

Abstract
Glioblastoma (GBM) is a deadly brain cancer, and all attempts to control it have failed so far. However, the future looks bright, as we now know the molecular landscape of GBM through the work of The Cancer Genome Atlas (TCGA) program. GBMs exhibit significant inter- and intra-tumoral heterogeneity, and to control this type of tumor, a personalized approach is required. One target, whose gene is amplified and mutated in a large number of GBMs, is the epidermal growth factor receptor (EGFR). But all attempts to target it have been unsuccessful. We attribute the reason for this failure to the molecular heterogeneity of EGFR in GBM, as well as to the poor brain penetration of previously tested EGFR-Tyrosine Kinase Inhibitors (EGFR-TKIs). In this review, we discuss the molecular heterogeneity of EGFR and provide rational preclinical and clinical guidelines for testing AZD9291, a third generation, irreversible EGFR-TKI with both a high affinity for EGFRvIII and excellent brain penetration.
Keywords: Glioblastoma, EGFR, heterogeneity, targeted, therapies, TCGA
INTRODUCTION
Glioblastoma (GBM) is among the most aggressive and common primary brain tumors in adults. GBM patients have extremely poor prognoses [1], and no current treatments (including surgery, radiation, and chemotherapy) have been able to extend median survival beyond fifteen months [2]. An additional drawback of the current standard of care is that the treatments lead to a poor quality of life. Therefore, there is an urgent need to develop alternate therapies that are not only more effective than current therapies, but are also better tolerated by patients. Therapies meeting these criteria can be developed if there is greater understanding of the molecular pathways that drive GBM growth. Fortunately, this information has now become available by the recently completed detailed molecular characterization of GBM by The Cancer Genome Atlas (TCGA) program [3]. These efforts led to the broad classification of GBMs into four subtypes (classical, mesenchymal, proneural, and neural), but it is clear that further heterogeneity exists within each subtype [3–5]. Each GBM subtype is characterized by specific molecular aberrations. A notable molecular abnormality seen in a large number of GBMs (>57%) is in the gene for epidermal growth factor receptor (EGFR) [3].
Although a key role for EGFR in GBM has been known for some time, it is TCGA’s detailed molecular characterization of GBMs that has truly highlighted the extensive molecular abnormalities present within the EGFR gene in GBM [3, 6]. These abnormalities include gene amplification, several point mutations in the extracellular domain, deletions in the extracellular domain, and insertions in the intracellular domain. A well-characterized molecular alteration in the extracellular domain of EGFR, which is present in over 20% of GBM patients, is the deletion of 267 amino acids (aa 30 to aa 297). This deletion mutant of EGFR is known as EGFRvIII [7, 8], and it differs from the wild-type EGFR in that it does not bind its ligand EGF and has a constitutively active tyrosine kinase domain [9–11]. Molecular alterations in the intracellular domain of EGFR have also been discovered. These include fusion between EGFR and SEPT14 genes [12] as well as tandem duplication of the kinase domain [13, 14]. Given that EGFR in GBMs has mutations both in its extracellular as well as intracellular domains, it is not justified to say that mutations in the intracellular domain of EGFR are present in lung cancer, but not in GBM [15]. Thus, the diversity of molecular changes seen in the EGFR gene in GBMs is enormous, and to develop successful therapies, this diversity must be considered in the development of therapies targeted at EGFR.
