45RA−) and TEMRA (CCR7−CD45RA+) CD4+CD3+ T cells were measured in healthy controls (n=16), IL-12Rβ2-Q138X patients (n=2, one measured twice), one IL-23R-C115Y patient (technical triplicates) and rL-12Rβ1−/− patients (n=5). The values shown are the percentage of total CD4+ T cells. On the right, frequencies of four subsets of CD4+ memory T cells (CD3+CD45RA−): Th1 (CCR6−CCR4−CXCR3+), Th1* (CCR6+CCR4−CXCR3+), Th17 (CCR6+CCR4+CXCR3−) and Th2 (CCR6−CCR4+CXCR3−) were measured in the same individuals as in the left panel. Error bars indicate the SEM. P values for the comparison of healthy controls with IL-12Rβ1-deficient patients in Mann-Whitney U-tests are shown. Formal statistical comparisons including IL- 12Rβ2-Q138X and IL-23R-C115Y patients were not appropriate, due to the extremely small number of individuals (2 and 1, respectively). B) Naïve CD4+ T cells from healthy controls, two IL-12Rβ2-Q138X patients (P1 and P2), one IL-23R-C115Y patient (P4) and four IL- 12Rβ1 -deficient patients were either left unpolarized (Th0) or were polarized under Th1 conditions (TAE+IL-12), Th17 conditions (TAE+IL-1/IL-6/IL-21/IL-23/TGF-β), or Th2 conditions (TAE+IL-4). The production of IFN-γ, IL-17F and IL-10 was measured with cytometric bead arrays, in cell culture supernatants, after 5 days. C) Naïve and memory CD4+ T cells from healthy controls, two IL-12Rβ2-Q138X (P1 and P2), one IL- 23R-C115Y (P4) and four IL-12Rβ1-deficient patients were stimulated with TAE beads, and cytokine production was measured 5 days later. Data for the production of IFN-γ, TNF and IL-2 are shown in the upper panels and for IL-17A, IL-17F and IL-10 in the lower panels.
In vitro Th differentiation in IL-12Rβ2 and IL-23R deficiency
We then investigated the effects of IL-12 and IL-23 signaling defects on the generation of CD4+ T-cell subsets upon stimulation with polarizing cytokines in vitro. Naïve (CD45RA+CCR7+) CD4+ T cells from healthy donors, IL-12Rβ2-, IL-23R-, and IL- 12Rβ1-deficient patients were purified and cultured under Th0 (non-polarizing) or “Th1”-, “Th2”- or “Th17”-polarizing conditions (36), and the production of various cytokines was measured in culture supernatants with cytometric bead arrays or by ELISA. “Th1” differentiation in vitro, which occurs in the presence of IL-12 and is assessed by monitoring IFN-γ production, was abolished in cells from IL-12Rβ2- and IL-12Rβ1-deficient patients, but intact in IL-23R-deficient patient cells (Fig. 3B). By contrast, “Th17” differentiation in vitro in the presence of IL-23, as assessed by monitoring IL-17F production, was maintained in IL-12Rβ2-deficient cells, but abolished in naïve CD4+ T cells from IL-23R-and IL-12Rβ1-deficient patients (Fig. 3B). Following “Th2” differentiation in vitro, which occurs in the presence of IL-4, cells of all genotypes produced similar amounts of IL-10 (Fig. 3B). Collectively, these data suggest that the cytokine-dependent in vitro differentiation of naïve CD4+ T cells into “Thi” and “Th17” cells is affected by IL-12Rβ2 deficiency and IL-23R deficiency, respectively, consistent with previous findings (13). We probed genotype-dependent effects on the generation of cytokine-producing CD4+ T cells in vivo further, by purifying naïve (CD45RA+CCR7+) and memory (CD45RA-) CD4+ T cells from healthy donors, IL-12Rβ2-, IL23R- or IL-12Rβ1-deficient patients and culturing them with polyclonal TCR-activating ligands (Th0 conditions) (36). As expected, IL- 12Rβ1- and IL23R-deficient cell cultures displayed only low levels of IL-17A and IL-17F production (Fig. 3C). Similar amounts of IL-10 were again produced by all cells tested. Surprisingly, the production of IFN-γ, TNF, and IL-2 by memory CD4+ T cells was reduced not only by IL-12Rβ1 or IL-12Rβ2 deficiency, but also by IL-23R deficiency, possibly due to T cell-intrinsic functional defects or to the low frequency of Th1 and/or Th1* cells in the three groups of patients.
