

Abstract
Rationale:
Hyper-IgE syndrome (HIES) is a rare primary immunodeficiency presenting as two forms including autosomal dominant HIES (AD-HIES) and autosomal recessive HIES (AR-HIES), which are mainly caused by mutations in STAT3 and DOCK8, respectively. To date, only about 500 cases have been reported worldwide including 37 cases in China. The spectrum and prevalence of mutations and molecular pathogenesis in HIES remain poorly understood.
Patient concerns:
Here we reported two Chinese children presenting clinical manifestations of HIES.
Diagnosis:
Based on medical history, clinical manifestations, and laboratory findings, a diagnosis of HIES was made for both children. Targeted next-generation sequencing (NGS) identified a novel heterozygous deletion of 15 bp (c.1960_1974del, p.G654_D658del or alternatively c.1966_1980del, and p.G656_D660del), and a recurrent missense mutation (c.1144C>T, p.R382W) in STAT3 in the two patients, respectively.
Interventions:
The two patients have been given the successful treatment of skin infections with cefaclor.
Outcomes:
Both patients have been under follow-up for more than 6 months, with no signs of recurrent infections.
Lessons:
Our results extend the spectrum of STAT3 mutations associated with ADHIES and highlight the value of targeted NGS in confirming diagnosis of genetic disorders.
Keywords: autosomal dominant, Chinese, hyper-IgE syndrome, STAT3 mutations, targeted next-generation sequencing
1. Introduction
Hyper-IgE syndrome (HIES) is a rare primary immunodeficiency characterized by a remarkably high level of serum IgE accompanied by eczema, recurrent skin and pulmonary infections, and connective tissue and skeletal abnormalities.[1] This disease can present as two forms, including autosomal dominant HIES (AD-HIES, OMIM 147060) and autosomal recessive HIES (AR-HIES, OMIM 243700). AD-HIES is mainly caused by mutations in the signal transducer and activator of transcription3 gene (STAT3)[2] and usually associated with distinct connective tissue and skeletal abnormalities, whereas AR-HIES is mainly caused by mutations in the dedicator of cytogenesis 8 gene (DOCK8),[3] with a high susceptibility to skin infections. Although mutations in other genes, including the tyrosine kinase 2 gene (TYK2)[4] and phosphoglucomutase 3 gene (PGM3),[5] have also been identified in sporadic cases of HIES, their roles in HIES remain unclear. Despite having been recognized for a half century, the prevalence of HIES remains not well known, with only about 500 cases reported worldwide[1,6–9]; the spectrum of mutations and molecular pathogenesis in HIES are also still poorly defined. Here we report two pediatric cases of HIES associated with novel and recurrent STAT3 mutations.
2. Methods
2.1. Ethical approval and consent for publication
The study was approved by the ethics committee of the Medical Faculty, University of Sichuan. Written informed consents were obtained from the parents of the patients before blood samples were taken.
2.2. Mutation analysis
To confirm and further clarify the diagnosis, we first performed a targeted capture next-generation sequencing (NGS) to screen potential mutations, followed by confirmation by Sanger sequencing. Peripheral blood samples were obtained from both patients and their family members as well as 100 unrelated healthy Chinese individuals as controls. Genomic DNA was extracted from peripheral blood samples using the QIAamp DNA Mini Kit (Qiagen Inc., Hilden, Germany) according to the manufacturer's instructions. DNA samples from both patients were used for targeted capture NGS, which was performed commercially (MyGenostics, Beijing, China). Targeted capture was conducted using the custom GenCap enrichment kit, which allowed capturing exons of STAT3, DOCK8, and TYK2 genes, and an additional 33 genes whose mutations are associated with immune system disorders.