PREVIOUS EFFORTS TO TARGET EGFR IN GBM HAVE “FAILED”
Given the importance of EGFR and its variant EGFRvIII in GBM biology, extensive efforts have been made to target it. In fact, several agents (antibodies as well as small molecules) targeting EGFR in glioblastomas have already been tested in patients. However, targeting EGFR has not been effective so far [16, 17]. There are several potential reasons. Firstly, it is possible that the EGFR inhibitors blocked EGFR, but the downstream proteins involved in EGFR signaling were not blocked; this was found to be the case with the drug gefitinib [18]. This scenario, i.e. receptor blockade without downstream signaling blockade, suggests that an EGFR blockade is activating alternate growth pathways [5]. Secondly, the failed trials included all GBM patients [19, 20], not just patients with EGFR activation. Thirdly, the tested agents (mainly erlotinib, gefitinib, and lapatinib) had poor brain penetration. We conclude that the failure of previous clinical trials to target EGFR was due to one or more of these flaws in the study design. Unfortunately, testing of EGFR-TKI under poorly designed clinical trials continues. This is exemplified by a recent study testing the irreversible EGFR-TKI, afatinib, in recurrent GBM patients [21]. This study has two fatal flaws: 1) instead of only including patients with activated EGFR or EGFRvIII, the study included all GBM patients; 2) afatininb was never tested in animal models bearing intracranial xenografts, and it remains unknown if afatinib actually reaches the brain, engages the target in the tumor, and inhibits downstream EGFR signaling in vivo. To be fair, the authors themselves pointed out these two major faults [21], but this does not help the tarnished image of EGFR blockers. The fact remains that it will inevitably be classified as another “failed” clinical trial of EGFR blockers in GBM patients.
Therefore, it is clear that previous attempts to target EGFR in GBMs were flawed. Moving forward, we need to make two changes in our strategy of targeting EGFR in GBM: 1) we need to select an EGFR-TKI that crosses the blood-brain barrier, reaches the brain at concentrations required to inhibit EGFR, engages the EGFR, and inhibits downstream signaling; and 2) test this EGFR-TKI in GBMs expressing a defined EGFR genotype (e.g. wild-type EGFR with gene amplification or EGFRvIII). Furthermore, it should be confirmed that the selected EGFR genotype has elevated kinase activity. This is important because, as we have shown [22], presence of EGFR gene amplification does not correspond to elevated EGFR kinase activity. Taking these steps, no matter how painstaking and intricate they may be, are critical to mending the tarnished image of EGFR blockers and to finding a successful treatment for GBM.
AZD9291, A SUITABLE EGFR-TKI, TO TARGET EGFR IN GBM
AZD9291, developed by AstraZeneca to target a specific EGFR mutant in lung cancer, is an oral irreversible inhibitor of epidermal growth factor receptor (EGFR) kinase [23, 24]. A key feature of AZD9291 is that it also has high affinity for EGFRvIII and also inhibits HER2 [23]. Importantly, animal studies have shown that AZD9291 has good brain penetration; it exhibits 5–25 fold higher exposure in brain tissue compared to plasma, and approximately 10 fold greater exposure than gefitinib [23]. Thus, AZD9291 is an appropriate EGFR-TKI to test in a subset of GBM patients expressing EGFRvIII
TARGETING EGFRVIII WITH AZD9291
Given that AZD9291 binds to EGFRvIII irreversibly with high affinity, we have proposed its evaluation in a subset of GBM patients expressing EGFRvIII. There are two main reasons to evaluate AZD9291 in EGFRvIII-expressing GBM: 1) EGFRvIII occurs in a large fraction of GBM patients [25] and methods to detect it are available in CLIA-certified laboratories; and 2) recent data indicate that EG-FRvIII is present in glioblastoma stem cells (GSCs) [25]. Importantly, over 80% of GSCs that expressed EGFRvIII also co-expressed CD133, a prominent biomarker of GSCs [26], and GSCs expressing both EGFRvIII and CD133 had higher self-renewal and tumor-initiating properties [25]. It should be pointed out that discovery of EGFRvIII in GSCs makes it an ideal target to halt GBM recurrence since GBM recurrence is believed to be initiated by the presence of GSCs in the residual tumor left behind from surgical resection [27–29].
