

Abstract
Background:
A parallel downregulation of brain-derived neurotrophic factor (BDNF) and somatostatin (SST), a marker of inhibitory Y-amino-butyric acid (GABA) interneurons which target pyramidal cell dendrites, has been reported in several brain areas of subjects with major depressive disorder (MDD), and rodent genetic studies suggests they are linked and both contribute to the illness. However, the mechanism by which they contribute to the pathophysiology of the illness has remained elusive.
Methods:
With qPCR, we determined the expression level of BDNF transcript variants and synaptic markers in the prefrontal cortex (PFC) of MDD patients and matched controls (n=19/group) and of C57BL/6J mice exposed to chronic stress or control conditions (n=12/group). We next suppressed BDNF transcripts with long 3’ untranslated region (L-3’-UTR) using small hairpin RNA (shRNA) and investigated changes in cell morphology, gene expression and behavior.
Results:
L-3’-UTR containing BDNF mRNAs, which migrate to distal dendrites of pyramidal neurons, are selectively reduced and highly correlated with SST expression in the PFC of MDD subjects. A similar downregulation occurs in mice submitted to chronic stress. We next show that Bdnf L-3’-UTR knockdown is sufficient to induce (i) dendritic shrinkage in cortical neurons, (ii) cell-specific MDD-like gene changes (including Sst downregulation), and (iii) depressive-/anxiety-like behaviors. The translational validity of the Bdnf L-3’-UTR shRNA-treated mice was confirmed by significant crossspecies correlation of changes in MDD-associated gene expression.
Conclusions:
These findings provide evidence for a novel MDD-related pathological mechanism linking local neurotrophic support, pyramidal cell structure, dendritic inhibition and mood regulation.
Keywords: BDNF, dendrite, SST, major depressive disorder, GABA, stress
INTRODUCTION
MDD is characterized by persistent low mood and/or anhedonia, and cognitive impairment. These symptoms are associated with dysfunction of brain networks involved in cognition, emotion and reward (1). The PFC plays a role in cognition and emotion regulation and is affected by stress (2). Consistent with functional impairment and atrophy (3–5), postmortem studies reported decreased neuronal body size, spine loss, and reduced cellular density in the PFC of MDD subjects (6,7).
Decreased GABAergic neurotransmission in MDD has been revealed by transcranial magnetic stimulation (8) and proton magnetic resonance spectroscopy (9,10). Decreased expression of GABA- related genes has been observed in corticolimbic areas of MDD patients(11–14). Downregulation of SST, a molecular marker of dendritic-targeting interneurons was consistently found, whereas parvalbumin (PVALB), the marker of perisomatic-targeting interneurons, remained relatively unaffected by disease, although see Tripp et al (14). Downregulation of SST is also observed in other neurological diseases (15), suggesting a vulnerability of GABA interneurons coexpressing this marker. The underlying mechanism is unknown, however, chronic stress and corticosterone exposure induced endoplasmic reticulum stress in these neurons (16). Another mechanism could be the loss of neurotrophic support. MDD and prolonged stress reduce BDNF expression (17,18), which might compromise structural integrity of various cells(19,20). GABAergic cells do not produce BDNF and rely on supply from other cell populations, mostly pyramidal neurons (21–23). In MDD, SST reduction is accompanied by BDNF and/or NTRK2 (also known as TRKB) downregulation (11,14) and genetic studies in mice revealed that SST expression is highly dependent on BDNF (11,14,24), together suggesting a pathogenic mechanism linking BDNF and GABA.
Local action of BDNF is closely associated with neural plasticity. In addition to its autocrine action regulating dendritic and synaptic plasticity of pyramidal cells (25,26), BDNF can alter the presynaptic GABAergic system in a paracrine manner (23,27). In a previous transcriptome analysis of human PFC, we found that the expression of GABRA5 (gamma-aminobutyric acid A receptor, alpha 5), a subunit of GABA-A receptor enriched adjacent to dendritic GABAergic synapses and that mediates the function of SST neurons (28), showed the highest correlation with BDNF levels among all investigated genes (29). This suggests that local BDNF supply may play a role in maintaining the function of SST neurons on pyramidal cell dendrites. Interestingly, BDNF transcripts are present in dendrites (30–32) and cellular localization of BDNF transcripts corresponds with phospho-NTRK2 immunostaining (30), suggesting that BDNF may be locally translated and act on dendrites. Studies showed that dendritic BDNF is implicated in stress-related mood disorders: dendritic BDNF transcripts are decreased by stress (33–35), and increased by antidepressant treatment (36). Knockdown of dendritic BDNF impairs structural integrity of primary hippocampal neurons (30,37), replicating the effects of chronic stress.
Here, we investigated changes in dendritic BDNF transcripts in the dorsolateral PFC (dlPFC) of MDD subjects and medial PFC (mPFC) of mice exposed to chronic stress, and tested if such changes were linked to dendritic-targeting interneuron markers. Using shRNA, we then knocked-down dendritic Bdnf mRNA in primary neuronal culture and in mouse mPFC to assess whether reduced dendritic Bdnf expression was sufficient to induce MDD- and stress-related phenotypes. We predicted that decreased local neurotrophic support contributes to selective disturbance in dendritic structure and in dendritic- targeting GABAergic neurons, as a putative mechanism leading to mood dysregulation.
METHODS AND MATERIALS
A complete description can be found in the Supplementary Information.
Human postmortem subjects
Postmortem brains were collected during autopsies conducted at the Allegheny County Medical Examiner’s Office (Pittsburgh, PA) after consent from next of kin. For all cases, a committee of experienced clinicians makes consensus DSM-IV diagnoses using information obtained from clinical records, toxicology exam and standardized psychological autopsy (38). Individuals were screened for the absence of neurodegenerative disorders by neuropathological examination(39–41). All procedures were approved by the University of Pittsburgh’s Committee for the Oversight of Research and Clinical Trials Involving the Dead and Institutional Review Board for Biomedical Research. After careful examination of clinical and technical parameters, 19 pairs of MDD and control subjects were selected (Table S1).
Animals and unpredictable chronic mild stress (UCMS)
9–10 week old C57BL/6J male mice were divided into two groups and submitted to control housing condition or UCMS consisting of a 7-week regimen of pseudo-random unpredictable mild stressors. The progression of the UCMS syndrome was monitored weekly by assessing coat state and weight changes for each mouse. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Behavioral testing and emotionality z-score
Tests for depressive- and anxiety-like phenotypes included the Elevated Plus Maze (EPM), Open Field (OF), novelty suppressed feeding (NSF) and Cookie Tests (CT). To assess behavioral consistency across tests, we integrated emotionality-related measures across tests as described earlier (42).