Mycobacterium-specific IFN-γ production is compromised in Th cells
We assessed the potential effects of deficiencies of IL-12Rβ1, IL-12Rβ2, or IL-23R on the induction of Mycobacterium-specific CD4+ T-cell responses in vivo, by analyzing memory CD4+ T cells from healthy controls and patients of each genotype (37). Both BCG- vaccinated and -unvaccinated individuals have been shown to robustly produce IFN-γ in response to BCG (38) and mycobacteria-derived antigens (39). Multiple T-cell lines were generated from sorted CD4+CD45RA-CCR6- cells (containing Th1 and Th2 cells and known to be enriched in viral antigen-reactive T cells) and CD4+CD45RA-CCR6+ cells (containing Th1* and Th17 cells known to be enriched in bacterial and fungal antigen- reactive T cells, respectively) separately (40). Following polyclonal stimulation, both CCR6+ and CCR6- T cells produced IFN-γ in similar amounts for healthy controls and all patients (Fig. S5A). By contrast, IL-17A and IL-22 were produced mostly by CCR6+ cells for healthy controls and an IL-12Rβ2-deficient patient, but not for IL-12Rβ1- and IL-23R- deficient patients (Fig. S5A), consistent with the notion that IL-23 is an important inducer of the production of IL-17A and IL-22 by CD4+ T cells (41). In the same experiment, the production of IL-4 was not affected by any of the mutations tested (Fig. S5A). All T-cell lines were then screened in vitro for recall responses to influenza virus, respiratory syncytial virus (RSV), BCG, and Mycobacterium tuberculosis (MTB). Several T-cell lines from both healthy donors and patients proliferated in response to the antigens tested (Fig. S5B). As expected, the frequency of influenza- and RSV-reactive cells was higher in CCR6- T-cell lines, whereas the frequency of BCG- and MTB-reactive cells was higher in CCR6+ T-cell lines. (Fig. S5B-C). No major differences in the frequency of precursor cells specific for the tested antigens (Fig. S5C) were observed, but genotype-dependent defects were observed in the production of cytokines by proliferating T cells. The production of IFN-γ, but not of IL-4, by influenza virus- or RSV-reactive CCR6- T cells was impaired in IL-12Rβ2- and IL-12Rβ1-deficient patients, but not in IL-23R-deficient patients (Fig. S5D). Interestingly, the levels of IFN-γ production by BCG- and MTB-responding CCR6+ T cells were low in IL-12Rβ1-, IL-12Rβ2-, and IL-23R-deficient patients (Fig. 4A, left panels). IL-17A was produced by only a few healthy donor CCR6+ T cells in response to BCG and MTB, precluding comparisons between genotypes (Fig. 4A). In the same cells, IL-22 production was low in IL-12β1- and IL-23R-deficient patients, but not in IL-12Rβ2- deficient patients (Fig 4A), providing further evidence of a functional IL-23R deficiency. However, the role of human IL-22 in immunity to infection remains undetermined. Interestingly, CD4+ T cells from Il23a–/– mice showed severely defective IL-17 and IL-22 production, mildly reduced IFN-γ production, and increased mycobacterial growth, when compared with WT mice following M. tuberculosis infection (42). Yet, Il17ra−/− and Il22- /- mice did not differ from WT mice in their control of M. tuberculosis growth (42), unlike Ifng−/− mice (43) suggesting that IL-23-dependent IFN-γ production is involved in this setting. Other studies have shown that IL-23 augments local IFN-γ production in the lung and limits pulmonary M. tuberculosis replication in mice (44). Overall, these data indicate that, in the absence of either IL-12 or IL-23 signaling, Mycobacterium-specific IFN-γ production is compromised in CCR6+ memory CD4+ Th1* cells, a cellular phenotype common to inherited ROR-γ/ROR-γT (36) and SPPL2A deficiencies (Kong X-F., Martinez-Barricarte R., Nature Immunology, in press).
Figure 4: Specific defects in the IL-12- and IL-23-dependent generation of an IFN-γ immune response in patients with novel homozygous mutations of IL12RB2 or IL23R.
