

ABSTRACT
A new modality in antibody engineering has emerged in which the antigen affinity is designed to be pH dependent (PHD). In particular, combining high affinity binding at neutral pH with low affinity binding at acidic pH leads to a novel antibody that can more effectively neutralize the target antigen while avoiding antibody-mediated antigen accumulation. Here, we studied how the in vivo pharmacokinetics of the superantigen, Staphylococcal enterotoxin B (SEB), is affected by an engineered antibody with pH-dependent binding. PHD anti-SEB antibodies were engineered by introducing mutations into a high affinity anti-SEB antibody, 3E2, by rational design and directed evolution. Three antibody mutants engineered in the study have an affinity at pH 6.0 that is up to 68-fold weaker than the control antibody. The pH dependency of each mutant, measured as the pH-dependent affinity ratio (PAR – ratio of affinity at pH 7.4 and pH 6.0), ranged from 6.7–11.5 compared to 1.5 for the control antibody. The antibodies were characterized in mice by measuring their effects on the pharmacodynamics and pharmacokinetics (PK) of SEB after co-administration. All antibodies were effective in neutralizing the toxin and reducing the toxin-induced cytokine production. However, engineered PHD antibodies led to significantly faster elimination of the toxin from the circulation than wild type 3E2. The area under the curve computed from the SEB PK profile correlated well with the PAR value of antibody, indicating the importance of fine tuning the pH dependency of binding. These results suggest that a PHD recycling antibody may be useful to treat intoxication from a bacterial toxin by accelerating its clearance.
Keywords: monoclonal antibody, pH-dependent antibody, Staphylococcal enterotoxin B, antigen pharmacokinetics
Introduction
Monoclonal antibodies (mAb) represent the fastest growing class of drugs in clinical pipelines.1,2 Therapeutic targets are wide ranging, and include cell surface proteins, extracellular ligands, soluble proteins and peptides in serum.3,4 A mAb can be tailored to bind the target molecule with high affinity in order to modulate signaling, relocation, or degradation. Many of these functions benefit from high affinity interaction because it allows the same activity to be achieved using a lower dose. However, regardless of the binding affinity, a traditional antibody can suppress at most a stoichiometric amount of the antigen. If the target is produced continuously or has a high level of synthesis, therapeutic antibodies need to be administered frequently, and in high doses, to ensure that the antibody always exists in stoichiometric excess. There are technical challenges in developing high dose (i.e., high concentration) antibody therapeutics. A high dosing frequency is also inconvenient to the patient and raises the overall cost of treatment.
Toward addressing these challenges, recent therapeutic mAb developments have included the engineering of antibodies that bind the antigen in a pH-dependent (PHD) manner.5–7 In particular, a PHD antibody that binds the target antigen tightly at a neutral pH but weakly at an acidic pH has been shown to be useful in treating conditions caused by excessive production of a pathological molecule. The benefits of pH-dependent binding derives from the ability of the antibody to bind a target and facilitate its endolysosomal degradation by releasing it in the acidic environment of the endosome. An antibody molecule in the blood is nonspecifically taken up by endothelial cells in fluid-phase pinocytosis. Once in the endosome, the molecule is either sorted to the lysosome for degradation, or recycled to the surface by binding to membrane-bound neonatal Fc receptor (FcRn).8 When a conventional (pH-independent) antibody binds an antigen in the serum, the antigen avoids endolysosomal degradation since the antigen is recycled to the cell surface along with the antibody (Figure 1). On the other hand, an antigen that is bound to a PHD antibody is released in the acidic environment of the endosome so that it is sorted to the lysosome for degradation while the antibody is recycled to the blood. The recycled antibody can bind and neutralize another antigen. Therefore, a PHD antibody makes use of a natural clearance pathway to catalytically eliminate (i.e., “catch and release”) antigen from the circulation, thus creating a net flow of the antigen from the circulation to the lysosome.
Figure 1.
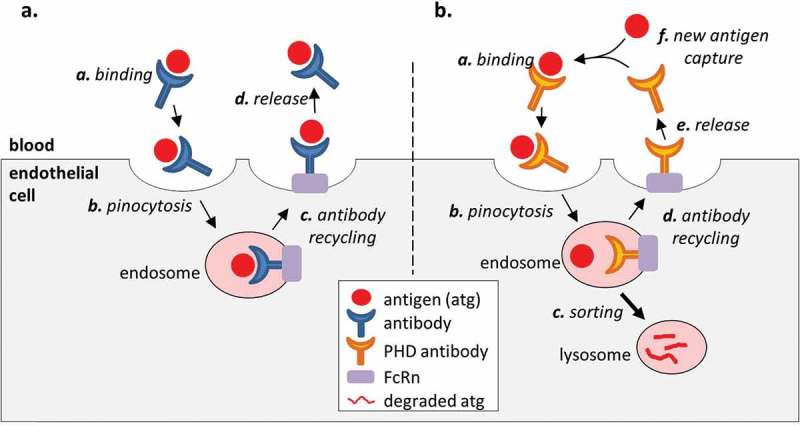
Effect of antibody binding on the antigen PK. (a). A conventional high affinity antibody increases the serum stability of the antigen because the antigen remains bound to the antibody in the endosome and is recycled to the serum along with the antibody. (b). An engineered PHD antibody releases the bound antigen in the acidic environment of the endosome to allow endolysosomal degradation of the molecule while the antibody is returned to the serum. This creates a net flow of the antigen from the serum to the lysosome and increases the rate of antigen elimination compared to a high affinity antibody with pH independent binding.
Numerous antibodies have been engineered with pH-dependent binding properties, and they target both soluble and surface bound antigens (IL-6R, IL-6, PCSK9, TNF, and CXCL10), with indications primarily focused on autoimmune diseases and treatment of high cholesterol.9,10 In this study, we present the first application of a PHD antibody in the treatment of infectious pathogens by engineering antibodies with pH-dependent binding to the bacterial superantigen Staphylococcal enterotoxin B (SEB). SEB is secreted by methicillin-resistant Staphylococcus aureus (MRSA), and is a well-known and sometimes fatal cause of sepsis in patients. MAbs against SEB has been engineered to inhibit SEB activity in vitro11–16 and in vivo,17,18 and there are also some promising leads in developing vaccines against the molecule.19,20 Still, there are challenges in developing an antibody-based treatment, and to our knowledge, no antibody-based therapy has been approved for clinical use to date. One factor that contributes to the difficulty of developing an antibody therapy is the high rate of SEB production during active MRSA infection, which requires a prohibitively high dose of a conventional neutralizing antibody. The serum concentration of SEB is increased further through antibody-mediated stabilization, making it progressively more difficult to deliver effective treatment.