Recent studies indicate that GBMs not only exhibit molecular diversity between patients (inter-tumoral heterogeneity) [3], but also within a single tumor (intra-tumoral heterogeneity) [5, 30, 31]. Therefore, the question arises whether a single agent can prevent recurrence of GBM. The answer is no for many cases of GBM, but we believe that EGFRvIII-expressing GBMs may represent a special case, and their recurrence may be substantially delayed by blocking EG-FRvIII. This is because EGFRvIII is present in GSCs, and EGFR-TKI therapy, which should be started right after the surgical removal of the bulk of the tumor, would either kill EGFRvIII-expressing GSCs or substantially slow their growth and hence tumor recurrence. Below we describe a rational preclinical and clinical testing protocol to evaluate the effectiveness of AZD9291 to halt the growth of EG-FRvIII-expressing GBM.
PRECLINICAL GBM MODELS EXPRESSING EG-FRVIII
For preclinical testing of AZD9291, two GBM models that express EGFRvIII are being used: 1) GSCs that express EGFRvIII and 2) patient-derived glioblastoma xenografts (PDGXs) implanted intracranially in immunocompromised mice. GSCs expressing EGFRvIII have been isolated directly from a primary tumor [25, 32] or from a PDGX developed from a tumor expressing EGFRvIII [33]. GSCs isolated from EGFRvIII-expressing primary tumors retain the expression of EGFRvIII [25]. Similarly, we have found that GSCs isolated from EGFRvIII-expressing PDGX also retain the expression of EGFRvIII (Kwatra MM. unpublished data). Since it is likely that EGFRvIII-expressing GBMs will exhibit differences in the extent of EGFRvIII kinase activation, we are testing AZD9291’s ability to inhibit the growth of EGFRvIII-expressing GSCs with varying levels of EGFRvIII kinase activation [32]. Finally, EGFRvIII-expressing GSCs, both control and treated, are being subjected to proteomic analysis using Reverse Phase Protein Arrays (RPPA) [34]; this analysis provides information on the phosphorylation status of several signaling molecules, including those involved in EGFR signaling [22]. Thus, RPPA data on control and AZD9291-treated GSCs will assess the effectiveness of AZD9291 to inhibit downstream signaling. In addition, RPPA data can also show if the blockade of EGFRvIII with AZD9291 is activating alternate growth pathways.
Assuming that AZD9291 is effective in blocking the growth of EGFRvIII-expressing GSCs, the next step in its preclinical development will be to test it in animal models. For these studies, patient-derived xenograft (PDX) models are commonly used as they retain the molecular characteristics of the parent tumor [35, 36]. Our group has developed a panel of twenty PDGXs and characterized them according to their EGFR abnormalities, as well as TCGA-subtyping (Table 1) [22]. As Table 1 shows, EGFR gene amplification was seen in nine PDGX, with three expressing EGFRvIII. Proteomic analysis of these twenty PDGX using RPPA revealed elevated EGFR phosphorylation at Y1068, a measure of EGFR activation [37], in seven PDGX (Fig. (1). It is key to note that the increased phosphorylation of EGFR at Y1068 observed in seven PDGX originates from four different EGFR genotypes as shown in Table 1 and Fig. (1; 1) in three PDGX, the EGFR is EGFRvIII (PDGXs #3,4, and 8); 2) in two PDGX, the EGFR is of wild-type with gene amplification (PDGXs #2 and #7); 3) in one PDGX, the EGFR is truncated (PDGX #9); and 4) in one PDGX, the EGFR is wild-type with no gene amplification (PDGX #20). The four molecular subtypes of EGFR, all with increased phosphorylation at Y1068, are likely to have different signaling properties, underscoring the importance of segregating GBMs according to their EGFR genotype.
Table 1.