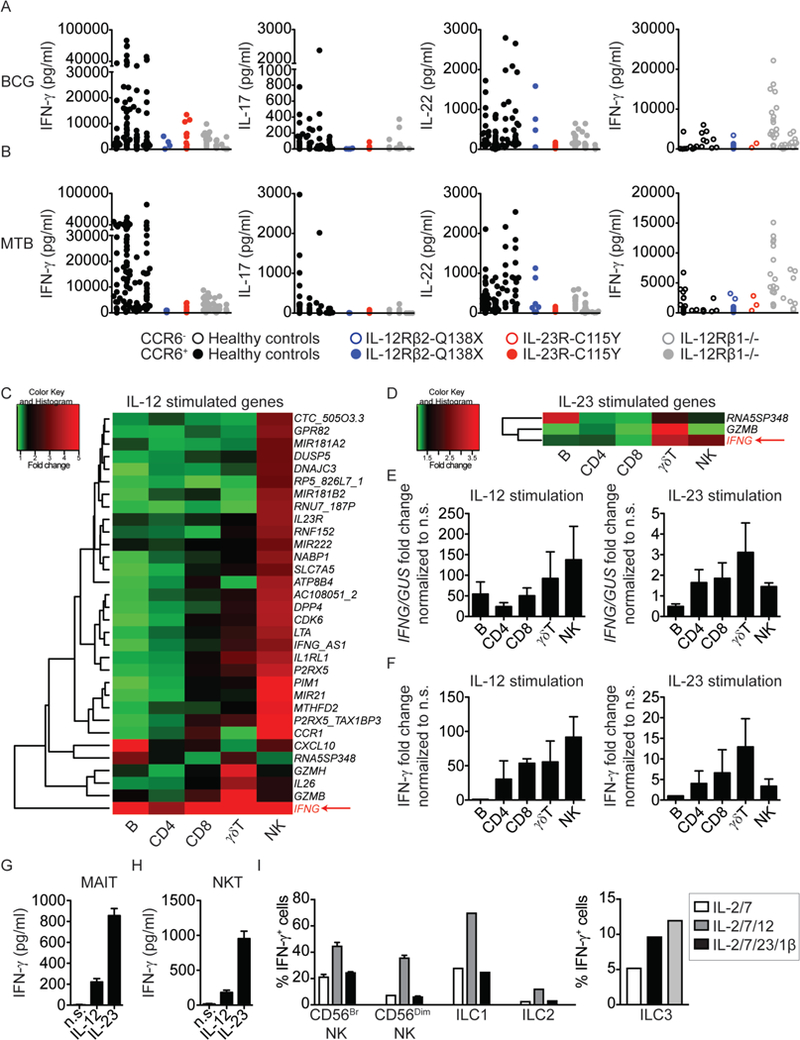
A, B) Multiple CCR6+ or CCR6− memory CD4+ T-cell lines were generated by the polyclonal stimulation of sorted peripheral blood subsets from healthy controls (n=4), one IL-12Rβ2-Q138X patient, one IL-23R-C115Y patient, and three IL-12Rβ1-deficient patients. Lines were screened for reactivity with peptide pools covering antigens from BCG (upper panel) and MTB (lower panel). BCG- and MTB-reactive CD4+CCR6+ T-cell lines from each individual were selected and the cytokines accumulating in the culture supernatant were determined with a Luminex machine. Each dot on the graph corresponds to a value for a single antigen-reactive T-cell line. CCR6+ T-cell lines are shown as closed circles, and CCR6− cell lines are shown as open circles. C, D) Microarray heat map of isolated B, CD4+ T, CD8+ T, γδ+ T and NK cells stimulated with IL-12 (C) or IL-23 (D) for 6 h. The data shown are the fold-induction relative to non-stimulated (n.s.) cells. The most commonly upregulated gene, IFNG, is highlighted in the lower right corner of each heat map. E) IFNG induction by isolated B, CD4+ T, CD8+ T, γδT, and NK cells from 5 healthy controls, upon stimulation with IL-12 or IL-23 for 6 h, was assessed by qPCR, and the data were normalized relative to n.s. cells. F) IFN-γ levels in the supernatants from the cells used in (E) were analyzed by ELISA and represented as a fold-change, relative to n.s. cells. G, H) Sorted MAIT cells (G) or NKT cells (H) (>95% pure) were left unstimulated, or stimulated with rhIL-12 (20 ng/mL) or rhIL-23 (100 ng/mL) for 6 h. Cell culture supernatants were harvested and used for IFN-γ determination in a multiplex cytokine assay. I) NK cells, ILC1, or ILC2 were sorted, by FACS, from blood samples from healthy donors and cultured in the presence of the indicated cytokines for 24 h. Total ILCs were gated on viable CD45+Lin− (CD3−CD4−CD5−TCRαβ−TCRγδ−CD14−CD19−) CD7+ cells. NK cells were identified as CD56bright and CD56dim, ILC2 as CD56−CD127+CRTh2+ and ILC1 as CD56−CD127+CD117−CRTh2−. IFN-γ levels were determined by intracellular staining. ILC3 were sorted by FACS from the tonsillar tissues of healthy donors, by gating on viable CD45+ Lin−CD7+CD117+NKp44+ cells. ILC3 were cultured for 4 days in the presence of the indicated cytokines, and IFN-γ was then determined by intracellular staining.