The PHD antibodies presented herein were engineered in a two-step process that includes rational engineering based on histidine substitutions and affinity maturation by yeast surface display. The mutants were first expressed and screened as antibody single-chain variable fragments (scFvs) on the yeast surface before full-length antibodies were constructed and tested in vitro and in mice. Selective loss of affinity at pH 6.0 was demonstrated by affinity measurements (22–68-fold loss in affinity at pH 6.0 compared to wild type). The measured kinetics of dissociation suggests up to 92% loss of bound SEB in 15 min at pH 6.0 (a timescale relevant in endolysosomal clearance), indicating that a large fraction of bound SEB will be subject to endolysosomal degradation. The engineered and control antibodies were tested in a co-injection mouse model, which showed that the potential SEB exposure is reduced when using a PHD antibody, with a linear correlation observed between the pH dependency of an antibody and the exposure of the host to SEB. Our study thus describes a robust approach to targeted immunotoxicotherapy by simultaneously neutralizing and eliminating the toxic bacterial antigen with neutralizing PHD antibody.21
Results
Targeting interfacial residues for site-specific histidine substitution
Using the crystal structure of a previously reported antibody 3E2 Fab in complex with SEB,14 we selected 20 antibody residues that are within 4.5 Å of SEB, and targeted them for histidine mutation (Figure 2(a)). The antibody inhibits the activity of SEB by interfering with its interaction with MHC II on antigen-presenting cells (Fig. S1). Because of their proximity to the bound ligand, mutating the interfacial residues is likely to affect how the antibody interacts with SEB. The resulting 20 single point mutants as well as wild type 3E2 were expressed as scFv on the yeast surface by joining the heavy (VH) and light chain (VL) variable domains through a flexible Gly-Ser linker. Nine mutants were successfully displayed, whereas the rest either did not express or had a significantly reduced expression level, suggesting that the mutations perturb folding.22 The stably displayed mutants were then tested for SEB binding (Figure 2(b)). Seven of the nine expressing mutants showed a higher mean fluorescence intensity (MFI) at pH 7.4 than at pH 6.0, i.e., stronger SEB binding at pH 7.4. Mutant 13 (m13) containing VH-Y124H was selected from this pool because it bound SEB efficiently at pH 7.4 and exhibited clearly reduced binding at pH 6.0. m13 was further tested by measuring the equilibrium dissociation constant KD. The affinity of the mutant was nearly 100-fold lower at pH 6.0 compared to wild type (Figure 2(c), Table 1). However, the mutation also reduced the affinity at pH 7.4 by 20 fold.
Figure 2.
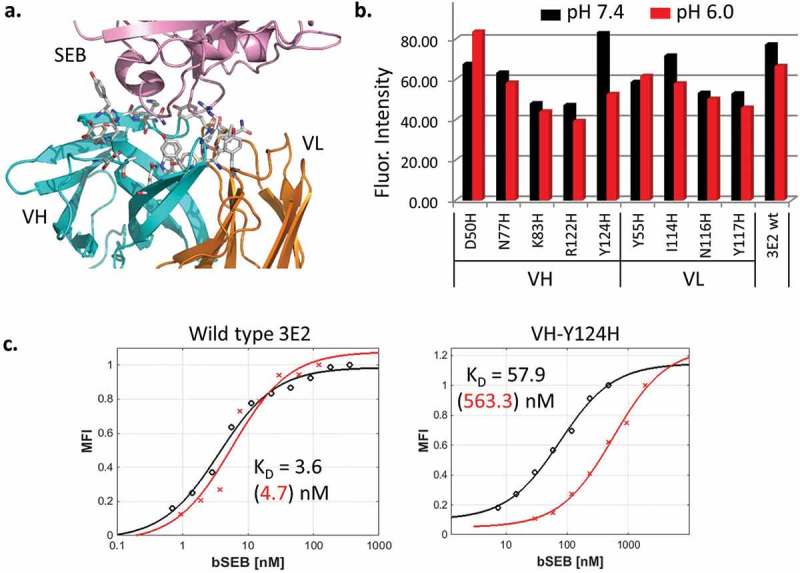
(a). SEB (pink) bound to wt anti-SEB 3E2 Fab (PDB: 3W2D). Heavy chain (VH) and light chain (VL) variable domains are colored cyan and orange, respectively. The antibody residues within 4.5 Å of SEB are shown as sticks. (b). 3E2 scFv mutants containing interfacial histidine mutations were displayed on the yeast surface and analyzed by flow cytometry for SEB binding. The amount of biotinylated SEB bound was measured using SAPE. The mean fluorescence intensity of the displaying population is plotted. The labeling was performed at pH 7.4 and 6.0. The binding at pH 6.0 is lower for the VH-Y124H mutant (m13). (c). The equilibrium binding affinity KD of wild type 3E2 and VH-Y124H was measured by yeast surface display. The representative binding curves at pH 7.4 (black) and pH 6.0 (red) and their fitted values are shown.
Table 1.
Amino acid position and residue substitution after three rounds of library screening, MFI of expression tag, affinities at pH 7.4 and pH 6.0 and the ratio R of KD at pH 6.0/KD at pH 7.4. CDR residues are shaded in gray. WT residues correspond to the amino acids in 3E2. L1 – L24 were selected after sorting a yeast surface library of scFv that was constructed using m13 as the template. The other mutants were designed by combining the mutations identified from the sorting.
| VH |
VL |
||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDR |
1 |
2 |
3 |
1 |
3 |
||||||||||||||||||||||||
| Res. No |
21 |
33 |
39 |
42 |
46 |
48 |
50 |
51 |
56 |
59 |
72 |
73 |
88 |
100 |
124 |
136 |
25 |
32 |
40 |
43 |
52 |
53 |
56 |
63 |
117 |
MFI |
KD (7.4) |
KD (6.0) |
R |
| wt |
Q |
S |
T |
V |
S |
T |
D |
F |
I |
F |
Y |
S |
I |
Q |
Y |
T |
V |
I |
T |
M |
S |
S |
L |
S |
Y |
1336 |
3.6 |
4.7 |
1.3 |
| m13 | H | 1452 | 75.9 | 563.3 | 7.4 | ||||||||||||||||||||||||
| L1 | A | A | A | H | H | A | P | P | 3040 | 37.6 | 244.2 | 6.5 | |||||||||||||||||
| L2 | P | H | I | 2368 | 22.9 | 649.9 | 28.4 | ||||||||||||||||||||||
| L4 | L | H | A | P | 2738 | 42.2 | 606.2 | 14.4 | |||||||||||||||||||||
| L5 | H | H | 2630 | 28.2 | 141.8 | 5.0 | |||||||||||||||||||||||
| L6 | V | A | H | 1643 | 22.1 | 255.5 | 11.6 | ||||||||||||||||||||||
| L9 | H | R | H | A | 2184 | 32.6 | 132.7 | 4.1 | |||||||||||||||||||||
| L10 | V | V | H | 3496 | 24.7 | 296.9 | 12.0 | ||||||||||||||||||||||
| L12 | P | H | I | 3672 | 42.6 | 567.4 | 13.3 | ||||||||||||||||||||||
| L14 | P | S | 2841 | 3.0 | 8.5 | 2.8 | |||||||||||||||||||||||
| L16 | G | S | H | 2571 | 30.4 | 343.8 | 11.3 | ||||||||||||||||||||||
| L17 | L | H | M | 3273 | 22.0 | 76.4 | 3.5 | ||||||||||||||||||||||
| L18 | H | A | H | 2410 | 35.3 | 82.5 | 2.3 | ||||||||||||||||||||||
| L21 | H | P | 3360 | 24.5 | 63.0 | 2.6 | |||||||||||||||||||||||
| L22 | V | H | H | 2866 | 15.5 | 54.0 | 3.5 | ||||||||||||||||||||||
| L23 | A | V | H | 1803 | 28.9 | 282.6 | 9.8 | ||||||||||||||||||||||
| L24 | H | P | 2983 | 51.4 | 76.9 | 1.5 | |||||||||||||||||||||||
| L6.R | V | R | A | H | 2606 | 13.3 | 80.0 | 6.0 | |||||||||||||||||||||
| L6.H | V | H | A | H | 2159 | 19.7 | 59.9 | 3.0 | |||||||||||||||||||||
| L6 + 21 | V | A | H | P | 3370 | 26.0 | 205.9 | 7.9 | |||||||||||||||||||||
| L6 + 5 | V | A | H | H | 2003 | 30.1 | 251.1 | 8.3 | |||||||||||||||||||||
| L22 + 5 | V | H | H | H | 3330 | 23.0 | 66.4 | 2.9 | |||||||||||||||||||||
Library generation and screening
Since high affinity antigen binding is important during therapy, we performed a directed evolution study to improve the binding affinity of m13 and regain some of the lost binding affinity. A random library with a diversity of 2 × 107 unique variants was created by polymerase chain reaction (PCR). Mutations were introduced by supplementing the PCR mix with mutagenic nucleotide analogs, dPTP and 8-oxo-dGTP, and varying the number of cycles.23 The assembled PCR inserts containing a mutated scFv gene were combined with the expression vector by homologous recombination, and the displayed library of mutants were screened and sorted via fluorescence-activated cell sorting (FACS).
