

Abstract
Ribonuclease 1 (RNase 1) is the most prevalent human homolog of the archetypal enzyme, RNase A. RNase 1 contains sequons for N-linked glycosylation at Asn34, Asn76, and Asn88 and is N-glycosylated at all three sites in vivo. The effect of N-glycosylation on the structure and function of RNase 1 is unknown. By using an engineered strain of the yeast Pichia pastoris, we installed a heptasaccharide (Man5GlcNAc2) on the side chain of Asn34, Asn76, and Asn88 to produce the authentic triglycosylated form of human RNase 1. As a glutamine residue is not a substrate for cellular oligosaccharyltransferase, we used strategic asparagine-to-glutamine substitutions to produce the three diglycosylated and three monoglycosylated forms of RNase 1. We found that the N-glycosylation of RNase 1 at any position attenuates its catalytic activity but enhances both its thermostability and its resistance to proteolysis. N-Glycosylation at Asn34 generates the most active and stable glycoforms, in accord with its sequon being highly conserved among vertebrate species. These data provide new insight on the biological role of the N-glycosylation of a human secretory enzyme.
TOC Graphic
INTRODUCTION
Pancreatic-type ribonucleases (ptRNases; EC 3.1.27.5) are a family of secretory endoribonucleases that enable a variety of biological functions in vertebrates by catalyzing the cleavage of RNA. Members of this family are characterized by their small size and high conformational stability.1,2 Insights into the structure and function of ptRNases have emerged from a bovine prototype—RNase A. The mass production of RNase A as a byproduct of the meatpacking industry provided seminal information on the biological chemistry of proteins,3–8 including the first enzymatic reaction mechanism.9
The “A” in RNase A refers not to an adenine nucleobase, but to RNase A being the predominant ribonuclease in the cow pancreas.10,11 RNase A is unglycosylated, whereas “RNase B”, “RNase C”, and “RNase D” are N-glycosylated forms of RNase A.12,13 The glycan is added to the side chain of Asn34 residue by oligosaccharyltransferase, which acts on Asn-Xaa-Ser/Thr sequons,14–17 where Xaa represents any residue other than proline.18 RNase A has only one sequon.
The most prevalent human homolog of RNase A is RNase 1 (UniProtKB P07998), which is present in many bodily fluids19,20 and circulates at ~0.5 µg/mL.21 In RNase 1, Asn34, Asn76, and Asn88 are in Asn-Xaa-Ser/Thr sequons (Figure 1A).22 An analysis of RNase 1 in human serum has shown that each of these sites is N-glycosylated in vivo.23 (There are no apparent C-, O-, or S-linked glycans.) The three sites of human RNase 1 are, however, N-glycosylated differentially depending on tissue- and cell-type.24–26 Moreover, Asn34 is glycosylated in nearly all isolates, whereas Asn76 and Asn88 are occupied in half or a small fraction of isolates, respectively.27 These occupancies are consistent with the observed preference for the glycosylation of Asn-Xaa-Thr rather than Asn-Xaa-Ser sequences.28
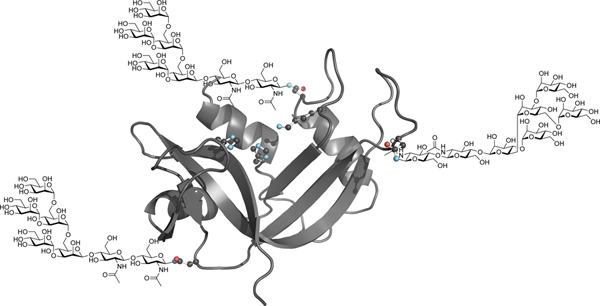
Figure 1.