RNA extraction and real-time quantitative polymerase chain reaction (qPCR)
For human studies, gray matter of dlPFC were collected in Trizol and further purified with Qiagen RNeasy micro kit (QIAGEN, Valencia, CA, USA). For mice, mPFC punches were taken and processed with Qiagen RNeasy micro kit. To generate cDNA, total RNA was mixed with qScript cDNA supermix(Quanta BioSciences, Gaithersburg, MD, USA). PCR products were amplified in triplets on a Mastercycler real-time PCR machine(Eppendorf, Hamburg, Germany).
shRNA, DNA constructs and Adeno-associated viral (AAV) vector
shRNA sequence targeting L-3’-UTR of mouse Bdnf was adapted from (37). Lentiviral vectors containing shRNA against Bdnf L-3’-UTR or scrambled shRNA, and eGFP reporter gene were purchased from GeneCopeia (Rockville, MD, USA). AAV9 vectors containing Bdnf L-3’-UTR shRNA- GFP or scrambled shRNA were commercially prepared (Virovek, Hayward, CA, USA).
Primary culture, transfection and fluorescent-activated cell sorting (FACS) of mouse cortical neurons
Primary mouse neurons (Thermo Fisher Scientific; Waltham, MA, USA; A15585) were cultured in Neurobasal media (Thermo Fisher Scientific; 21103) supplemented with B27 (Thermo Fisher Scientific; 17504), GlutaMAX™-I (Thermo Fisher Scientific; 35050). At 7 days in vitro (DIV7), transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific; L3000015). GFP-expressing cells were collected by FACS into Qiagen RNeasy lysis buffer.
Immunostaining and image analysis
After 5 days of transfection, cells were fixed with 4% paraformaldehyde (PFA) and stained with rabbit anti-GFP (Life Technologies, Carlsbad, CA, USA; A21311) and mouse anti-MAP2 (Sigma-aldrich, St. Louis, MO, USA; M9942). The morphology of dendrites was automatically traced and analyzed by Imaris software (Bitplane AG, Concord, MA, USA). To quantify BDNF protein in AAV-infused region, immunostaining was performed with rabbit anti-BDNF (abcam; ab108319) and mouse anti-GFP (Santa Cruz Biotechnology, Dallas, TX, USA; sc-9996).
Animals and stereotaxic surgery
9–10 week old C57BL/6J male mice were bilaterally injected with AAV9-Bdnf L-3’-UTR shRNA-GFP or with AAV9-scrambled shRNA-GFP into the mPFC (A/P +2.0 mm, M/L ±0.4 mm, D/V −2.0 mm). Behavioral tests were performed before and after UCMS exposure. For gene expression analysis, mice received Bdnf L-3’-UTR shRNA in one hemisphere and scrambled shRNA in the other for intra-subject comparison.
Fluorescent in situ hybridization (FISH) and laser capture microdissection (LCM)
AAV-infused mice were perfused and lightly fixed with 1 mL of 4% PFA. Mouse brains were sectioned and thaw-mounted onto a polyethylene naphthalate-membrane slides (Leica Microsystems, Concord, ON, Canada). Sst- and Pvalb-expressing neurons were visualized by FISH (RNAscope, Advanced Cell Diagnostics, Newark, CA, USA). Briefly, brain sections were permeabilized with protease and antisense probe targeting Sst and Pvalb, preamplifiers, amplifiers, and Atto 550 conjugated-probes were serially hybridized at 40°C. After dehydration, ~200 Sst+ or Pvalb+ cells were collected using a LMD7 system (Leica Microsystems) and processed for RNA extraction.
Statistical analysis
Gene expression differences between MDD and control subjects were determined by analysis of covariance (ANCOVA) using SPSS (SPSS, Inc., Chicago, IL, USA). To determine covariates to include in gene-specific models, nominal factors were tested as main factors by ANOVA, scale covariates were tested by Pearson correlation, and repeated measures were corrected by modified Holm-Bonferroni test. ANCOVA models including significant co-factors were then applied. For cell culture and animal studies, statistical significance between two groups was determined with Student’s t-test. Repeated measures ANOVA was performed to determine interaction between shRNA and stress, effect of stress or shRNA on coat state over time.
RESULTS
Parallel downregulation of dendritic BDNF transcripts and dendritic targeting interneuron markers in the PFC of human subjects with MDD
The BDNF gene is composed of at least nine exons and makes diverse transcript variants using a combination of alternative splicing and polyadenylation sites (Figure S1A). The spatial segregation of BDNF mRNA is encoded by different untranslated regions; transcript variants with exon 2, 6 and the L-3’-UTR can migrate to distal dendrites whereas mRNAs containing exon 1, 4 and the short 3’ UTR are restricted to the soma and proximal dendrites (30,43,44). We used qPCR to quantify BDNF expression, focusing on transcript variants with known cellular localization, its receptor NTRK2, and synaptic function-related genes whose expressions are closely linked to BDNF levels in human PFC (29). Primers targeting BDNF protein coding sequence (CDS) were included to measure pan-BDNF level. Several BDNF variants displayed nominal changes, but only L-3’-UTR was significantly reduced in MDD samples (F=6.98, p=0.012; Figure 1A). The dendritic localization of BDNF L-3’-UTR was confirmed in mouse primary cortical neurons and hippocampal tissue(Figure S1B-C). Expression changes were not observed for NTRK2 isoforms and excitatory synaptic-related genes, however, inhibitory synaptic genes displayed disease-related downregulation (Figure 1B-E). Interestingly, genes for all three dendritic-targeting interneuron markers (SST, NPY, CORT) and for dendritically-localized GABRA5 were decreased in MDD (SST: F=15.03, p=4.6E-04; NPY: F=10.56, p=0.003; CORT: F=19.76, p=8.5E-05;GABRA5: F=10.18, p=0.003), whereas expression of PVALB, the molecular marker of perisomatic-targeting interneurons, was not affected (F=1.94, p=0.173).
Figure 1. MDD-related changes in BDNF transcript variants and synaptic markers in human dlPFC.
