

Abstract
Objective:
Worldwide, most new HIV infections occur through mucosal exposure. Immunoglobulin M (IgM) is the first antibody class generated in response to infectious agents; IgM is present in the systemic circulation and in mucosal fluids as secretory IgM. We sought to investigate for the first time the role of IgM in preventing AIDS virus acquisition in vivo.
Design:
Recombinant polymeric monoclonal IgM was generated from the neutralizing monoclonal IgG1 antibody 33C6-IgG1, tested in vitro, and given by passive intrarectal (i.r.) immunization to rhesus macaques (RMs) 30 min before i.r. challenge with simian-human immunodeficiency virus (SHIV) that carries an HIV-1 envelope gene.
Results:
In vitro, 33C6-IgM captured virions more efficiently and neutralized the challenge SHIV with a 50% inhibitory molar concentration (IC50) that was 1 log lower than that for 33C6-IgG1. The IgM form also exhibited significantly higher affinity and avidity compared to 33C6-IgG1. After i.r. administration, 33C6-IgM prevented viremia in four out of six rhesus macaques after high-dose intrarectal SHIV challenge. Five out of six RMs given 33C6-IgG1 were protected at a five times higher molar concentration compared to the IgM form; all untreated controls became highly viremic. RMs passively immunized with 33C6-IgM with breakthrough infection had notably early development of autologous neutralizing antibody responses.
Conclusion:
Our primate model data provide the first proof-of-concept that mucosal IgM can prevent mucosal HIV transmission and have implications for HIV prevention and vaccine development.
Keywords: IgM, R5 SHIV, HIV neutralization, mucosal transmission, virion capture
Introduction
Worldwide, ~90% of all HIV infections occur through mucosal exposure and almost always involve CCR5 (R5)-tropic HIV strains that are relatively difficult to neutralize (tier 2). After acute infections, IgM is the first antibody (Ab) class to respond. IgM is the only Ab present in all vertebrates [1]. It is required for the maturation of IgG responses [2], regulation of B cell development [3], modulating inflammatory responses [4], agglutination of pathogens, and clearance of apoptotic cells via complement activation [5]. IgM exists as dimer on the surface of B cells, forming the B-cell receptor [6]. Plasma IgM is mainly pentameric and contains the joining (J)-chain [7]. At mucosal sites, IgM is produced locally by plasma cells in the lamina propria. After its production, IgM binds to the polymeric immunoglobulin receptor (pIgR) expressed on the basolateral surface of the epithelial barrier to form pIgR–IgM complexes. The latter are transported across the epithelial monolayer in transcytotic vesicles and released at the luminal side through proteolytic cleavage of pIgR. This process results in the release of secretory component (SC) that remains associated with IgM, thus generating secretory IgM (SIgM). The role of IgM in preventing HIV transmission is currently unknown.
Preclinical vaccine efficacy studies rely on nonhuman primate models, especially Indian-origin rhesus macaques (RMs). However, because the envelope of the simian immunodeficiency virus (SIV) is so divergent that Abs against one do not recognize the other, simian–human immunodeficiency viruses (SHIVs) have been constructed; these chimeras carry HIV env in a SIVmac239 backbone. The R5 SHIV/RM model is used to assess the protective potential of recombinant human anti-HIV Env monoclonal antibodies (mAbs) by passive immunization.
Here we sought to test whether recombinant monoclonal IgM given mucosally could prevent infection of RMs after i.r. SHIV challenge. Two thirds of 33C6-IgM-treated RMs were completely protected, and in those with breakthrough infection, autologous neutralizing Abs appeared earlier compared to untreated controls. Our data reveal for the first time the protective potential of mucosal anti-HIV IgM.
Methods
Cell lines, reagents and virus
TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, from J.C. Kappes, X. Wu and Tranzyme Inc. SHIV-1157ip gp120 was prepared as described [8]. SHIV-1157ipEL-p stock (grown in RM peripheral blood mononuclear cells (PBMC)) had a p27 concentration of 792 ng/ml and 7.8 × 105 50% tissue culture infectious doses (TCID50)/ml (measured in TZM-bl cells).
Preparation of 33C6 mAbs and in vitro assays, including surface plasmon resonance (SPR) and dynamic light scattering (DLS)
We previously described the production of 33C6-IgG1 mAb [9]; 33C6-IgM mAb was prepared and tested by ELISA, neutralization, avidity, SPR, DLS and virion capture assays as described in the Supplemental Digital Content.
