

Abstract
Rationale:
Obesity increases morbidity and mortality in acute illnesses such as sepsis and septic shock. We showed previously that the early/hyper-inflammatory phase of sepsis is exaggerated in obese mice with sepsis; sirtuin 2 (SIRT2) modulates sepsis-inflammation in obesity. Evidence suggests that obesity with sepsis is associated with increased oxidative stress. It is unknown whether exaggerated hyper-inflammation of obesity with sepsis modulates the SIRT2 function in return. We showed recently that SIRT6 oxidation during hyper-inflammation of sepsis modulates its glycolytic function. This study tested the hypothesis that increased oxidative stress and direct SIRT2 oxidation exaggerate hyper-inflammation in obesity with sepsis.
Methods:
Using spleen and liver tissue from mice with diet induced obesity (DIO) we studied oxidized vs. total SIRT2 expression during hyper- and hypo-inflammation of sepsis. To elucidate the mechanism of SIRT2-oxidation (specific modifications of redox sensitive cysteines) and its effect on inflammation, we performed site-directed mutations of redox-sensitive cysteines Cys221 and Cys224 on SIRT2 to serine (C221S and C224S), transfected HEK293 cells with mutants or WT SIRT2, and studied SIRT2 enzymatic activity and NFĸBp65 deacetylation. Finally, we studied effect of SIRT2 mutation on LPS-induced inflammation using RAW 264.7 macrophages.
Results:
In an inverse relationship, total SIRT2 decreased while oxidized SIRT2 expression increased during hyper-inflammation and SIRT2 was unable to deacetylate NFĸBp65 with increased oxidative stress of obesity with sepsis. Mechanistically, both the mutants (C221S and C224S) show decreased 1) SIRT2 enzymatic activity, 2) deacetylation of NFĸBp65, and 3) anti-inflammatory activity in response to LPS vs. WT SIRT2.
Conclusion:
Direct oxidation modulates SIRT2 function during hyper-inflammatory phase of obesity with sepsis via redox sensitive cysteines.
Keywords: Obesity, Sepsis, Septic shock, hyper-inflammation, oxidative stress
Summary statement:
Cysteine thiols regulate SIRT2 during hyper-inflammation of obesity with sepsis
INTRODUCTION:
Sepsis and septic shock are the leading causes of death in non-coronary intensive care units with increasing incidence worldwide. With over 100 clinical trials failed to decrease sepsis-related mortality [35], currently there are no specific therapies to treat sepsis. Sepsis and septic shock are the most expensive conditions in the United States with over $23 billion in healthcare expenditure in 2013[50]. Sepsis-inflammation transitions its phenotype from early/hyper-inflammatory to a late/hypo-inflammatory phase [4, 17]. Previously, we reported hyper-inflammatory and hypo-inflammatory phases in mice [55, 63].
Obesity not only increases incidence of chronic conditions such as hypertension, dyslipidemia, type 2 diabetes mellitus, cancer, cognitive dysfunction in elderly, obesity also increases morbidity and resource utilization in critically ill patients including sepsis [41, 13, 6]. Obese individuals are prone to sepsis compared to lean counterparts[62]. Impact of obesity on sepsis-related mortality remains controversial; however literature strongly supports the notion that obesity increases morbidity, ICU and hospital length of stay and in turn the cost of care in critically ill patients including sepsis[51, 46, 43, 47, 1]. Using a mouse model of obesity with sepsis, we found that obesity exaggerates the hyper-inflammatory and prolongs hypo-inflammatory phase of sepsis via modulation of cytokines/adipokines[52, 60, 65, 58, 64, 63].
Sirtuins play a critical role in transition of sepsis phenotype from hyper- to hypo-inflammation [32, 31, 55, 30]. Sirtuins, a highly conserved family of proteins, are critically important in the metabolic and immune response of cells [48, 26, 57]. The seven mammalian sirtuins (SIRT1–7) are dispersed among different cell compartments and in some cases move from one compartment to another. SIRTs 1, 6, and 7 are primarily nuclear; SIRT 3,4, 5 are mitochondrial and SIRT2 is best known for its cytosolic location[16]. However, SIRT2 translocates to the nucleus and controls epigenetic programming of inflammation and metabolism [29, 39].
Obesity is a sirtuin deficient state; sirtuin deficiency is implicated in chronic inflammation with obesity [37, 34]. Although associated with overall sirtuin deficiency, SIRT2 is the most abundant of sirtuins present in obese adipose tissue [27]. We have reported that SIRT2 modulates microvascular inflammation in obese mice with sepsis; SIRT2 directly deacetylates and deactivates NFkB p65[63]. We have also reported that mice with SIRT2 deficiency show exaggerated while those with SIRT2 over-expression show attenuated hyper-inflammatory phase of sepsis [5]. Thus, together, these data indicate that SIRT2 modulates sepsis-related inflammatory response. It is unclear however, whether sepsis itself modulates SIRT2 function in return. This project’s goal was to study how sepsis-inflammation regulates SIRT2 function.
Reactive oxygen species (ROS) or nitrogen derived species (RNS) generation during hyper-inflammation of sepsis injures DNA and proteins [15], particularly in mitochondria. It is less well understood whether and how ROS and RNS species might control inflammation programming by contributing to signal transduction of inflammatory pathways rather than disrupting cell structure and function. For example, H2O2 clearly directs cell signaling in many pathways and phenotypes of human physiology, including inflammation [14]. Literature suggests that ROS regulate the deacetylase activity of SIRT1 in vitro [23]. All members of the sirtuin family contain a highly-conserved Zinc region surrounded by a cysteine CXXC motif [8]. Recent reports indicate oxygen or nitrogen derived species (e.g., nitrosation) regulate SIRT1 deacetylase activity by repositioning of the tetra thiolate subdomain away from the rest of the catalytic domain thereby directly disrupting the NAD+ and acetyl-lysine-binding sites [25]. Direct oxidation of sirtuins is also implicated in regulating SIRT1 and SIRT6 enzymatic and metabolic activity in vitro; whether direct oxidation of sirtuins affects their ability to modulate inflammation and immunity remains unknown [68, 23, 33]. Moreover, the functional consequences of direct oxidation of SIRT2 are unknown as well.
Obesity with sepsis increases oxidative stress during hyper-inflammation [61]. Moreover, obesity increases oxidant stress of immune cells in its basal state [12]. Here, we hypothesized that the cysteine-coordinated Zn2+ cofactor in SIRT2 regulates its deacetylase function, especially in acute (sepsis) on chronic (obesity) inflammation in obesity with sepsis. We find that during hyper-inflammation of sepsis, there is an inverse relationship between total SIRT2 and oxidized SIRT2 expressions in vivo in mouse model of obesity with sepsis. We also show that specific redox sensitive cysteines located near the Zn2+ cofactor in SIRT2 play a critical role in regulating expression of the key inflammatory and immune mediators. Mechanistically, mutation of redox-sensitive cysteines, Cys221 or Cys224 decreases SIRT2’s enzymatic activity and its ability to deacetylate NFkB p65, which amplifies expression of the key pro-inflammatory mediators TNF-α, IL-6 and IL1-β. Thus direct oxidation of SIRT2 cysteine thiols contributes to the exaggerated hyper-inflammatory phase of obesity with sepsis.
MATERIALS AND METHODS:
Animals:
All the experiments were in accordance with NIH guidelines and approved by IACUC at Wake Forest School of Medicine. The C57Bl/6J (WT) (6–8 weeks of age) mice, diet induced obesity (DIO) mice, mice on control diet (CTRL mice); DIO and CTRL mice 13–15 weeks of age were purchased from Jackson laboratories. DIO mouse model is created by feeding C57BL/6 mice with high fat diet (D12492, Research Diets Inc., 60%: the diet induced obesity diet: DIO); corresponding age matched control mice were fed with low fat diet (D12450B, Research Diets Inc., 10%: Control diet: CTRL) or for 7–9 weeks. DIO/CTRL diet feeding was initiated at 6 weeks of age.