Three-dimensional structure of human RNase 1. (A) The side chains of the asparagine residues within N-glycosylation sequons (Asn34, Asn76, and Asn88) and the active-site residues (His12, Lys41, and His119) are shown explicitly. The image was created with Protein Data Bank entry 1z7x, chain Z22 and the program PyMOL from Schrödinger (New York, NY). (B) Electrostatic potential map of the surface of human RNase 1. The image was created as in Figure 1A. (C) Structure of the core heptasaccharide, Man5GlcNAc2, that is appended to asparagine residues. (D) Macroscopic glycoforms of human RNase 1 generated by strategic asparagine→glutamine substitution. The three-letter shorthand is used to name each variant.
The consequences of the glycosylation of human RNase 1 are unknown.29 A phylogenetic analysis reveals multiple N-glycosylation sequons, many of which occur in rapidly evolving segments of the enzyme (Figure 2).30 Of the potential N-glycosylation sites, Asn34 is conserved most widely, and the analysis of isolates confirms that this site is N-glycosylated in a range of diverse species.31,32 The specific triglycosylation pattern observed in human RNase 1 appears to have evolved in more recent hominids, with only orangutans lacking Asn88. Accordingly, all three N-glycosylation sites are worthy of consideration.
Figure 2.

Putative N-glycosylation sites in RNase 1 homologs across an evolutionary spectrum. Circles indicate an asparagine residue within an N-glycosylation sequon. The neighbor-joining phylogenetic tree is adapted from ref. 30 and shows bootstrap values >30. Species in red lack an N-glycosylation sequence. The Bos taurus (cow) entry is for RNase A; bovine brain ribonuclease has a sequon at residue 62 but not residue 34.33
In this work, we present an extensive biochemical analysis of human RNase 1 with authentic glycosylation. First, we establish a heterologous production system that generates high yields of individual RNase 1 glycoforms. Then, we measure the biochemical characteristics of each glycoform, including catalytic activity, conformational stability, and resistance to proteolysis. The ensuing data provide insight into the impact of glycosylation on RNase 1 and, specifically, the potential evolutionary significance of glycosylation at Asn34.
EXPERIMENTAL PROCEDURES
Strategy for Producing Human RNase 1 Glycoforms.
Previous studies of human RNase 1 have relied on the aglycosylated enzyme produced by heterologous expression in Escherichia coli. Detailed investigations of N-glycosylated RNase 1 have been hindered by the intrinsic heterogeneity of isolated RNase 1. This heterogeneity arises from both the differential utilization of particular N-glycosylation sites (macroheterogeneity) and the variable glycan composition at each site (microheterogeneity). RNase 1 glycoforms have been produced by recombinant DNA technology in Chinese hamster ovary cells34 and Saccharomyces cerevisiae,35 but such systems suffer from the microheterogeneity and macroheterogeneity of human isolates, as well as the presence of non-human glycans.
Recently, a highly engineered strain of methylotrophic Pichia pastoris yeast has emerged as an ideal platform for glycoprotein biosynthesis.36 The Pichia Glycoswitch® System has been altered by a disruption in the gene encoding for a mannosyltransferase as well as the addition of heterologous genes encoding enzymes that append human-like glycans.37,38 The Superman 5 (M5) strain produces large quantities of protein with a Man5GlcNAc2 heptasaccharide that is identical to the core N-linked glycosylation structure in human proteins, attached to Asn-Xaa-Ser/Thr sequences.
With its three sequons, RNase 1 has seven possible macroscopic glycoforms (Figure 1D). Even though replacing an asparagine residue with glutamine is a conservative substitution, Gln-Xaa-Ser/Thr sequences are not substrates for oligosaccharyltransferase.15–17 Accordingly, the production of a particular glycoform of RNase 1 can be designated by replacing an asparagine codon with a glutamine codon.
Hence, we chose to use the M5 strain of P. pastoris and strategic aspargine→glutamine substitutions to generate the glycoforms of RNase 1.
Materials.
The M5 strain of P. pastoris was from Research Corporation Technologies (Tucson, Arizona). The BL21(DE3) strain of Escherichia coli cells was from Novagen (Madison, WI).