Passive immunization and mucosal SHIV-1157ipEL-p challenge
All primate studies were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the U.S.A (see Supplemental Digital Content). RMs were randomized into groups (n = 6/group). Groups 1 and 2 were given i.r. mAbs at a total dose of 1.25 mg in 2.1 ml of PBS. Group 1 RMs received 33C6-IgM (1.26 nmol); Group 2 received 33C6-IgG1 (8.45 nmol). Group 3 (controls) received 2.1 ml of PBS i.r. only.
Thirty min after mAb or PBS administration, RMs were atraumatically challenged i.r. with 31.5 50% animal infectious doses (AID50) of the R5 clade C SHIV-1157ipEL-p [10]. Plasma samples for mAb detection and viral load determination were obtained on the day of SHIV challenge and prospectively thereafter. Plasma viral RNA levels were measured as described [11].
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 5 for Windows (GraphPad Software Inc.). The time-to-peak viremia was analyzed by Kaplan-Meier analysis using the log-rank test with Holm-Sidak adjusted two-sided p-values.
Results
Generation of class-switched mAb 33C6-IgM
We previously identified and produced mAb 33C6-IgG1 [9]. To class-switch the latter to IgM, we cloned the heavy and light variable gene fragments in-frame with the human μ and λ chain constant regions, respectively. To express 33C6-IgM, we cotransfected the resulting vector constructs with the human J chain precursor expression plasmid [8] into Expi293 cells. We purified 33C6-IgM from filtered culture supernatant with thiophilic resin affinity binding and cation exchange chromatography. The presence of polymeric 33C6-IgM was confirmed with denaturing, non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot analysis (Fig. 1a) and by DLS (Fig. 1b, and Supplemental Digital Content).
Figure 1.
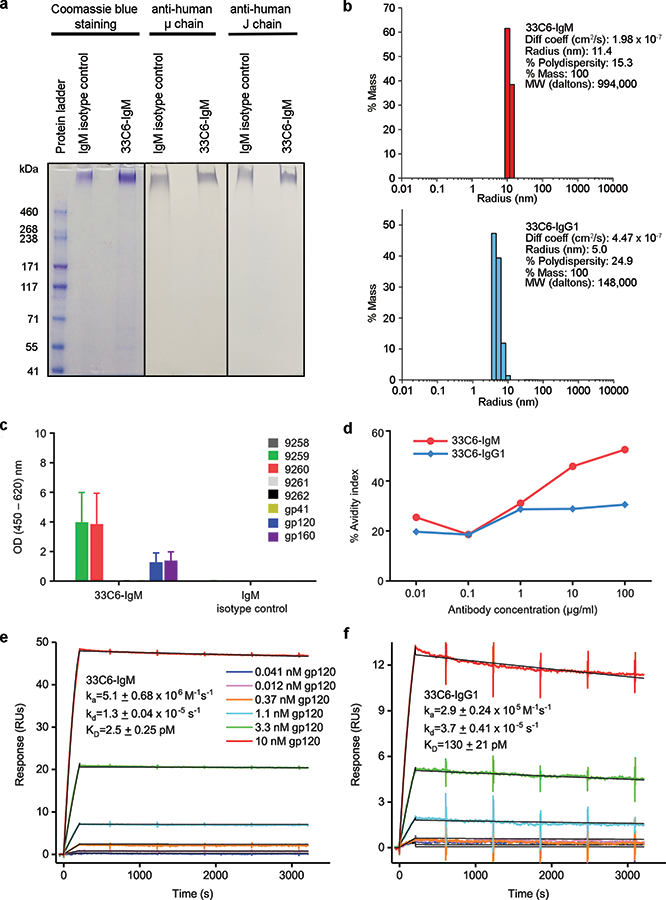
33C6 mAb characteristics. (a) SDS-PAGE and western blot analysis for 33C6-IgM and 33C6-IgG1. (b) Dynamic light scattering assay to determine particle size of 33C6-IgM and 33C6-IgG1. Data are representative of 4 independent experiments. (c) 33C6-IgM binding to consensus HIV clade C peptides representing V3 and to Env proteins. (d) Binding avidity. Data are representative of two independent experiments. (e, f) Binding affinities of captured antibodies for solution phase SHIV-1157ip gp120, with representative concentration series of SPR sensorgrams ranging from 41 pM - 10 nM gp120 are shown. Global fits to a 1:1 binding model are overlaid in black. Average ka, kd, and KD with standard errors from 3 replicates are indicated.
