

Abstract
Purpose
Leiomyosarcoma (LMS) is a soft-tissue sarcoma characterized by multiple copy number alterations (CNAs) and without common recurrent single-nucleotide variants. We evaluated the feasibility of detecting circulating tumor DNA (ctDNA) with next-generation sequencing in a cohort of patients with LMS whose tumor burden ranged from no evidence of disease to metastatic progressive disease.
Patients and Methods
We evaluated cell-free DNA in plasma samples and paired genomic DNA from resected tumors from patients with LMS by ultra-low passage whole-genome sequencing. Sequencing reads were aligned to the human genome and CNAs that were identified in cell-free DNA and tumor DNA by ichorCNA software to determine the presence of ctDNA. Clinical data were reviewed to assess disease burden and clinicopathologic features.
Results
We identified LMS ctDNA in 11 (69%) of 16 patients with disease progression and total tumor burden greater than 5 cm. Sixteen patients with stable disease or low disease burden at the time of blood draw were found to have no detectable ctDNA. Higher ctDNA fraction of total cell-free DNA was associated with increasing tumor size and disease progression. Conserved CNAs were found between primary tumors and ctDNA in each case, and recurrent CNAs were found across LMS samples. ctDNA levels declined after resection of progressive disease in one case and became detectable upon disease relapse in another individual patient.
Conclusion
These results suggest that ctDNA, assayed by a widely available sequencing approach, may be useful as a biomarker for a subset of patients with uterine and extrauterine LMS. Higher levels of ctDNA correlate with tumor size and disease progression. Liquid biopsies may assist in guiding treatment decisions, monitoring response to systemic therapy, surveying for disease recurrence, and differentiating benign and malignant smooth muscle tumors.
INTRODUCTION
Leiomyosarcoma (LMS) is a malignant neoplasm derived from smooth muscle that represents one of the most common subtypes of soft-tissue sarcoma.1,2 Although the majority of LMS cases are sporadic, predisposing factors include Li-Fraumeni syndrome, hereditary retinoblastoma, and radiation exposure.3,4 There are no oncogenic single-nucleotide variants (SNVs) that characterize LMS, although loss of tumor suppressors, including TP53, RB1, and PTEN, are commonly observed, as are multiple copy number alterations (CNAs).5-8 LMS is frequently a clinically aggressive disease, and patients are at high risk for local and metastatic relapse after initial complete resection.9,10 Efforts to improve outcomes for patients would benefit from more reliable indicators of high-risk disease and biomarkers of response to therapy.
Detection of circulating tumor DNA (ctDNA) has emerged as a new approach for identifying oncogenic mutations, measuring disease burden, clinical prognostication, and assessing tumor response to therapy.11,12 Most ctDNA assays have been developed to detect SNVs that are highly recurrent in many types of carcinomas.13 Although the lack of recurrent SNVs in LMS limits efforts at targeted sequencing, the numerous CNAs that are characteristic of this disease represent an ideal target for detection. A next-generation sequencing approach using ultra-low passage whole-genome sequencing (ULP-WGS) can detect CNAs in cell-free DNA, which indicates the tumor fraction of ctDNA present in blood.14,15 Previous studies have shown that ctDNA can be detected in cell-free DNA samples that are sequenced at a minimum coverage of 0.1× across the whole genome.14 Sequencing for ULP-WGS uses the standard Illumina next-generation sequencing platform without modifications or special adaptions (Illumina, San Diego, CA). The ichorCNA algorithm is used to detect megabase-scale CNAs from ULP-WGS data in which ctDNA comprise as little as 3% of the total cell-free DNA extracted from a plasma sample.14
In the current study, we evaluated plasma from patients with uterine and extrauterine LMS for the presence of ctDNA using ULP-WGS. Paired resected tumors from each patient were also sequenced when available (29 of 30 patients), which enabled the identification and comparison of CNAs between primary tumors and ctDNA. We related the tumor fraction of cell-free DNA with the clinical status of the patient’s disease. Finally, we found that longitudinal measurements of ctDNA declined with tumor resection in one patient and in another patient became detectable at the time of disease recurrence. These results suggest that monitoring ctDNA may have clinical utility in establishing diagnosis, estimating prognosis, measuring treatment response, and performing surveillance for relapse in patients with LMS.
PATIENTS AND METHODS
Patients
Patients with LMS who had previously provided written consent for enrollment in an institutional review board–approved sample banking and research protocol, including collection of clinical data, and who underwent surgery and treatment at Brigham and Women’s Hospital and Dana-Farber Cancer Institute were reviewed for inclusion in the evaluated cohort. Inclusion required a diagnosis of LMS by pathologic review of a surgically resected specimen. We identified 30 patients with at least one plasma sample, 29 of which had a matched tumor sample that was available for profiling. We also identified an additional eight patients with only tumor samples. Clinical, radiologic, and pathologic data were obtained from the medical record. All evaluated patients with plasma samples ultimately experienced either local or metastatic disease recurrence.