The sorting schedule was developed to optimize high affinity binding at pH 7.4 and rapid dissociation at pH 6.0 (Fig. S2). First, yeast cells were incubated with unmodified “cold” SEB at a high saturating concentration to occupy all SEB binding sites. This was followed by a short wash at pH 6.0 (15–30 min) to induce dissociation of bound SEB and create new SEB binding sites. Since the proteins in the endosome are recycled or moved to the lysosome in 5–15 min, an antibody that releases bound antigen on a similar time scale is more likely to induce antigen degradation.24,25 Finally, the cells were incubated with biotinylated “hot” SEB and labeled with streptavidin-phycoerythrin (SAPE) for detection and sorting. Successive rounds of sorting under increasingly more stringent labeling conditions (i.e., shorter wash duration at pH 6.0 and lower “hot” SEB concentrations) were used to enrich the library for the desired combination of binding characteristics.
The cells were labeled with anti-FLAG antibody to impose selection based on expression of mutant antibodies. The fraction of the mutants that bind SEB was low before the sort, but increased steadily during successive rounds of sorting, as can be seen from an increase in the PE signal (Figure 3(a)). After three rounds of sorting, the selected cells were separated on a plate and 24 clones were harvested and sequenced. The mutations in the selected clones were found to be distributed throughout the variable domains (Figure 4(a)).
Figure 3.
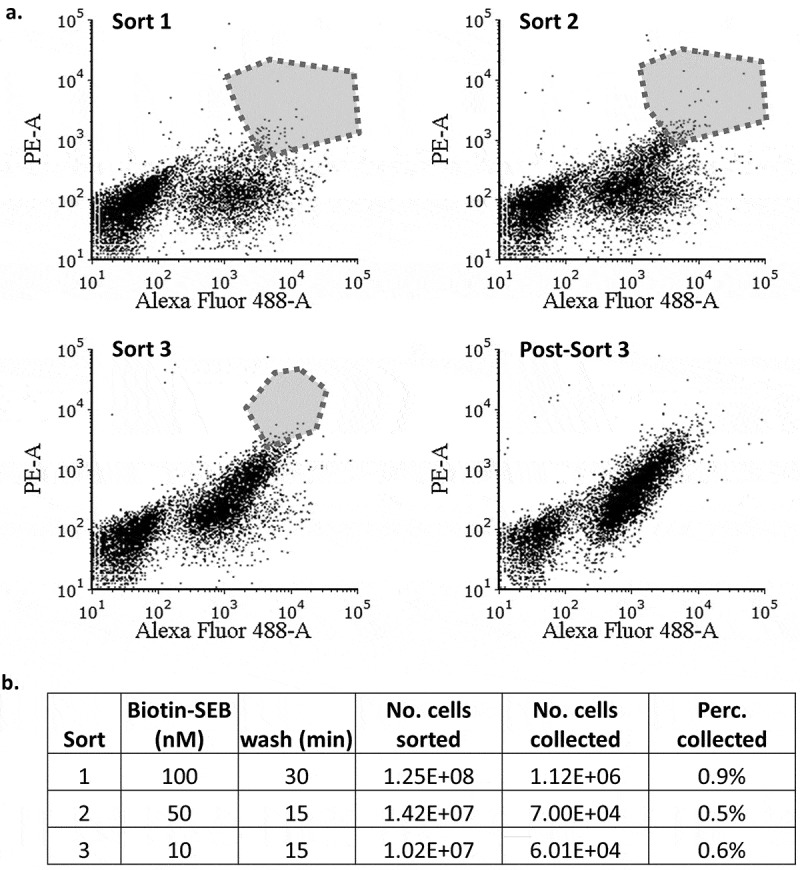
Fluorescence-activated cell sorting of m13 scFv library expressed on the yeast surface. (a). Cells were first incubated with unbiotinylated SEB, washed in PBST at pH 6.0, and finally incubated with biotinylated SEB at pH 7.4. Bound SEB was detected with SAPE. The cells were also labeled with anti-FLAG antibody and anti-mouse antibody-FITC to impose selection based on the scFv expression. The polygon represents the pool collected during each sorting. (b). The labeling and washing conditions during each round of sorting. The “percentage collected” refers to the number of cells collected divided by the number of cells sorted.
Figure 4.
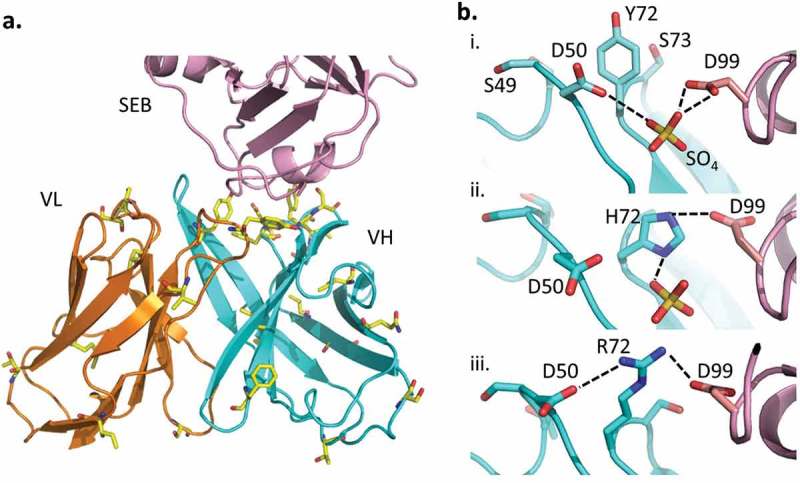
(a). Mutations found in the clones selected from the yeast library are shown as yellow sticks. (b). The interaction between VH residue 72 and SEB(D99) was modeled. i. VH-Y72 (wild type) does not interact directly with SEB(D99), but ii. VH-Y72H, which is found in several selected mutants, may contribute to improved affinity by forming a hydrogen bond (dotted line). iii. VH-Y72R may stabilize the interaction further through electrostatic interaction. Minor adjustments of surface residues allow the formation of a network of salt bridges (dotted lines), involving VH-D50, VH-R72 and SEB(D99).
Characterizing the selected clones
Each sequenced DNA plasmid was transfected back into yeast and expressed in order to confirm the selection and measure the binding affinities at pH 7.4 and 6.0. In all selected mutants, the pH 7.4 affinity was improved over the parent clone, m13 (Table 1). Additionally, in the mutants L2, L4, and L12, the pH 6.0 affinity was further reduced compared to m13. We evaluated the quality of pH-dependent binding by computing the PAR value, i.e., KD at pH 6.0 divided by KD at pH 7.4. A higher value of PAR indicates lower binding affinity at pH 6.0 than at pH 7.4. Seven mutants from the library screen showed a higher PAR value than m13. Additionally, all of the library mutants examined displayed higher stability than wild type 3E2 or m13, based on the level of expression.26 Additional sequences were created by combining the mutations found among the selected clones (Table 1). The VH-Y72H mutation improved the affinity by nearly 5 fold compared to m13 (clone L22), possibly by forming a hydrogen bond with SEB D99 (Figure 4(b)). Modeling showed that mutating VH-Y72 to R may lead to a different arrangement of the surface residues, resulting in a more stable complex (Figure 4(b)). We thus introduced the VH-Y72R mutation in the L6 background. The resulting L6.R bound SEB with higher affinity at both pH 7.4 and 6.0.