Lymphocyte subsets respond differently to IL-12 and IL-23
However, the mycobacterial disease in the patients studied cannot be entirely dependent on impaired effector functions of CD4+ T cells alone, as patients with HLA-II deficiency due to mutations of CIITA, RFXANK, RFXAP, or RFX5, with very low frequencies of functional CD4+ αβ T cells, are not prone to clinical disease caused by weakly virulent mycobacteria (45). Indeed, BCG or environmental mycobacterial disease of genetic origin in patients without isolated or syndromic MSMD, has been documented only in patients with severe combined immunodeficiency (SCID), who lack all types of endogenous T cells (46). An additional lack of NK cells in SCID patients seems to underlie more frequent and severe forms of mycobacterial disease (unpublished), although isolated NK deficiency does not confer a predisposition to mycobacterial disease (47–49). We therefore assessed mRNA levels for IL12RB1, IL12RB2, and IL23R in various leukocyte subsets, and assessed the consequences of IL-12 and IL-23 signaling in each of these cell types. Human IL12RB1 mRNA production has been reported in many types of human leukocytes, the lowest levels being reported in B cells and monocytes and the highest levels in NK cells (50). IL12RB2 and IL23R display a high degree of sequence identity and probably arose through gene duplication (12), but they nevertheless have different patterns of expression, with IL12RB2 mRNA levels highest in γδ T cells and NK cells, whereas IL23R mRNA levels are highest in γδ T cells and only moderate in CD4+ and CD8+ αβ T cells (50). We confirmed these findings (Fig. S6A), and stimulated purified B, CD4+ αβ T, CD8+ αβ T, γδ T, and NK cells from healthy donors with IL-12 or IL-23 for 6 h, before assessing the genome-wide transcriptional consequences by mRNA microarray analysis. Cytokine-specific and cell type-specific patterns were observed (Tables S2 and S3). Strikingly, IFNG transcripts were the most consistently induced across cell types and stimuli (Fig. 4C-D). This result was validated by qPCR (Fig 4E) and ELISA for IFN-γ on cell culture supernatants (Fig. 4F). We expanded this study to include MAIT, NKT cells, and ILCs, all of which have been reported to be potent IFN-γ producers (reviewed in (51, 52)). MAIT and NKT cells clearly responded to each cytokine by producing IFN-γ, but they responded more strongly to IL-23 (Fig. 4G-H). ILC1 and ILC2 isolated from peripheral blood responded to IL-12, but not IL-23, by producing IFN-γ (Fig. 4H). ILC3 isolated from tonsillar tissue clearly responded to IL-23 and IL-12 by producing IFN-γ (Fig. 4I). Finally, the stimulation of PBMCs from healthy donors or IL-12Rβ1-deficient patients with M. bovis BCG together with IL-12 or IL-23 revealed that IL-12 strongly potentiated the IFN-γ response to BCG, but that IL-23 also increased IFN-γ production, albeit to a lesser extent (Fig. S6B-D).