PDGX Characteristics.
| PDGX # | ID # | Mouse Passage # | Dx | Sex/Age | TCGA Subtype | TP53 | PTEN | TERT | EGFR* | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| vIII | Wt | Amplified | |||||||||
| 1 | 08–0308 | 10 | GBM | M/76 | C | Wt | Wt | C228T | − | + | Y |
| 2 | 08–0500 | 14 | GBM | M/75 | C | Mut | Mut | C250T | − | + | Y |
| 3 | 08–0549 | 14 | GBM | M/62 | C | Wt | Wt | C228T | + | + | Y |
| 4 | 09–0155 | 20 | GBM | F/52 | C | Wt | Wt | C228T | + | + | Y |
| 5 | 09–0211 | 26 | GBM | F/53 | C | Wt | Wt | C228T | − | + | N |
| 6 | 09–0394 | 33 | GBM | M/73 | C | Wt | Mut | C228T | − | + | N |
| 7 | 09–0584 | 7 | GBM | M/64 | C | Wt | Mut | C228T | − | + | Y |
| 8 | 10–0171 | 12 | GBM | M/54 | C | Wt | Wt | C228T | + | + | Y |
| 9 | 08–0430 | 11 | GBM | M/59 | M | Wt | Wt | C250T | − | + | Y‡ |
| 10 | 08–0679 | 21 | GBM | F/47 | M | Wt | Mut | C250T | − | + | Y |
| 11 | 09–0337 | 19 | GBM | M/52 | M | Wt | Mut | C228T | − | + | N |
| 12 | 09–0362 | 39 | GBM | M/50 | M | Mut | Mut | C228T | − | + | N |
| 13 | 09–0477 | 27 | GBM | M/73 | M | Wt | Mut | C228T | − | + | N |
| 14 | 09–0500 | 21 | GBM | M/58 | M | Wt | Mut | C228T | − | + | N |
| 15 | 09–0627 | 9 | GBM | M/61 | M | Wt | Mut | C228T | − | + | Y |
| 16 | 11–0040 | 11 | GBM | M/65 | M | Mut | Mut | C250T | − | + | N |
| 17 | 08–0478 | 13 | GBM | M/62 | P | Mut | Mut | C250T | − | + | N |
| 18 | 08–0499 | 20 | GBM | M/66 | P | Mut | Mut | C250T | − | + | N |
| 19 | 08–0624 | 11 | GBM | M/63 | P | Wt | Mut | C228T | − | + | N |
| 20 | 10–0021 | 12 | GBM | F/82 | P | Wt | Wt | C250T | − | + | N |
Mut – Mutation; Wt – Wild-type; TCGA – The Cancer Genome Atlas; C – Classical; M – Mesenchymal; P – Proneural; TP53 – Tumor Protein p53; PTEN - Phosphatase and Tensin homolog; TERT – Telomerase Reverse Transcriptase; EGFR – Epidermal Growth Factor Receptor.
EGFR qPCR data expressed as relative quantity (RQ; not shown), fold difference in expression measured against normal brain RNA as described in the methods section. Those having an RQ > 3 were considered to be amplified. None of the PDGX showed mutations in Isocitrate Dehydrogenase1 or 2.
Presence of transcript and amplification was observed only with primers specific for the carboxyl-end, consistent with a truncated variant detected by western blotting. Taken from [22] with permission.
Fig. (1).
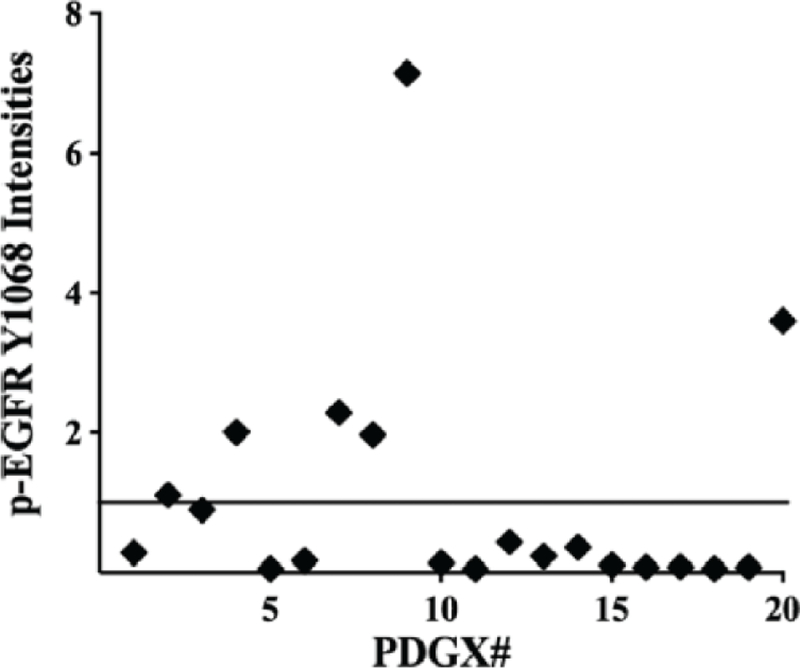
Expression of epidermal growth factor receptor (EGFR) phosphorylated at tyrosine 1068 (pEGFRY1068) in patient-derived glioblastoma xenografts (PDGX). The expression levels of p-EGFRY1068 were obtained by reverse phase protein array (RPPA) analysis of 20 PDGX shown in Table 1. Taken from [22] with permission.