Antibody generation
Three mutants (L2, L6, and L6.R) and wild type 3E2 were selected for further testing in vitro. Each construct was expressed as full-length IgG in Chinese hamster ovary (CHO) cells and purified using immobilized protein G. The purified antibody was checked using SDS-PAGE (Fig. S3). The equilibrium binding affinity of each antibody was then measured by enzyme-linked immunosorbent assay (ELISA) (Table 2). The measured affinity for wild type 3E2 (0.17 nM) closely matches a previously reported value (0.58 nM).14 The PAR values obtained by measuring the KD at pH 7.4 and 6.0 remained largely unchanged from the estimates obtained using yeast displayed scFv, although the affinity was higher for full-length antibodies (Table 2).27,28 The affinity measurement was repeated by surface plasmon resonance (SPR), which showed similar pH-dependent binding by engineered antibodies (Table S1).
Table 2.
Full length antibody affinity measured by ELISA at pH 7.4 and 6.0. Standard deviations are from triplicate well measurements. PAR is defined as (KD at pH 6.0)/(KD at pH 7.4).
| KD at pH 7.4 (nM) | KD at pH 6.0 (nM) | PAR | |
|---|---|---|---|
| WT | 0.17 ± 0.01 | 0.25 ± 0.02 | 1.5 |
| L2 | 1.48 ± 0.18 | 17.00 ± 1.84 | 11.5 |
| L6 | 0.62 ± 0.07 | 7.11 ± 0.91 | 11.5 |
| L6.R | 0.81 ± 0.11 | 5.44 ± 0.76 | 6.7 |
We performed a dissociation study by ELISA to evaluate whether the engineered PHD antibodies release SEB on a physiologically relevant timescale. To this end, each antibody was immobilized on an ELISA plate and saturated with 1 µM SEB at pH 7.4. The complex was then washed at pH 7.4 or 6.0 for 15 min and the remaining bound SEB was quantified. Comparison of the remaining SEB after a pH 6.0 vs 7.4 wash revealed a marked decrease in bound SEB for the three engineered antibodies at pH 6.0 (Figure 5). In contrast, bound SEB dissociated similarly at both pH from wild type 3E2 during the same time period. This experiment demonstrates a potential for PHD antibody to quickly release SEB in an acidic environment of the endosome on a timescale that is relevant to in vivo endocytic trafficking.
Figure 5.
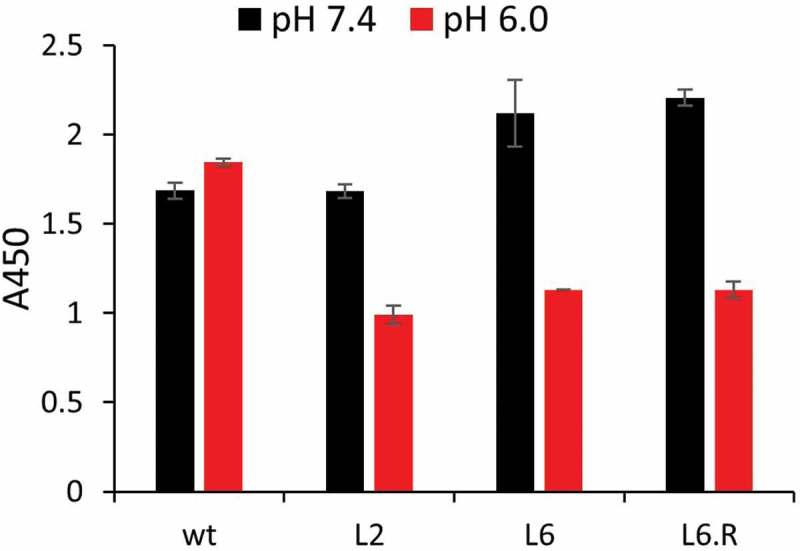
Dissociation study of full-length anti-SEB antibody. Wt, L2, L6, or L6.R was immobilized on ELISA surface and then incubated with a saturating concentration of biotinylated SEB. After washing in PBST pH 6.0 or 7.4 for 15 min, bound SEB was detected using streptavidin-HRP.
PHD antibodies retain neutralizing properties in vivo
Altering the antibody interface can affect its function, including its ability to neutralize the target molecule. The ability to inhibit SEB-induced cytokine release is essential to the therapeutic effects of engineered PHD antibodies and needs to be preserved. We tested if L2, L6 and L6.R are able to neutralize SEB intoxication in vivo by injecting mice with each antibody and SEB. The SEB activity was quantified by measuring the levels of interleukin (IL)-2, IL-6 and interferon gamma (IFNγ) (Figure 6). Three hours post co-injection was chosen as a monitoring point because the cytokine production reaches a relative maximum at this time point in the absence of a treatment antibody, i.e., SEB alone. Following co-injection of each of the three antibodies, the cytokine production was significantly lower than in untreated animals, and statistically similar to the level observed with wild type 3E2. Therefore, the engineered antibody variants continue to neutralize SEB in vivo, despite their newly acquired ability to bind the target in a pH-dependent manner.
Figure 6.
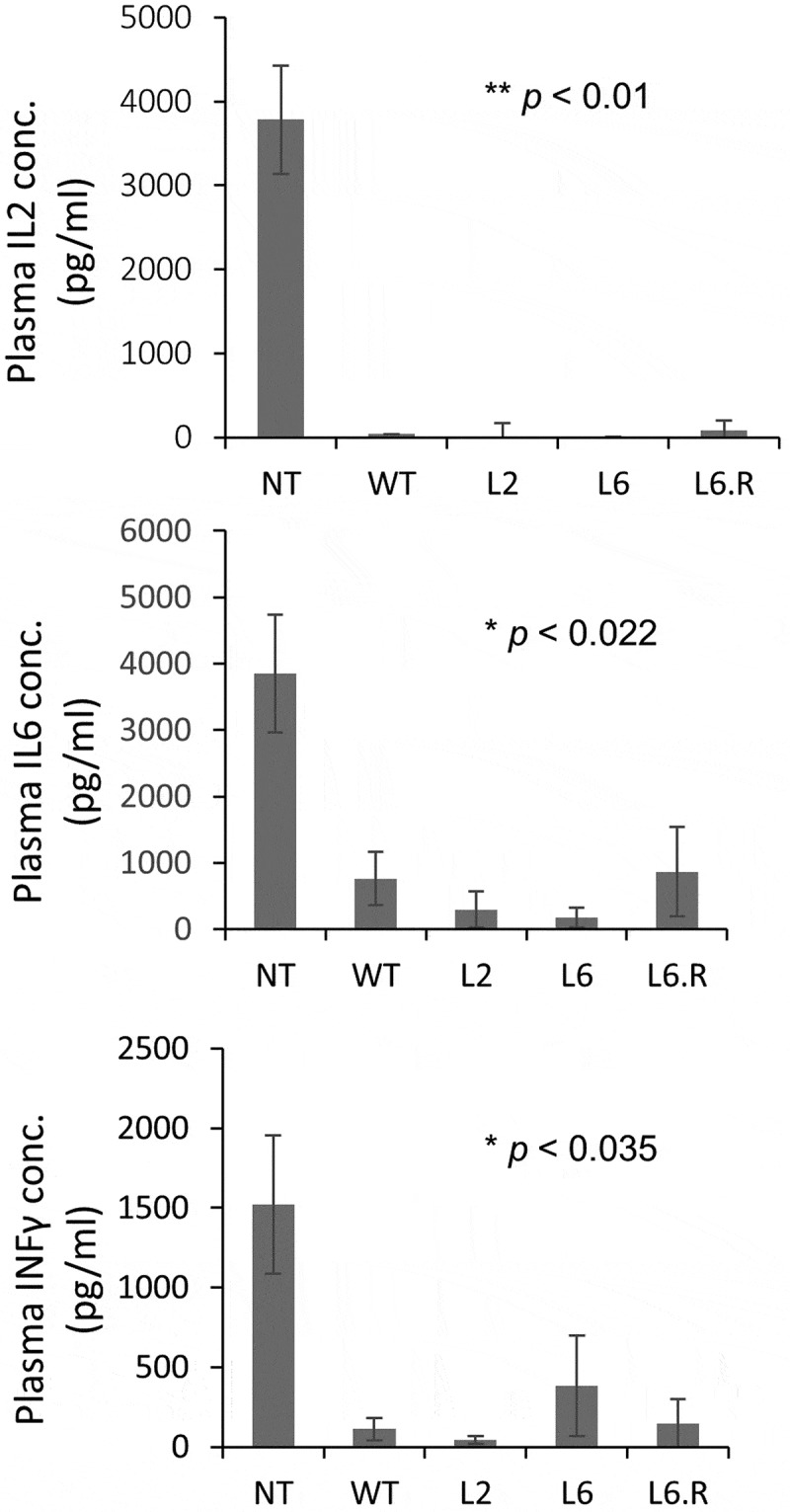
The concentrations of inflammatory cytokines IL-2, IL-6 and IFNγ in plasma were measured 3 hr after SEB injection, either alone (NT, no treatment) or together with various treatment antibodies. The untreated mice had the highest concentrations for all three cytokines (n = 3). The co-injection of SEB (i.p.) and antibodies (i.v.) reduced the cytokine production, indicating that the antibodies are all capable of neutralizing the toxicity of SEB. For each measured cytokine, pair-wise two-tailed comparisons were made between NT and each antibody treatment. The maximum p value is indicated. No statistical significance was observed among various antibody treatments.