33C6-IgM recognized and bound to the conserved V3 loop crown of HIV Env as expected based upon the known epitope specificity of 33C6-IgG1 [9] (Fig. 1c). 33C6-IgM bound with greater avidity to SHIV-1157ip gp120 than 33C6-IgG1 (Fig. 1d), confirming the known superior avidity of IgM. Surface plasmon resonance (SPR) analysis revealed that 33C6-IgM bound to SHIV-1157ip gp120 with a KD of 2.5 pM (Fig. 1e). In contrast, 33C6-IgG1 had a KD of 130 pM (Fig. 1f), indicating that the IgM class bound significantly tighter than the IgG1 isotype of mAb 33C6. The on-rate for 33C6-IgM was 18-fold faster than that of the IgG1 version, and the IgM off-rate was 2.8 times slower than that of 33C6-IgG1. Together, these parameters account for the much tighter binding of the IgM isoform to soluble gp120 of the challenge virus.
33C6-IgM neutralized and captured SHIV in vitro
To assess the potential of 33C6-IgM to protect RMs against mucosal SHIV challenge – we tested the neutralization of SHIV-1157ipEL-p, the intended challenge virus strain, by 33C6-IgM and the IgG1 isotype by TZM-bl assay. This R5 clade C tier 1 SHIV strain had been used to demonstrate complete cross-clade protection of RMs by the human mAb HGN194 [12]; the latter has a similar epitope specificity as the 33C6 mAbs. 33C6-IgM neutralized SHIV-1157ipEL-p 10x better compared to 33C6-IgG1 (Fig. 2a). Of note, the pentameric IgM contains 5x more antigen binding sites compared to IgG1 at the same molar concentration, which might explain the greater neutralization potency of 33C6-IgM.
Figure 2.
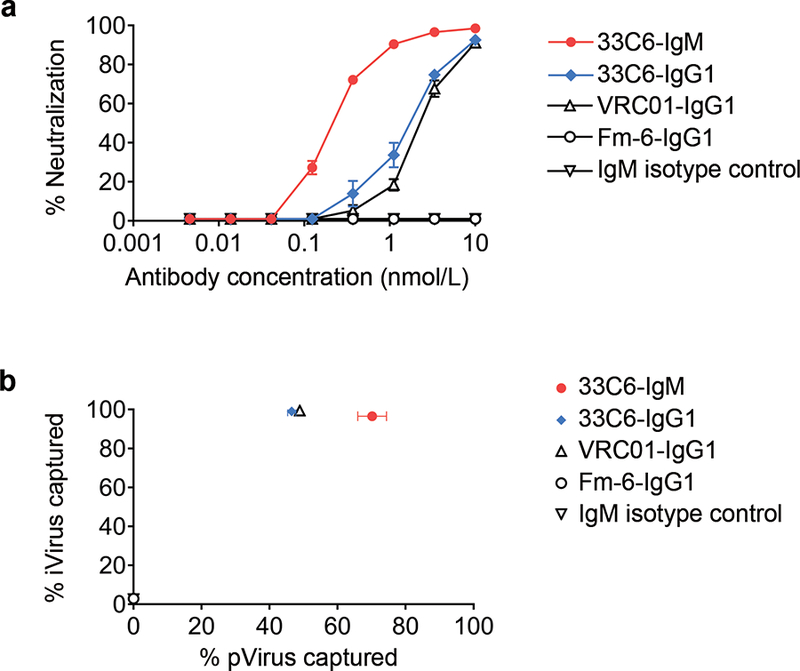
Neutralization and virion capture by 33C6 mAbs. (a) Neutralization of the challenge virus, SHIV-1157ipEL-p, by 33C6-IgM and 33C6-IgG1. (b) Capture of physical virus (pVirus) and infectious virus (iVirus) particles by 33C6-IgM and 33C6-IgG1. Data are representative of two independent experiments. Error bars, mean ± SEM. VRC01-IgG1 was used as positive control, while Fm-6-IgG1 and IgM isotype control were used as negative controls. Because a different secondary anti-IgM capture antibody was required for 33C6-IgM to capture cell-free virus using Protein G micro-beads, virion capture by 33C6-IgM could not be compared directly to that of 33C6-IgG1.