Sample Preparation
Patients had one or serial venous blood draws collected for research in EDTA tubes (Becton Dickinson, Franklin Lakes, NJ), and plasma was isolated within 4 hours of collection and stored at −80°C, as previously described.16 A QIAamp Circulating Nucleic Acid Kit (Qiagen, Venlo, the Netherlands) was used to isolate cell-free DNA from 3.4 mL to 5.0 mL of frozen plasma. Genomic DNA from LMS tumors was isolated from formalin-fixed, paraffin-embedded (FFPE) tissue using a QIAamp DNA FFPE Tissue kit (Qiagen) or fresh frozen tumors using a QIAamp DNA Mini-Kit (Qiagen).
ULP-WGS and ctDNA Quantification
Extracted DNA was quantified using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA). High-molecular-weight DNA contamination of cell-free DNA was determined by Bioanalyzer (Agilent Technologies, Santa Clara, CA) and size selection was performed if necessary (AMPure XP beads; Beckman Coulter, Brea, CA). DNA that was extracted from FFPE or fresh frozen tumors was fragmented (Covaris) to 250 bp and purified with AMPure XP beads. Up to 40 ng of cell-free DNA, 100 ng of DNA from fresh frozen tissue, and 200 ng of DNA from FFPE tissue were used for KAPA Hyper library preparation (Kapa Biosystems, Wilmington, MA). Libraries were assessed for quality by Bioanalyzer followed by quantification using the MiSeq Nano flow cell (Illumina). Barcoded libraries were pooled and we performed sequencing on a HiSeq 2500 in rapid run mode (Illumina) to a targeted coverage of 0.2× (actual range, 0.15× to 0.30×). Seventeen of 37 tumors were previously sequenced for a separate research project at higher coverage (range, 1.1× to 2.1×). Sequencing was performed after Bioruptor sonication (Diagenode) of formaldehyde-fixed fresh frozen sample, with library preparation using a ThruPLEX DNA sequencing kit (Rubicon) and sequencing on an Illumina NextSeq 500. This higher sequencing coverage did not alter downstream analyses or data interpretation.
Sequencing results were demultiplexed, aligned, and processed using Picard, Burrows-Wheeler Alignment tool,17 and Genome Analysis Toolkit.18,19 To assess for CNAs in tumor and cell-free DNA and determine tumor fraction or percentage of ctDNA in cell-free DNA, we used ichorCNA software14 with manual curation of results as necessary to confirm tumor percentages. Previous studies have demonstrated that this technique can be used to identify and quantitate ctDNA that constitutes as little as 3% of the cell-free DNA in a sample.14
Copy Number Analysis
Copy number segments and estimates of tumor fraction and ploidy were generated by ichorCNA. The log2 ratio values of segments were adjusted for tumor fraction and ploidy such that the data were consistent across samples. GISTIC2.020 was used to determine gene-level copy number analyses. For copy neutral segments predicted by ichorCNA, the log2 ratio was set to zero. The amplification/deletion log2 ratio threshold used for GISTIC was 0.3. Significant gains and losses were determined with a false discovery rate of 0.25. This analysis was performed separately on tumor resection samples, including the eight samples for which plasma was not available, and on plasma samples that were positive for detectable levels of ctDNA.
Statistical Analysis
Group comparisons between patients with LMS with active or indolent disease at the time of plasma collection was performed by nonparametric Mann-Whitney test, and we performed Pearson correlation coefficient between tumor fraction or cell-free DNA and tumor burden using GraphPad Prism (version 7.0; GraphPad Software, La Jolla, CA). One was added to integer values before log2 transformation to generate non-negative values for data representation.
RESULTS
Detection of LMS ctDNA
We retrospectively identified 30 patients who were diagnosed with metastatic LMS who had plasma banking performed at variable time points in their treatment history between January 2007 and November 2017. Two of these patients had plasma samples banked at two separate time points for a total of 32 plasma samples. Median age at diagnosis was 51 years, most patients were female, the most common primary tumor location was the uterus (n = 16) followed by the retroperitoneum (n = 8), and most tumors were originally of intermediate or high grade (Table 1). Twenty-nine of 30 patients had a tumor resection sample available for comparison with cell-free DNA.
Table 1.
Clinical Characteristics of 30 Leiomyosarcoma Cases With Plasma Samples

In review of clinical data at the time of plasma banking, the cases could be divided into active disease or indolent disease groups on the basis of tumor volume and evidence of disease progression. The active disease group consisted of 16 patients and was defined by having a total tumor burden greater than 5 cm in greatest diameter and progressive disease at the time of blood draw on the basis of imaging or clinical determination. In contrast, 16 patients in the indolent disease group had stable disease at the time of blood draw and/or tumor burden less than 5 cm. Among the active disease group, 11 (69%) of 16 patients had detectible ctDNA, including patients with both uterine and extrauterine primary tumors. None of the samples obtained from the indolent disease group had detectible ctDNA (Fig 1A).
Fig 1.