Cecal ligation and puncture:
Cecal ligation and puncture (CLP) procedure was performed as described before [55]. Briefly, mice were anesthetized using isoflurane (2–3 l/min Isoflurane and O2 mixture). Anterior abdominal wall and peritoneal incision (1.5 cm in length) were made, cecum was isolated, ligated (1 cm of cecum) and punctured two times with 22 gauge needle twice (CLP 22.2 model), and contents were returned into the abdomen. Abdomen was closed in two layers (peritoneum and skin) and mice were allowed to wake up. Rigorous monitoring of mice for pain and distress was completed as described previously. Mice were humanely euthanized at indicated times under isoflurane anesthesia, liver and spleen tissue collected and splenocytes were isolated immediately as described before [63].
In vivo cysteine oxidation assay:
We used injection of Biotin-1, 3-cyclopentanedione (BP1), a selective protein sulfenylation probe (gift from our collaborator Dr. Furdui) to track cysteine oxidation in vivo in obese mice with sepsis [44, 33]. Three groups of mice were used to perform this assay (n=3/group), control, hyper-inflammation (6h CLP) and hypo-inflammation (24h CLP). Mice were anesthetized and cannulated via carotid artery and jugular vein followed by intravenous injection of BP1 (25 mg/kg body weight). BP1 was allowed to circulate for 30 minutes before initiation of isovolemic blood exchange as described in literature [54, 60]. Liver tissue was obtained post-blood exchange and preserved in OTC medium for frozen section as described previously [63, 5].
Immunohistochemistry:
Liver tissue was collected from DIO mice without CLP (control) or during hyper and hypo-inflammatory phases of sepsis and stored in OTC medium for frozen section and immunohistochemistry for SIRT2 expression was performed as described before [63]. Briefly, liver tissue was harvested fixed frozen sections of tissue were stained using SIRT-2 antibody (Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA), Cy™3-conjugated labeled secondary antibody for SIRT-2 was purchased from Jackson Immuno Research Laboratories, Inc. (West Grove, PA, USA). Virtual images were captured as described previously [56]. Representative image was shown in the Figure 1A. Image analysis of IHC in three mice in each group was performed using Image J software and shown in Figure 1B. To image BP1-labeled protein sulfenylation in tissue from BP1 injected mouse, we used streptavidin-AF594 (Molecular Probes S11227 2mg/ml 1:100) antibody (since the probe is already biotinylated) and DAPI.
Figure 1: Total SIRT2 expression in the liver tissue is decreased during hyper-inflammatory and increased during hypo-inflammatory phase of obese with sepsis:
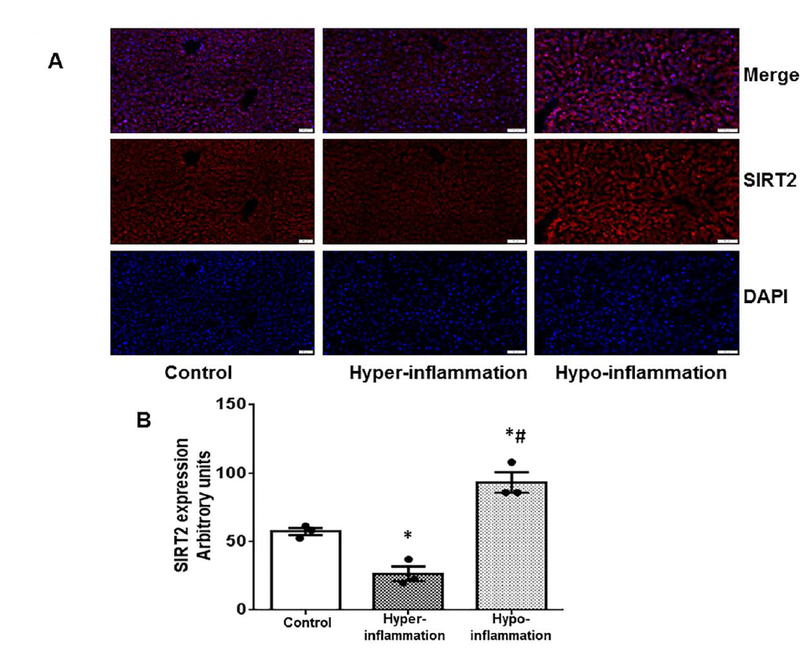
We studied the SIRT2 expression in the liver tissue of diet induced obesity mice with sepsis using immunohistochemistry. The representative images are shown in Figure 1A while quantitative assessment of image analysis from three different mice is shown in Figure 1B. SIRT2 expression significantly decreased during the hyper-inflammatory phase (6h CLP) of sepsis and increased significantly during the hypo-inflammatory phase (24h post-CLP) of sepsis compared to control (without sepsis) in DIO mice (* p<0.05 vs. control; # p<0.05 vs. hyper-inflammation).
Cell lines used:
RAW264.7 (ATCC® TIB-71™: RAW) and HEK 293 (ATCC® CRL- 1573™: HEK) cells were obtained from ATCC. Cells were cultured in DMEM containing 10% heat inactivated FBS, 100 units/ml penicillin, and 100 mg/ml streptomycin at 37°C and 5% CO2. Early passage (2–10) cultures were used in all experiments.
Mouse tissue and splenocyte oxidation:
To evaluate the expression of oxidized SIRT2 during hyper- and hypo-inflammatory response of sepsis, we used whole spleen and liver tissue as well as splenocytes from diet induced obesity (DIO) mice with sepsis. Splenocytes (n= 6 ×106) were isolated at indicated time points after CLP and lysed in 600 μL lysis buffer (25mM HEPES, 100mM NaCl, 1% NP-40, 0.1% SDS, 1mM EDTA, 100mM N-Ethyl Maleimide: NEM) with shaking for 25 minutes at 50oC. Lysates were spun down to remove insoluble material. Supernatant was collected and passed through a Zeba column (Thermo Scientific) to remove unbound NEM. Freshly prepared 60 μL of 500mM DTT solution was added to each sample and samples were incubated for 20 minutes at room temperature. Samples were passed through a Zeba column to remove excess DTT. An aliquot of 40 μL was removed from sample and saved as an input control. Biotin conjugated maleimide (Thermo Scientific) was added to remaining sample at a final concentration of 2mM and samples were labeled for 2 hours in the dark. Samples were then passed through a Zeba column to remove unbound maleimde. Labeled samples were incubated overnight with magnetic streptavidin beads at 4oC. Beads were collected and were washed extensively. Washed beads were boiled for 5 minutes in reducing sample buffer and then boiled samples were loaded onto a gel along with unlabeled samples as an input control. Gels were blotted with anti SIRT2 (Santa Cruz). Tissue was incubated for 10 minutes in 100mM NEM to block free thiols and then snap frozen. Tissue was thawed on ice and homogenized in 1% SDS supplemented with 10mM NEM and protease inhibitors. Homogenized tissue was incubated with shaking at 50°C for 25 minutes. Lysed tissues were sonicated and then passed through a Zeba column to remove unbound NEM. Protein concentration was determined on lysates and equal amounts of protein from each lysate were then labeled with Biotin conjugated maleimide as above. Two different methods were utilized to detect SIRT2 oxidation in whole tissue. Some samples were then incubated overnight with Streptavidin magnetic beads and immunoblotted with anti-SIRT2 antibody and other samples were immunoprecipitated overnight with anti-SIRT2 antibody followed by immunoblotting with anti-Biotin antibody.
Western blot:
Western blot experiments were performed as described previously[66]. Cells were treated as above and lysed in a modified RIPA buffer (Cell Signaling Technology, Cat# 9806). To check SIRT2 protein expression level, protein lysate was resolved by SDS-PAGE. The membrane was incubated with primary antibody overnight in cold room, and subsequently with Alexa Fluor® 680-IgG Fraction Monoclonal Mouse Anti-Rabbit IgG (Jackson ImmunoResearch Lab, Cat# 211–622–171) or Alexa Fluor® 680-AffiniPure Goat Anti-Mouse IgG ((Jackson ImmunoResearch Lab, Cat# 115–625–174)). The images were developed with an Odyssey Infrared Imager System (Li-COR Biosciences). Bands signal was quantified with software Odyssey V3.0. The plot was completed with GrapPad Prism 6. SIRT2 antibody was from Santa Cruz Biotechnology, Cat# sc-20966. Acetylated p65 antibody was from Cell Signaling Technology, Cat# 3045S. GAPDH antibody was from OriGene, Cat# TA802519. Flag antibody was from Sigma, Cat# F1804. Licor Odyssey software was used to quantitate WB blots where indicated.