Next, we performed virion capture assays with the two 33C6 mAbs since we had shown previously that virion capture correlated with protection against mucosal SHIV-1157ipEL-p challenge [8], the virus strain as that used in the current study. The IgM form depleted physical virus particles by 75% and infectious virions by 96% (Fig. 2b); compared to 33C6-IgG1, the IgM form depleted more physical particles. Importantly, both isoforms removed almost all infectious virions.
33C6-IgM protected RMs against high-dose mucosal SHIV challenge
RMs were passively immunized i.r. with 33C6-IgM (Group 1) or 33C6-IgG1 (Group 2); Group 3 (control) RMs were given i.r. PBS only (Fig. 3). Thirty min later, all RMs were challenged i.r with 31.5 50% animal infectious doses (AID50) of SHIV-1157ipEL-p, and plasma viral RNA (vRNA) levels were monitored for 12 weeks. Four out of six (67%) RMs in Group 1 (33C6-IgM) (Fig. 4a), and five out of six (83%) RMs in Group 2 (33C6-IgG1) (Fig. 4b) remained aviremic. In contrast, all control RMs became systemically infected by week 2 with a median peak viremia of 106 vRNA copies/ml (Fig. 4c); the three RMs with breakthrough infection in Groups 1 and 2 had peak vRNA levels >106 copies/ml. The time to peak viremia was analyzed by Kaplan-Meier analysis using the log-rank test with Holm-Sidak adjusted two-sided p-values (Fig. 4d). Passively immunized RMs in Groups 1 and 2 had delayed peak viremia compared to Group 3 controls (Group 1, p = 0.009; and Group 2, p = 0.043). Of note, the 1.25 mg mAb dose used in this study contains 5-fold fewer IgM than IgG1 molecules. Thus, at equimolar concentrations, IgM might be better than IgG1. Taken together, data demonstrate that IgM is as effective as IgG1 in protecting against mucosal SHIV acquisition.
Figure 3.
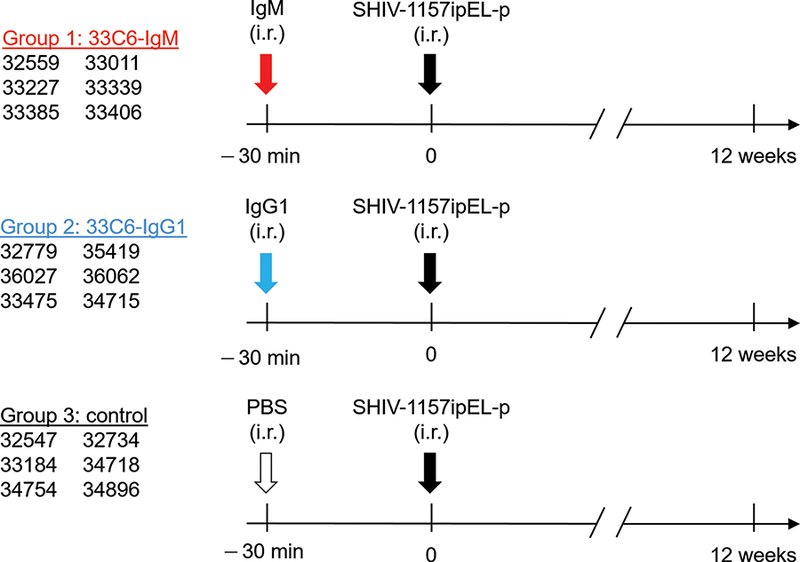
Study design. Rhesus macaques were randomized into three groups (n = 6 per group). Red arrow, Group 1 received 33C6-IgM intrarectally (i.r.); blue arrow, Group 2 was given 33C6-IgG1 i.r.; empty arrow, Group 3 received only phosphate-buffered saline (PBS) i.r.; black arrow, high-dose i.r. SHIV-1157ipEL-p challenge with 31.5 50% animal infectious doses (31.5 AID50). 33C6-IgM, 33C6-IgG1 or PBS was given 30 min before the virus challenge.
Figure 4.