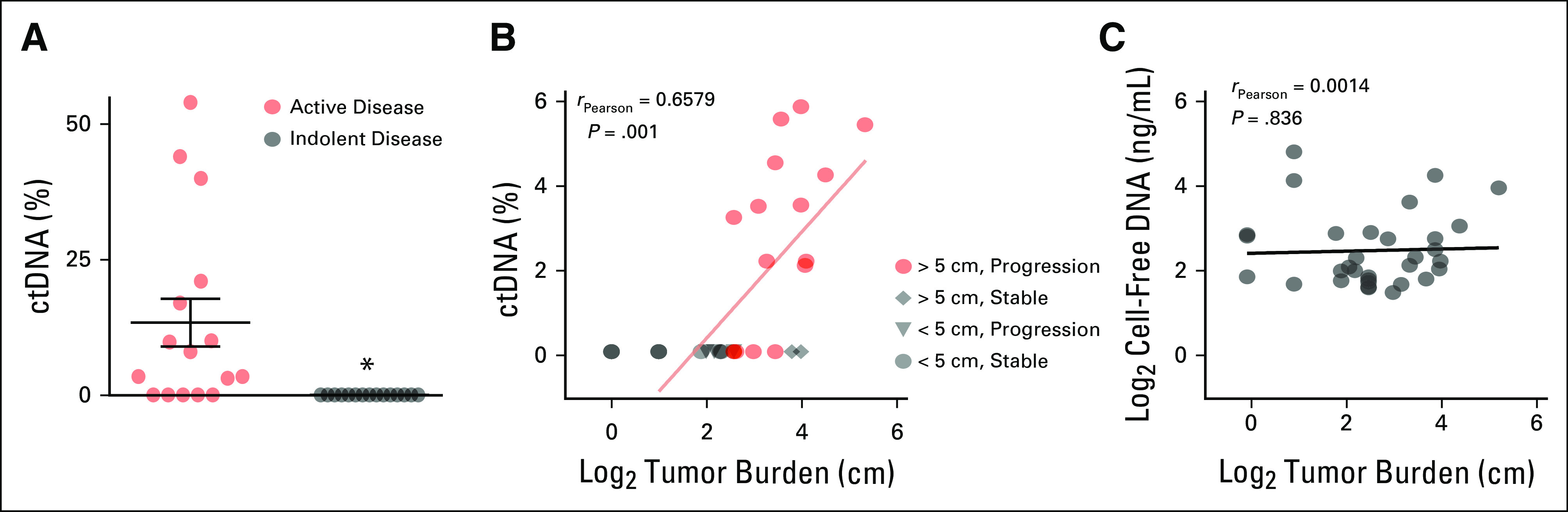
Identification of circulating tumor DNA (ctDNA) in plasma from patients with leiomyosarcoma (LMS). (A) Plot of percent ctDNA in active disease (n = 16) and indolent disease (n = 16) LMS groups. The active disease group consists of patients with tumors > 5 cm in size and with progressive disease at the time of blood draw as indicated by computed tomography (CT) scan or clinical report. The indolent disease group consists of patients with tumors < 5 cm in size and/or no evidence of disease progression by CT scan. Groups were compared using Mann-Whitney test. (*) P < .001. (B) Scatterplot of log2 tumor fraction and tumor burden. Labels indicate the active disease subgroup (red) with tumor size > 5 cm and progressive disease (n = 16), or indolent disease subgroups (gray) with tumor size > 5 cm and stable disease (n = 2, diamond), tumor size < 5 cm and progressive disease (n = 4; triangle), and tumor size < 5 cm and stable disease (n = 10; circle). Tumor burden was determined by adding the diameters of the tumor lesions reported on CT scan at the time of ctDNA assessment. Pearson correlation coefficient between tumor fraction and tumor burden is shown for all patients with measurable disease. (C) Scatterplot of log2 total extracted cell-free DNA from all plasma samples (n = 32) and tumor burden. Pearson correlation coefficient across all samples is shown.
When comparing the amount of ctDNA—measured as a fraction of total cell-free DNA—in the plasma sample to the volume of tumor burden reported by computed tomography scan, there was a significant association between higher ctDNA levels and increasing tumor burden (Fig 1B). In contrast, there was no correlation between the amount of total cell-free DNA extracted from the plasma sample and tumor burden (Fig 1C). Several patients in the indolent disease group of similarly high tumor burden produced no detectible ctDNA despite significant tumor volume. Taken together, these data demonstrate that ctDNA is detectible using a ULP-WGS approach in the majority of patients with LMS with a tumor burden of greater than 5 cm and progressive disease.
CNA Concordance Between LMS Tumors and ctDNA
In the 11 plasma samples with detectible levels of ctDNA, there was a high concordance of CNAs between the tumor sample and ctDNA (Figs 2A-2D), with blood collection occurring less than 2 years apart from resection of the matched tumor specimen. In cases of high ctDNA with lower tumor fraction in the surgical sample, CNAs were more pronounced in cell-free DNA (Fig 2A). In contrast, with low tumor fraction, many CNAs observed in the surgical sample could not be readily identified (Figs 2D and 2E). These results are consistent with those of prior publications that demonstrated agreement between plasma and tumor CNAs21 and further underscores the diversity of chromosomal gains and losses observed across LMS tumors.
Fig 2.