Discussion
Collectively, these data shed light on the respective contributions of IL-12 and IL- 23 to human immunity to Mycobacterium and Candida, through the discovery and characterization of AR complete IL-12Rβ2 and IL-23R deficiencies in two multiplex MSMD kindreds without CMC. The surprising absence of CMC in IL-23R-deficient P4 and P5, as in most patients with complete IL-12Rβ1 deficiency, may be due to the incomplete penetrance of IL-23 deficiency for poor IL-17A/IL-17F production, or the incomplete penetrance of low levels of IL-17 production for CMC, or both. Indeed, all known forms of isolated or syndromic CMC disrupt IL-17A/F immunity (16, 18–22). These data also reveal the specific transcriptional consequences of IL-12 vs. IL-23 signaling in human B, CD4+ αβ T, CD8+ αβ T, γδ T, MAIT, NKT, NK cells and ILCs, with a notable convergence on IFN-γ induction (Table S4). Clinically, these results add IL12RB2 and IL23R to the list of 11 other genes (IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, NEMO, CYBB, IRF8, ISG15, TYK2, SPPL2A) mutated in MSMD patients, the products of which are all involved in IFN-γ-mediated immunity (4, 5, 36). Importantly, patients with deleterious mutations in IL17F, IL17RA, IL17RA, or ACT1 have CMC without mycobacterial disease immunity (16, 18–22). Ouresults therefore reveal that both IL-12- and IL-23-dependent signaling pathways play critical roles in human anti- mycobacterial immunity, via the induction of IFN-γ. Indeed, our findings show that human IL-12 and IL-23 are partly, but not totally redundant with each other, for IFN-γ-dependent anti-mycobacterial immunity. These findings in natural conditions in humans corroborate and extend previous experiments performed in mice (42).
IL-12- and IL-23-dependent induction of IFN-γ may occur either within the same cell type, or differentially across different cell types, and IL-23 may provoke IFN-γ by direct or indirect means (which may include IL-17- and IL-22-dependent and -independent processes). The absence of response to both these molecules, as in patients with complete IL-12Rβ1 deficiency, underlies MSMD with incomplete but high penetrance, reaching an estimated 65% by five years of age and 80% in adults (4). It was not possible to calculate the penetrance of IL-12Rβ2 and IL-23 deficiencies, but the data from our population and family genetic studies strongly suggest that penetrance is much lower in both these deficiencies, accounting for the diagnosis of IL-12Rβ1 deficiency in a hundred times as many kindreds to date. This notion is strongly supported by our accompanying report that as many as 1/1,000 Europeans homozygous for TYK2 P1104A have normal responses to IL-12, and impaired responses to IL-23, rendering them vulnerable to tuberculosis and, more rarely, to MSMD (Boisson-Dupuis S, Ramirez-Alejo N., et al.). The primary, essential function of both IL-12 and IL-23 in humans is to induce IFN-γ and control mycobacteria and other intramacrophagic pathogens, perhaps due to the duplication of the loci encoding both these cytokines and their receptors. The contribution of IL-23 to IL-17 anti-fungal immunity is more modest and may be more recent in terms of evolution, as both the IL-23R-deficient patients described here, and most IL-12Rβ1-deficient patients described to date are not susceptible to Candida.
Supplementary Material
Acknowledgments
We thank the patients and their families for their collaboration, both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions and support, Y. Nemirovskaya, E. Anderson, M. Woolett, L. Amar and D. Papandrea for administrative support, and J. Gleeson (UCSD) for sharing WES data from individuals of middle eastern descent. The data presented in the manuscript are tabulated in the main paper and the Supplementary Online Materials. The WES data are available from the Sequence Read Archive (www.ncbi.nlm.nih.gov/sra) [accession numbers to be added when available]. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the National Institute of Allergy and Infectious Diseases (NIAID) grant numbers 5R37AI095983, R01AI089970 and K99AI127932, the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) of the National Institutes of Health grant number UL1TR001866, The Rockefeller University, the St. Giles Foundation, the European Research Council (ERC-2010-AdG-268777 and grant no. 323183), Institut National de la Santé et de la Recherche Médicale, Paris Descartes University, the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and by the French National Research Agency (ANR) under the “Investissement d’avenir” program (grant ANR-10-IAHU-01), ANR- TBPATHGEN (grant ANR-14-CE14–0007-01), ANR-IFNPHOX (grant ANR13-ISV3– 0001-01), ANR-GENMSMD (grant ANR16-CE17–0005-01). This work was supported by NIAID award #U19AI118626 (to AS and FS). JM was funded by the Canadian Institutes of Health Research, The NIH Translational Science Award (CTSA) program (#UL1 TR000043), the Swiss National Science Foundation (grant n. IZKOZ3_173586), The Charles H. Revson Foundation, and the NIAID (1K99AI127932–01A1). RMB was supported by the European Molecular Biology Organization (EMBO). NRA was supported by the National Council of Science and Technology of Mexico (CONACYT, 264011) and the Stony Wold-Herbert Fund Fellowship Grant. YI was supported by the AXA Research Fund. SGT, EKD and CSM are supported by research grants and fellowships from the National Health and Medical Research Council of Australia (SGT, CSM, EKD) and the Office of Health and Medical Research of the State Government of NSW Australia (CSM). AS is supported by NIH research grant HHSN272200900044C. JB is supported by SRC2017. The Institute for Research in Biomedicine and F.S. are supported by the Helmut Horten Foundation.