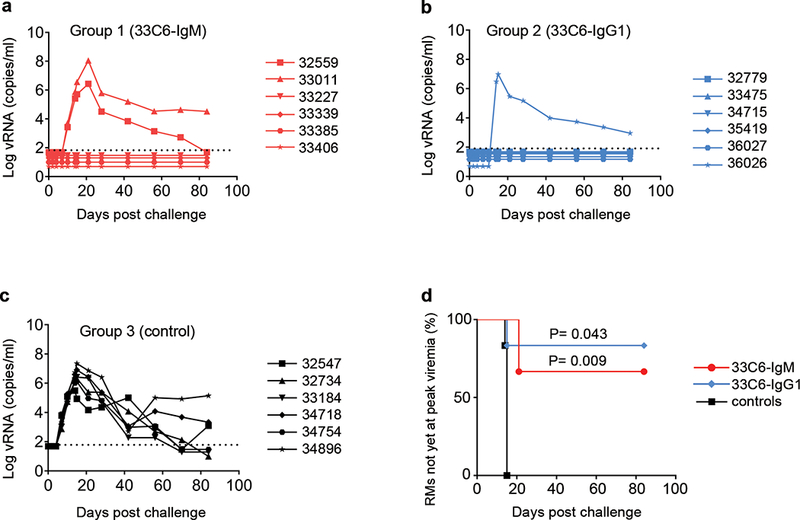
Plasma viral loads in macaques challenged with SHIV-1157ipEL-p. Plasma viral RNA (log vRNA copies/ml). (a) Group 1 (33C6-IgM); (b) Group 2 (33C6-IgG1); (c) Group 3 (controls). Black dotted line, limit of viral RNA detection (50 copies/ml). (d) Kaplan-Meier analysis of time until peak viremia. Log-rank test was used to determine significance.
33C6-IgM treatment accelerated the induction of anti-SHIV neutralizing antibodies
Next, we sought to examine the development of SHIV-specific antibodies in RMs with breakthrough infection. All infected RMs seroconverted (Fig. 5a); in passively immunized RMs of Groups 1 and 2 that became infected, anti-Env antibodies became detectable with delays.
Figure 5.
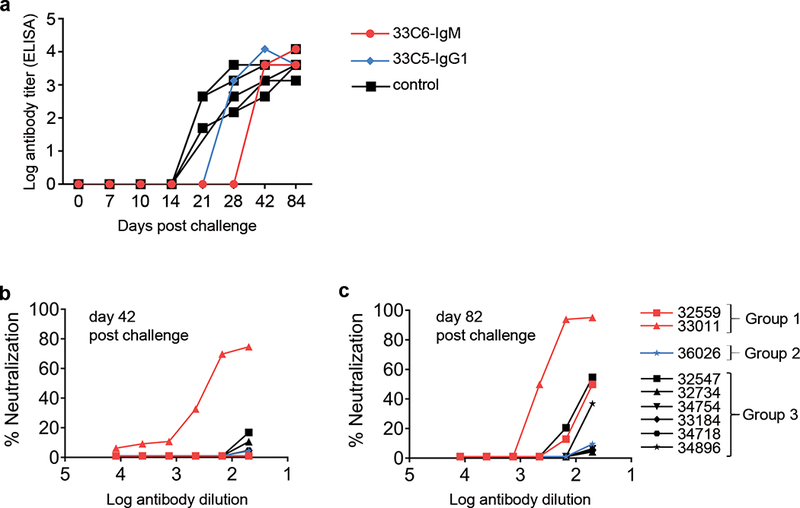
Titers of anti-SHIV plasma antibodies and their neutralization profiles against the challenge virus, SHIV-1157ipEL-p. (a) Titers of gp120-binding antibodies in plasma from rhesus macaques (RMs) with breakthrough infection. (b) In vitro neutralization of SHIV-1157ipEL-p of day 42 and (c) day 84 RM plasma samples. Data are representative of two independent experiments; each sample was analyzed in duplicate.
We then tested plasma samples from all RMs with systemic SHIV infection for neutralizing antibodies against the SHIV-1157ipEL-p challenge virus (Fig. 5b, c). Among day 42 plasma samples, neutralizing antibodies were only seen in one RM, animal 33011 treated with 33C6-IgM (Fig. 5b). By day 84, plasma from both of the infected, IgM-treated RMs neutralized the challenge SHIV by ≥50% (Fig. 5c). In contrast, neutralizing antibodies were present in only one out of six controls, indicating a trend for earlier development of autologous neutralizing antibodies in the IgM-treated RMs with breakthrough infection (p = 0.1). We conclude that mucosally administered IgM protects against high-dose mucosal SHIV challenge and may accelerate the development of autologous neutralizing antibodies in case virus acquisition is not prevented.