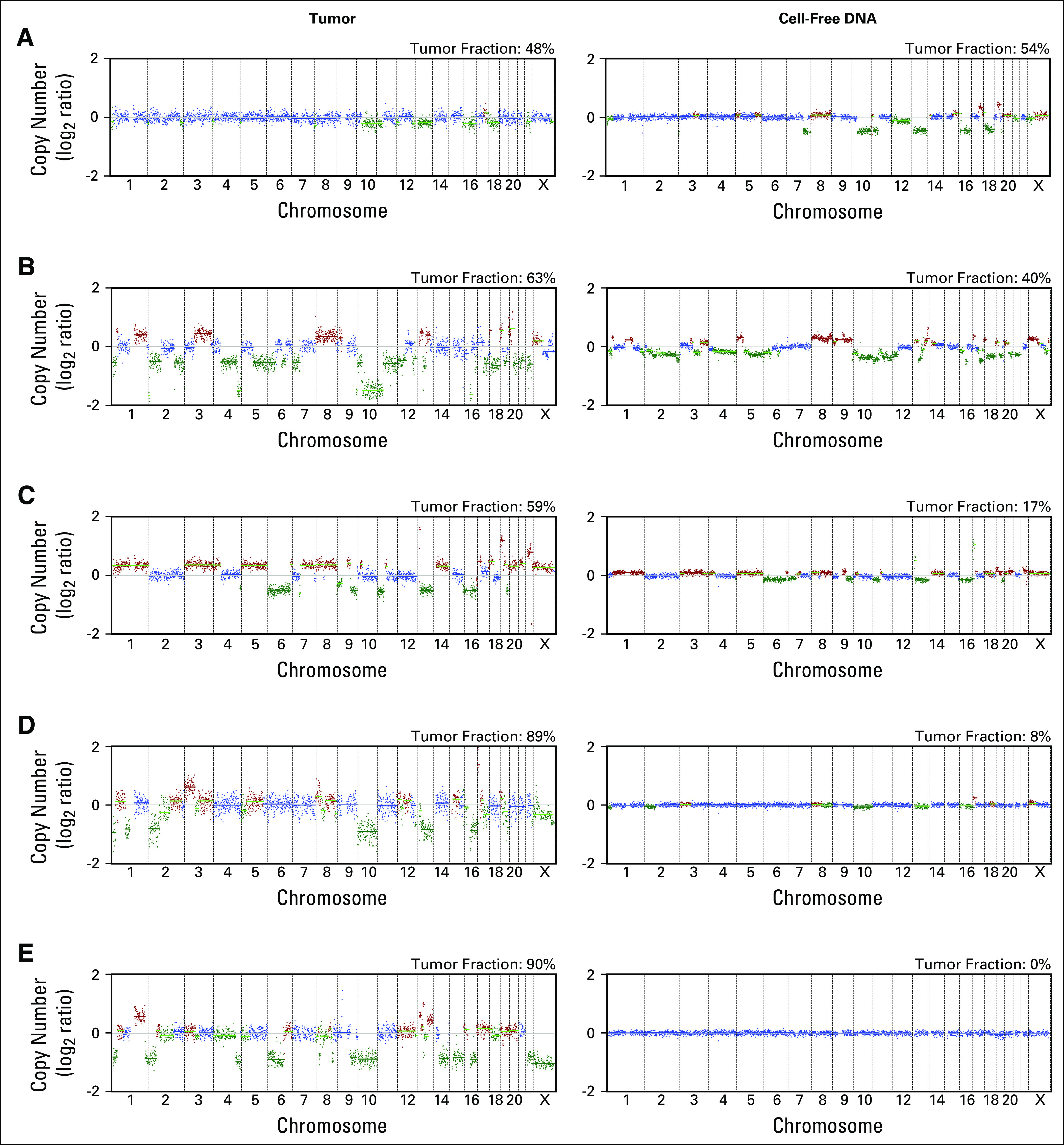
Concordant copy number alterations in leiomyosarcoma tumors and circulating tumor DNA. (A-E) Copy number plots generated from ultra-low passage whole-genome sequencing of five representative tumor surgical (left panels) and plasma (right panels) sample pairs. The x-axis indicates chromosome and y-axis copy number (log2 ratio). Tumor fraction is indicated for each plot.
We used GISTIC2.020 to characterize recurrent gene-level CNAs in tumor and cell-free DNA in this LMS cohort. Overall, recurrent CNAs from tumors in this cohort are remarkably similar to those found in prior studies.5,8 The most significantly amplified genomic region was found on chromosome 17p, which includes the transcriptional regulator MYOCD (copy gained in 20 of 37 tumors). This gene has previously been reported as significantly amplified in LMS and is associated with smooth muscle differentiation.22 Additional putative oncogenic and LMS-associated genes that were recurrently amplified include HDGF in 20, MYC in 14, CTHRC1 in 17, TNFRSF19 in 10, and MYH2 in 16 of 37 tumors (Fig 3A). Copy gains in these genes were also observed in a portion of cell-free DNA samples with detectable ctDNA (MYOCD in four, HDGF in six, MYC in eight, CTHRC1 in nine, TNFRSF19 in three, and MYH2 in four of 11 ctDNA-positive samples); however, the number of evaluable samples was too small for any copy gains of these genes to reach statistical significance (Fig 3B). From tumor samples, recurrent deletions in tumor suppressors were found, which are characteristic of LMS,5,8,23 including PTEN deletions in 27, RB1 in 27, and TP53 in 11 of 37 tumors profiled (Fig 3C). Deletions that involved these genes were also detected in ctDNA (PTEN in eight, RB1 in six, and TP53 in two of 11 ctDNA-positive samples), but sample size was too small for many of these deletions to reach significance (Fig 3D). As described in previous studies, gains and losses of these genes were caused by events that ranged from focal (megabase scale) to whole-chromosomal CNAs. Thus, ULP-WGS is capable of detecting recurrent CNAs characteristic of LMS, and these methods may be helpful in identifying gene-level CNAs in ctDNA.