3. Case report
3.1. Case 1
The patient was an 8-year-old girl born after full-term gestation. She developed pneumonia at age of 2 months and thereafter suffered from recurrent upper respiratory tract infections, bronchitis, and pneumonia at a frequency of 2–3 times per year, which required intravenous antibiotic treatment. At age of 1 year, she started experiencing eczematous dermatitis, which spread gradually from the head to the rest of the body and became worse in the recent 2 years. At the age of 4 years, she was diagnosed as pulmonary abscess and porosis. Both of her parents and two brothers appeared healthy. She was referred to our hospital due to severe eczema on her scalp. Physical examination revealed a prominent forehead and a broad nasal bridge (Fig. 1). Scattered or aggregated erythema and papules with erosions, exudation, or scabs were noted on the skin of the upper part of the body, predominantly the face, ears, and scalp. In addition, she retained two primary teeth and had flexion deformity of the distal interphalangeal joints of both index fingers. A complete blood count showed all normal except for an increased eosinophil count to 0.59 × 109/L (reference range: 0.02–0.52 × 109/L). Liver and renal function tests were normal. Immunologic assessment found a significantly elevated serum IgE concentration of >3000 IU/mL (normal range: 0.1–150 IU/mL) while all other parameters were normal, including the levels of serum IgG, IgM, and IgA antibodies, and circulating B and T cell populations. Chest computed tomography displayed residual pulmonary infection and cavitation. X-ray examination demonstrated osteoporosis in the left knee joint. She received a score of 48 based on the National Institutes of Health (NIH) HIES scoring system (threshold: 40).[8]
Figure 1.
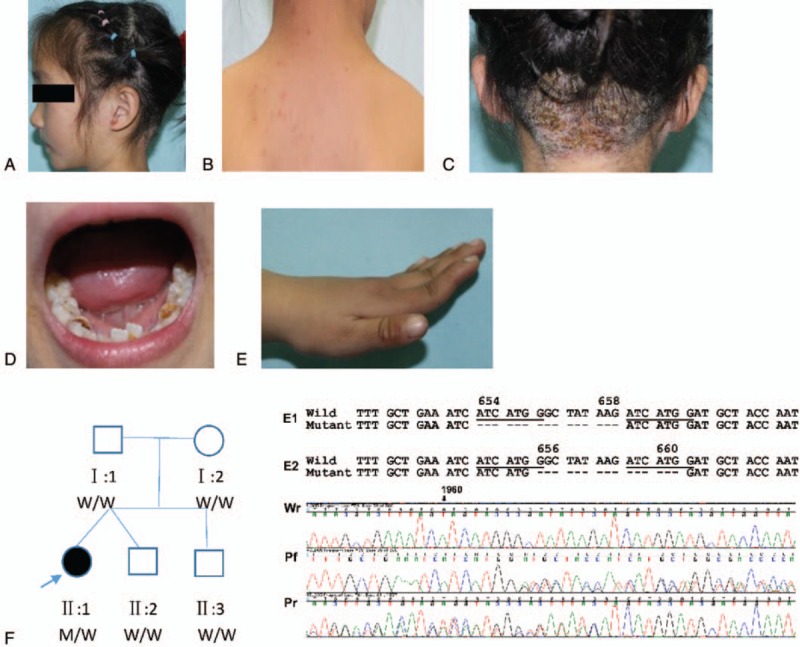
Clinical features and pedigree of case 1. (A and B) Eczema on the head and back. (C) Multiple scars of Staphylococcus aureus infection on the scalp. (D) Retained primary teeth. (E) Hyperextensibility of metacarpophalangeal joints. (F) Pedigree of the patient's family (left) and their STAT3 sequencing results (right). In the pedigree map, squares indicate male, circle indicates female, solid symbol indicates affected individual, open symbols indicate unaffected individuals, and the arrow indicates proband. W, wild-type; M, mutant. The deletion mutation could be caused by two possible events as shown in E1 and E2. Shown on the bottom are partial chromatograms of Sanger sequencing results of the patient from both forward (Pf) and reverse (Pr) directions and her father from reverse direction (Wr). Sequencing chromatograms from the patient showed overlapping peaks representing the wild-type and mutant alleles. The nucleotide and codon positions indicated in the figure are relative to the wild-type sequence of the human STAT3 gene (NM_139276). A novel heterozygous deletion of 15 bp in exon 21 of STAT3 (c.1960_1974del or alternatively c.1966_1980del) is indicated as hyphens. The deletion led to a loss of 5 amino acids between codon positions 654 and 658 (p.G654_D658del) or alternatively between positions 656 and 660 (p.G656_D660del).