Synthetic oligonucleotides were from Integrated DNA Technologies (Coralville, IA). Poly(cytidylic acid) was from Sigma–Aldrich (St. Louis, MO). SYPRO Orange Protein Gel Stain was from Life Technologies (Grand Island, NY). All other chemicals and reagents were of commercial reagent grade or better, and were used without further purification.
Aqueous solutions were made with water that was generated with an arium Pro water purification system from Sartorius (Bohemia, NY) and had resistivity ≥18 MΩ·cm–1. Phosphate-buffered saline (PBS) was 10 mM Na2HPO4 and 1.8 mM KH2PO4 buffer, pH 7.4, containing NaCl (137 mM) and KCl (2.7 mM). Buffered glycerol-complex medium (BMGY) was 100 potassium phosphate buffer, pH 5.0, containing peptone (2% w/v), yeast extract (1% w/v), yeast nitrogen base without amino acids (1.34% w/v), glycerol (1% w/v), and biotin (0.4 µg/mL). Buffered methanol-complex medium (BMMY) was 100 mM potassium phosphate buffer, pH 5.0, containing peptone (2% w/v), yeast extract (1% w/v), dextrose (2% w/v), yeast nitrogen base without amino acids (1.34% w/v), methanol (1% v/v), and biotin (0.4 µg/mL). BMGY and BMMY were sterilized by autoclaving prior to use. Yeast extract peptone–dextrose solution (YPD) was 100 mM potassium phosphate buffer, pH 5.0, containing peptone (2% w/v), yeast extract (1% w/v), dextrose (2% w/v), and agar (2% w/v). The solution was sterilized by autoclaving, poured into 60 mm × 15 mm plates, and allowed to cool to room temperature. Dialysis was performed with 3.5-kDa MWCO tubing from Spectrum Labs (Rancho Dominguez, CA). A Spectra Multicolor Broad Range Protein Ladder from Thermo Fisher Scientific (Waltham, MA) was used a molecular weight standard for SDS–PAGE.
Instrumentation.
Chromatography was conducted using an ÄKTA Pure system from GE Healthcare Life Sciences (Piscataway, NJ), and the results were analyzed with the UNICORN Control System. HiTrap SP HP, HiTrap SP Sepharose FF, HiTrap ConA 4B, and HiLoad® 26/600 Superdex® 75 pg columns for protein purification were from GE Healthcare Life Sciences.
Protein concentrations were determined with a NanoVue Plus spectrophotometer from GE Healthcare Life Sciences by absorbance at 280 nm using Beer’s law, ε = 0.53 mL·mg–1·cm–1, and the molecular mass of the unglycosylated protein.
Differential scanning fluorimetry (DSF),39 which requires the monitoring of fluorescence during thermal denaturation, was performed with a ViiA 7 Real-Time PCR system from Applied Biosystems (Foster City, CA). Denaturation data were obtained with ViiA 7 version 2.0 software and analyzed further with Protein Thermal Shift version 1.4 software, both from Applied Biosystems.
The intact molecular mass of RNase 1 glycoforms was determined by MALDI–TOF mass spectrometry using a microflex LRF instrument from Bruker (Billerica, MA) and by ESI mass spectrometry using a 6530 Accurate-Mass Q-TOF LC/MS from Agilent (Santa Clara, CA).
Conditions.
All procedures were performed in air at ambient temperature (~22 ∞C) and pressure (1.0 atm) unless indicated otherwise.
Plasmid Preparation for Expression in P. Pastoris.