Fig 3.
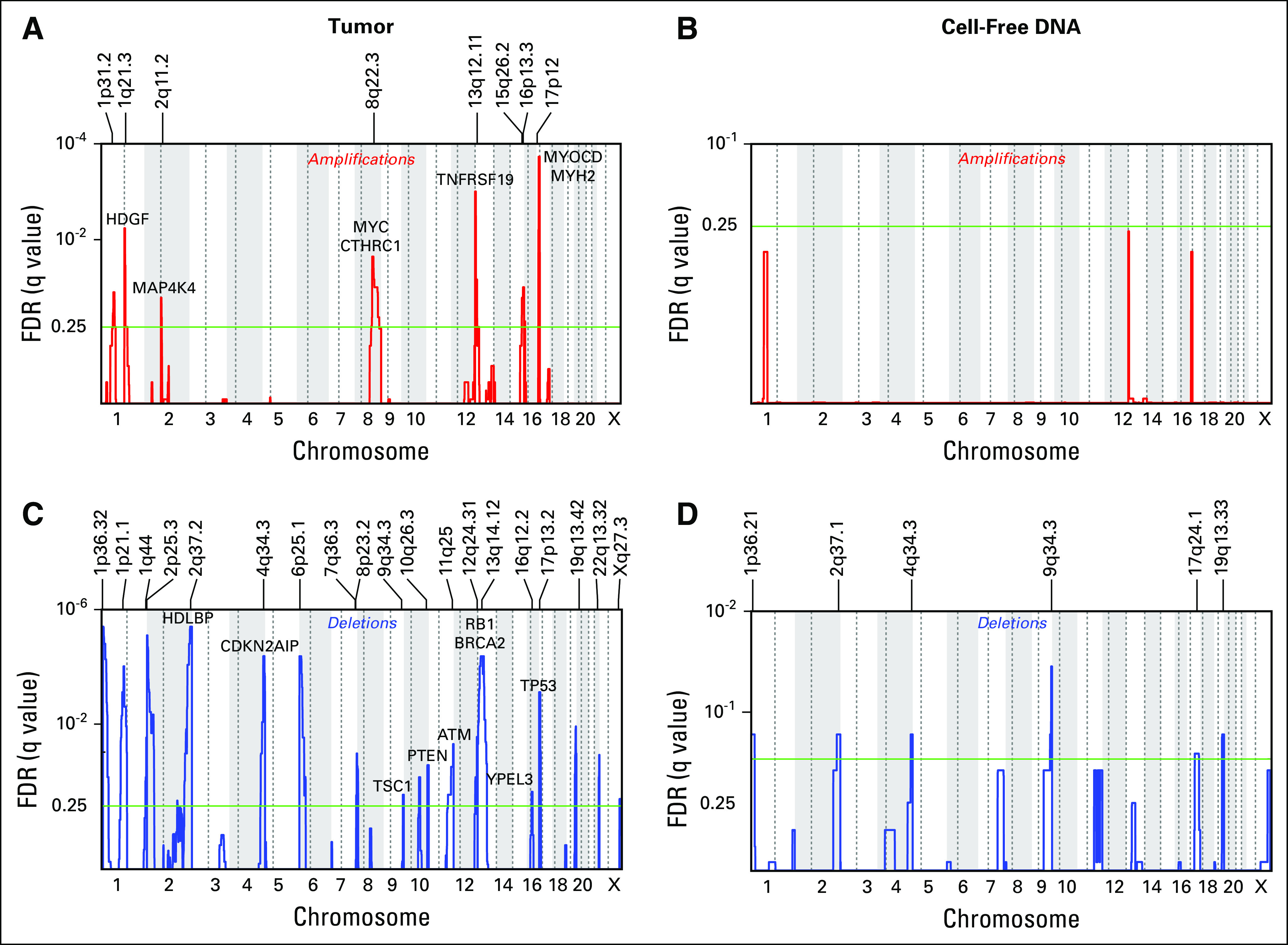
Recurrent copy number alterations in leiomyosarcoma (LMS). (A-D) GISTIC2.0 analysis identifying recurrent focal amplified (A and B) and deleted (C and D) regions in LMS tumors (n = 37; left panels) and cell-free DNA samples with detectable circulating tumor DNA (n = 11; right panels). LMS-associated genes and/or putative oncogenes are labeled in panel A, whereas putative tumor suppressor genes are labeled in panel C. The x-axis indicates chromosome and y-axis false discovery rate (FDR). The green line indicates an FDR of 0.25.
ctDNA Longitudinally Correlates With Disease Status
To determine whether ctDNA levels change in agreement with longitudinal disease status, we evaluated serial plasma samples in two patients after either resection of localized LMS or with disease recurrence after surgery. In the patient who underwent resection of locally recurrent disease, ctDNA detected preoperatively was eliminated 5 weeks after surgery (Fig 4A). In contrast, in the patient with disease recurrence 32 months after surgical resection, cell-free DNA tumor fraction increased from undetectable postoperatively to detectable at the time of disease recurrence (Fig 4B). These data demonstrate that ctDNA correlates in individual patients with disease burden over time in this small cohort and that such liquid biopsies may represent a valuable diagnostic tool to support evidence of LMS recurrence or disease burden.