Discussion
Here we give the first report of preventing SHIV infection with a mucosally administered, anti-HIV 33C6-IgM. The majority of the passively immunized RMs was completely protected. In IgM-treated RMs with breakthrough infection, there was a trend for earlier development of autologous neutralizing Abs than in virus-only controls. 33C6-IgM neutralized SHIV, captured physical virus particles and depleted infectious virions in vitro, suggesting potential protective mechanisms. Our data give proof-of-concept that IgM can be an effective first-line of defense at mucosal barrier.
33C6-IgM targets a protruding element of HIV Env, the conserved V3 loop crown that is easily accessible in the challenge virus. Consequently, 33C6-IgM potently neutralized the challenge SHIV and not only captured most of the physical virions present in the stock, but also removed close to 100% of the infectious particles. The IgG1 isotype was equally effective in eliminating infectious virions by capture. These characteristics can be explained by the binding profiles for 33C6-IgM and 33C6-IgG1 as assessed by avidity indices and SPR analyses; by both measures, 33C6-IgM showed tighter binding. The affinity of 33C6-IgM for the soluble challenge virus gp120 was extraordinarily high with a KD in the low picomolar range indicating that binding was 52-fold tighter than binding of the IgG1 isoform. A large part of this difference in affinity is also likely due to avidity effects, which may be even greater for binding to virions, where the presence of multiple copies of gp120 would allow a larger number of simultaneous interactions with the multiple binding sites on the IgM mAb. Such powerful avidity effects may contribute substantially to the greater neutralization ability of 33C6-IgM compared to the IgG1 isoform.
The dual action – direct neutralization and efficient infectious virion capture – is likely the underlying basis for the protection we observed in vivo for both mAb forms of 33C6; the difference in the degree of protection between 33C6-IgM and its IgG1 counterpart was not significant. We interpret our data that pentameric IgM can efficiently trap incoming virus by crosslinking and prevent mucosal transmission through immune exclusion.
The rules for HIV/SHIV immune exclusion in the mucosal compartment remain to be determined. We have demonstrated that intra-luminal administration of mAbs of different Ig classes prevents SHIV transmission; this includes recombinant monoclonal IgM, IgG, and dimeric IgA (dIgA) [8, 13–15]. Other groups have focused on vaginal administration of anti-HIV IgG1 neutralizing mAbs [16, 17]. The passive mucosal immunizations with human dIgA1 and dIgA2 isotypes involved mAbs with almost identical epitope specificity as that of 33C6-IgM, namely HGN194 [12]. Both series of mAbs target the conserved gp120 V3 loop crown in R5-tropic HIV strains. HGN194 was isolated as an IgG1 from an infected person harboring an HIV clade AG circulating recombinant form. In contrast, 33C6-IgG1 was initially cloned from a single memory B cell from a RM with chronic clade C SHIV infection; mAb 33C6-IgG1 specifically recognized a conformational mimotope representing the V3 loop [9]. An unanswered question is whether mAbs targeting recessed epitopes can be effective for immune exclusion. Likewise, it remains to be determined whether mAbs need to be neutralizing in order to capture infectious virions.
Anti-HIV neutralizing mAbs have been class-switched from IgG to IgM, such as the IgG1 mAbs 2F5, 4E10, and 2G12. MAbs 2F5 and 4E10 target epitopes in the membrane proximal external region (MPER) of HIV gp41 known to be difficult to access (reviewed in [18]). Class switching from IgG1 to IgM resulted in loss of neutralizing activity [19–21]. In contrast, the IgM isoform of 2G12, a mannose-dependent neutralizing Ab with epitopes located on the glycan shield [22], not only retained the ability to neutralize HIV but actually neutralized the virus up to 28-fold-more efficiently in PBMC cultures than the corresponding IgG1 isoform [20]. The contrasting results obtained with anti-MPER and anti-glycan mAbs can be explained by differential epitope accessibility. The 33C6 mAbs used in our study target the readily accessible, conserved V3 loop crown epitope of gp120 [12]. This translated into efficient neutralization and virion capture by IgM and protection of RMs against a mucosal SHIV challenge.