References
- 1.Casanova J-L, Human genetic basis of interindividual variability in the course of infection. Proc. Natl. Acad. Sci. 112, 201521644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casanova J-L, Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. 1, 201521651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casanova J-L, Abel L, Genetic dissection of immunity to mycobacteria: The Human Model. Annu. Rev. Immunol. 20, 581–620 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL, Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin. Immunol. 26, 454–470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreins etal AY., Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 212, 1641–1662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Beaucoudrey L et al. , Revisiting Human IL-12RP1 Deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 89, 381–402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fieschi C et al. , Low Penetrance, Broad Resistance, and Favorable Outcome of Interleukin 12 Receptor β1 Deficiency. J. Exp. Med. 197, 527–535 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boisson-Dupuis S et al. , Il-12rβ1 deficiency in two of fifty children with severe tuberculosis from IRN, MAR, and TUR. PLoS One. 6, 1–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Vosse E et al. , IL-12Rβ1 deficiency: Mutation update and description of the IL12RB1 variation database. Hum. Mutat. 34, 1329–1339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altare F et al. , Interleukin-12 receptor beta1 deficiency in a patient with abdominal tuberculosis. J. Infect. Dis. 184, 231–236 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Presky DH et al. , A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc. Natl. Acad. Sci. U. S. A. 93, 14002–7 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parham C et al. , A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12R 1 and a Novel Cytokine Receptor Subunit, IL-23R. J. Immunol. 168, 5699–5708 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Teng MWL et al. , IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Prando C et al. , Inherited IL-12p40 Deficiency. Medicine (Baltimore). 92, 109–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Beaucoudrey L et al. , Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J. Exp. Med. 205, 1543–1550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puel A et al. , Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science (80-. ). 332, 65–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ouederni M et al. , Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin. Infect. Dis. 58, 204–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beziat V et al. , A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci. Immunol. in press (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boisson B et al. , An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 39, 676–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lévy R et al. , Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl. Acad. Sci. 113, E8277–E8285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Vinh DC, Casanova JL, Puel A, Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr. Opin. Microbiol. 40, 46–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling Y et al. , Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 212, 619–631 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bloch Y et al. , Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shares Receptor IL- 12Rb1. 48, 45–58.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belkadi A et al. , Whole-exome sequencing to analyze population structure, parental inbreeding, and familial linkage. Proc. Natl. Acad. Sci. 113, 6713–6718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kircher M, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itan Y, Shang L, Boisson B, The mutation significance cutoff (MSC): gene-level thresholds for variant-level predictions Yuval. Nat. Methods. 13, 109–110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Paus RA, Geilenkirchen MA, Van Riet S, Van Dissel JT, Van De Vosse E, Differential expression and function of human IL-12RB2 polymorphic variants. Mol. Immunol. 56, 380–389 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Vogt G et al. , Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J. Exp. Med. 205, 1729–1737 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eilertson KE, Booth JG, Bustamante CD, SnIPRE: Selection Inference Using a Poisson Random Effects Model. PLoS Comput. Biol. 8 (2012), doi: 10.1371/journal.pcbi.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altshuler DM et al. , An integrated map of genetic variation from 1,092 human genomes. Nature. 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itan Y et al. , The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. 112, 13615–13620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB, Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 9 (2013), doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deschamps M et al. , Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am. J. Hum. Genet. 98, 5–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van de Vosse E, de Paus RA, van Dissel JT, Ottenhoff THM, Molecular complementation of IL-12Rbeta1 deficiency reveals functional differences between IL-12Rbeta1 alleles including partial IL-12Rbeta1 deficiency. Hum Mol Genet. 