In targeted NGS, the patient showed wild-type sequences in all targeted genes except for a heterozygous deletion of 15 bp in exon 21 of STAT3 (c.1960_1974del or alternatively c.1966_1980del). Based on alignment of NGS raw reads, this deletion could be caused by two possible events (Fig. 1F, E1 and E2). The alternative mutation could be caused by slippage during DNA synthesis due to the presence of an indirect repeat unit of ATCATGG (Fig. 1F underlined). Of note, the exact deletion position cannot be determined by any currently available methods while the resulting mutant allele remains the same in sequence, which was predicted to give rise to an in-frame deletion of 5 amino acids between positions 654 and 658 (p.G654_D658del) or alternatively between positions 656 and 660 (p.G656_D660del) (Fig. 1F). A search of the literature, GenBank, and Human Gene Mutation Database (http://www.hgmd.org/) did not find the same mutation, indicating it as a novel mutation. This novel mutation was further confirmed by PCR using the primers (forward primer 5′-TCTCAAATCCCATCGGTCAC-3′ and reverse primer 5′-TGTGACAGGTAAGACCCAGATC-3′), which were designed by the available online Primer 5. The amplified 392 bp PCR product was directly sequenced in an ABI Prism 3100 Automated Sequencer, using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems, Foster City, CA). The mutation was absent in other members of the patient's family and 100 ethnically matched control individuals. The deleted amino acids are located in a hydrophobic pocket in the SH2-domain of STAT3, which limits the mobility of amino acids 689–716.[10] The loss of 5 amino acids in this pocket are likely to affect the nuclear translocation of STAT3, thus leading to dysfunction of STAT3. All these findings support the causative role of this deletion in HIES. Clearly, further studies are needed to confirm its impact on the function of STAT3.
3.2. Case 2
The patient was a 7-year-old boy born to healthy parents with a full-term gestation. He had a history of eczema and recurrent respiratory tract infections during his early infancy. The latter disease continued to occur at least twice a year, accompanied with painless abscesses on the scalp. As a result, he received intravenous antibiotic treatment multiple times per year. At ages of 1 and 4 years, he received surgery to remove pulmonary cysts due to life-threatening infectious complications. At age of 2 years, he developed chronic onychomycosis in both thumbs. None of his family members and relatives had a history of HIES. Upon admission to our hospital due to abscess on his scalp, physical examination found an apparent scoliosis of the cervical spine (>20 degree, Fig. 2), without any sign of pathologic bone fractures, or hyperextensible joints. Other positive signs included facial asymmetry with hemihypertrophy, broad nasal bridge, high-arched palate, and retained primary teeth. Blood and immunological tests were all normal except for a significant increase in serum IgE concentration of 6320 IU/mL. Chest X-ray showed residual lung infection. He received a score of 68 according to the NIH HIES scoring system.
Figure 2.
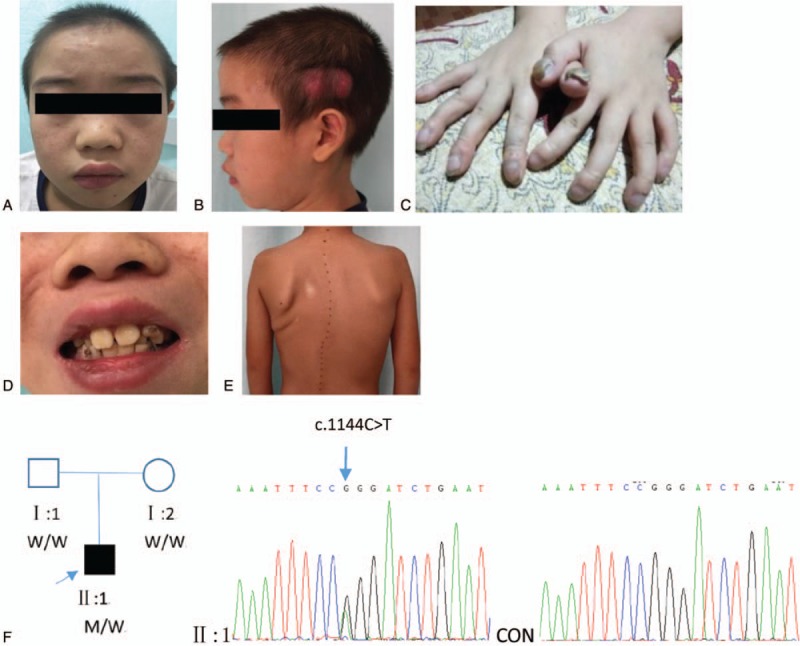
Clinical features and pedigree of case 2. (A) Eczema on the face. (B) Scars of Staphylococcus aureus infection on the scalp. (C) Onychomycosis on both thumbs. (D) Retained primary teeth. (E) Scoliosis. (F) Pedigree of the patient's family (left) and partial chromatograms of Sanger sequencing from the patient (middle) and control (right). A previously reported missense mutation (c.1144C>T, p.R382W) in exon 13 of STAT3 was identified as a heterogeneous mutation, which resulted in an arginine-to-tryptophan change at position 382. Squares indicate male, circle indicates female, solid symbol indicates affected individual, open symbols indicate unaffected individuals, and the arrow indicates proband. W, wild-type allele; M, mutation c.1144C>T.