While EGFR genotyping is the first step in patient selection, it needs to be followed by testing EGFR phosphorylation at Y1068. Why this is important is clear from the data presented in Table 1 and Fig. (1). While Table 1 shows that five PDGX express wild-type EGFR with gene amplification (PDGX#1, 2, 7, 10, and 15), only two PDGX have elevated EGFR phosphorylation (PDGX #2 and 7). Thus, selection of patients based on EGFR gene amplification alone would include patients whose EGFR is not activated and hence unlikely to respond to AZD9291.
To test the effectiveness of AZD9291 in animal models, immunocompromised mice with intracranial expression of EGFRvIII will be used. This is important because although animal models carrying subcutaneous GBM xenografts have been used to test therapies against GBMs [38], the use of orthotopic models is desirable and is more common [32, 39]. To consider AZD9291 as an efficacious agent to target EGFRvIII-expressing GBM, it should significantly (>25%) prolong the overall survival of animals bearing intracranial transplants of EGFRvIII-expressing GBMs. Finally, the tumors isolated from both control and AZD9291-treated animals will be analyzed using RPPA to determine the effectiveness of AZD9291 to inhibit EGFRvIII-mediated signaling, as well as to identify if the drug treatment is causing the activation of alternate growth pathways. This information will help to develop combination therapies that prevent the development of resistance to the selected EGFR-TKI [40].
It should be mentioned that while PDGXs retain the molecular features of the parent tumor, the response of intracranially transplanted PDGXs with activated EGFRvIII to AZD9291 might not be similar to what one would observe in humans. There may be several reasons for this, and the most probable is that PDGXs are in immune-deficient mice, whereas GBM patients are immunocompetent. These and other drawbacks of PDGX models are well documented [35, 36, 41]. Given these drawbacks of the PDGX models, the go/no-go decision for phase 1 studies will be based on the effect AZD9291 on GSCs rather than animals models. This makes sense, especially for AZD9291 because its safety in humans has already been demonstrated [42].
PHASE I AND PHASE II TESTING OF AZD9291 IN EGFRVIII-EXPRESSING GBMS
If AZD9291 is effective in prolonging the survival of animals bearing intracranial transplants of EGFRvIII-expressing tumors, the next step is to test it in GBM patients under phase I and phase II studies. The goal of phase I studies is to determine the dose of AZD9291 to be used in phase II studies. A 3+3 phase 1 cohort design is generally used [43]. To be included in the study, the resected tumor from newly diagnosed GBM patients is tested for EGFRvIII expression by RT-PCR performed by a CLIA-certified laboratory (e.g. the Pathology and Molecular Medicine laboratory at Henry Ford Health System (http://www.henryford.com/body.cfm?id=50927)). The safety and toxicity of combining AZD9291 with radiation and concurrent temozolomide can be assessed, and the maximum tolerated dose can be determined. Assuming that safety and tolerability are acceptable, the phase II trial can be started at the established dose. The design of a phase II trial that recruits patients having EGFRvIII-expressing GBM is shown in Fig. (2).
Fig. (2).