14, 3847–3855 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Wilson RP et al. , STAT3 is a critical cell-intrinsic regulator of human unconventional T cell numbers and function. J. Exp. Med. 212, 855–864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okada S, Markle JG, Deenick EK, Mele F, Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science (80-.). 349, 606–613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geiger R, Duhen T, Lanzavecchia A, Sallusto F, Human naive and memory CD4+ T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J. Exp. Med. 206, 1525–1534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinberg J et al. , Bacillus Calmette Guerin triggers the IL-12/IFN-γ axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur. J. Immunol. 34, 3276–3284 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Carpenter C et al. , A side-by-side comparison of T cell reactivity to fifty-nine Mycobacterium tuberculosis antigens in diverse populations from five continents. Tuberculosis. 95, 713–721 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sallusto F, Heterogeneity of Human CD4+ T Cells Against Microbes. Annu. Rev. Immunol. 34, 317–334 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Kastelein RA, Hunter CA, Cua DJ, Discovery and Biology of IL-23 and IL- 27: Related but Functionally Distinct Regulators of Inflammation. Annu. Rev. Immunol. 25, 221–242 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Khader SA et al. , IL-23 Is Required for Long-Term Control of Mycobacterium tuberculosis and B Cell Follicle Formation in the Infected Lung. J. Immunol. 187, 5402–5407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flynn JL, An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178, 2249–2254 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Happel KI et al. , Pulmonary interleukin-23 gene delivery increases local T-cell immunity and controls growth of Mycobacterium tuberculosis in the lungs. Infect. Immun. (2005), doi: 10.1128/IAI.73.9.5782-5788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanna S, Etzioni A, MHC class I and II deficiencies. J. Allergy Clin. Immunol. 134, 269–275 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Casanova J-L, Jouanguy E, Lamhamedi S, Blanche S, Fischer A, Immunological conditions of children with BCG disseminated infection. Lancet. 346, 581 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Gineau L et al. , Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. 122, 821–832 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cottineau J et al. , Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J. Clin. Invest. 127, 1991–2006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vély F et al. , Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 17, 1291–1299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chognard G et al. , The dichotomous pattern of IL-12R and IL-23R expression elucidates the role of IL-12 and IL-23 in inflammation. PLoS One. 9 (2014), doi: 10.1371/journal.pone.0089092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Godfrey DI, Uldrich AP, Mccluskey J, Rossjohn J, Moody DB, The burgeoning family of unconventional T cells. Nat. Immunol. 16 (2015), doi: 10.1038/ni.3298.1114. [DOI] [PubMed] [Google Scholar]
- 52.Klose CSN, Artis D, Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 17, 765–74 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Grant AV et al. , Accounting for genetic heterogeneity in homozygosity mapping: Application to mendelian susceptibility to mycobacterial disease. J. Med. Genet. 48, 567–571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purcell S et al. , PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abecasis GR, Cherny SS, Cookson WO, Cardon LR, Merlin — Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Li H, Durbin R, Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mckenna A et al. , The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 0–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K, Li M, Hakonarson H, ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Byun M et al. , Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 207, 2307–2312 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cingolani P et al. , A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biesinger B et al. , Stable growth transformation of human T lymphocytes by Herpesvirus saimiri. Proc. Natl. Acad. Sci. 89, 3116–3119 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinez-Barricarte R et al. , Curr. Protoc. Immunol, in press, doi: 10.1002/cpim.15. [DOI] [PubMed] [Google Scholar]
- 63.Moncada-Velez M et al. , Partial IFN-yR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. 122, 2390–2401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma C et al. , Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 119, 3997–4008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acosta-Rodriguez EV et al. , Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 8, 639–646 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Lefkovits I, Waldmann H, Limiting dilution analysis of cells of the immune system. 5, 38–204 (1979). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.