Fig 4.
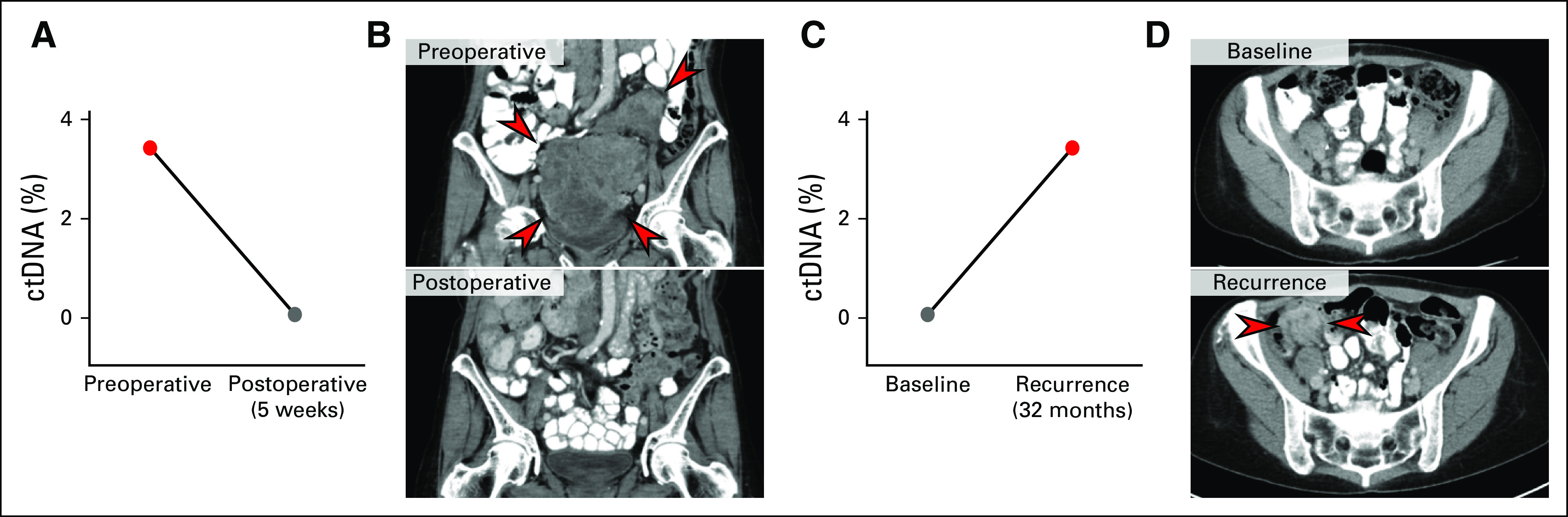
Leiomyosarcoma circulating tumor DNA (ctDNA) correlates with changes in disease burden. (A and B) Change in tumor fraction in the preoperative and postoperative setting, with images indicating tumor burden at each time point. (C and D) Change in tumor fraction after unifocal recurrence of disease, with images indicating tumor burden at each time point. The interval between surgical date and cell-free DNA collection is indicated.
DISCUSSION
In the current study, we analyzed cell-free DNA from the plasma of patients with LMS for evidence of ctDNA. Stratifying patients by tumor burden and active versus indolent disease, we found that the majority of patients with active disease have detectable ctDNA. A significant association between tumor burden and the amount of ctDNA was identified, which suggests that larger and actively growing tumors are more likely to release tumor DNA into circulation. We were unable to detect ctDNA in any tumor less than 5 cm in size, regardless of evidence of disease progression, which may represent a technologic sensitivity threshold. In addition, we were unable to detect ctDNA in patients with significant disease burden but with a lack of evidence of disease progression. This presumably arises from reduced levels of apoptosis or necrosis in tumors without active growth, which may be exploited to determine the therapeutic benefit of antineoplastic therapies. DNA from tumor resections and associated ctDNA demonstrated preserved CNAs, and recurrent CNAs were found across LMS samples. We further found that ctDNA levels decline with resection of disease and increase with disease recurrence in individual patients, which indicates that these methods may be useful for supporting a diagnosis of disease recurrence or response to therapy.