For the patient, targeted capture NGS revealed wild-type sequences in all targeted genes except for a heterozygous missense mutation in exon 13 of STAT3 (c.1144C>T, p.R382W), which is one of the most common mutations associated with AD-HIES.[2]
After successful treatment of skin infections with cefaclor, both patients have been under follow-up for more than 6 months, with no signs of recurrent infections.
4. Discussion
DNA sequencing analysis supports the diagnosis of both children as AD-HIES. Furthermore, we searched the PubMed and China Science and Technology Journal (CSTJ) databases to identify research articles in English or Chinese language published on or before July 2, 2018. Search was implemented by using the following keywords: “Hyper IgE syndrome,” “Job syndrome,” “STAT3,“ and ”DOCK8”. In addition, we checked the reference lists from retrieved articles for any supplementary relevant publications.
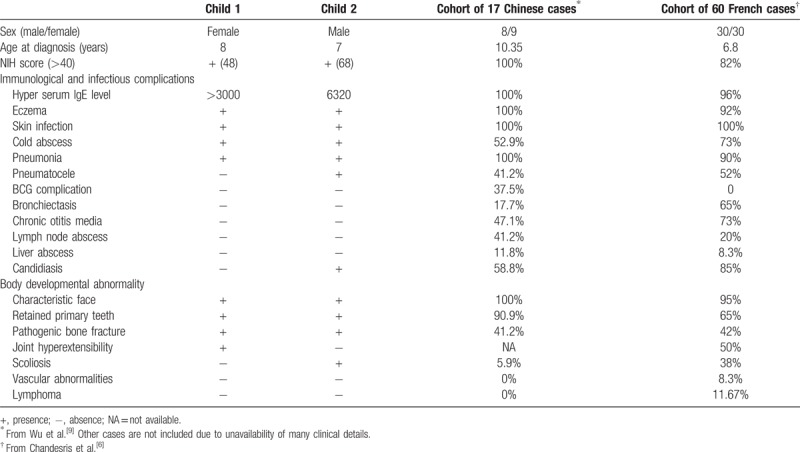
Combining these two cases with the 37 cases reported in the literature[9,11] gives a total number of AD-HIES cases to 39 in China. These two cases shared many clinical characteristics of AD-HIES though case 2 showed a significantly higher IgE level, more serious and widespread infections (in skin, lung, and nail), and severe developmental impairment (scoliosis) compared to case 1 (Table 1). Of note, the pulmonary infection in case 2 was life-threatening and required surgery; such severe complication has been never reported in HIES patients. Consistent with the previous notion of a lower prevalence of chronic otitis media and bronchiectasis among AD-HIES patients in China than other countries,[9] neither patients in our study presented with these complications (Table 1). In addition, our study also supports the previous observation of an absence or a low incidence of lymphoma, vascular abnormality, and scoliosis in AD-HIES from China.[9]
Table 1.
Main clinical characteristics of AD-HIES in two children in this report compared to previous reports.