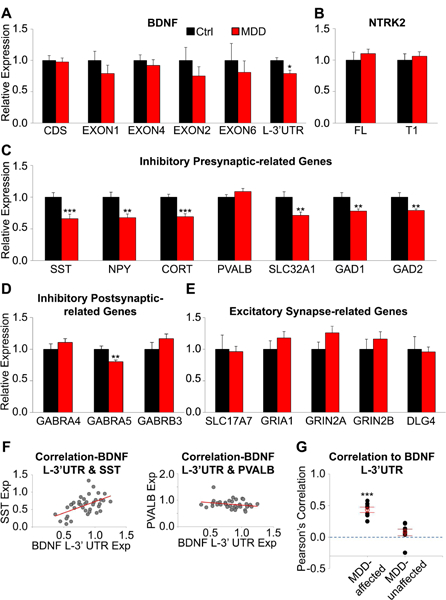
(A) Relative expression level (MDD/control) of BDNF transcripts, (B)NTRK2 isoforms, and (C-E) selected synapse-related genes (n=19 subjects/group). BDNF EXON1 and 4 are associated with localization in soma and proximal dendrite. BDNF EXON2, 6 and L-3’-UTR are associated with localization in distal dendrites.(F) Pearson’s correlation between BDNF L-3’-UTR and SST, and PVALB. BDNF L-3’-UTR is positively correlated with SST, a dendritic targeting interneuron marker, but not with PVALB, a marker of perisomatic targeting interneurons. (G) Overall correlation between BDNF L-3’- UTR and MDD-affected or unaffected synaptic markers. Significant positive correlations between BDNF L-3’-UTR level and MDD-associated gene expression changes suggest a contribution of dendritic BDNF to transcriptome alterations. *, p<0.05; **, p<0.01; ***, p<0.001. Data are represented as mean ± SEM.
Despite the absence of BDNF messenger in interneurons (Figure S2), BDNF L-3’-UTR level showed positive correlations to MDD-affected genes such as SST (r=0.53, p=0.001), but not to genes unaffected by disease such as PVALB (r=−0.25, p=0.131;Figure 1F). The average correlation values between BDNF L-3’-UTR and MDD-affected or unaffected genes were 0.43±0.11 (p=5.7E-05) and 0.08±0.15 (p=0.178; Figure 1G), respectively. No other BDNF transcript variants including total BDNF showed significant correlation to MDD-affected genes (CDS: r=−0.14±0.07, p=0.104; EXON1: r=- 0.09±0.04, p=0.082; EXON4; r=−0.13±0.09, p=0.222; EXON2: r=0.11±0.10, p=0.310; EXON6: r=- 0.09±0.09, p=0.320).
Consistent with previous finding (13), female MDD subjects showed greater SST reduction compared to male subjects (Table S2). However, correlation between BDNF-L-3’-UTR and SST was comparable in both sex (male: r=0.51, female: r=0.48). We observed age-related reduction of BDNF L- 3’-UTR,SST and CORT transcripts in controls, however, such age-effects were less robust and not significant in MDD, consistent with prior findings in other cohorts(13,14). Other parameters including antidepressants or suicide were not consistently associated with the expression of all tested genes.
Together, selective downregulation in the expression of dendritic BDNF and dendritic-targeting interneuron markers was observed in the dlPFC of MDD subjects. The high correlation between expressions of BDNF L-3’-UTR and specific GABA-related genes suggests that reduced dendritic BDNF may be associated with functional alterations of dendritic-targeting GABAergic neurons.
Chronic stress downregulates dendritic Bdnf transcript in the rodent mPFC
Next, we tested whether chronic stress, a major risk factor for MDD, was sufficient to induce MDD-like gene expression changes in mice. UCMS increased the latency to bite on day 2 of the cookie test (p=0.03; Figure 2A) and the food pellet in the NSF test (p=0.03; Figure 2B). The effect of stress was variable in other tests (Figure S3). To assess behavioral consistency across tests, we derived behavioral z-scores (16,42) and confirmed that UCMS-exposed mice exhibited higher depression/anxiety-related behaviors, denoted as behavioral emotionality, compared to controls (p=0.003; Figure 2C).
Figure 2. Chronic stress-related changes of anxiety-/depressive-like behaviors and expression of Bdnf transcript variants and Sst in mouse mPFC.
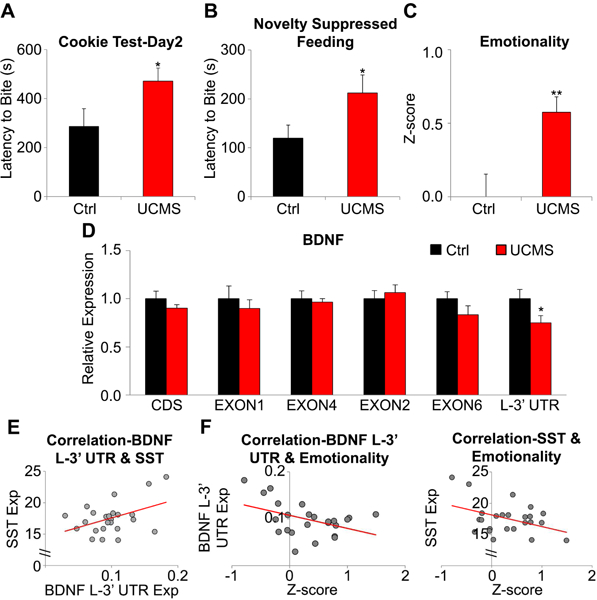
(A) Stress-associated behavioral change in the cookie test (increased latency to bite cookie), (B) novelty suppressed feeding (increased latency to bite food pellet). (C) Increased depressive-/anxiety-like phenotype in stressed mice compared to controls. (D) Relative expression level (stress/control) of Bdnf transcript variants. (E) Positive correlation between Bdnf L-3’-UTR and Sst suggests a functional link between the two genes in mouse brain. (F) Behavioral emotionality was negatively correlated to Bdnf L-3’-UTR (left) and Sst (right). n=12/group; *p<0.05, **p<0.01. Data are represented as mean ± SEM.
We found a significant reduction of Bdnf L-3’-UTR in the mPFC of UCMS-exposed mice (p=0.029; Figure 2D). This expression change was not observed for other genes (Figure S4). Despite an absence of difference at the group level, there was a positive correlation between Bdnf L-3’-UTR and Sst (r=0.43, p=0.036; Figure 2E) and negative correlations between behavioral emotionality and both Bdnf L-3’-UTR (r=−0.43, p=0.036) and Sst (r=−0.41, p=0.047; Figure 2F). In comparison, Pvalb expression was not correlated with either Bdnf L-3’-UTR level (r=−0.23, p=0.281) or emotionality (r=0.17, p=0.420).