Sequencing of SNVs in cell-free DNA has proven clinical utility in detecting resistance mutations to targeted therapies and directing treatment strategies.24 Emerging use of whole-genome sequencing to identify and quantify CNAs from tumor-derived DNA circulating in patients with cancer has been evaluated in a number of cancer types.14,25-27 This noninvasive approach can be useful in the genomic characterization of malignancy and provide diagnostic and prognostic information relevant for clinical care.15,21,28
In a recent study of patients with LMS, a highly customized LMS-specific assay (Cancer Personalized Profiling by Deep Sequencing, or CAPP-Seq) was designed to detect selected SNVs and combined with CNA analysis to identify and quantify ctDNA in patients with progressive disease.29 In this study, the investigators found that approximately 20% of patients (two of nine) had tumors that did not have somatic events that were detectable by the assay. In contrast, we found that tumors from 100% of cases had CNAs that were detectable by ULP-WGS, which suggests that ULP-WGS may be more broadly applicable for patients with LMS. Conversely, CAPP-Seq was able to detect ctDNA in six of seven baseline samples of patients who were eligible for study (86%) and had a sensitivity of 68% among all samples tested from their cohort. In comparison, our study detected ctDNA in 11 (69%) of 16 patients with active disease. Although these numbers are small, this suggests that CAPP-Seq may have a higher sensitivity for ctDNA, which might be expected given the deep sequencing coverage used in this assay. It remains unclear what assay features will have the highest clinical utility in sarcomas, and we expect that different assays may be beneficial for different clinical indications. For example, our recent study in osteosarcoma demonstrated that pretreatment ctDNA levels detected by ULP-WGS are prognostic,28 whereas a more sensitive assay, such as CAPP-Seq, may be advantageous in the setting of disease surveillance.29
There are several potential clinical uses of ctDNA in the diagnosis and management of patients with LMS. First, ctDNA may be able to differentiate benign smooth muscle neoplasms, such as leiomyoma, from LMS.30,31 This knowledge would principally inform surveillance of suggestive lesions and considerations of operative strategies for uterine tumors that could potentially harbor LMS.32 Second, there is significant clinical uncertainty regarding which patients with LMS derive benefit from adjuvant chemotherapy and radiation.10,33,34 Should ctDNA levels bear prognostic significance for tumors that are at highest risk of recurrence or identify the presence of residual disease, their measurement may help guide clinical decisions regarding adjuvant therapies. Finally, ctDNA levels may be a useful indicator of response to systemic therapy and provide an early indication for switching or intensifying treatment regimens used in this disease.3,35 Together with technologic improvements in sensitivity and throughput, these initial reports that identify ctDNA in LMS may quickly evolve to transform clinical practice. Larger prospective studies will be needed to determine the optimal approaches for adapting ctDNA assays to the clinical care of patients with LMS.
ACKNOWLEDGMENT
The authors are indebted to the patients and families who donated clinical samples that enabled this research.
Footnotes
Supported by the American Society of Oncology Conquer Cancer Foundation Young Investigator Award (M.L.H.), National Institutes of Health Grant No. K08 CA188073-01A1 (B.D.C.), Friends of Dana-Farber Cancer Institute (B.D.C.), the Catherine England Leiomyosarcoma Fund (S.G.), and The Jill Effect (S.G.).
Presented at the 2018 Annual Meeting of the American Society of Clinical Oncology.
AUTHOR CONTRIBUTIONS
Conception and design: Matthew L. Hemming, Kelly Klega, Anwesha Nag, Suzanne George, Brian D. Crompton
Financial support: Matthew L. Hemming, Brian D. Crompton
Administrative support: Brian D. Crompton
Provision of study material or patients: Matthew L. Hemming, Chandrajit P. Raut, Brian D. Crompton
Collection and assembly of data: Matthew L. Hemming, Kelly Klega, Kate E. Acker, Jessica L. Andersen, Edwin Thai, Anwesha Nag, Aaron R. Thorner, Suzanne George, Brian D. Crompton
Data analysis and interpretation: Matthew L. Hemming, Kelly Klega, Justin Rhoades, Gavin Ha, Anwesha Nag, Aaron R. Thorner, Chandrajit P. Raut, Suzanne George, Brian D. Crompton
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Matthew L. Hemming
No relationship to discloses
Kelly Klega
No relationship to discloses
Justin Rhoades
No relationship to discloses
Gavin Ha
No relationship to discloses
Kate E. Acker
No relationship to discloses
Jessica L. Andersen
No relationship to discloses
Edwin Thai
No relationship to discloses
Anwesha Nag
No relationship to discloses
Aaron R. Thorner
No relationship to discloses
Chandrajit P. Raut
No relationship to discloses
Suzanne George
Stock and Other Ownership Interests: Abbott Laboratories, AbbVie (I), Allergan (I)
Consulting or Advisory Role: Blueprint Medicines, Deciphera, AstraZeneca, Blueprint Medicines, Bayer, Eli Lilly
Research Funding: Pfizer (Inst), Novartis (Inst), Bayer (Inst), Ariad Pharmaceuticals (Inst), Blueprint Medicines (Inst), Deciphera (Inst)
Patents, Royalties, Other Intellectual Property: UpToDate
Expert Testimony: Bayer
Other Relationship: Research to Practice
Brian D. Crompton
Employment: Mersana (I), Shire (I)
Stock and Other Ownership Interests: Mersana (I), Shire (I)
REFERENCES
- 1.Toro JR, Travis LB, Wu HJ, et al. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the Surveillance, Epidemiology and End Results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119:2922–2930. doi: 10.1002/ijc.22239. [DOI] [PubMed] [Google Scholar]