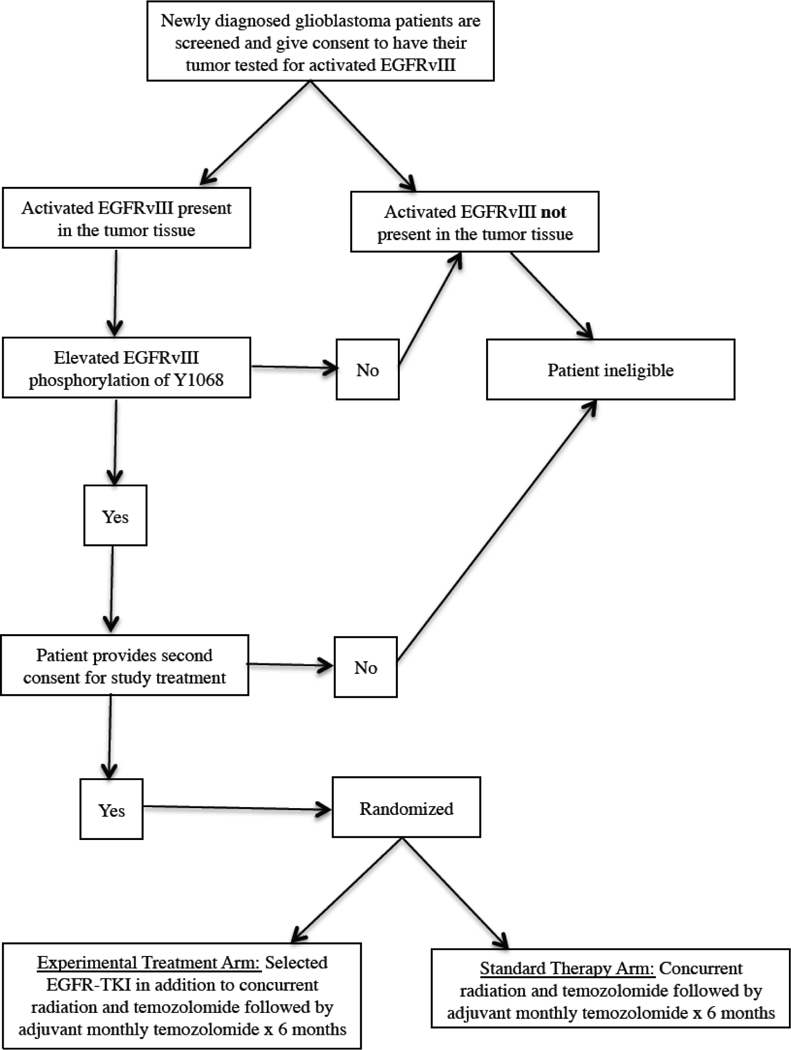
Patient selection based on EGFRvIII genotype.
While a blood-based biomarker reflecting the efficacy of an EGFR-TKI is not yet established, blood samples will be taken to measure the plasma concentration of AZD9291, as it may vary depending on the genotypes of each patient’s drug-metabolizing enzymes. For pharmacodynamics studies, peripheral blood mononuclear cells (PBMCs) from the blood collected before the initiation of the therapy and at monthly intervals after therapy initiation can be used to discover new biomarkers reflecting response to the therapy. The use of PBMCs to assess neuronal function has been described [44], and “OMICS” approaches, such as DNA microarray, proteomics and metabolomics can be used for discovering biomarkers reflecting a response to AZD9291 [45]. A recent study reported serum levels of carcinoembryonic antigen (CEA) correlating with EGFR-TKI response in lung cancer patients [46], and a similar situation may occur in GBM patients. The effectiveness of AZD9291 can also be assessed by measuring plasma levels of three proteins: glial fibrillary acidic proteins (GFA), chitinase-2-like protein (YKL-40), and insulin-like growth factor binding protein 2 (IGFBP2), as these proteins appear to be the biomarkers of GBM [47, 48].