A cDNA that encodes N34Q/N76Q/N88Q RNase 1 (QQQ) was synthesized from synthetic DNA oligonucleotides by Gibson assembly40 using the kit from New England Biolabs (Ipswich, MA) (Figure S1). The synthetic gene was inserted into the pPICZ expression vector, which has a neomycin phosphotransferase (NeoR)/G418 resistance gene and was from Thermo Fisher Scientific. The vector encodes DNA for an RNase 1 glycoform fused to the α-mating factor signal sequence with expression under the control of a strong methanol-inducible promoter. cDNAs coding RNase 1 glycoforms were made by site-directed mutagenesis of the QQQ pPICZα plasmid using synthetic oligonucleotides (Figure S2) and the Phusion site-Directed Mutagenesis Kit from Thermo Fisher Scientific. After target-plasmid amplification, the parent template plasmid was degraded by digestion with 1 unit of DpnI restriction endonuclease from New England Biolabs, which was added to the PCR reaction mixture. The resulting nuclease-resistant target plasmid was transfected into E. coli. Plasmids were isolated from the resulting colonies, and the sequence of the cDNA that encodes RNase 1 was assessed for the desired modification.
Transformation and Selection of RNase 1 Glycoform Expression Plasmids.
In preparation for transformation into P. pastoris, plasmids were linearized by digestion with 1 unit of PmeI restriction endonuclease from New England Biolabs at 37 °C overnight. The aqueous DNA solution was washed with 1 volume of 25:24:1 phenol/chloroform/isoamyl alcohol that was saturated with 10 mM Tris–HCl buffer, pH 8.0. The aqueous (i.e., upper) layer containing the linearized DNA was transferred to a new tube, and 1/10th volume of 3 M sodium acetate, pH 5.0, was added. A twofold excess of isopropanol was added, and the resulting solution was then subjected to centrifugation at 14,000g at 4 °C for 1 h to pellet the DNA. After spinning, the supernatant was removed by careful decantation, and the DNA pellet was resuspended in ethanol (70% v/v) to remove excess salts. The solution was subjected to centrifugation at 14,000g at 4 °C for 1 h, and the supernatant was removed by careful decantation. Excess ethanol was evaporated by allowing the DNA pellet to sit at room temperature for 10 min. The pure linearized DNA was resuspended in water.
P. pastoris strain M5 was transformed by the addition of linearized DNA (1 µg) to an aliquot of electrocompetent P. pastoris cells, followed by incubation on ice for 5 min. After transfer to an ice-cold 2-mm electroporation cuvette, cells were pulsed at 1.5 kV with an electroporator from Bio-Rad Laboratories (Hercules, CA). Cells were resuspended immediately in an ice-cold 1.0-mL solution of sorbitol (1.0 M) and allowed to recover for 2 h at 30 °C without shaking. After recovery, 200 μL of resuspended cells were spread on YPD-agar plates containing G418 (0.50 mg/mL) from Thermo Fisher Scientific and incubated at 30 °C for 2 days. To test for the ability of transformed cells to secrete RNase 1 into the culture medium, colonies were used to inoculate 10 mL of BMGY. The resulting culture was allowed to grow for 48 h, and then subjected to centrifugation. The resulting pellet was suspended in 5 mL of BMMY. Aliquots of methanol (each 1% v/v) were added to the culture at intervals of 12 h during an incubation period of 60 h. The supernatants were collected and the level of ribonucleolytic activity was assessed by using a zymogram assay as described below.
Production and Purification of RNase 1 Glycoforms from P. pastoris.
Colonies producing a high level of ribonucleolytic activity were used to inoculate 10 mL of BMGY. After shaking at 250 rpm for 48 h at 30 °C, the starter culture was used to inoculate 200 mL of BMGY in a 2-L baffled shaker flask. After growth for another 48 h at 30 °C, the large-scale cultures were subjected to centrifugation at 5,000 rpm for 10 min. The cell pellet was resuspended in 100 mL of BMMY containing EDTA (0.5 mM) and allowed to grow for 60 h at 30 °C, with aliquots of methanol (each 1% v/v) added every 12 h to induce gene expression. The culture was subjected to centrifugation at 5,000 rpm for 10 min. The supernatant was concentrated and exchanged into 50 mM sodium acetate buffer, pH 5.0, using a Vivaflow 200 tangential flow concentrator from Sartorius (Bohemia, NY). After a further five-fold dilution in 50 mM sodium acetate buffer, pH 5.0, the sample was applied to a HiTrap SP Sepharose FF cation-exchange column and eluted with a linear gradient of NaCl (0–1.0 M). The eluted fractions were pooled and dialyzed overnight against 5 L of 20 mM Tris–HCl buffer, pH 7.4. containing NaCl (0.50 M), MnCl2 (1 mM), and CaCl2 (1 mM). RNase 1 glycoforms were purified from the retentate by lectin-affinity chromatography using a HiTrap ConA 4B column, which was eluted with a solution of methyl α-d-mannopyranoside (0.50 M). RNase 1 glycoforms were purified further by cation-exchange chromatography using a HiTrap SP HP column before being dialyzed against PBS.