- 2.Ducimetière F, Lurkin A, Ranchère-Vince D, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One. 2011;6:e20294. doi: 10.1371/journal.pone.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George S, Serrano C, Hensley ML, et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol. 2018;36:144–150. doi: 10.1200/JCO.2017.75.9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjerkehagen B, Smeland S, Walberg L, et al. Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta Oncol. 2008;47:1475–1482. doi: 10.1080/02841860802047387. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171:950.e28–965.e28. doi: 10.1016/j.cell.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otaño-Joos M, Mechtersheimer G, Ohl S, et al. Detection of chromosomal imbalances in leiomyosarcoma by comparative genomic hybridization and interphase cytogenetics. Cytogenet Cell Genet. 2000;90:86–92. doi: 10.1159/000015640. [DOI] [PubMed] [Google Scholar]
- 7.Larramendy ML, Kaur S, Svarvar C, et al. Gene copy number profiling of soft-tissue leiomyosarcomas by array-comparative genomic hybridization. Cancer Genet Cytogenet. 2006;169:94–101. doi: 10.1016/j.cancergencyto.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Chudasama P, Mughal SS, Sanders MA, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Commun. 2018;9:144. doi: 10.1038/s41467-017-02602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gladdy RA, Qin L-X, Moraco N, et al. Predictors of survival and recurrence in primary leiomyosarcoma. Ann Surg Oncol. 2013;20:1851–1857. doi: 10.1245/s10434-013-2876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hensley ML, Ishill N, Soslow R, et al. Adjuvant gemcitabine plus docetaxel for completely resected stages I-IV high grade uterine leiomyosarcoma: Results of a prospective study. Gynecol Oncol. 2009;112:563–567. doi: 10.1016/j.ygyno.2008.11.027. [DOI] [PubMed] [Google Scholar]
- 11.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 13.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324. doi: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klega K, Imamovic-Tuco A, Ha G, et al. Detection of somatic structural variants enables quantification and characterization of circulating tumor DNA in children with solid tumors. JCO Precis Oncol. doi: 10.1200/PO.17.00285. doi:10.1200/PO.17.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wollison BM, Thai E, Mckinney A, et al. Blood collection in cell-stabilizing tubes does not impact germline DNA quality for pediatric patients. PLoS One. 2017;12:e0188835. doi: 10.1371/journal.pone.0188835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stover DG, Parsons HA, Ha G, et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J Clin Oncol. 2018;36:543–553. doi: 10.1200/JCO.2017.76.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pérot G, Derré J, Coindre J-M, et al. Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Res. 2009;69:2269–2278. doi: 10.1158/0008-5472.CAN-08-1443. [DOI] [PubMed] [Google Scholar]
- 23.Cuppens T, Moisse M, Depreeuw J, et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int J Cancer. 2018;142:1230–1243. doi: 10.1002/ijc.31129. [DOI] [PubMed] [Google Scholar]
- 24.Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J Clin Oncol. 2016;34:3375–3382. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Roy N, Van Der Linden M, Menten B, et al. Shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin Cancer Res. 2017;23:6305–6314. doi: 10.1158/1078-0432.CCR-17-0675. [DOI] [PubMed] [Google Scholar]
- 26.Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4:162ra154. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heitzer E, Ulz P, Belic J, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5:30. doi: 10.1186/gm434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shulman DS, Klega K, Imamovic-Tuco A, et al. Detection of circulating tumour DNA is associated with inferior outcomes in Ewing sarcoma and osteosarcoma: A report from the Children’s Oncology Group. Br J Cancer. 2018;119:615–621. doi: 10.1038/s41416-018-0212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Przybyl J, Chabon JJ, Spans L, et al. Combination approach for detecting different types of alterations in circulating tumor DNA in leiomyosarcoma. Clin Cancer Res. 2018;24:2688–2699. doi: 10.1158/1078-0432.CCR-17-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehine M, Kaasinen E, Mäkinen N, et al. Characterization of uterine leiomyomas by whole-genome sequencing. N Engl J Med. 2013;369:43–53. doi: 10.1056/NEJMoa1302736. [DOI] [PubMed] [Google Scholar]
- 31.Mehine M, Kaasinen E, Heinonen H-R, et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc Natl Acad Sci USA. 2016;113:1315–1320. doi: 10.1073/pnas.1518752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raspagliesi F, Maltese G, Bogani G, et al. Morcellation worsens survival outcomes in patients with undiagnosed uterine leiomyosarcomas: A retrospective MITO group study. Gynecol Oncol. 2017;144:90–95. doi: 10.1016/j.ygyno.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Hensley ML, Wathen JK, Maki RG, et al. Adjuvant therapy for high-grade, uterus-limited leiomyosarcoma: Results of a phase 2 trial (SARC 005) Cancer. 2013;119:1555–1561. doi: 10.1002/cncr.27942. [DOI] [PubMed] [Google Scholar]
- 34.Reed NS, Mangioni C, Malmström H, et al. Phase III randomised study to evaluate the role of adjuvant pelvic radiotherapy in the treatment of uterine sarcomas stages I and II: An European Organisation for Research and Treatment of Cancer Gynaecological Cancer Group Study (protocol 55874) Eur J Cancer. 2008;44:808–818. doi: 10.1016/j.ejca.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Amant F, Coosemans A, Debiec-Rychter M, et al. Clinical management of uterine sarcomas. Lancet Oncol. 2009;10:1188–1198. doi: 10.1016/S1470-2045(09)70226-8. [DOI] [PubMed] [Google Scholar]