In summary, UCMS induced a selective downregulation in L-3’-UTR-containing Bdnf mRNAs in the mouse mPFC which was significantly correlated with behavioral emotionality and Sst levels. These results suggest a role for dendritic Bdnf in anxiety-/depressive-like measures via regulation of dendritic- targeting interneurons.
Knockdown of dendritic Bdnf transcript is sufficient to induce MDD- and stress-associated phenotypes
We next tested whether altered dendritic Bdnf expression was causal to MDD/stress-related dendritic shrinkage of cortical neurons. Using GFP as a marker (Figure 3A), we collected shRNA-expressing primary neurons with FACS and confirmed that shRNA treatment knocked down Bdnf L-3’-UTR expression (Figure 3B), without affecting total Bdnf levels. shRNA expression reduced the number of intersections in distal dendrites without changes in proximal dendrites (0 to ~30µm from soma) (Figure 3C,Table S3). The total length of all dendrites of shRNA-treated neurons was shorter than controls (p=0.009, Figure 3D) while the number of dendritic segments remained unchanged (p=0.91; Figure 3E).
Figure 3. Bdnf L-3’-UTR knockdown-induced structural changes of primary mouse cortical neurons.
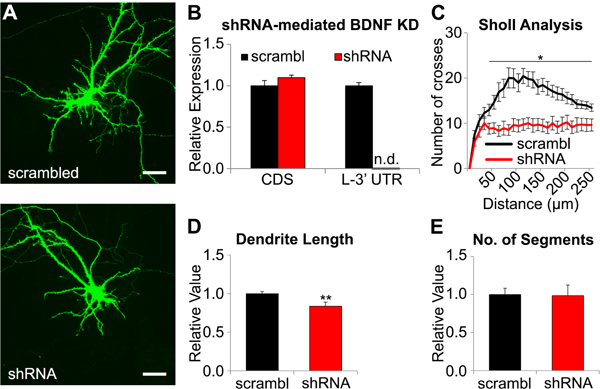
(A) Representative image of primary mouse cortical neuron expressing scrambled shRNA and Bdnf L-3’-UTR-targeting shRNA. Scale bar = 50μm (B) Relative expression (shRNA/scrambled) of Bdnf-CDS and Bdnf L-3’-UTR in shRNA expressing cells compared to controls. Expression of L-3’-UTR was not detectable(n.d.) after shRNA treatment. (C) Sholl analysis for shRNA-treated and control neurons showed that shRNA treatment disrupted integrity of distal dendrite. (D) Relative value of total length of dendrites of shRNA-treated and control neurons. (E) Relative value of number of dendritic segments of shRNA-expressing neurons compared to control. Total dendritic length was significantly decreased without changes in branch number (control: n=14, shRNA: n=10; *p<0.05, **p<0.01). Data are represented as mean ± SEM.
Next, we injected shRNA-expressing AAV in mouse mPFC (Figure 4A) to investigate whether low dendritic Bdnf expression is associated with behavioral changes. Behavioral tests were performed after recovery, then after UCMS (Figure 4B). Stress exposure did not affect body weight (Figure 4C), but induced a progressive worsening of the coat state (Figure 4D), indicating that both groups responded to UCMS (p=3.6E-28). Notably, shRNA-treated mice displayed worse coat state than the control group over time (p=0.002). shRNA-treated mice showed trend-level differences in the OF (p=0.052; Figure 4E) and in emotionality z-scores before UCMS (p=0.093; Figure 4F).
Figure 4. Effect of Bdnf L-3’-UTR knockdown and chronic stress on behavior.
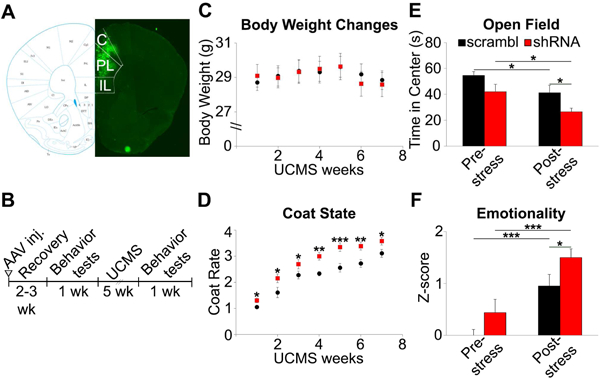
(A) Representative image of GFP expression following AAV injection in mPFC. The mouse mPFC map is from Paxinos and Franklin’s the mouse brain in stereotaxic coordinates (66). C: Cingulate cortex, PL: Prelimbic cortex, IL: Infralimbic cortex). (B) Schematic of the experimental design. (C) No body weight changes by chronic stress. (D) Progressive coat state changes with stress exposure. (E) shRNA-treated mice spent less time in the center of the open field apparatus after UCMS. (F) shRNA-treated mice group showed significantly higher emotionality z-scores than control after stress exposure (scrambled: n=9, shRNA: n=13; *p<0.05, **p<0.01, ***p<0.001). Data are represented as mean ± SEM.
After UCMS, shRNA-treated mice spent less time in the OF center (p=0.009;Figure 4E). Although other behavioral measures did not show significant group differences(Figure S5), shRNA-treated mice displayed higher emotionality compared to control vector-treated mice after UCMS (p=0.029; Figure 4F). There was no interaction between shRNA treatment and chronic stress on emotionality (p=0.763) or coat state (p=0.270).
To examine whether knockdown of dendritic Bdnf was sufficient to induce MDD-associated molecular changes, we infused AAV expressing Bdnf L-3’-UTR shRNA into one hemisphere and scrambled shRNA into the other (Figure 5A) and sacrificed mice at 2, 4, and 6 weeks after surgery. Gene expression changes were not found at post-surgery week 2.Bdnf L-3’-UTR was significantly downregulated at week 4 (p=0.013) and week 6 (p=0.030), whereas Sst level was reduced at week 6 only (p=0.002; Figure 5B). Sst and Bdnf L-3’-UTR expression showed positive correlation at week 6 (r=0.67, p=0.016), whereas Pvalb expression did not show such relationship (r=−0.05, p=0.866; Figure 5C), confirming the selective effect of Bdnf L-3’-UTR on Sst. In contrast to the knockdown yields observed in primary culture, total and EXON4+Bdnf mRNA levels were reduced by shRNA (p=0.009 and 0.035, respectively; Figure S6A), suggesting differences between in vitro and in vivo Bdnf transcripts regulation, and/or in vivo functional adaptations. Notably, total and EXON4+Bdnf levels were not correlated with Sst level (total; r=0.18, p=0.566; EXON4; r=0.39, p=0.205). The expression of other genes was not changed by shRNA treatment (Figure S6B-E). BDNF protein levels were also significantly reduced by shRNA treatment (91.4 ± 9.6%, p=0.037; Figure S6F).