SIRT2 oxidation effects on NFĸB p65 acetylation:
We reported previously, that SIRT2 deacetylates NFĸB p65 [63]. Here we studied the effect of pro-oxidant environment on deacetylation function of NFĸB p65. We used HEK 293 cells due to the fact that this cell line is known to have a high transfection efficiency. In our hands, the transfection efficiency was nearly 100% with single as well as multiple plasmid transfections. HEK293 cells were co-transfected with plasmid NFĸB p65, pCBP, and pSIRT2-Flag. Luperox® TBH70X, tert-Butyl hydroperoxide solution (Sigma, Cat# 458139, and final 10uM) was applied to cell culture for 12h after 36h of transfection. Total NFĸB p65 and ac- NFĸB p65 were blotted, and the ratio of ac- NFĸB p65/total NFĸB p65 signal was plotted. pcDNA3β-FLAG-CBP- HA was a gift from Tso-Pang Yao (Addgene plasmid # 32908) [69], pCMV4 NFĸB p65 was a gift from Warner Greene (Addgene plasmid # 21966) [3]. Wild-type plasmid SIRT2 Flag was a gift from Eric Verdin (Addgene plasmid # 13813) [38].
Cysteine mutation of SIRT2:
To study the effect of redox sensitive cysteines, Cys221 and Cys224, we mutated those two cysteines for further mechanistic studies.
The following primers were used to create point mutations.
S2F: 5’-ACCGAGCTCGGATCCGAATTCTATGGCAGAGCAGACCG-3’
S2R: 5’-CGGGTTTAAACGGGCCCTCTAGATTACTTGTCATCGTC-3’
C221S-F: 5’-GGTGACGCCCAAGAGTGAAGACTGTCAGAG-3’
C221S-R: 5’-CTCTGACAGTCTTCACTCTTGGGCGTCACC-3’
C224S-F: 5’-CCCAAGTGTGAAGACAGTCAGAGCCTGGTGAAG-3’
C224S-R: 5’-CTTCACCAGGCTCTGACTGTCTTCACACTTGGG-3’
All primers were purchased from Invitrogen. To develop point mutant plasmid pSIRT2 C221S (Cys 221 replaced with Ser), primers S2F/C221S-R and C221S-F/S2R were used to introduce point mutant in the PCR amplification of two fragments from wild-type plasmid SIRT2-Flag, followed by gel purification of PCR products, and then a second run PCR was completed using primers S2F/S2R to amplify the full-length of mutant SIRT2 coding sequence taking the gel purified PCR products as template. The final PCR products were digested with BamHI/XbaI and ligated into pcDNA3.1+ vector which is restricted with BamHI/EcoRI/XbaI, producing mutant plasmid pSIRT2 C221S (SIRT2 C221S). The mutant plasmid pSIRT2 C224S (SIRT2 C224S) was completed with the same method, but using primers C224S-F and C224-R. Flag tag was kept in both mutant plasmids. All plasmids were verified by sequencing.
SIRT2 enzymatic activity assay:
To explore the effects of point mutation on SIRT2 deacetylation activity, plasmid pcDNA3.1 (as control; denoted as “Empty vector”), wild-type SIRT2-Flag, mutant plasmids SIRT2 C221S and SIRT2 C224S were transfected into HEK293 cells. After 48 hours total protein lysate was collected in IP Lysis Buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1% Triton X-100%), followed by protein concentration quantification and adjustment. Protein lysate (1.4mg) was mixed with anti-FLAG M2 Magnet beads (Sigma, Cat# M8823), and incubated overnight at 4oC. The beads were washed with IP Washing Buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl), and then washed with SIRT Activity Buffer. The beads-binding SIRT2 activity was performed by incubating with SIRT activity buffer, Fluoro-Substrate Peptide, NAD, and Developer at 37oC with vigorous agitation (800rpm) for 2h in a shaker following the protocol of SIRT2 Activity kit (Abcam, Cat# ab156066). The supernatant was transferred into 96-well black plate to read fluorescence on a fluorometric plate reader (ex485/em528). The activity was corrected for respective SIRT2 (WT and mutant) protein expression.
SIRT2 mutation effects on NFĸBp65 acetylation:
HEK293 cells were co-transfected with CBP and NFĸBp65 plasmids, together with WT SIRT2 plasmid or C221S or C224S mutant plasmids, respectively. Total cell lysate was collected to blot with anti-acetylated NFĸBp65 and anti-Flag as described above. The acetylation activity of WT SIRT2 or cysteine mutants SIRT2 C221S and SIRT2 C224S was monitored by AC-NFĸB p65 which is corrected with anti-Flag signal. The fold change from WT SIRT2 is presented.
SIRT2 mutation effects on LPS-induced inflammation:
To assess the effect of cysteine mutations (C221S and C224S) on LPS-induced inflammation. We used RAW 264.7 macrophage cell line to study the effect of SIRT2 mutation on inflammatory response to LPS. While the transfection efficiency of RAW cells was good enough for single plasmid, in our hands, the co-transfection efficiency was not good. So we used RAW cells for single plasmid transfection only. Plasmid WT SIRT2, mutant plasmid SIRT2 C221S, and SIRT2 C224S were transfected into RAW264.7 cells, respectively. Cells were treated with normal saline (control) or LPS (final concentration of 100ng/mL) for 4h after the transfection of 48h. Total protein lysate was collected for western blotting with SIRT2 antibody. Total RNA was extracted (as described below) to determine the mRNA levels of inflammation cytokines. The final cytokine levels were corrected for respective anti-flag (WT SIRT2 or mutant SIRT2) signal.
RNA extraction and RT-PCR:
RNA extraction and RT-PCR were completed as described previously and modified [34]. Total RNA was isolated following the protocol of Tri-Reagent manufacture. The mRNA expression was detected by quantitative real time PCR with Luna® Universal Probe One-Step RT-qPCR Kit (NEB, E3006L). GAPDH gene expression was used to correct the target gene expression data. Relative quantification was calculated using the ΔΔ comparative threshold formula. All samples were run in quadruplicates to calculate average and SE value. TaqMan primer/probes were purchased from Invitrogen (Grand Island, NY). Mouse TNF-α, Mm00443258_m1; mouse IL1β, Mm00434228_m1; mouse GAPDH, Mm99999915_g1.
Statistical methods:
All data were analyzed using Graph Pad Prism 6.0 (Graph Pad Software, La Jolla, CA, USA); data presented mean± SEM. Analyses with more than two groups were analyzed using one-way ANOVA or twoway ANOVA with Tukey’s post hoc comparisons as appropriate. A p value of < 0.05 was designated as significant.
RESULTS:
Total SIRT2 expression in the liver tissue decreases during hyper-inflammatory and increases during hypo-inflammatory phase of obesity with sepsis:
We have reported previously that ob/ob mice showed increased SIRT2 expression during hypo-inflammatory phase of sepsis [63]. Here we studied the SIRT2 expression in the liver tissue of DIO mice with sepsis (CLP) using immunohistochemistry. As shown in the representative IHC image in Figure 1A and IHC image quantification from three different groups (n=3 each group) (Figure 1B), we observed that the SIRT2 protein expression decreased significantly during the hyper-inflammatory phase (6h post-CLP) of sepsis, while it increased significantly during the hypo-inflammatory phase (24h post-CLP) of sepsis compared to control (without sepsis) in DIO mice.