Modulation of the SEB pharmacokinetics
Studies have shown that antigens in serum are stabilized by the binding of high affinity antibody, resulting in a longer in vivo half-life.29–32 We measured the rate of SEB clearance in mice with or without wild type 3E2. The rate of SEB clearance was significantly slower in the mice treated with 3E2, indicating, as expected (see Ref. 33), that the toxin is stabilized by antibody binding (Figure 7(a)). On the other hand, when the mice were challenged with SEB and an engineered PHD antibody, there was a marked reduction in circulating SEB compared to 3E2, indicating that pH-dependent binding helps reduce the circulatory half-life of SEB (Figure 7(a)). The enhancement in the rate of SEB clearance was not caused by a change in the antibody PK, which remained largely unchanged by the engineering. Importantly, all antibodies remained at stoichiometric excess throughout the study (Figure 7(b)). To estimate the efficacy of each antibody for treating SEB intoxication, we computed the area under the curve (AUC) for different antibody treatments, which effectively represents the integrated exposure of the host to the toxin. AUC did not correlate with the affinity of the antibodies at either pH. However, a linear relationship was observed between AUC and the PAR values of the treatment antibodies, suggesting that the efficiency of antigen clearance is influenced by the ability of an antibody to bind the antigen in the blood and release it in the endosome (Figure 7(c)). Since eliminating SEB from the circulation should reduce SEB-induced toxicity, PHD antibodies may prove to be therapeutic in treating the symptoms caused by SEB exposure.
Figure 7.
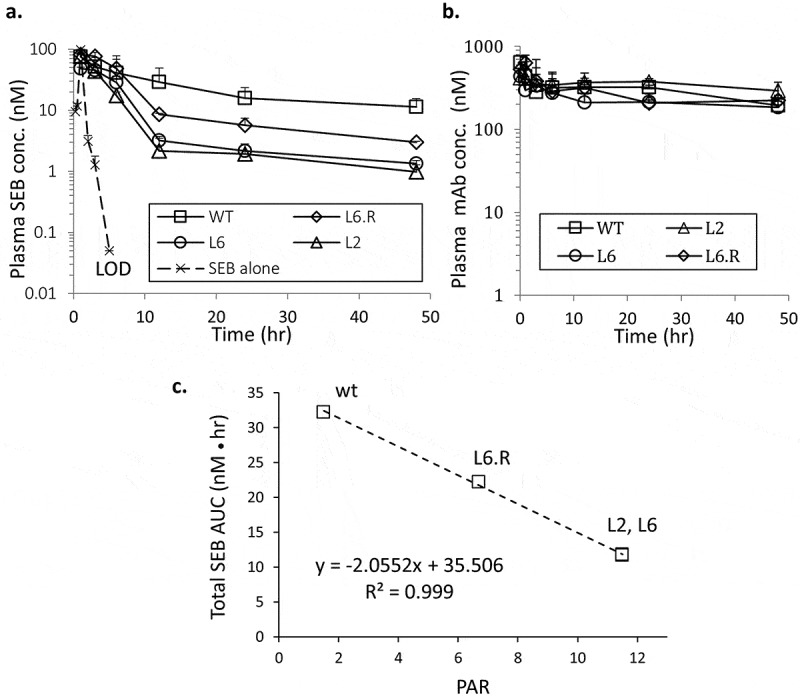
In vivo characterization of PHD antibodies. Mice were injected with 20 µg i.p. SEB immediately followed by 150 µg antibody i.v. (a). Total SEB concentration in plasma was determined from sandwich ELISA. The mean value is shown (n = 3 animals) for each time point. The standard deviations are represented by positive error bars. LOD, limit of detection. (b). Total antibody concentration in plasma was determined by ELISA at various time points. (c). A linear correlation between the SEB AUC (in units of nM●hr) and the PAR values of the antibodies is observed with a high Pearson correlation coefficient.
Discussion
In this study, we reported the engineering of PHD antibodies designed to facilitate the rate of clearance of the bacterial superantigen SEB in mice. Immediate and sustained neutralization of SEB is important to treat SEB intoxication and prevent cytokine storms that are incapacitating and can be fatal.34,35 The toxin is extremely stable against heat and chemical denaturation and will continue to exert its toxic effects unless it is actively neutralized or eliminated. An antibody, such as 3E2, neutralizes the toxin by binding to a biologically active region and interfering with its downstream biological processes.14 However, to date the conventional approach based on high affinity antibody treatment has not led to a US Food and Drug Administration-approved treatment against bacterial toxins, including SEB. One possible challenge for developing an antibody therapy is the target accumulation during active infection caused by antibody binding. Several recent studies have demonstrated that engineered PHD antibodies can neutralize overproduced endogenous ligands, such as inflammatory cytokines, without stabilizing them. Here we show that engineered PHD antibodies are similarly able to mitigate intoxication by an exogenously introduced bacterial toxin, while accelerating clearance of the molecule (Figure. 6 ad 7(a)). Our study provides a first example that a PHD antibody may be useful in immunotoxicotherapy.
For the purpose of developing therapeutic solutions based on PHD antibodies, it is encouraging to note that such antibodies may be engineered in a simple process that sequentially combines a rational design phase to achieve pH-dependent binding and a directed evolution study phase to achieve affinity maturation. We believe this to be a preferred alternative to using a biased library to simultaneously engineer pH-dependent binding while optimizing the affinity [e.g. Ref.7], especially because the challenges associated with assembling a large complex library are nontrivial. Rational design is used to introduce a histidine mutation based on structural knowledge of the antibody-antigen interaction (Figure 2(a)) and convert the initial pH independent interaction to one that varies with pH. Only a small number of residues need to be examined (~20) so that the study can be completed predictably in a finite amount of time. The individual point mutants can be displayed on the yeast surface and directly studied for pH-depending binding by fluorescent labeling, thus avoiding laborious and lengthy purification from mammalian cells.36 Any mutation that disrupts folding of the antibody and complicates downstream applications can be identified and eliminated at this stage since it results in reduced scFv expression on the yeast surface.