Tomaras et al. [23, 24] have examined specific antiviral IgM responses arising during acute HIV infection. IgM is the first antibody class to respond to infection or immunization. Thus, IgM responses have been used to diagnose new infections by various pathogens. Indeed, the very first free plasma antibodies detected in acute HIV infection involved IgM; these antibodies had autologous anti-gp41 specificity [23, 24]. They appeared as early as five days after plasma viremia became detectable and were also found as virion-IgM complexes. Mathematical modeling showed that the early anti-gp41 IgM responses did not affect plasma viremia and thus were not felt to benefit the host [23]. This important study focused on virus-specific IgM responses after the HIV transmission had already occurred. Here we give evidence that passive immunoprophylaxis with anti-HIV IgM before virus exposure can be protective.
Passive immunization is considered a classical tool in immunology to determine the causal relationship between antibodies and protection against infectious agents. This includes mAbs with well-characterized epitopes; as such, passive immunization data can serve as blue prints for immunogen selection and design. In general, induction of protective IgM responses has not been a defined goal for vaccine development due to the waning of early IgM responses that are replaced by IgG, IgA and other Ig class responses that arise by programmed class switching. Thus, IgM is not usually considered to play an important role in long-term immunity. Surprisingly, recent mouse model studies showed that IgM responses can be long-lived and contribute to long-term protection [25, 26]. The significance of these findings for vaccine development remains to be determined. However, Dorfmeier et al. [27] performed post-exposure vaccination and pathogenic RABV challenge studies within a short time window of 10 days. Their data pointed to the rapid induction of specific IgM responses as the key determinant of the vaccine-linked protection against lethal RABV challenge in the mice. In our passive immunization study with mAb 33C6-IgM, we also made the intriguing observation of unusually early appearance of SHIV-neutralizing antibody responses in the context of breakthrough infection despite passive IgM immunization. The underlying mechanism(s) responsible for the accelerated induction of virus-neutralizing antibody responses remain to be determined.
In summary, we demonstrated that recombinant, monoclonal IgM protects against mucosal SHIV acquisition. Our data have implications for vaccine development in general and anti-HIV/AIDS vaccines in particular.
Supplementary Material
Acknowledgments
RMR conceived and designed the project. SG, KT, VK, SKL designed and performed experiments and analyzed data. KT, VK, and SKL coordinated the primate studies. LW and EML designed, performed, and analyzed the SPR and DLS experiments. OSA, DH, AS, and EP assisted with experiments and data analysis. SJR performed the biostatistical analyses. SG, KT, and RMR wrote the manuscript; all coauthors contributed and gave input.
Support
We thank A. Nabbale and J. Esquivel for assistance with the preparation of this manuscript, A.M. Sholukh for discussions, S.-L. Hu for SHIV-1157ip gp120, W. Marasco for mAb Fm-6-IgG1, and J. Mascola for mAb VRC01 obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
This project was supported by National Institutes of Health grants R01 AI100703 and P01 AI048240 to R.M.R. This study used resources supported by the Southwest National Primate Research Center (SNPRC) grant P51 OD011133 from the Office of Research Infrastructure Programs, National Institutes of Health. The SPR studies were performed in the University of Texas Health Science Center at San Antonio (UTHSCSA) Center for Macromolecular Interactions, which is supported by the Cancer Therapy and Research Center through the National Cancer Institute P30 Grant CA054174, and Texas State funds provided through the UTHSCSA Office of the Vice President for Research.
Footnotes
Conflicts of Interest and source of funding: The authors declared no conflict of interest.