Plasmid Preparation for Expression in E. coli.
A cDNA that encodes Met(–1) RNase 1 in a pET22b(+) expression vector33 was used to generate wild-type RNase 1 (NNN) in E. coli (Figure S1). A synthetic cDNA that encodes the N34Q/N76Q/N88Q RNase 1 (QQQ) variant flanked by regions of sequence-identity near the T7 promoter and terminator found in the pET22b vector was obtained from Integrated DNA Technologies (Figure S1). Linear pET22b was prepared by PCR using primers that complement the DNA that encodes RNase 1 (5′-AAGCCCGAAAGGAAGCTGAGTTGGCTGCTG-3′ and 3′-AAACAAATTGAAATTCTTCCTCTATATGTA-5′). Gene and plasmid fragments were combined with Gibson assembly40 for expression in E. coli.
Production and Purification of RNase 1 from E. coli.
The N34Q/N76Q/N88Q variant and wild-type RNase 1 were purified from inclusion bodies as described previously.41 Briefly, induced cells were lysed at 19.0 kpsi with a benchtop cell disruptor from Constant Systems (Kennesaw, GA). After centrifugation at 10,500 rpm for 45 min, the resulting pellet was suspended in 20 mM Tris–HCl buffer, pH 8.0, containing guanidine–HCl (7 M), EDTA (10 mM), and dithiothreitol (0.10 M). This solution was diluted 10-fold by the slow addition of degassed 20 mM acetic acid, then subjected to centrifugation at 10,500 rpm for 45 min. The supernatant was then dialyzed overnight against 16 L of 20 mM acetic acid. After centrifugation at 10,500 rpm for 40 min, the supernatant was added dropwise to folding solution (which was 100 mM Tris–HCl buffer, pH 7.8, containing 100 mM NaCl, 1.0 mM reduced glutathione, and 0.2 mM oxidized glutathione) and allowed to fold for 5 days at 4 °C. The pH of the solution was adjusted to 5.0, and the solution was then concentrated to 10 mL by using an Amicon Stirred Cell concentrator from EMD Millipore (Billerica, Massachusetts) with Hydrostart® 10-kDa filters from Sartorius. Protein in the resulting solution was purified by gel-filtration chromatography using a HiLoad 26/600 Superdex 75 pg column and elution with 50 mM sodium acetate buffer, pH 5.0, containing NaCl (0.10 M) and sodium azide (0.05% w/v). The protein was purified further by cation-exchange chromatography using a HiTrap SP HP column before being dialyzed against PBS.
Zymogram Analysis of RNase 1 Glycoforms.
To verify the production of secreted, properly folded ribonucleases, zymogram assays were performed on conditioned medium using an assay similar to one reported previously.27 Briefly, a polyacrylamide gel containing SDS (0.1 % w/v), Tris (375 mM), and poly(cytidylic acid) (5.5 mg, incubated at 50 °C prior to addition) was cast. Aliquots of P. pastoris cultures were prepared with 2× Laemmli sample buffer, which was 68.5 mM Tris–HCl buffer, pH 6.7, containing glycerol (26.3% w/v), SDS (2.1% w/v), and bromophenol blue (0.01% w/v). After electrophoresis, the gel was washed twice with 10 mM Tris–HCl buffer, pH 7.5, containing isopropanol (20% v/v), and once with 10 mM Tris–HCl buffer, pH 7.5. Then, the gel was incubated overnight in 100 mM Tris–HCl buffer, pH 7.5, to allow the RNase 1 glycoform to refold within the gel. The following day, the gel was washed with 10 mM Tris–HCl buffer, pH 7.5, before staining with an aqueous solution of toluidine blue (0.2% w/v) for 10 min. The gel was rinsed with deionized water and destained in 10 mM Tris–HCl buffer, pH 7.5.