CONCLUSION
In summary, the molecular characterization of a large number of GBMs by TCGA has identified numerous somatic mutations in EGFR. These genetic changes in EGFR gene affect GBM growth differentially. Therefore, any attempt to target EGFR in GBMs must segregate GBM patients in terms of their EGFR genotype. A failure to select GBM patients in terms of their EGFR genotype in previous attempts to target EGFR may be the cause of not observing any response to EGFR-TKI and thus “failed” trials. Additionally, the preclinical testing should involve testing in GSCs (isolated from GBMs carrying the selected EGFR genotype) and animal models carrying the xenografts of GBMs with the selected EGFR genotype. This example of the study of the drug AZD9291 can perhaps guide future preclinical and clinical research of targeted therapies for GBMs, so a successful treatment can replace the insufficient standard of care.
ACKNOWLEDGEMENTS
These studies were supported by an NIH/NCATS grant #1 UH2 TR001370-01.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- [1].Louis DN, et al. , The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol, 2007. 114(2): p. 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stupp R, et al. , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med, 2005. 352(10): p. 987–96. [DOI] [PubMed] [Google Scholar]
- [3].Brennan CW, et al. , The somatic genomic landscape of glioblastoma. Cell, 2013. 155(2): p. 462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Little SE, et al. , Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res, 2012. 72(7): p. 1614–20. [DOI] [PubMed] [Google Scholar]
- [5].Snuderl M, et al. , Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell, 2011. 20(6): p. 810–7. [DOI] [PubMed] [Google Scholar]
- [6].Maire CL and Ligon KL, Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro Oncol, 2014. 16(suppl 8): p. viii1–viii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ekstrand AJ, et al. , Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res, 1991. 51(8): p. 2164–72. [PubMed] [Google Scholar]
- [8].Frederick L, et al. , Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res, 2000. 60(5): p. 1383–7. [PubMed] [Google Scholar]
- [9].Huang HS, et al. , The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem, 1997. 272(5): p. 2927–35. [DOI] [PubMed] [Google Scholar]
- [10].Nagane M, et al. , A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res, 1996. 56(21): p. 5079–86. [PubMed] [Google Scholar]
- [11].Prigent SA, et al. , Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J Biol Chem, 1996. 271(41): p. 25639–45. [DOI] [PubMed] [Google Scholar]
- [12].Frattini V, et al. , The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet, 2013. 45(10): p. 1141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Furgason JM, et al. , Whole genome sequencing of glioblastoma multiforme identifies multiple structural variations involved in EGFR activation. Mutagenesis, 2014. 29(5): p. 341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gallant JN, et al. , EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer That Is Clinically Responsive to Afatinib. Cancer Discov, 2015. 5(11): p. 1155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vivanco I, et al. , Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov, 2012. 2(5): p. 458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brandes AA, et al. , Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res, 2008. 14(4): p. 957–60. [DOI] [PubMed] [Google Scholar]
- [17].Karpel-Massler G, et al. , Therapeutic inhibition of the epidermal growth factor receptor in high-grade gliomas: where do we stand? Mol Cancer Res, 2009. 7(7): p. 1000–12. [DOI] [PubMed] [Google Scholar]
- [18].Hegi ME, et al. , Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther, 2011. 10(6): p. 1102–12. [DOI] [PubMed] [Google Scholar]
- [19].Haas-Kogan DA, et al. , Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst, 2005. 97(12): p. 880–7. [DOI] [PubMed] [Google Scholar]
- [20].Chakravarti A, et al. , RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys, 2013. 85(5): p. 1206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reardon DA, et al. , Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol, 2014. [DOI] [PMC free article] [PubMed]
- [22].Brown KE, et al. , Proteomic profiling of patient-derived glioblastoma xenografts identifies a subset with activated EGFR: Implications for drug development. J Neurochem, 2015. [DOI] [PMC free article] [PubMed]