Our study shows that a targeted tyrosine to histidine mutation can achieve pH-dependent binding while maintaining key antibody-antigen interactions. This finding may be expected since the mutation replaces one aromatic polar side chain with another, and thus constitutes a conservative substitution. On the other hand, mutating a tyrosine at the interface is likely to modify the antibody-antigen interaction because tyrosine residues are known to play important roles in antigen binding.37 Together, these observations suggest that mutating an interfacial tyrosine of an antibody to histidine has a high probability of converting pH-independent antigen binding to a pH-dependent interaction, while minimizing structural and functional perturbations. In support of this hypothesis, we were able to introduce a single tyrosine to histidine mutation at the interface of another human antibody-antigen complex and engineer a pH-dependent interaction with the PAR value of 4.2. Since the complementary-determining regions (CDR) of an antibody can be predicted based on sequence, targeted histidine mutations can be introduced even when a high resolution complex structure is not available, although the efficiency of an approach based on CDR sequence analysis remains to be tested in future studies.
The FACS protocol used in this study for affinity maturation, which was modified from Schroter et al.,6 overcomes the difficulty of simultaneously achieving two objectives that are at odds with each other, i.e., increasing the affinity at one pH while lowering the affinity at another pH, because increasing or decreasing the affinity at one pH tends to have a similar ramification at another pH. We optimized the rate of dissociation at acidic pH, rather than the equilibrium binding affinity, because the antibody-antigen complex stays only briefly in the endosome before the antibody is recycled, and therefore rapid dissociation of bound antigen is more relevant to the serum stability of the antigen.
The affinity of selected scFv correlates with the affinity of full-length antibody purified from CHO, demonstrating that scFv is a suitable platform for investigating pH-dependent properties. The affinity of full-length antibody is often higher than the corresponding scFv because of its higher conformational rigidity, which minimizes entropic penalty incurred during binding.27,28 The absolute values of the binding parameters measured by ELISA differ from those obtained by SPR (Table 2 and S1). These techniques are complementary and both are used widely in affinity measurements, but they sometimes measure different subsets of interaction, which result in differences in reported affinity values.38 The reasons for the difference in our measurements are not immediately obvious, but importantly, the relative strength of binding for various antibodies, e.g., KD of each antibody normalized by the KD of wild type, are consistent across both methodologies at both neutral and acidic pH.
The binding kinetics measured by SPR allow us to interpret the SEB pharmacokinetics (PK) data based on the changes in kinetic constants. For example, 67–92% of bound SEB would dissociate from engineered antibodies in 15 min at pH 6.0 (i.e., in the endosome), while the rest remains bound and returns to the serum. On the other hand, 86% of SEB recycles with wild-type antibody. Consistent with the proposed model of how a PHD antibody is expected to work, all three engineered PHD antibodies accelerated the rate of SEB clearance compared to wild type. While there is a small difference in pH 7.4 affinity, the total antibody concentration in plasma remains nearly 100 fold above the KD at all time (Figure 7(b)), and more than 99.5% of all SEB in the blood is expected to be bound to an antibody. As shown in Figure 6, PHD antibodies did not show a significant increase in cytokine response, indicating that SEB in plasma mostly exists in complex with antibody. The difference in the SEB PK is therefore a result of how the complex responds to a change in the chemical environment as it enters the endosome.
Our study suggests that testing the antibodies with a range of affinity and PAR values may be important to optimize their in vivo properties. For example, the total and free SEB concentrations are the lowest for L2 and L6, which do not have the lowest KD at either pH (wild type has the lowest value, whereas L6 and L6.R have similar values). In this regard, the reduction in the SEB AUC correlated most closely with the combination of the affinities at neutral and acidic pH, quantified here as PAR. A strong linear correlation (R2 = 0.912) was also observed between AUC and the kd at pH 6.0, indicating that the rate of dissociation plays an important role in the antigen PK. As such, the PAR value and the dissociation rate at pH 6.0 may be useful parameters for optimizing the antibodies to modulate the target PK, although the limited scope of the current study does not allow rigorous testing of the accuracy of this prediction.
Recycling of the antibody in the endosome is mediated by the membrane-bound FcRn. Recycled antibody dissociates from FcRn at the cell surface because of the low affinity of interaction at neutral pH.39 It was reported that the rate of antigen clearance can be further accelerated by mutations in the antibody Fc that increase the affinity of interaction at neutral pH.40 The Fc mutations can help overcome the bottleneck in antigen degradation by complementing the rate-limiting pinocytosis step with more efficient receptor-mediated endocytosis. The antibody-SEB complex may thus be cleared more rapidly if the antibody carries affinity-increasing Fc mutations to improve FcRn-mediated capture at the cell surface. However, if the recycled antibody is not released at the cell surface due to increased affinity, the impact of the antibody on SEB clearance may be different from the antibodies containing just PHD mutations. In particular, it remains to be determined whether an antibody containing both PHD and affinity-increasing Fc mutations will be ultimately more efficient than a PHD antibody in neutralizing and eliminating circulating SEB.
In summary, we have engineered PHD antibodies against the superantigen SEB through rational design and directed evolution. The resulting antibodies have an affinity at neutral pH that is similar to wild type, but a reduced affinity at acidic pH. At least part of the affinity loss is caused by a change in the rate of dissociation. The PHD antibody mutants were able to accelerate the rate of SEB clearance in vivo compared to the parent, pH-independent antibody. Because of its small size, SEB is eliminated most rapidly when no antibody was added. This may erroneously lead one to dismiss the benefits of an antibody-based treatment. However, even at a low concentration SEB can wreak havoc on the immune system by inducing the release of inflammatory cytokines. This is evident from the burst in inflammatory cytokine production following an SEB injection in the absence of a treatment antibody. Therefore, temporary exposure of the host to the toxin may be enough to cause a potentially dangerous cytokine storm. A pH-independent antibody, such as wild type 3E2, quickly neutralizes circulating toxin and delivers a therapeutic effect, although it also slows the rate of toxin elimination. A high affinity PHD antibody achieves both objectives, i.e., neutralize the toxin and also accelerate its elimination. This unique feature of a PHD antibody is expected to play an even more important role during active infection, where the toxin is continuously produced, and managing the antigen PK becomes critically important to effectively treat accumulating toxin. Future studies will evaluate the effect of PHD antibodies on the PK and pharmacodynamics of SEB to facilitate the development of an optimal dosing regimen for clinical translation, while expanding the use of engineered PHD antibodies to treat intoxication from other lethal toxins.
Materials and methods
Site directed mutagenesis and yeast surface display
The expression vector for the scFv consisting of the variable domains of heavy and light chains of anti-SEB antibody 3E2 was constructed by PCR using overlapping primers. The insert was digested with NheI and BamHI and ligated into pCT302 vector containing a FLAG tag on the C-terminus.41 Single histidine substitutions were introduced using mutagenic primers at twenty interfacial residue positions identified from the 3E2-SEB structure (PDB 3W2D). Saccharomyces cerevisiae strain EBY100 cells (Thermo Fisher Scientific) were then transformed with wild type and mutant expression vectors and selected on SD-CAA plate lacking Trp and Ura. Colonies were selected and inoculated in SD-CAA dropout medium and grown to OD600 = 4–6. The expression of scFv was induced by transferring 200 μl of cells into 2 ml galactose containing media, SG-CAA, and shaken at 300 rpm at 30°C for 14–20 hr. Expression was verified using anti-FLAG-FITC. The expressing mutants were assessed for SEB binding by incubating 5 × 105 cells in 50 μl with 500 nM biotinylated SEB for 1 hr at 20°C in phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST) either at pH 7.4 or 6.0. The cells were washed and SAPE was added for SEB detection. Fluorescence was analyzed by flow cytometry using BD LSRFortessa (Becton Dickinson).