Oxidized SIRT2 expression during hyper-inflammatory phase of sepsis in liver tissue of obese mice with sepsis:
Obesity with sepsis is associated with significantly increased oxidative stress [61]. Next we tracked the expression of oxidized proteins in liver tissue. We used our novel BP1 probe developed by Dr. Furdui. As shown in Figure 2A and B (fluorescent image quantification from three different samples) protein cysteine thiol oxidation in liver tissue increases during the hyper-inflammatory phase vs. control; in contrast, cysteine thiol oxidation decreased significantly during the hypo-inflammatory phase. Next, we assessed oxidized SIRT2 expression in liver tissue from control, hyper-inflammatory and hypo-inflammatory phase of sepsis using biotin switch assay. A representative image from Figure 2C and western blot image quantification from three different tissue samples (Figure 2D) show that similar to total protein oxidation (Figures 2A and B), SIRT2 oxidation in liver tissue increased significantly during hyper-inflammation vs. control and that during hypo-inflammation decreased significantly vs. hyper-inflammatory phase (down to baseline control level) of obese mice with sepsis.
Figure 2: Oxidized total protein and SIRT2 expression in liver and spleen during hyper- and hypo-inflammatory phase in obese with sepsis:
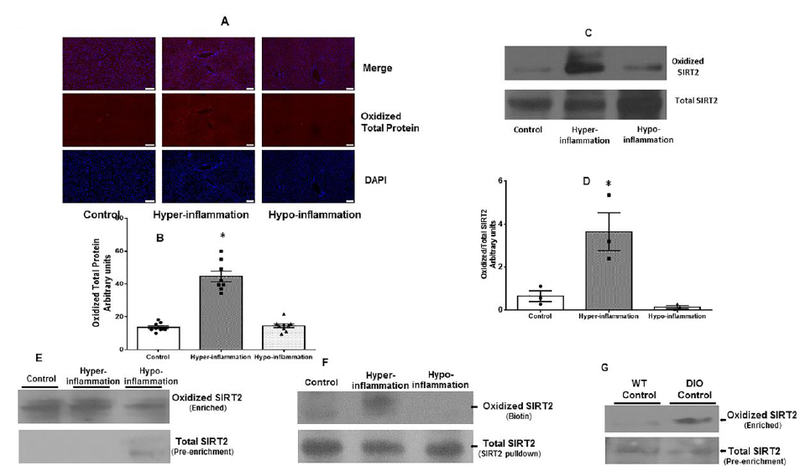
Figure 2A and 2B: We tracked cysteine oxidation using a selective protein sulfenylation probe (BP1) in vivo in three groups of mice under control, hyper-inflammatory and hypo-inflammatory phases of sepsis. The BP1 expression was probed using immunohistochemistry in the liver tissue. The representative images (Figure 2A) and image quantification from three different samples (Figure 2B) shows that the oxidized protein expression in the liver tissue increased significantly during hyper-inflammatory phase vs. control and it decreased significantly during hypo-inflammatory phase vs. hype-inflammation. Figure 2C and D: In separate groups of mice, we used biotin switch assay to study oxidized vs. total SIRT2 expression in liver tissue using western blot. Representative blots (Figure 2C) and image quantification from three samples per group (Figure 2D) shows that similar to total protein oxidation, oxidized SIRT2 expression increased significantly during hyper-inflammation vs. control; oxidized SIRT2 expression during hypo-inflammation decreased significantly during hypo-inflammatory phase vs. hyper-inflammation. (* p<0.05 vs. Control; # p<0.05 vs. hyper-inflammation). Figure 2E: Biotin switch assay of isolated splenocytes DIO mice revealed that oxidized SIRT2 expression increased during the hyper-inflammatory phase while it decreased during the hypo-inflammatory phase of sepsis while total SIRT2 expression was detectable only during the hypo-inflammatory phase. Figure 2F: Biotin switch assay of spleen tissue with immunoprecipitation for SIRT2 followed by western blot assay for biotin (oxidized protein) revealed that similar to splenocytes (figure 2E), there was increased oxidized SIRT2 expression during hyper-inflammation and decreased oxidized SIRT2 expression during hypo-inflammatory phase of sepsis. Figure 2G: Increased expression of oxidized SIRT2 was observed at baseline in DIO mice vs. WT (lean C57Bl/6) mice. Please note that the basal total SIRT2 expression in the splenocytes DIO mice was also low in the DIO vs. WT.
Oxidized SIRT2 expression increased during the hyper-inflammatory phase in immune cells in obese mice with sepsis:
To study the effect of oxidative stress during obesity with sepsis on direct SIRT2 oxidation in immune cells, we isolated splenocytes and whole spleen tissue from obese mice during hyper- (6 hours post-CLP) and hypo-inflammatory (24h post-CLP) phases of sepsis. Using biotin switch assay, we studied oxidized SIRT2 expression. We used two different strategies to study SIRT2 oxidation. In Figure 2E, we immunoprecipitated for biotin and probed for SIRT2 protein expression. Due to inability to detect sufficient SIRT2 protein in the pre-enrichment lysate, we immunoprecipitated for SIRT2 and then probed for biotin using anti-biotin antibody. As shown in representative images from three separate sets of experiments in Figure 2E and F we observed similar pattern in both the cases; oxidized SIRT2 expression increased during hyper-inflammatory and decreased during the hypo-inflammatory phase of sepsis while total SIRT2 expression increased/was detectable only during hypo-inflammation (Figure 2E). This pattern simulated to that seen in the liver tissue. Interestingly, the basal level of SIRT2 oxidation in obese mice without sepsis was higher than that in WT mice (Figure 2G).The data support that SIRT2 oxidation is increased in basal obese phenotype which further increases during hyper-inflammatory phase of sepsis. Together, data presented in Figures 1 and 2 indicate an inverse relationship between total and oxidized SIRT2 expression in mouse model of obesity and sepsis; decreased total SIRT2 protein expression with increased cysteine thiol SIRT2 oxidation during hyper-inflammatory phase of sepsis. This is then followed by decreased SIRT2 oxidation during hypo-inflammation.
Oxidative stress decreases SIRT2 function:
In order to further elucidate the effect of oxidized SIRT2 protein on sepsis-inflammation, we first studied whether the pro-oxidant environment influences deacetylation function of SIRT2. We showed previously, that the SIRT2 directly deacetylates and deactivates NFkB p65 [63]. Here, we studied the effect of pro-oxidant environment on SIRT2 deacetylation function by assessing acetylated NFkB p65 expression. We co-transfected HEK 293 cells with NFkB p65 and p300 (to increase baseline acetylation) with SIRT2 plasmids and treated with and without tert-butyl hydroperoxide (TBH) to induce oxidative stress. Total protein expression was studied using western blot for NFkB p65 acetylation (AC-NFkB p65 expression). As shown in Figure 3, acetylated NFkB p65 (AC-NFkB p65) expression was higher in TBH-treated cells than that in control (normal saline treated) cells; indicating that under oxidative stress conditions, SIRT2 is less efficient in deacetylating NFkB p65.
Figure 3: Oxidation decreases SIRT2 deacetylation function:
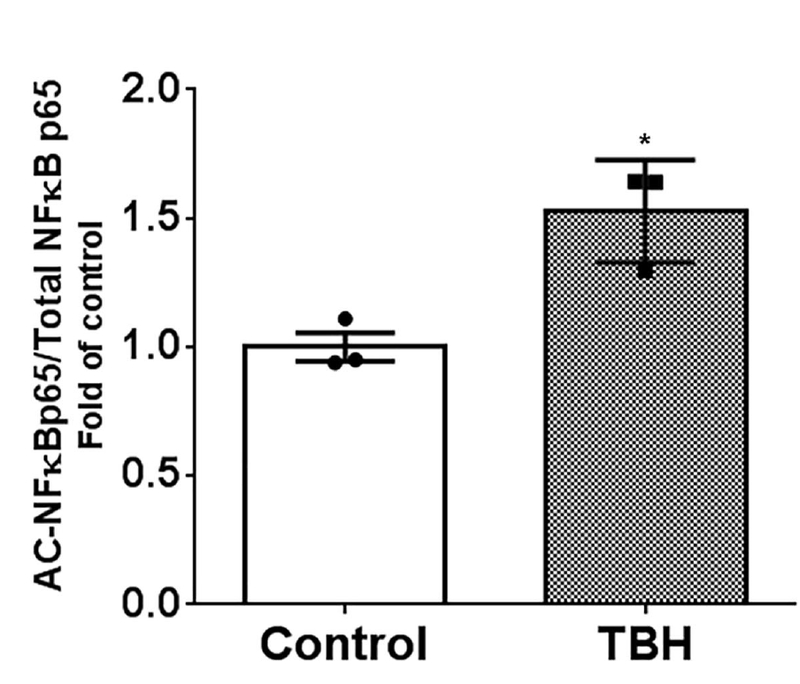
HEK cells were transfected with SIRT2+p65+p300 plasmids to increase p65 acetylation and treated with and without TBH to study acetylated NFĸBp65 (AC-NFĸBp65) expression. AC-NFĸBp65 expression significantly increased in TBH treated vs. untreated (control) cells (* p<0.05 vs. control).