Figure 5. Bdnf L-3’-UTR knockdown-related gene expression changes.
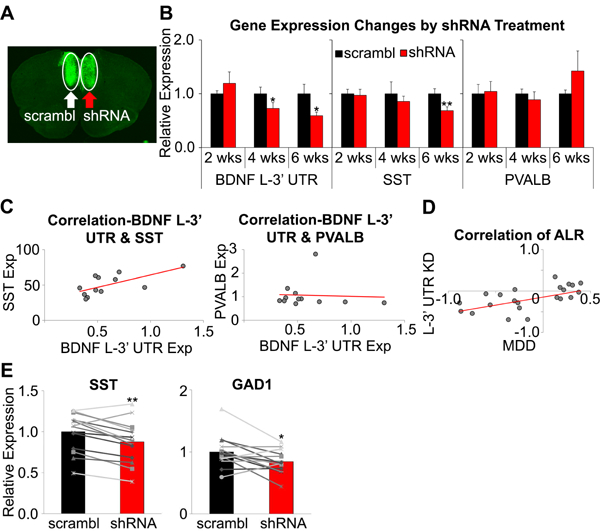
(A) Representative image of GFP expression following AAV injection in mPFC. (B) Expression changes in core genes at 2, 4, 6 weeks after surgery. Changes in Bdnf L-3’-UTR expression were observed from week 4, and Sst expression change was observed only in week 6. (C) Correlation between expression of Bdnf L-3’-UTR and Sst (left) or Pvalb (right) level at post-surgery week 6. Significant correlation to Bdnf L-3’-UTR level was observed in Sst, but not in Pvalb (n=6/group at each time point). (D) Similarity between MDD- and Bdnf L-3’-UTR knockdown-induced gene expression changes (ALR: average log ratio(MDD/control, BDNF L-3’-UTR KD/control)). (E)Sst and Gad1 in Sst+ cells were significantly downregulated by shRNA treatment (n=14/group; *p<0.05, **p<0.01, ***p<0.001). Data are represented as mean ± SEM.
To validate the translational value of our findings, we compared shRNA-induced changes of Bdnf-, synaptic function-related genes in mice to expression changes observed in MDD subjects compared to controls. We found a significant correlation(r=0.45, p=0.009; Figure 5D,Table S4), demonstrating that dendritic Bdnf knockdown in mice replicated gene expression changes observed in human MDD.
Finally, we collected Sst- or Pvalb-expressing cells from AAV-infused mPFC using laser microdissection to investigate whether Bdnf L-3’-UTR knockdown induced gene expression changes in a cell type-specific manner. Expression of Sst and Gad1 mRNAs was significantly reduced in Sst- positive cells by shRNA treatment (p=0.001 and p=0.014, respectively; Figure 5E), but Pvalb and Gad1 remained unchanged in Pvalb-positive interneurons (p=0.478 and p=0.217, respectively; Figure S6G).
In summary, Bdnf L-3’-UTR knockdown recapitulated MDD- and chronic stress-associated molecular, structural, behavioral phenotypes. Our observations that Sst reduction followed dendritic Bdnf knockdown, and that shRNA-treated mice showed transcriptome profiles similar to MDD suggest that MDD-related dendritic Bdnf downregulation is causally implicated in MDD-related GABAergic gene changes and associated symptoms(Figure 6).
Figure 6. Model of BDNF role in MDD- and stress-related molecular and behavioral phenotypes.
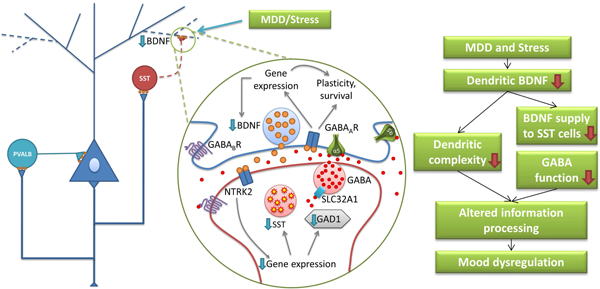
Results from the current study suggest a sequence of events associated with MDD-related brain changes. First, prolonged stress induces selective deficit of dendritic BDNF indicated by low BDNF L- 3’-UTR expression in the PFC. Second, reduced BDNF supply to dendritic compartments and GABA interneurons targeting distal dendrites of pyramidal cells leads to impaired function of dendritic-targeting interneurons (e.g. downregulation of SST and GABA synthesizing enzyme), disrupted dendritic integrity, altered filtering and processing of excitatory input onto pyramidal cell dendrites, and finally, mood dysregulation.
DISCUSSION
MDD is a devastating disease characterized by low mood, anhedonia, and cognitive deficits. Studies suggest that impaired neural plasticity and GABAergic function are associated with MDD, however, the mechanism underlying these two phenomena has not been clearly defined. In the current study, we report a selective downregulation of dendritic BDNF mRNA in the dlPFC of human MDD subjects and a parallel downregulation of dendritic-targeting interneuron markers without affecting a perisomatic targeting interneuron marker. Strong and selective correlations between BDNF L-3’-UTR and inhibitory synaptic genes were observed, suggesting that an MDD-related decrease in dendritic BDNF transcripts contributed to impairment of dendritic-targeting interneurons. However, human postmortem brains do not allow examining dynamic molecular changes induced by the disease, therefore our findings could be the results of the disease, not the cause. To test a putative causal link, we used animal models. The UCMS paradigm is known to reproduce phenotypes resembling human MDD(65), although note that it is a model of chronic stress-induced pathologies, rather than MDD itself. We observed a selective decrease in Bdnf L-3’-UTR in the mPFC of mice exposed to UCMS. Although Sst expression did not show significant group differences, it was positively correlated with Bdnf L-3’-UTR level and negatively to behavioral emotionality. The fact that Sst downregulation did not reach statistical significance may relate to differences in timescale; human subjects had suffered from MDD for months to years, whereas mice were exposed to mild stress for only seven weeks. Moreover, we performed animal experiments in male mice, and it is not known whether greater effects would be observed in female mice.