- [23].Cross DA, et al. , AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov, 2014. [DOI] [PMC free article] [PubMed]
- [24].Politi K, Ayeni D, and Lynch T, The Next Wave of EGFR Tyrosine Kinase Inhibitors Enter the Clinic. Cancer Cell, 2015. 27(6): p. 751–3. [DOI] [PubMed] [Google Scholar]
- [25].Emlet DR, et al. , Targeting a Glioblastoma Cancer Stem Cell Population Defined by EGF Receptor Variant III. Cancer Res, 2013. [DOI] [PMC free article] [PubMed]
- [26].Beier D, et al. , CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res, 2007. 67(9): p. 4010–5. [DOI] [PubMed] [Google Scholar]
- [27].Bayin NS, Modrek AS, and Placantonakis DG, Glioblastoma stem cells: Molecular characteristics and therapeutic implications. World J Stem Cells, 2014. 6(2): p. 230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng L, Bao S, and Rich JN, Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol, 2010. 80(5): p. 654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matsuda K, et al. , Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci Rep, 2012. 2: p. 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Szerlip NJ, et al. , Intratumoral eterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A, 2012. 109(8): p. 3041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Soeda A, et al. , The evidence of glioblastoma heterogeneity. Sci Rep, 2015. 5: p. 7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Van Brocklyn JR, et al. , Aurora-A Inhibition Offers a Novel Therapy Effective against Intracranial Glioblastoma. Cancer Res, 2014. 74(19): p. 5364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Higgins DM, et al. , Brain tumor stem cell multipotency correlates with nanog expression and extent of passaging in human glioblastoma xenografts. Oncotarget, 2013. 4(5): p. 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Akbani R, et al. , Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: a workshop report: the RPPA (Reverse Phase Protein Array) society. Mol Cell Proteomics, 2014. 13(7): p. 1625–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Siolas D and Hannon GJ, Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res, 2013. 73(17): p. 5315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tentler JJ, et al. , Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol, 2012. 9(6): p. 338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wheeler S, et al. , Tumor epidermal growth factor receptor and EGFR PY1068 are independent prognostic indicators for head and neck squamous cell carcinoma. Clin Cancer Res, 2012. 18(8): p. 2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Poteet E, et al. , Reversing the Warburg effect as a treatment for glioblastoma. J Biol Chem, 2013. 288(13): p. 9153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Luchman HA, et al. , Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin Cancer Res, 2014. 20(22): p. 5756–67. [DOI] [PubMed] [Google Scholar]
- [40].Bernards R, A missing link in genotype-directed cancer therapy. Cell, 2012. 151(3): p. 465–8. [DOI] [PubMed] [Google Scholar]
- [41].Mattie M, et al. , Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia, 2013. 15(10): p. 1138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Janne PA, et al. , AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med, 2015. 372(18): p. 1689–99. [DOI] [PubMed] [Google Scholar]
- [43].Reardon DA, et al. , A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res, 2013. 19(4): p. 900–8. [DOI] [PubMed] [Google Scholar]
- [44].Liew CC, et al. , The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med, 2006. 147(3): p. 126–32. [DOI] [PubMed] [Google Scholar]
- [45].Meyer UA, Zanger UM, and Schwab M, Omics and drug response. Annu Rev Pharmacol Toxicol, 2013. 53: p. 475–502. [DOI] [PubMed] [Google Scholar]
- [46].Pan JB, Hou YH, and Zhang GJ, Correlation between efficacy of the EGFR tyrosine kinase inhibitor and serum tumor markers in lung adenocarcinoma patients. Clin Lab, 2014. 60(9): p. 1439–47. [DOI] [PubMed] [Google Scholar]
- [47].Preusser M, Neuro-oncology: a step towards clinical blood biomarkers of glioblastoma. Nat Rev Neurol, 2014. 10(12): p. 681–2. [DOI] [PubMed] [Google Scholar]
- [48].Gallego Perez-Larraya J, et al. , Diagnostic and prognostic value of preoperative combined GFAP, IGFBP-2, and YKL-40 plasma levels in patients with glioblastoma. Cancer, 2014. [DOI] [PubMed]