Redox sensitive cysteine thiols control SIRT2 function:
Next, we studied the mechanism of how oxidation on cysteine thiols could directly affect SIRT2. Four cysteines flank an evolutionarily conserved Zinc tetra thiolate motif which locate near the NAD+ binding site in all SIRT family members [23, 49]. Cysteine sites are already implicated in the tetra-thiolate zinc-binding motif of SIRT1 [23] and SIRT6 [33]. To answer the question of whether oxidative stress decreased SIRT2 deacetylation function through the modifications of 4 redox sensitive cysteines of SIRT2 protein we mutated two of the four cysteines which are conserved in the tetra-thiolate Zinc-binding domain of SIRT2, namely Cys221 and Cys224, to serine to develop plasmids SIRT2 C221S and SIRT2 C224S respectively. We then transfected HEK 293 cells with pcDNA3 control (Empty vector), normal/wild type SIRT2 (WT SIRT2), C221S SIRT2 and C224S SIRT2 plasmids and studied enzymatic activity of SIRT2. We observed that, as expected, there was significantly increased SIRT2 activity with WT SIRT2 transfection vs. empty vector control (Figure 4A) group. Cells transfected with both the mutants, namely SIRT2 C221S and SIRT2 C224S, showed significantly lower SIRT2 enzymatic activity compared with WT SIRT2. This data indicates that C221S and C224S mutations may be responsible for modulation of SIRT2 activity.
Figure 4: Cysteines Cys221 and Cys224 mutation affects SIRT2 function Figure 4A:
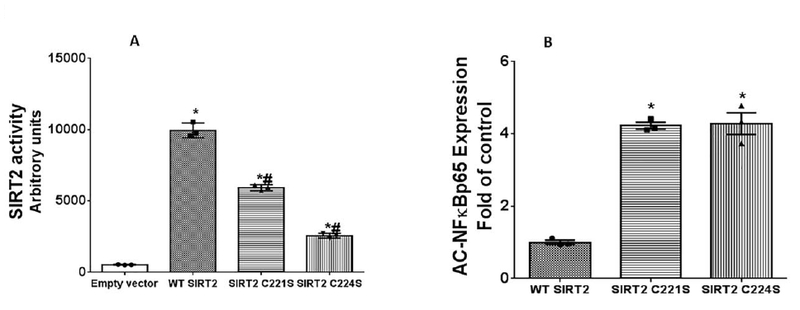
HEK cells were transfected with control (pcDNA: Empty vector), WT SIRT2 (without mutation), SIRT2 C221S and SIRT2 C224S plasmids and SIRT2 function studied using enzymatic assay (Figure 4A). WT SIRT2 transfected cells showed significantly increased SIRT2 function vs. empty vector. SIRT2C221S and SIRT2C224S transfected cells showed significantly decreased SIRT2 function vs. WT SIRT2. (* p<0.05 vs. empty vector; # p<0.05 vs. WT SIRT2). Figure 4B: Cysteines Cys221 and Cys224 mutation affects SIRT2-deacetylation function: HEK cells were transfected with WTSIRT2, SIRT2C221S and SIRT2C224S along with NFĸBp65 and p300 plasmids and acetylated NFkB p65 expression was examined. SIRT2C221S and SIRT2 C224S transfected groups showed significantly increased acetylated NFĸBp65 (AC-NFĸBp65) expression (decreased deacetylation) vs. WTSIRT2 (*p<0.05 vs. WTSIRT2).
To further define SIRT2 deacetylation-function in C221S or C224S mutants, we transfected HEK 293 cells with WTSIRT2, SIRT2C221S or SIRT2C224S plasmids along with CBP and NFkB p65, and assessed acetylated NFĸBp65 (AC-NFĸBp65) expression. As shown in Figure 4B, both SIRT2 C221S and SIRT2 C224S transfected cells showed significantly higher expression of acetylated NFĸBp65 vs. WT SIRT2. These data suggested that both mutations decreased SIRT2 deacetylation function.
Cysteine mutation affects biological function of SIRT2:
To further assess whether cysteine mutation of SIRT2 impact the immune response, we transfected RAW macrophage cells with WT SIRT2, SIRT2 C221S and SIRT2 C224S plasmids, and stimulated with LPS to determine TNF-α, IL-1β, IL-6 and IL-10 mRNA expression during hyper-inflammation 4h post-LPS. Figure 5 A, B and C show significantly higher TNF-α, IL-1β and IL-6 mRNA expression in response to LPS in cells with SIRT2 C221S and SIRT2 C224S mutants vs. WT SIRT2 transfected-cells. Interestingly, we also show increased IL-10 mRNA levels in the mutant transfected groups compared to WT SIRT2 in response to LPS. These data together suggests that cysteine thiol oxidation of SIRT2 contributes to the exaggerated hyper-inflammatory response during obesity with sepsis.
Figure 5: Cysteines Cys221 and Cys224 mutations affect LPS-induced inflammation:
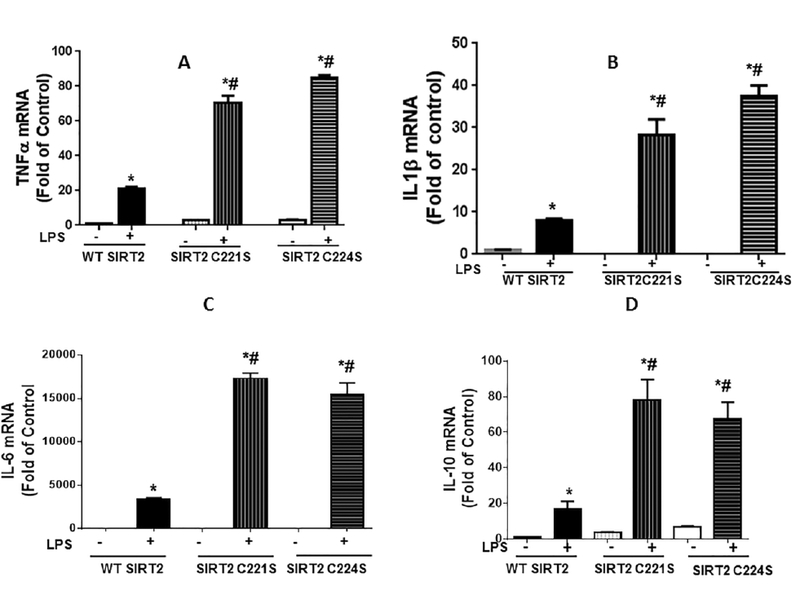
RAW 264.7 (RAW) cells were transfected with WTSIRT2, SIRT2C221S and SIRT2C224S and stimulated with and without LPS for four hours. LPS treatment significantly increased TNF-α (Figure 5A), IL-1β (Figure 5B), IL-6 (Figure 5C) and IL-10 (Figure 5D) mRNA expression vs. control in all three groups (WT, SIRT2C221S and SIRT2C224S). However there was significantly higher TNF-α, IL-1β, IL-6 and IL-10 mRNA expression in C221S and C224S transfected cells vs. WT SIRT2. (* p<0.05 vs. control; # p< 0.05 vs. WT SIRT2).