Knockdown of Bdnf L-3’-UTR replicated MDD-like structural, behavioral and molecular phenotypes. We observed a trend for elevated anxiety-/depression-like behaviors in shRNA treated mice at baseline, and a significant elevation after UCMS in shRNA-treated animals compared to controls. Following shRNA-induced Bdnf knockdown, Sst expression was reduced and showed positive correlation to Bdnf L-3’-UTR level. These progressive molecular changes were consistent with non-significant group differences in baseline emotionality at post-surgery week 2–3 and with the earlier UCMS-induced fur degradation in shRNA-treated mice. Moreover, cell-specific analyses suggest that Sst+ cells are more vulnerable than Pvalb+ interneurons to low dendritic Bdnf as revealed by reduced Gad1 expression. Notably, although the mouse studies were inspired by findings in human postmortem brains, species differences may limit their interpretability. Whilst not all MDD-associated gene expression changes are recapitulated, we observed a positive correlation between Bdnf L-3’-UTR shRNA-induced gene expression changes in mice and those observed in human MDD brain samples. Hence the collective results suggest a novel mechanism underlying the pathophysiology of MDD, linking local neurotrophic support, pyramidal cell structure and dendritic inhibition (Figure 6).
Contribution of low dendritic BDNF to impaired structural integrity and function in the dlPFC of MDD subjects
Previous studies showed that about 50% of Bdnf transcripts had L-3’-UTR (31,45). Knockdown of Bdnf L-3’-UTR in mice was sufficient to induce stress- and MDD-associated morphological changes in cortical neurons. In agreement with a human postmortem study reporting shortened dendritic processes without changes in the number of processes in basilar dendrites of pyramidal neurons in dlPFC of subjects with psychiatric illnesses including MDD (46) and with animal studies showing stress-induced structural changes mainly occurring on the distal portion of apical dendrites (4,19,20,47,48), shRNA targeting Bdnf L-3’-UTR induced retraction of distal dendrites of primary cortical neurons.
Animal studies show that dendritic Bdnf transcripts are closely linked to stress-related mental illness(33–35). Human genetic studies also support the idea that the dendritic action of BDNF is essential to mood regulation. The most common single nucleotide polymorphism in human BDNF, resulting in Valine to Methionine substitution at codon 66 (Val66Met), has been implicated in defective fear extinction (49), and increased susceptibility to psychiatric diseases (50). Mice harboring the Met allele exhibit impaired dendritic transport of Bdnf mRNA (51,52), reduced activity-dependent Bdnf secretion (53), impaired synaptic plasticity in mPFC (54), and elevated anxiety-like behavior which is not normalized by fluoxetine (55). Our finding that emotionality is significantly correlated to Bdnf L-3’-UTR expression provides evidence that dendritic Bdnf is important for the function of mood regulatory systems.
Then how can the loss of dendritic BDNF contribute to functional abnormalities? First, defects in activity-dependent BDNF supply might play a role. A recent study found that over 2,000 transcripts are localized in dendrites and/or axons, maybe for faster on-demand protein supply by local synaptic translation (32). Not only does L-3’-UTR+Bdnf mRNA preferentially exist in dendrites (31), it stays dormant in resting state, yet binds to a polyribosome responding to neuronal activity (45). Given that MDD may be conceived as a disease of maladaptive processes, impaired activity-dependent neuroplasticity may underlie the inability to cope with stress. A second possible mechanism could be through reduced GABAergic transmission. Knockdown of dendritic Bdnf mRNA and/or blockade of dendritic transport downregulate GABAergic receptors and GABA synthesizing enzymes (56,57). In addition, Sst acts as inhibitory neuropeptide and Sst knockout is sufficient to induce depressive-/anxiety-related behavior in mice (16). These data imply that the depressive-like behavioral outcome may derive at least in part from low inhibitory control.
Close link between pyramidal cell dendritic BDNF transcripts and dendritic-targeting interneurons and its implication for the pathology of MDD
SST and PVALB are expressed in distinct classes of cortical interneurons. Multiple lines of evidence imply that BDNF-NTRK2 signaling plays a crucial role in interneuron development and maintenance of GABAergic function. Our studies have consistently found that low BDNF function is closely linked to reduced expression of GABA-related genes observed in the brains of MDD patients (11,14,24). As NTRK2 is predominantly expressed in PVALB+ interneurons compared to other interneuron populations (21,22), higher BDNF dependency of SS7 than PVALB raised questions about underlying mechanisms. Our current study suggests that selectivity of MDD/stress-induced effects may be achieved through reduced dendritic BDNF function, leading to altered neuronal plasticity, behavior and impaired function of dendritic-targeting interneurons.
To date, it is not clearly defined how dysfunction of SSt+ interneurons contribute to the pathophysiology of mood disorders (58). In a previous study, we reported that acute inactivation of SS7 neurons in mPFC can increase anxiety- and depressive-like behavior (59). The mPFC, the target of our study, has excitatory projections to amygdala to produce fear/stress response (60). Hence low BDNF L- 3’-UTR-mediated dysfunction of SSt neurons may result in increased mPFC pyramidal neuron baseline activity leading to amygdala hyperactivity. In addition, distal dendrites mainly receive inputs from other cortical areas and regulation of dendritic excitability is crucial for functional computations (reviewed in Larkum (61)). Considering that dendritic excitation mainly occurs in actively-behaving animals, one can assume that proper control of dendritic activity is crucial for regulation of active behavior. For instance, Sst+ interneurons in barrel cortex are inactivated during active whisking which leads to increased excitability for the sensorimotor stimuli(62). During contextual fear learning, Sst+ interneurons in the basolateral amygdala are inactivated by multisensory environmental context, whereas an aversive event inactivates both Pvalb+ and Sst+ interneurons (63,64). Hence, loss of dendritic inhibition may result in increased signal-to-noise ratio and failure to discriminate different stimuli(58).
Combining current knowledge and our experimental results, we propose a biological model linking low BDNF, structural changes and GABAergic dysfunction, three phenomena commonly observed in the PFC of MDD subjects and stressed animals (Figure 6). MDD and stress may disturb dendritic BDNF, which leads to low neurotrophic supply to dendrites and dendritic targeting interneurons. Reduced BDNF signaling may contribute to decreased dendritic complexity of pyramidal neurons and low inhibitory function via reduced GAD (i.e. low GABA production) and SSt expression in SSt+ cells, finally culminating in altered information processing by cortical microcircuits, leading to behavioral symptoms of MDD.