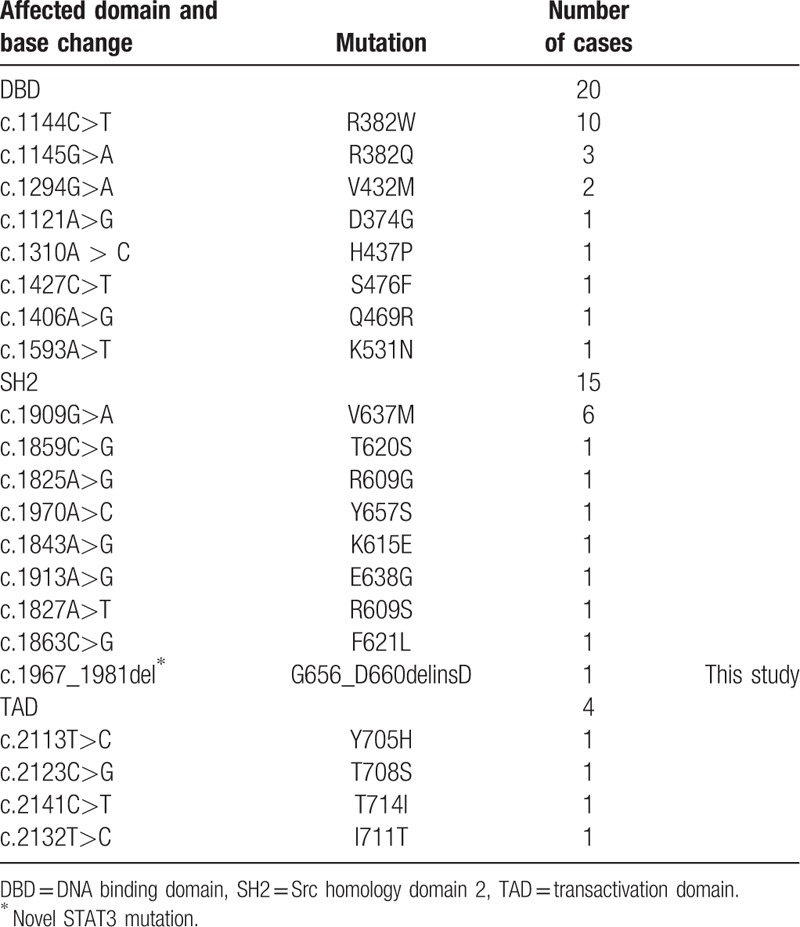
STAT3 was one of a protein family that is responsible for mediating the expression of a variety of genes during cell stimuli, and thus plays a key role in many cellular processes such as cell growth and apoptosis. The structure of STAT3 included three major region: the N-terminal region (1–355), the central region (355–555), and the C-terminal region (555–770). The central region was the canonical DNA binding domain (DBD) and the C-terminal region was the linker-SH2 domain (SH2).[12] The SH2 domain is required for STAT3 phosphorylation and subsequent nuclear translocation. A hydrophobic pocket is formed in the SH2 domain by Leu706, Thr714, Val637, Tyr640, and Tyr657, which limits the mobility of amino acids 689–716. The changes in these amino acids bring about the loss of this hydrophobic pocket which could lead to the greater mobility of the STAT3 monomer. Thus, the stability of STAT3 is compromised, which leads to its subsequent disruption.[13] The identified mutation in case 1 was also located in SH domain and the deletion contained amino acid Tyr657. Therefore, we supposed that the deletion could impact the stability of STAT3 and affect the function of STAT3. The functional studies of the deletion are necessary to illustrate the hypothesis. The DBD of STAT3 is highly conserved among different species in its amino acid sequence. The mutants identified in the DBD domain, including R382W, displayed dominant-negative effects.[14] After analyzing the mutations of the 39 patients, we found that 20 patients (51.28%) carried the mutations in DBD domain, 15 (38.46%) in SH2 domain, and 4 (10.26%) in TAD domain (Table 2). All of the mutations were missense heterozygous mutations, except for the novel deletion identified in the present study. Among DBD mutations, R382W/Q is the most common reported mutation (13/20, 65%). V637M is the hotspot mutation of SH2 mutations (6/15, 40%). Considering clinical manifestation and genotype together, we found that scoliosis and onychomycosis are the two less reported clinical symptoms among Chinese HIES patients with STAT3 mutations-associated phenotype. Six patients were described with onychomycosis and seven patients with scoliosis. For the 13 patients with the R382W/Q mutation, only one had onychomycosis and the other one had scoliosis. In the present study, case 2 had onychomycosis and scoliosis at the same time and had the more serious clinical manifestations than that of case 1. We supposed that the discrepancy of the clinical phenotypes between the two children might result from a difference in the location of their mutations. Because of the low case finding rate in mainland Chinese, only 39 Chinese HIES patients with STAT3 deficiency have been reported and more cases are needed for further analysis.