DISCUSSION:
The main objective of this project was to study the effect of increased oxidative stress on SIRT2 expression and activity during exaggerated hyper-inflammation of obesity with sepsis [61, 64]. We have reported previously, that obesity exaggerates the hyper-inflammatory phase of sepsis and prolongs the hypo-inflammatory phase of sepsis and SIRT2 plays a critical role in doing so [64]. Here we aimed to study how obesity with sepsis modulates SIRT2 during the exaggerated hyper-inflammation. Specific features of the current project that expand the concept that SIRT2 regulates inflammation during obesity include the following: 1) there is an inverse relationship between total and oxidized SIRT2 expression; decreased total SIRT2 and increased oxidized SIRT2 expression during hyper-inflammatory phase of obesity with sepsis. 2) SIRT2 deacetylation function is decreased (increased fraction of acetylated NFkBp65) with oxidative stress 3) site-directed mutation of two redox sensitive cysteine sites (creating “oxidation-mimics” of SIRT2) in the tetra-thiolate Zinc-binding domain in SIRT2, Cys221 and Cys224, significantly decrease SIRT2 enzymatic activity, 4) Cys221 and Cys224 mutations significantly alter the biological activity of SIRT2 and modulate cytokine expression.
Sepsis and septic shock are the leading causes of death in non-coronary intensive care units worldwide [36]. Literature indicates that sepsis phenotype transitions from early hyper-inflammation to a late hypo-inflammatory phase [4, 18]. We have tracked these phases using “endotoxin tolerance” used extensively to study late/hypo-inflammation in cell model of sepsis. Specifically, we used cecal ligation and puncture to induce sepsis. Subsequently, we challenged these sepsis mice with lipopolysaccharide (LPS: endotoxin) as a “second hit” at different time points after CLP. We then studied microvascular inflammation using leukocyte adhesion to map response to endotoxin vs. normal saline stimulation (after CLP) at every time point. Leukocyte adhesion in the microcirculation was studied as a microvascular inflammatory marker since it is the rate-determining step in the inflammatory response [24]. We showed that the lean mice undergo three distinct phases. The hyper-inflammatory phase when there is significant increase in leukocyte adhesion in response to second-hit LPS, a hypo-inflammatory phase when leukocyte adhesion in response to LPS is blunted vs. normal saline stimulation (endotoxin tolerance). Mice surviving for 72h post-CLP showed that there was return of LPS response vs. normal saline stimulation indicating resolution phase [55]. Mechanistically, we showed that SIRTs 1, 3, and 6 are critical in modulating the phase switch from hyper- to hypo-inflammation in lean mice [32, 53, 31, 30].
Interestingly, using the same assay in obese mice with sepsis, we reported that the mice undergo exaggerated hyper-inflammatory and prolonged hypo-inflammatory phases with significant decrease in survival compared to lean mice[54, 59, 65, 64, 63]. Furthermore, we showed that the SIRT2 deficiency exaggerates while sustained increase in SIRT2 expression decreases sepsis hyper-inflammation [63, 5]. In the current project, we used the same model of obesity with sepsis (CLP) with established time points for hyper- and hypo-inflammatory phases to study total vs. oxidized SIRT2 expression in tissue from obese with sepsis mice. Furthermore, to elucidate the mechanism of oxidation of SIRT2 (i.e. specific redox sensitive cysteine sites for oxidation), we studied the effect of mutations of two of the four redox sensitive cysteines on enzymatic and biological function of SIRT2.
Sirtuins depend on NAD+ to control their expression and function [21]. A major function of SIRTs is to control cell-stress responses by using both epigenetic and posttranslational mechanisms. Mechanisms of how NAD+ generation and availability control the deacetylase functions of SIRTs is well understood [20]. Deacetylase activities of SIRTs are linked to a broad range of physiologic and pathophysiologic phenotypes. Among these are: cell development and differentiation, acute and chronic inflammation, obesity with inflammation, diabetes, atherosclerosis, Alzheimer’s disease, and the aging process, even without its co-morbidities [20, 53, 63, 5]. Our laboratory described how nuclear SIRTs 1, 2 and 6 and mitochondrial SIRT3 contribute to extreme and often lethal states of acute systemic inflammation as seen in sepsis [53, 57]. Importantly, inhibition of SIRT1 in lean mice and SIRT2, but not SIRT1 in obese mice reversed the hypo-inflammatory phase of sepsis [55, 63]. The molecular processes that inform SIRT protein deacetylase enzymatic activity proximal to its homeostasis protection of cells and organs during homeostasis protection are unknown.
In this work, we identified that direct redox effects on SIRT2 in the obese phenotype inform the inflammatory response of sepsis and defined the molecular mechanistic pathway that uses the conserved Zinc cysteine tetra thiolate motif of SIRT2 as informers of the deacetylase property of nuclear SIRT2. Specifically, direct and reversible oxidation of zinc tetrad cysteine thiols controls deacetylase function of SIRT2 to then control deacetylation/acetylation of NFkBp65 to support transcription of TNF-α, IL-1β and IL-6. This study did not expand to SIRT2 regulation of other pro-inflammatory mediators, but it seems likely a pro-inflammatory set of genes with NFkB consensus sites join TNF-α in initiating inflammation after endotoxin is sensed by macrophages and regulated by epigenetic chromatin modifications. We also studied whether cysteine mutations affect the anti-inflammatory cytokine [7, 28] IL-10 mRNA function in response to LPS in our study. SIRT2 mutants show significantly increased IL-10 mRNA in response to LPS. Literature indicates that SIRT2 affects other transcription pathways such as MAP kinase and CREB as well [10, 67, 11, 22]. Evidence suggests that in bacterial sepsis, IL-10 modulates inflammatory response, which in turn is regulated by CREB [2]. While we do not understand the exact mechanism at this time, we can only speculate that the increased IL-10 expression through MAP kinase/CREB pathway along with the TNF-α, IL-1β and IL-6 expression is a part of cytoprotective response to inflammation. However, the specific mechanisms of SIRT2 control of IL-10 need to be further elucidated. Moreover, we have only studied NFκBp65 deacetylation by SIRT2. The effect of SIRT2 on other transcription factors needs further elucidation as well.
Site-specific phosphorylation of SIRT2 modulates/controls its deacetylation function (activation vs. inhibition) during mitosis [40, 42, 9, 45]. This project did not investigate the association between phosphorylation and oxidation which merits further research. It is possible that the hypo-inflammatory/immunosuppressive phase of sepsis is associated with reduced SIRT2 state leading to protein stabilization/reactivation of SIRT2 function. We have not explored the role of specific reductases that may reactivate the SIRT2 protein function and this needs further elucidation.
This is the first time, to our knowledge, that SIRT2 activity and function are shown to be modulated by direct redox signaling during oxidative stress, similar to SIRT1 and SIRT6. REF-1 regulates enzymatic activity of SIRT1 in vitro. Direct oxidation of SIRT6, especially cysteine thiol 144, affects glucose metabolism in cells [23, 19, 33]. Here we show that cysteine oxidation not only modulates the enzymatic activity of SIRT2 but also modulates hyper-inflammatory response of sepsis. A plausible new unifying concept is that direct redox control over key homeostasis guarding Sirtuin family with the conserved cysteine thiol Zinc associated tetrad informs inflammation reprogramming on redox axis of oxidation and reduction of cysteine thiol network. A potential extension of this concept is that a cross talking network of protein associated and functional cysteine thiols may form a nexus that coordinates to fuel substrate selection to satisfy bioenergy needs of pro-inflammatory and immune-repressor cell functions to drive inflammation on its course back to homeostasis. This model, if true, predicts that failure to progress along balanced oxidation and reduction leads to a block in homeostasis retrieval pathways and causes chronic inflammatory stress responses.
In summary, this study opens the door to further examining the role of the redox code that is controlled by specific cysteine thiols on SIRT2 and other family members in a disease-specific context, such as obesity with sepsis.
ACKNOWLEDGEMENTS:
This work was supported by NIH grants: Vidula T. Vachharajani: R01GM099807; Charles E McCall: 1) R01AI065791, 2) R01AI079144.