References
- 1.Fellah JS, Wiles MV, Charlemagne J, Schwager J. Evolution of vertebrate IgM: complete amino acid sequence of the constant region of Ambystoma mexicanum mu chain deduced from cDNA sequence. Eur J Immunol 1992,22:2595–2601. [DOI] [PubMed] [Google Scholar]
- 2.Yates JL, Racine R, McBride KM, Winslow GM. T cell-dependent IgM memory B cells generated during bacterial infection are required for IgG responses to antigen challenge. J Immunol 2013,191:1240–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker N, Ehrenstein MR. Cutting edge: selection of B lymphocyte subsets is regulated by natural IgM. J Immunol 2002,169:6686–6690. [DOI] [PubMed] [Google Scholar]
- 4.Zhang M, Austen WG Jr., Chiu I, Alicot EM, Hung R, Ma M, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A 2004,101:3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res 2010,20:34–50. [DOI] [PubMed] [Google Scholar]
- 6.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009,139:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randall TD, Brewer JW, Corley RB. Direct evidence that J chain regulates the polymeric structure of IgM in antibody-secreting B cells. J Biol Chem 1992,267:18002–18007. [PubMed] [Google Scholar]
- 8.Watkins JD, Sholukh AM, Mukhtar MM, Siddappa NB, Lakhashe SK, Kim M, et al. Anti-HIV IgA isotypes: differential virion capture and inhibition of transcytosis are linked to prevention of mucosal R5 SHIV transmission. AIDS 2013,27:F13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sholukh AM, Mukhtar MM, Humbert M, Essono SS, Watkins JD, Vyas HK, et al. Isolation of monoclonal antibodies with predetermined conformational epitope specificity. PLoS One 2012,7:e38943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddappa NB, Watkins JD, Wassermann KJ, Song R, Wang W, Kramer VG, et al. R5 clade C SHIV strains with tier 1 or 2 neutralization sensitivity: tools to dissect env evolution and to develop AIDS vaccines in primate models. PLoS One 2010,5:e11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann-Lehmann R, Swenerton RK, Liska V, Leutenegger CM, Lutz H, McClure HM, et al. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res Hum Retroviruses 2000,16:1247–1257. [DOI] [PubMed] [Google Scholar]
- 12.Corti D, Langedijk JP, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM, et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One 2010,5:e8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watkins JD, Siddappa NB, Lakhashe SK, Humbert M, Sholukh A, Hemashettar G, et al. An anti-HIV-1 V3 loop antibody fully protects cross-clade and elicits T-cell immunity in macaques mucosally challenged with an R5 clade C SHIV. PLoS One 2011,6:e18207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sholukh AM, Watkins JD, Vyas HK, Gupta S, Lakhashe SK, Thorat S, et al. Defense-in-depth by mucosally administered anti-HIV dimeric IgA2 and systemic IgG1 mAbs: complete protection of rhesus monkeys from mucosal SHIV challenge. Vaccine 2015,33:2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruprecht RM, Lakhashe SK. Antibody-mediated immune exclusion of HIV. Curr Opin HIV AIDS 2017,12:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veazey RS, Shattock RJ, Pope M, Kirijan JC, Jones J, Hu QX, et al. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. NatMed 2003,9:343–346. [DOI] [PubMed] [Google Scholar]
- 17.Moog C, Dereuddre-Bosquet N, Teillaud JL, Biedma ME, Holl V, Van Ham G, et al. Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol 2014,7:46–56. [DOI] [PubMed] [Google Scholar]
- 18.Burton DR, Mascola JR. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol 2015,16:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunert R, Wolbank S, Stiegler G, Weik R, Katinger H. Characterization of molecular features, antigen-binding, and in vitro properties of IgG and IgM variants of 4E10, an anti-HIV type 1 neutralizing monoclonal antibody. Aids Research and Human Retroviruses 2004,20:755–762. [DOI] [PubMed] [Google Scholar]
- 20.Wolbank S, Kunert R, Stiegler G, Katinger H. Characterization of human class-switched polymeric (immunoglobulin M [IgM] and IgA) anti-human immunodeficiency virus type 1 antibodies 2F5 and 2G12. J Virol 2003,77:4095–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein K, Veazey RS, Warrier R, Hraber P, Doyle-Meyers LA, Buffa V, et al. Neutralizing IgG at the portal of infection mediates protection against vaginal simian/human immunodeficiency virus challenge. J Virol 2013,87:11604–11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, et al. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 1996,70:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol 2008,82:12449–12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomaras GD, Haynes BF. HIV-1-specific antibody responses during acute and chronic HIV-1 infection. Curr Opin HIV AIDS 2009,4:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bohannon C, Powers R, Satyabhama L, Cui A, Tipton C, Michaeli M, et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat Commun 2016,7:11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Racine R, McLaughlin M, Jones DD, Wittmer ST, MacNamara KC, Woodland DL, et al. IgM production by bone marrow plasmablasts contributes to long-term protection against intracellular bacterial infection. J Immunol 2011,186:1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorfmeier CL, Shen S, Tzvetkov EP, McGettigan JP. Reinvestigating the role of IgM in rabies virus postexposure vaccination. J Virol 2013,87:9217–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.