Table 2.
Mutations of STAT3 identified in Chinese cases with AD-HIES.
In summary, we report here two pediatric cases of AD-HIES caused by novel and recurrent STAT3 mutations. The identification of the novel mutation extends the spectrum of STAT3 mutations associated with AD-HIES. This report highlights the necessity to be aware of genetic disorders associated with recurrent infections and abnormal development among children and illustrates the value of targeted capture NGS in confirming diagnosis of genetic disorders.
Acknowledgments
We are grateful to all the participants in this study, especially Jessie J. Ma who is a high school student.
Author contributions
Formal analysis: Ying Deng.
Investigation: Ying Deng, Tong Li, Xiaoqin Xie, Dengmei Xia, Jessie J. Ma, Li Ding, Hongmei Xiang.
Methodology: Ying Deng, Jessie J. Ma.
Project administration: Wei Li.
Resources: Tong Li.
Supervision: Wei Li.
Writing – original draft: Ying Deng.
Writing – review & editing: Ying Deng.
Footnotes
Abbreviations: AD = autosomal dominant, AR = autosomal recessive, DOCK8 = dedicator of cytogenesis 8 protein, HIES = hyper-IgE syndrome, NIH = National Institutes of Health, PGM3, phosphoglucomutase 3, SH2 = Src homology domain 2, STAT3 = signal transducer and activator of transcription 3, TYK2 = tyrosine kinase 2.
YD and TL contributed equally.
JJM participated as a volunteer.
The data collection was funded by “National State Research Program of China” (ID: 81573050).
The authors state no conflict of interest.
References
- [1].Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N Engl J Med 1999;340:692–702. [DOI] [PubMed] [Google Scholar]
- [2].Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007;357:1608–19. [DOI] [PubMed] [Google Scholar]
- [3].Engelhardt KR, McGhee S, Winkler S, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol 2009;124:1289–302. e1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Minegishi Y, Saito M, Morio T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 2006;25:745–55. [DOI] [PubMed] [Google Scholar]
- [5].Zhang Y, Yu X, Ichikawa M, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol 2014;133:1400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chandesris MO, Melki I, Natividad A, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012;91:e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Renner ED, Rylaarsdam S, Anover-Sombke S, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol 2008;122:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Woellner C, Gertz EM, Schaffer AA, et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol 2010;125:424–32. e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu J, Chen J, Tian ZQ, et al. Clinical manifestations and genetic analysis of 17 patients with autosomal dominant hyper-IgE syndrome in Mainland China: new reports and a literature review. J Clin Immunol 2017;37:166–79. [DOI] [PubMed] [Google Scholar]
- [10].Alcantara-Montiel JC, Staines-Boone T, Lopez-Herrera G, et al. Functional characterization of two new STAT3 mutations associated with hyper-IgE syndrome in a Mexican cohort. Clin Genet 2016;89:217–21. [DOI] [PubMed] [Google Scholar]
- [11].Fan H, Huang L, Yang D, et al. Pediatric hyperimmunoglobulin E syndrome: a case series of 4 children in China. Medicine (Baltimore) 2018;97:e0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998;394:145–51. [DOI] [PubMed] [Google Scholar]
- [13].Bocchini CE, Nahmod K, Katsonis P, et al. Protein stabilization improves STAT3 function in autosomal dominant hyper-IgE syndrome. Blood 2016;128:3061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007;448:1058–62. [DOI] [PubMed] [Google Scholar]