The plasmids were gifted to us by Addgene: pcDNA3β-FLAG-CBP- HA was a gift from Tso-Pang Yao (Addgene plasmid # 32908), pCMV4 NFĸB p65 was a gift from Warner Greene (Addgene plasmid # 21966). Wild-type plasmid SIRT2 Flag was a gift from Eric Verdin (Addgene plasmid # 13813)
ABBREVIATIONS:
- CLP
Cecal ligation and puncture
- CTRL
Control diet mice
- Cys
Cysteine
- DIO
Diet induced obesity
- LPS
Lipopolysaccharide
- NAD
Nicotinamide dinucleotide
- NFĸB
Nuclear factor kappa B
- Ob/ob
B6.Cg-Lepob/J
- PM
Peritoneal macrophages
- SIRT
Sirtuin
- SIRT1
Sirtuin 1
- SIRT2
Sirtuin 2
- TBH
tertiary-Butyl Hydroperoxide
- WT
Wild type (lean)
- BP1
Biotin-1,3-cyclopentanedione
- NEM
N-Ethyl Maleimide
Footnotes
AUTHOR CONTRIBUTIONS: Concept and design:
VTV, CF; Data collection: XW, NB, DL, VTV; Data Analysis and interpretation: VTV, XW, CEM, CF; generating the manuscript: VTV, XW, CEM, CF.
CONFLICT OF INTEREST STATEMENT:
The authors declare that the submitted work was not carried out in the presence of any personal, professional or financial relationships that could potentially be construed as a conflict of interest.
REFERENCES.
- 1.Arabi YM, Dara SI, Tamim HM, Rishu AH, Bouchama A, Khedr MK, Feinstein D et al. 2013. Clinical characteristics, sepsis interventions and outcomes in the obese patients with septic shock: an international multicenter cohort study. Crit Care 17 (2):R72. doi: 10.1186/cc12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aziz M, Holodick NE, Rothstein TL, and Wang P. 2017. B-1a Cells Protect Mice from Sepsis: Critical Role of CREB. J Immunol 199 (2):750–760. doi: 10.4049/jimmunol.1602056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, Stein B, and Greene WC. 1992. The 65-kDa subunit of human NF-kappa B functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc Natl Acad Sci U S A 89 (5):1875–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL et al. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306 (23):2594–2605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buechler N, Wang X, Yoza BK, McCall CE, and Vachharajani V. 2017. Sirtuin 2 Regulates Microvascular Inflammation during Sepsis. J Immunol Res 2017:2648946. doi: 10.1155/2017/2648946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calle EE, Rodriguez C, Walker-Thurmond K, and Thun MJ. 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine 348 (17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 7. Chang J, Kunkel SL, and Chang CH. 2009. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci U S A 106 (43):18327–18332. doi: 10.1073/pnas.0905815106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L, Feng Y, Zhou Y, Zhu W, Shen X, Chen K, Jiang H, and Liu D. 2010. Dual role of Zn2+ in maintaining structural integrity and suppressing deacetylase activity of SIRT1. J Inorg Biochem 104 (2):180–185. doi: 10.1016/j.jinorgbio.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 9. Dryden SC, Nahhas FA, Nowak JE, Goustin AS, and Tainsky MA. 2003. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 23 (9):3173–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eo SH, Choi SY, and Kim SJ. 2016. PEP-1-SIRT2-induced matrix metalloproteinase-1 and −13 modulates type II collagen expression via ERK signaling in rabbit articular chondrocytes. Exp Cell Res 348 (2):201–208. doi: 10.1016/j.yexcr.2016.09.024. [DOI] [PubMed] [Google Scholar]
- 11. Eo SH, Kim DW, Choi SY, Kim HA, and Kim SJ. 2015. PEP-1-SIRT2 causes dedifferentiation and COX-2 expression via the MAPK pathways in rabbit articular chondrocytes. Exp Cell Res 339 (2):351–359. doi: 10.1016/j.yexcr.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 12. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, and Shimomura I. 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114 (12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goncalves Damascena K, Batisti Ferreira C, Dos Santos Teixeira P, Madrid B, Goncalves A, Cordova C, de Toledo Nobrega O, and Pimentel Ferreira A. 2017. Functional capacity and obesity reflect the cognitive performance of older adults living in long-term care facilities. Psychogeriatrics 17 (6):439–445. doi: 10.1111/psyg.12273. [DOI] [PubMed] [Google Scholar]
- 14. Groeger G, Quiney C, and Cotter TG. 2009. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid Redox Signal 11 (11):2655–2671. doi: 10.1089/ARS.2009.2728. [DOI] [PubMed] [Google Scholar]
- 15. Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, and Piantadosi CA. 2007. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. American journal of respiratory and critical care medicine 176 (8):768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haigis MC, and Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hotchkiss RS, and Karl IE. 2003. The pathophysiology and treatment of sepsis. The New England journal of medicine 348 (2):138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 18. Hotchkiss RS, and Opal S. 2010. Immunotherapy for sepsis--a new approach against an ancient foe. The New England journal of medicine 363 (1):87–89. doi: 10.1056/NEJMcibr1004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu S, Liu H, Ha Y, Luo X, Motamedi M, Gupta MP, Ma JX, Tilton RG, and Zhang W. 2015. Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic Biol Med 79:176–185. doi: 10.1016/j.freeradbiomed.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Imai S, and Guarente L. 2010. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends in pharmacological sciences 31 (5):212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Imai S, and Guarente L. 2014. NAD+ and sirtuins in aging and disease. Trends Cell Biol 24 (8):464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeong SG, and Cho GW. 2017. The tubulin deacetylase sirtuin-2 regulates neuronal differentiation through the ERK/CREB signaling pathway. Biochem Biophys Res Commun 482 (1):182–187. doi: 10.1016/j.bbrc.2016.11.031. [DOI] [PubMed] [Google Scholar]
- 23. Jung SB, Kim CS, Kim YR, Naqvi A, Yamamori T, Kumar S, Kumar A, and Irani K. 2013. Redox factor-1 activates endothelial SIRTUIN1 through reduction of conserved cysteine sulfhydryls in its deacetylase domain. PLoS One 8 (6):e65415. doi: 10.1371/journal.pone.0065415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jung U, Norman KE, Scharffetter-Kochanek K, Beaudet AL, and Ley K. 1998. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J Clin Invest 102 (8):1526–1533. doi: 10.1172/JCI119893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalous KS, Wynia-Smith SL, Olp MD, and Smith BC. 2016. Mechanism of Sirt1 NAD+-dependent Protein Deacetylase Inhibition by Cysteine S-Nitrosation. J Biol Chem 291 (49):25398–25410. doi: 10.1074/jbc.M116.754655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, and Salminen A. 2013. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal 25 (10):1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 27. Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, Mirtschink P et al. 2012. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 26 (3):259–270. doi: 10.1101/gad.180406.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li YP, Huang J, Huang SG, Xu YG, Xu YY, Liao JY, Feng X, Zhang XG, Wang JH, and Wang J. 2014. The compromised inflammatory response to bacterial components after pediatric cardiac surgery is associated with cardiopulmonary bypass-suppressed Toll-like receptor signal transduction pathways. J Crit Care 29 (2):312 e317–313. doi: 10.1016/j.jcrc.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 29. Lin J, Sun B, Jiang C, Hong H, and Zheng Y. 2013. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem Biophys Res Commun 441 (4):897–903. doi: 10.1016/j.bbrc.2013.10.153. [DOI] [PubMed] [Google Scholar]
- 30. Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, and McCall CE. 2015. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 290 (1):396–408. doi: 10.1074/jbc.M114.566349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu TF, Vachharajani VT, Yoza BK, and McCall CE. 2012. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem 287 (31):25758–25769. doi: 10.1074/jbc.M112.362343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, and McCall CE. 2011. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem 286 (11):9856–9864. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Long D, Wu H, Tsang AW, Poole LB, Yoza BK, Wang X, Vachharajani V, Furdui CM, and McCall CE. 2017. The Oxidative State of Cysteine Thiol 144 Regulates the SIRT6 Glucose Homeostat. Sci Rep 7 (1):11005. doi: 10.1038/s41598-017-11388-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mariani S, Di Rocco G, Toietta G, Russo MA, Petrangeli E, and Salvatori L. 2017. Sirtuins 1–7 expression in human adipose-derived stem cells from subcutaneous and visceral fat depots: influence of obesity and hypoxia. Endocrine 57 (3):455–463. doi: 10.1007/s12020-016-1170-8. [DOI] [PubMed] [Google Scholar]