In conclusion, our results contribute to our understanding of BDNF function under control and disease-states, and provide evidence that MDD-related downregulation of dendritic BDNF contributes to selective impairment of dendritic-targeting interneurons in MDD subjects, and that maintaining the integrity of the BDNF-NTRK2 system is critical for mood regulation.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institute of Mental Health (NIMH) MH077159 (ES).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURES
David Lewis currently receives investigator-initiated research support from Pfizer, and in 2012–2014 served as a consultant in the areas of target identification and validation and new compound development to Autifony, Bristol-Myers Squibb, Concert Pharmaceuticals, and Sunovion. Etienne Sibille is co-inventor on a US provisional patent application that covers compounds modulating the function of GABA neurons. The other authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Kupfer DJ, Frank E, Phillips ML (2012):Major depressive disorder: new clinical, neurobiological, and treatment perspectives.Lancet.379:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McEwen BS,Morrison JH (2013): The Brain on Stress: Vulnerability and Plasticity of the Prefrontal Cortex over the Life Course. Neuron.79:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drevets WC (2000):Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression.Prog Brain Res.126:413–431. [DOI] [PubMed] [Google Scholar]
- 4.Liston C, Miller MM,Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. (2006):Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci.26:7870–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan HY,Callicott JH,Weinberger DR (2007):Dysfunctional and compensatory prefrontal cortical systems, genes and the pathogenesis of schizophrenia.Cereb Cortex. 17 Suppl 1:i 171–181. [DOI] [PubMed] [Google Scholar]
- 6.Kang HJ, Voleti B, Hajszan T,Rajkowska G,Stockmeier CA,Licznerski P, et al. (2012):Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med.18:1413–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajkowska G (2000):Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry.48:766–777. [DOI] [PubMed] [Google Scholar]
- 8.Levinson AJ,Fitzgerald PB, Favalli G,Blumberger DM, Daigle M,Daskalakis ZJ (2010):Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry.67:458–464. [DOI] [PubMed] [Google Scholar]
- 9.Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OAC, et al. (1999):Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Arch Gen Psychiat.56:1043–1047. [DOI] [PubMed] [Google Scholar]
- 10.Sanacora G,Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. (2004):Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression.Arch Gen Psychiat.61:705–713. [DOI] [PubMed] [Google Scholar]
- 11.Guilloux JP,Douillard-Guilloux G, Kota R, Wang X, Gardier AM,Martinowich K, et al. (2012):Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry.17:1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibille E, Morris HM, Kota RS, Lewis DA (2011):GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol.14:721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tripp A, Kota RS, Lewis DA, Sibille E (2011):Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis.42:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tripp A, Oh H, Guilloux JP,Martinowich K, Lewis DA, Sibille E (2012):Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am J Psychiatry.169:1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin LC, Sibille E (2013):Reduced brain somatostatin in mood disorders: a common pathophysiological substrate and drug target? Front Pharmacol.4:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin LC, Sibille E (2015):Somatostatin, neuronal vulnerability and behavioral emotionality. Mol Psychiatry.20:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duman RS,Monteggia LM (2006):A neurotrophic model for stress-related mood disorders. Biol Psychiatry.59:1116–1127. [DOI] [PubMed] [Google Scholar]
- 18.Dunham JS, Deakin JF,Miyajima F,Payton A,Toro CT (2009): Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. Journal of psychiatric research.43:1175–1184. [DOI] [PubMed] [Google Scholar]
- 19.Radley JJ, Rocher AB,Rodriguez A,Ehlenberger DB, Dammann M, McEwen BS, et al. (2008):Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol.507:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, et al. (2004):Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience.125:1–6. [DOI] [PubMed] [Google Scholar]
- 21.Cellerino A, Maffei L, Domenici L (1996):The distribution of brain-derived neurotrophic factor and its receptor trkB in parvalbumin-containing neurons of the rat visual cortex. The European journal of neuroscience.8:1190–1197. [DOI] [PubMed] [Google Scholar]
- 22.Gorba T, Wahle P (1999):Expression of TrkB and TrkC but not BDNF mRNA in neurochemically identified interneurons in rat visual cortex in vivo and in organotypic cultures. The European journal of neuroscience.11:1179–1190. [DOI] [PubMed] [Google Scholar]
- 23.Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T (2007):A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci.27:7234–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glorioso C, Sabatini M, Unger T,Hashimoto T,Monteggia LM, Lewis DA, et al. (2006):Specificity and timing of neocortical transcriptome changes in response to BDNF gene ablation during embryogenesis or adulthood. Mol Psychiatry.11:633–648. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Chang X, She L, Xu D, Huang W, Poo MM (2015):Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J Neurosci.35:8384–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka J, Horiike Y,Matsuzaki M, Miyazaki T,Ellis-Davies GC, Kasai H (2008):Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines.Science.319:1683–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohba S, Ikeda T, Ikegaya Y,Nishiyama N, Matsuki N, Yamada MK (2005):BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition.Cereb Cortex.15:291–298. [DOI] [PubMed] [Google Scholar]
- 28.Ali AB, Thomson AM (2008):Synaptic alpha 5 subunit-containing GABAA receptors mediate IPSPs elicited by dendrite-preferring cells in rat neocortex. Cereb Cortex.18:1260–1271. [DOI] [PubMed] [Google Scholar]
- 29.Oh H, Lewis DA, Sibille E (2016):The Role of BDNF in Age-Dependent Changes of Excitatory and Inhibitory Synaptic Markers in the Human Prefrontal Cortex. Neuropsychopharmacology.41:3080–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baj G, Leone E, Chao MV,Tongiorgi E (2011): Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc Natl Acad Sci U S A.108:16813–16818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An JJ, Gharami K, Liao GY, Woo NH, Lau AG,Vanevski F,et al. (2008): Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell.134:175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cajigas IJ, Tushev G, Will TJ, tom Dieck S,Fuerst N,Schuman EM (2012): The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron.74:453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luoni A, Berry A,Calabrese F, Capoccia S,Bellisario V, Gass P, et al. (2014):Delayed BDNF alterations in the prefrontal cortex of rats exposed to prenatal stress: preventive effect of lurasidone treatment during adolescence. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology.24:986–995. [DOI] [PubMed] [Google Scholar]