Library construction and sorting
A scFv containing a rationally designed PHD mutation VH-Y124H, or m13, was used as a template to assemble a random library. Mutations were introduced by performing PCR under mutagenic conditions. To introduce random mutations throughout the gene, mutagenic nucleotide analogs 8-oxo-dGTP and dPTP (TriLink Biotechnologies) were added at 2, 20, or 200 µM along with conventional dNTP, and 5, 10, and 20 cycles PCR amplification was performed using Taq (New England Biolabs). The resulting PCR products were inserted into pCT302 by homologous recombination. The transformed EBY100 cells were grown as previously described. Serial dilutions of transformed cells were grown on plates to estimate the number of unique transformants. Cells were expanded to achieve a 10-fold coverage of the expected diversity. The library was sorted for a combination of high/low affinity SEB binding at pH 7.4/pH 6.0. The cell sorting protocol was as follows: 1) Incubate with 1 µM unmodified SEB for 1 hr at room temperature (20°C); 2) Wash in pH 6.0 PBST for 15–30 min; 3) Label with biotinylated SEB (10–100 nM) and 1 µM anti-FLAG for 1 hr at 4°C; 4) Label with SAPE and with anti-mouse antibody-FITC. The cells were sorted on a BD FACS Aria II cell sorting system (Roswell Park Cancer Institute) by gating for positive PE and positive FITC signals. The sorted cells were expanded and subjected to additional rounds of sorting under progressively more stringent labeling conditions, i.e., a shorter pH 6.0 wash and a lower biotinylated SEB concentration. After the third and final sort, 24 clones were selected for sequencing and analysis.
Determination of scFv KD by yeast display
3E2, m13, and the selected scFv mutants on the yeast surface were tested for SEB binding at pH 7.4 and 6.0. Biotinylated SEB was serially diluted in pH 7.4 or 6.0 PBST, and allowed to incubate with 5 × 105 cells at room temperature for 1 hr. SAPE was added for detection and the cells were analyzed by flow cytometry. The MFI of the displaying population was measured and fitted against the SEB concentration to compute the equilibrium dissociation constants KD, i.e., the concentration where the MFI is 50% of the maximum value.42
Full-length IgG construction
The PHD scFv clones selected by yeast display were PCR amplified and ligated into the IGK-FRT expression vector (Thermo Fisher Scientific) so that full-length antibody containing the engineered variable domains can be expressed in mammalian cells. The engineered expression vector was transfected into CHO cells using Lipofectamin 3000. Positive colonies were selected over 10 days in the presence of hygromycin. Cells were then grown in CD CHO AGT expression medium with supplemental glutamine, D+ glucose, and Pen/Strep. Transfected cells were grown for a period of two to three weeks, at which point the media was harvested and secreted antibody was purified over a protein G column (GE Healthcare) on the NGC chromatography system (Bio-Rad). The antibodies were buffer exchanged into PBS and the concentration was determined using NanoDrop Spectrophotometer (Thermo Fisher Scientific).
KD determination of full-length antibody by ELISA
One μg/ml purified recombinant antibody was anchored overnight at 4°C on a Nunc MaxiSorp flat-bottom 96-well plate. The plate was washed three times in PBST and then blocked using 1% bovine serum albumin in PBST for 1.5 hr at room temperature under orbital shaking. The plate was then rinsed three times in PBST and a serial dilution of SEB starting with 0.5 μg/ml (Toxin Technology) was added across the plate in a pH 7.4 or 6.0 buffer for 1 hr at room temperature. A polyclonal rabbit anti-SEB antibody (Sigma, S9008-1VL) was conjugated to horseradish peroxidase (HRP; Thermo, 31489) to generate the ELISA detection reagent. The polyclonal anti-SEB antibody-HRP (1:2000 dilution) was added as the detection reagent for 30 min. The plate was washed and 1-Step Ultra TMB-ELISA Substrate Solution (Thermo Fisher Scientific) was added for quantification. The plate was analyzed on a FilterMax F5 96-well plate reader (Molecular Devices).
Cytokine response and detection
The observed guidelines for animal use and care in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the University at Buffalo. In each treatment group (wt, L2, L6, and L6.R), three male BALB/c mice (Jackson Laboratories) were administered with 150 µg antibody intravenously (i.v.), followed by an injection of 20 µg SEB i.p. The SEB alone group, i.e., not treated (NT), received sterile PBS i.v. Blood was collected from each mouse at 3 hr post administration. Each cytokine (IL-2, IL-6, and INFγ) was assayed independently via an ELISA kit (Mabtech) and detected using a FilterMax F5 96-well plate reader.
SEB and antibody PK measurement in vivo
Male BALB/c mice were administered with a single dose of 20 µg SEB i.p. and 150 µg antibody i.v. Five mice were used for each treatment group and blood was collected in staggered sampling so that three animals can be analyzed at each time point. Total antibody and total SEB were quantified by ELISA in triplicates. To quantify total SEB, polyclonal rabbit anti-SEB (Sigma, S9008-1VL) was anchored on a Nunc MaxiSorp flat-bottom 96-well plate and diluted plasma was added to capture SEB. Bound SEB was detected with a monoclonal mouse anti-SEB (Toxin Technology, MB344) and a secondary anti-mouse antibody-HRP conjugate (Bethyl Labs, A90-337P). To quantify total antibody, polyclonal goat anti-human IgG Fc antibody (Bethyl Labs, A80248A) was anchored on the plate and diluted plasma was added to each well. Captured antibody was detected with polyclonal donkey anti-human IgG Fc conjugated with HRP (Bethyl Labs, A80-304P). 1-Step Ultra TMB-ELISA Substrate Solution was added as a substrate and the color change was stopped with 2M sulfuric acid.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases [AI138195] and the National Institute for General Medical Sciences [GM114179].