- 35. Marshall JC 2014. Why have clinical trials in sepsis failed? Trends Mol Med 20 (4):195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 36. Mayr FB, Yende S, and Angus DC. 2014. Epidemiology of severe sepsis. Virulence 5 (1):4–11. doi: 10.4161/viru.27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moschen AR, Wieser V, Gerner RR, Bichler A, Enrich B, Moser P, Ebenbichler CF, Kaser S, and Tilg H. 2013. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J Hepatol 59 (6):1315–1322. doi: 10.1016/j.jhep.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 38. North BJ, Marshall BL, Borra MT, Denu JM, and Verdin E. 2003. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11 (2):437–444. [DOI] [PubMed] [Google Scholar]
- 39. North BJ, and Verdin E. 2007. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS One 2 (8):e784. doi: 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. North BJ, and Verdin E. 2007. Mitotic regulation of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J Biol Chem 282 (27):19546–19555. doi: 10.1074/jbc.M702990200. [DOI] [PubMed] [Google Scholar]
- 41. Oguri M, Fujimaki T, Horibe H, Kato K, Matsui K, Takeuchi I, and Yamada Y. 2017. Obesity-related changes in clinical parameters and conditions in a longitudinal population-based epidemiological study. Obes Res Clin Pract 11 (3):299–314. doi: 10.1016/j.orcp.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 42. Pandithage R, Lilischkis R, Harting K, Wolf A, Jedamzik B, Luscher-Firzlaff J, Vervoorts J et al. 2008. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J Cell Biol 180 (5):915–929. doi: 10.1083/jcb.200707126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Prescott HC, Chang VW, O’Brien JM Jr., Langa KM, and Iwashyna T. 2014. Obesity and 1-Year Outcomes in Older Americans With Severe Sepsis. Crit Care Med doi: 10.1097/CCM.0000000000000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, and Furdui CM. 2011. Simple synthesis of 1,3-cyclopentanedione derived probes for labeling sulfenic acid proteins. Chem Commun (Camb) 47 (32):9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramakrishnan G, Davaakhuu G, Kaplun L, Chung WC, Rana A, Atfi A, Miele L, and Tzivion G. 2014. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J Biol Chem 289 (9):6054–6066. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robinson MK, Mogensen KM, Casey JD, McKane CK, Moromizato T, Rawn JD, and Christopher KB. 2015. The relationship among obesity, nutritional status, and mortality in the critically ill. Crit Care Med 43 (1):87–100. doi: 10.1097/CCM.0000000000000602. [DOI] [PubMed] [Google Scholar]
- 47. Ross PA, Newth CJ, Leung D, Wetzel RC, and Khemani RG. 2016. Obesity and Mortality Risk in Critically Ill Children. Pediatrics 137 (3):e20152035. doi: 10.1542/peds.2015-2035. [DOI] [PubMed] [Google Scholar]
- 48. Schug TT, and Li X. 2011. Sirtuin 1 in lipid metabolism and obesity. Ann Med 43 (3):198–211. doi: 10.3109/07853890.2010.547211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sherman JM, Stone EM, Freeman-Cook LL, Brachmann CB, Boeke JD, and Pillus L. 1999. The conserved core of a human SIR2 homologue functions in yeast silencing. Mol Biol Cell 10 (9):3045–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Torio CM, and Moore BJ. 2006. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2013: Statistical Brief #204. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Rockville (MD). [PubMed] [Google Scholar]
- 51. Trivedi V, Bavishi C, and Jean R. 2015. Impact of obesity on sepsis mortality: A systematic review. J Crit Care 30 (3):518–524. doi: 10.1016/j.jcrc.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 52. Vachharajani V, Cunningham C, Yoza B, Carson J Jr., Vachharajani TJ, and McCall C. 2012. Adiponectin-deficiency exaggerates sepsis-induced microvascular dysfunction in the mouse brain. Obesity (Silver Spring) 20 (3):498–504. doi: 10.1038/oby.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vachharajani V, Liu T, and McCall CE. 2014. Epigenetic coordination of acute systemic inflammation: potential therapeutic targets. Expert Rev Clin Immunol 10 (9):1141–1150. doi: 10.1586/1744666X.2014.943192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vachharajani V, Russell JM, Scott KL, Conrad S, Stokes KY, Tallam L, Hall J, and Granger DN. 2005. Obesity exacerbates sepsis-induced inflammation and micro vascular dysfunction in mouse brain. Microcirculation 12 (2):183–194. [DOI] [PubMed] [Google Scholar]
- 55. Vachharajani VT, Fu Liu T, Brown CM, Wang X, Buechler NL, Wells JD, Yoza BK, and McCall CE. 2014. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol. doi: 10.1189/jlb.3MA0114-034RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vachharajani VT, Liu T, Brown CM, Wang X, Buechler NL, Wells JD, Yoza BK, and McCall CE. 2014. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol 96 (5):785–796. doi: 10.1189/jlb.3MA0114-034RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, and McCall CE. 2016. Sirtuins Link Inflammation and Metabolism. J Immunol Res 2016:8167273. doi: 10.1155/2016/8167273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vachharajani V, Vital S, and Russell J. 2010. Modulation of circulating cell-endothelial cell interaction by erythropoietin in lean and obese mice with cecal ligation and puncture. Pathophysiology : the official journal of the International Society for Pathophysiology / ISP 17 (1):9–18. doi: 10.1016/j.pathophys.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 59. Vachharajani V, Vital S, Russell J, and Granger DN. 2007. Hypertonic saline and the cerebral microcirculation in obese septic mice. Microcirculation 14 (3):223–231. doi: 10.1080/10739680601139153. [DOI] [PubMed] [Google Scholar]
- 60. Vachharajani V, Vital S, Russell J, Scott LK, and Granger DN. 2006. Glucocorticoids inhibit the cerebral microvascular dysfunction associated with sepsis in obese mice. Microcirculation 13 (6):477–487. doi: 10.1080/10739680600777599. [DOI] [PubMed] [Google Scholar]
- 61. Vieira AA, Michels M, Florentino D, Nascimento DZ, Rezin GT, Leffa DD, Fortunato JJ et al. 2015. Obesity promotes oxidative stress and exacerbates sepsis-induced brain damage. Curr Neurovasc Res 12 (2):147–154. [DOI] [PubMed] [Google Scholar]
- 62. Wang HE, Griffin R, Judd S, Shapiro NI, and Safford MM. 2013. Obesity and risk of sepsis: a population-based cohort study. Obesity (Silver Spring) 21 (12):E762–769. doi: 10.1002/oby.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang X, Buechler NL, Martin A, Wells J, Yoza B, McCall CE, and Vachharajani V. 2016. Sirtuin-2 Regulates Sepsis Inflammation in ob/ob Mice. PLoS One 11 (8):e0160431. doi: 10.1371/journal.pone.0160431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang X, Buechler NL, Yoza BK, McCall CE, and Vachharajani V. 2016. Adiponectin treatment attenuates inflammatory response during early sepsis in obese mice. J Inflamm Res 9:167–174. doi: 10.2147/JIR.S119021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang X, Buechler NL, Yoza BK, McCall CE, and Vachharajani VT. 2015. Resveratrol attenuates microvascular inflammation in sepsis via SIRT-1-Induced modulation of adhesion molecules in ob/ob mice. Obesity (Silver Spring) 23 (6):1209–1217. doi: 10.1002/oby.21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X, Cao Q, Yu L, Shi H, Xue B, and Shi H. 2016. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 1 (19):e87748. doi: 10.1172/jci.insight.87748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu D, Lu W, Wei Z, Xu M, and Liu X. 2018. Neuroprotective Effect of Sirt2-specific Inhibitor AK-7 Against Acute Cerebral Ischemia is P38 Activation-dependent in Mice. Neuroscience 374:61–69. doi: 10.1016/j.neuroscience.2018.01.040. [DOI] [PubMed] [Google Scholar]
- 68. Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, McComb ME, Costello CE, Cohen RA, and Bachschmid MM. 2010. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid Redox Signal 13 (7):1023–1032. doi: 10.1089/ars.2010.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhao X, Sternsdorf T, Bolger TA, Evans RM, and Yao TP. 2005. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol 25 (19):8456–8464. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]