- 34.Luoni A, Macchi F, Papp M, Molteni R, Riva MA (2015):Lurasidone Exerts Antidepressant Properties in the Chronic Mild Stress Model through the Regulation of Synaptic and Neuroplastic Mechanisms in the Rat Prefrontal Cortex.Int J Neuropsychoph. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berry A, Panetta P, Luoni A,Bellisario V, Capoccia S, Riva MA, et al. (2015):Decreased Bdnf expression and reduced social behavior in periadolescent rats following prenatal stress.Developmental psychobiology.57:365–373. [DOI] [PubMed] [Google Scholar]
- 36.Baj G,D’Alessandro V, Musazzi L, Mallei A, Sartori CR,Sciancalepore M, et al. (2012):Physical Exercise and Antidepressants Enhance BDNF Targeting in Hippocampal CA3 Dendrites: Further Evidence of a Spatial Code for BDNF Splice Variants.Neuropsychopharmacology.37:1600–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orefice LL,Waterhouse EG,Partridge JG,Lalchandani RR, Vicini S, Xu B (2013):Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J Neurosci.33:11618–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glantz LA, Austin MC, Lewis DA (2000):Normal cellular levels of synaptophysin mRNA expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry.48:389–397. [DOI] [PubMed] [Google Scholar]
- 39.Glantz LA, Lewis DA (1997):Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch GenPsychiatry.54:660–669. [DOI] [PubMed] [Google Scholar]
- 40.Sweet RA, Bergen SE, Sun Z, Sampson AR, Pierri JN, Lewis DA (2004):Pyramidal cell size reduction in schizophrenia: evidence for involvement of auditory feedforward circuits. BiolPsychiatry.55:1128–1137. [DOI] [PubMed] [Google Scholar]
- 41.Sibille E, Arango V, Galfalvy HC,Pavlidis P,Erraji-BenChekroun L, Ellis SP, et al. (2004):Gene expression profiling of depression and suicide in human prefrontal cortex.Neuropsychopharmacology.29:351–361. [DOI] [PubMed] [Google Scholar]
- 42.Guilloux JP, Seney M, Edgar N, Sibille E (2011):Integrated behavioral z-scoring increases the sensitivity and reliability of behavioral phenotyping in mice: Relevance to emotionality and sex. J Neurosci Methods.197:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterhouse EG, Xu BJ (2009):New insights into the role of brain-derived neurotrophic factor in synaptic plasticity.Mol Cell Neurosci.42:81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vicario A, Colliva A, Ratti A,Davidovic L, Baj G, Gricman L, et al. (2015):Dendritic targeting of short and long 3’ UTR BDNF mRNA is regulated by BDNF or NT-3 and distinct sets of RNA-binding proteins. Front Mol Neurosci.8:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lau AG, Irier HA, Gu JP, Tian DH, Ku L, Liu GL, et al. (2010):Distinct 3 ‘ UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor(BDNF). P Natl Acad Sci USA.107:15945–15950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glantz LA, Lewis DA (2000):Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia.Arch Gen Psychiatry.57:65–73. [DOI] [PubMed] [Google Scholar]
- 47.Bloss EB, Janssen WG, McEwen BS,Morrison JH (2010): Interactive effects of stress and aging on structural plasticity in the prefrontal cortex. J Neurosci.30:6726–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook SC, Wellman CL (2004):Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J Neurobiol.60:236–248. [DOI] [PubMed] [Google Scholar]
- 49.Soliman F, Glatt CE, Bath KG, Levita L, Jones RM,Pattwell SS, et al. (2010):A Genetic Variant BDNF Polymorphism Alters Extinction Learning in Both Mouse and Human.Science.327:863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gatt JM,Nemeroff CB,Dobson-Stone C, Paul RH, Bryant RA,Schofield PR, et al. (2009):Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatr.14:681–695. [DOI] [PubMed] [Google Scholar]
- 51.Chiaruttini C, Vicario A, Li Z, Baj G, Braiuca P, Wu Y, et al. (2009):Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation.Proc Natl Acad Sci U S A.106:16481–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mallei A, Baj G, Ieraci A, Corna S, Musazzi L, Lee FS, et al. (2015):Expression and Dendritic Trafficking of BDNF-6 Splice Variant are Impaired in Knock-In Mice Carrying Human BDNF Val66Met Polymorphism. Int J Neuropsychopharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Egan MF, Kojima M,Callicott JH,Goldberg TE,Kolachana BS,Bertolino A, et al. (2003):The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell.112:257–269. [DOI] [PubMed] [Google Scholar]
- 54.Pattwell SS, Bath KG,Perez-Castro R, Lee FS, Chao MV, Ninan I (2012):The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci.32:2410–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. (2006):Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science.314:140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stein JM, Bergman W, Fang Y, Davison L,Brensinger C, Robinson MB, et al. (2006):Behavioral and neurochemical alterations in mice lacking the RNA-binding protein translin. J Neurosci.26:2184–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, et al. (2012):BDNF Promotes Differentiation and Maturation of Adult-born Neurons through GABAergic Transmission.Journal of Neuroscience.32:14318–14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fee C, Banasr M, Sibille E (2017):Somatostatin-Positive Gamma-Aminobutyric Acid Interneuron Deficits in Depression: Cortical Microcircuit and Therapeutic Perspectives. Biol Psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soumier A, Sibille E (2014):Opposing effects of acute versus chronic blockade of frontal cortex somatostatin-positive inhibitory neurons on behavioral emotionality in mice.Neuropsychopharmacology.39:2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vidal-Gonzalez I,Vidal-Gonzalez B, Rauch SL, Quirk GJ (2006):Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learning & memory.13:728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Larkum M (2013):A cellular mechanism for cortical associations: an organizing principle for the cerebral cortex.Trends Neurosci.36:141–151. [DOI] [PubMed] [Google Scholar]
- 62.Gentet LJ, Kremer Y,Taniguchi H, Huang ZJ, Staiger JF,Petersen CC (2012): Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex. Nat Neurosci.15:607–612. [DOI] [PubMed] [Google Scholar]
- 63.Tovote P, Fadok JP, Luthi A (2015):Neuronal circuits for fear and anxiety.Nat Rev Neurosci.16:317–331. [DOI] [PubMed] [Google Scholar]
- 64.Wolff SB,Grundemann J, Tovote P, Krabbe S, Jacobson GA, Muller C, et al. (2014):Amygdala interneuron subtypes control fear learning through disinhibition. Nature.509:453–458. [DOI] [PubMed] [Google Scholar]
- 65.Nollet M, Guisquet AML, Belzung C Models of depression: unpredictable chronic mild stress in mice. Current protocols in pharmacology.56561–65.65. 17. [DOI] [PubMed] [Google Scholar]
- 66.Franklin KBJ,Paxinos G (2008): The Mouse Brain in Stereotaxic Coordinates. Academic Press.<; /References> [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.