Abbreviations
- AUC
area under the curve
- MFI
mean fluorescence intensity
- PAR
pH-dependent affinity ratio
- PD
pharmacodynamics
- PHD
pH-dependent
- PK
pharmacokinetics
- scFv
single-chain variable fragment
- SEB
Staphylococcal enterotoxin B
Acknowledgments
This work was supported by the National Institutes of Health (R21 AI138195 to SP and DKS, R01 GM114179 to DKS). We acknowledge the use of the cell sorting facility at the Roswell Park Cancer Institute, Buffalo, NY.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Marshall MJE, Stopforth RJ, Cragg MS.. Therapeutic antibodies: what have we learnt from targeting CD20 and where are we going? Front Immunol. 2017;8:1245. doi: 10.3389/fimmu.2017.01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy PJ, Oliveira C, Granja PL, Sarmento B.. Monoclonal antibodies: technologies for early discovery and engineering. Crit Rev Biotechnol. 2018;38:394–408. doi: 10.1080/07388551.2017.1357002. [DOI] [PubMed] [Google Scholar]
- 3.Singh S, Kumar NK, Dwiwedi P, Charan J, Kaur R, Sidhu P, Chugh VK Monoclonal Antibodies: A Review. Curr Clin Pharmacol 2018;13:85-99. doi: 10.2174/1574884712666170809124728. [DOI] [PubMed] [Google Scholar]
- 4.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203-7. [DOI] [PubMed] [Google Scholar]
- 6.Schroter C, Gunther R, Rhiel L, Becker S, Toleikis L, Doerner A, Becker J, Schönemann A, Nasu D, Neuteboom B, et al. A generic approach to engineer antibody pH-switches using combinatorial histidine scanning libraries and yeast display. mAbs. 2015;7:138–151. doi: 10.4161/19420862.2014.985993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonvin P, Venet S, Fontaine G, Ravn U, Gueneau F, Kosco-Vilbois M, Proudfoot AE, Fischer N. De novo isolation of antibodies with pH-dependent binding properties. mAbs. 2015;7:294–302. doi: 10.1080/19420862.2015.1006993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu YB, Dammer EB, Ren RJ, Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4:18. doi: 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem. 2012;287:11090–11097. doi: 10.1074/jbc.M111.319764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devanaboyina SC, Lynch SM, Ober RJ, Ram S, Kim D, Puig-Canto A, Breen S, Kasturirangan S, Fowler S, Peng L, et al. The effect of pH dependence of antibody-antigen interactions on subcellular trafficking dynamics. mAbs. 2013;5:851–859. doi: 10.4161/mabs.26389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutta K, Varshney AK, Franklin MC, Goger M, Wang X, Fries BC. Mechanisms mediating enhanced neutralization efficacy of staphylococcal enterotoxin B by combinations of monoclonal antibodies. J Biol Chem. 2015;290:6715–6730. doi: 10.1074/jbc.M114.630715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, Devi VS, Stavale E, Warfield KL, Zeitlin L, et al. Synthetic human monoclonal antibodies toward staphylococcal enterotoxin B (SEB) protective against toxic shock syndrome. J Biol Chem. 2012;287:25203–25215. doi: 10.1074/jbc.M112.364075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong C, Neoh HM, Nathan S. Targeting staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins (Basel). 2016;8. doi: 10.3390/toxins8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia T, Liang S, Wang H, Hu S, Sun Y, Yu X, Han J, Li J, Guo S, Dai J, et al. Structural basis for the neutralization and specificity of Staphylococcal enterotoxin B against its MHC Class II binding site. mAbs. 2014;6:119–129. doi: 10.4161/mabs.27106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varshney AK, Wang X, Scharff MD, MacIntyre J, Zollner RS, Kovalenko OV, Martinez LR, Byrne FR, Fries BC. Staphylococcal enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J Infect Dis. 2013;208:2058–2066. doi: 10.1093/infdis/jit421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drozdowski B, Zhou Y, Kline B, Spidel J, Chan YY, Albone E, Turchin H, Chao Q, Henry M, Balogach J, et al. Generation and characterization of high affinity human monoclonal antibodies that neutralize staphylococcal enterotoxin B. J Immune Based Ther Vaccines. 2010;8:9. doi: 10.1186/1476-8518-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larkin EA, Stiles BG, Ulrich RG. Inhibition of toxic shock by human monoclonal antibodies against staphylococcal enterotoxin B. PLoS One. 2010;5:e13253. doi: 10.1371/journal.pone.0013253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varshney AK, Wang X, Cook E, Dutta K, Scharff MD, Goger MJ, Fries BC. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J Biol Chem. 2011;286:9737–9747. doi: 10.1074/jbc.M110.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boles JW, Pitt ML, LeClaire RD, Gibbs PH, Torres E, Dyas B, Ulrich RG, Bavari S. Generation of protective immunity by inactivated recombinant staphylococcal enterotoxin B vaccine in nonhuman primates and identification of correlates of immunity. Clin Immunol. 2003;108:51–59. [DOI] [PubMed] [Google Scholar]
- 20.Asensi GF, de Sales NF, Dutra FF, Feijo DF, Bozza MT, Ulrich RG, Miyoshi A, de Morais K, Azevedo VADC, Silva JT, et al. Oral immunization with Lactococcus lactis secreting attenuated recombinant staphylococcal enterotoxin B induces a protective immune response in a murine model. Microb Cell Fact. 2013;12:32. doi: 10.1186/1475-2859-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherrmann J-M. Antibody treatment of toxin poisoning recent advances. J Toxicol. 1994;32:363–375. [DOI] [PubMed] [Google Scholar]
- 22.Pepper LR, Cho YK, Boder ET, Shusta EV. A decade of yeast surface display technology: where are we now? Comb Chem High Throughput Screen. 2008;11:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaccolo M, Gherardi E. The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 beta-lactamase. J Mol Biol. 1999;285:775–783. doi: 10.1006/jmbi.1998.2262. [DOI] [PubMed] [Google Scholar]
- 24.Huotari J, Helenius A. Endosome maturation. Embo J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thilo L, Stroud E, Haylett T. Maturation of early endosomes and vesicular traffic to lysosomes in relation to membrane recycling. J Cell Sci. 1995;108(Pt 4):1791–1803. [DOI] [PubMed] [Google Scholar]
- 26.Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol. 1999;292:949–956. doi: 10.1006/jmbi.1999.3130. [DOI] [PubMed] [Google Scholar]
- 27.Killikelly A, Zhang HT, Spurrier B, Williams C, Gorny MK, Zolla-Pazner S, Kong X-P. Thermodynamic signatures of the antigen binding site of mAb 447-52D targeting the third variable region of HIV-1 gp120. Biochemistry. 2013;52:6249–6257. doi: 10.1021/bi400645e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bostrom J, Haber L, Koenig P, Kelley RF, Fuh G. High affinity antigen recognition of the dual specific variants of herceptin is entropy-driven in spite of structural plasticity. PLoS One. 2011;6:e17887. doi: 10.1371/journal.pone.0017887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakraborty A, Van LM, Skerjanec A, Floch D, Klein UR, Krammer G, Sunkara G, Howard D. Pharmacokinetic and pharmacodynamic properties of canakinumab in patients with gouty arthritis. J Clin Pharmacol. 2013;53:1240–1251. doi: 10.1002/jcph.162. [DOI] [PubMed] [Google Scholar]
- 30.Fetterly GJ, Aras U, Meholick PD, Takimoto C, Seetharam S, McIntosh T, de Bono JS, Sandhu SK, Tolcher A, Davis HM, et al. Utilizing pharmacokinetics/pharmacodynamics modeling to simultaneously examine free CCL2, total CCL2 and carlumab (CNTO 888) concentration time data. J Clin Pharmacol. 2013;53:1020–1027. doi: 10.1002/jcph.140. [DOI] [PubMed] [Google Scholar]
- 31.Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, Demattos RB. Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin Neuropharmacol. 2010;33:67–73. doi: 10.1097/WNF.0b013e3181cb577a. [DOI] [PubMed] [Google Scholar]
- 32.Xiao JJ, Krzyzanski W, Wang YM, Li HY, Rose MJ, Ma M, Wu Y, Hinkle B, Perez-Ruixo JJ. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. Aaps J. 2010;12:646–657. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. Isotype switching increases efficacy of antibody protection against staphylococcal enterotoxin B-induced lethal shock and Staphylococcus aureus sepsis in mice. MBio. 2014;5:e01007–14. doi: 10.1128/mBio.01007-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fraser JD. Clarifying the mechanism of superantigen toxicity. PLoS Biol. 2011;9:e1001145. doi: 10.1371/journal.pbio.1001145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fries BC, Varshney AK. Bacterial Toxins-Staphylococcal Enterotoxin B. Microbiol Spectr 2013;1:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.VanAntwerp JJ, Wittrup KD. Fine affinity discrimination by yeast surface display and flow cytometry. Biotechnol Prog. 2000;16:31–37. doi: 10.1021/bp990133s. [DOI] [PubMed] [Google Scholar]
- 37.Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol. 2006;357:100–114. doi: 10.1016/j.jmb.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 38.Heinrich L, Tissot N, Hartmann DJ, Cohen R. Comparison of the results obtained by ELISA and surface plasmon resonance for the determination of antibody affinity. J Immunol Methods. 2010;352:13–22. doi: 10.1016/j.jim.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 40.Yang D, Giragossian C, Castellano S, Lasaro M, Xiao H, Saraf H, Hess Kenny C, Rybina I, Huang Z-F, Ahlberg J, et al. Maximizing in vivo target clearance by design of pH-dependent target binding antibodies with altered affinity to FcRn. mAbs. 2017;9:1105–1117. doi: 10.1080/19420862.2017.1359455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 42.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1:755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.