Treatment with Peptide-N-glycosidase F.
To assess N-linked glycosylation, RNase 1 glycoforms were denatured by boiling for 10 min in an aqueous solution of SDS (0.5% w/v) and dithiothreitol (40 mM). The denatured proteins were incubated with peptide-N-glycosidase F (PNGase F) (5 U/mL) for 24 h at 37 °C in 50 mM sodium phosphate buffer, pH 7.4, containing NP-40 (1% v/v), SDS (0.25% w/v), and dithiothreitol (20 mM). Products were analyzed by SDS–PAGE.
Assays of Protein Phosphorylation.
To probe for phosphorylation of proteins produced by P. pastoris, RNase 1 glycoforms were denatured and subjected to SDS–PAGE. The ensuing gel was fixed, washed, and stained following the protocol for Pro-Q Diamond Phosphoprotein Gel Stain from Thermo Fisher Scientific. Protein bands were observed by excitation at 520 nm and emission at 605 nm with an Amersham Imager 600 from GE Healthcare. The gel was then stained with Coomassie blue to assess total protein content.
Assays of Single-Stranded Ribonucleolytic Activity.
The catalytic activity of each RNase 1 glycoform was evaluated by measuring the initial velocity of the cleavage of a single-stranded fluorogenic substrate42 in a 96-well plate (Corning) at 25 °C. A solution of 6-FAM–dArU(dA)2–6-TAMRA (0.2 µM) in 0.10 M Tris–HCl buffer, pH 7.5, containing NaCl (0.10 M) was added to each well. After baseline fluorescent readings were recorded, an RNase 1 glycoform was added (final concentration: 50 pM), and the initial velocity of substrate turnover was measured by the increase in fluorescence over time. After 8 min, substrate cleavage was saturated by the addition of RNase A to 5 µM. Values of kcat/KM were determined as described previously,43 and represent the mean of at least three independent experiments.
Analysis of Double-Stranded RNA Degradation.
Double-stranded RNA degradation was evaluated by using a fluorescent hairpin substrate, 5,6-FAM–CGATC(rU)ACTGCAACGGCAGTAGATCG, as described previously.33 This substrate has a single RNA nucleotide near the fluorophore at its 5′ terminus. An aqueous solution of the substrate was annealed into a hairpin by heating to 95 °C for 3 min, and then slowly cooling to room temperature. Degradation was assessed in 0.10 M Tris–HCl buffer, pH 7.4, containing NaCl (0.10 M), the hairpin substrate (50 nM), and an RNase 1 (1 μM). After 1 min, the reaction was quenched by the addition of ribonuclease inhibitor protein (rRNasin; 40 units) from Promega (Madison, WI). The reaction products were subjected to electrophoresis on a 20% w/v native acrylamide gel at 10 mAmp. Formation of the cleavage product was monitored by excitation of FAM at 460 nm and emission at 525 nm with an Amersham Imager 600 from GE Healthcare. The gel was then stained with SYBR Gold from Invitrogen and imaged for total nucleic acid content.
Assays of Conformational Stability.
The conformational stability of each RNase 1 glycoform was assessed by DSF as described previously.30 Briefly, an RNase 1 glycoform was dissolved in PBS to a final concentration of 2 µg/µL. SYPRO Orange (which was obtained at a concentration of 5000×) was added to a concentration of 50×. The assay solution was heated from 10 to 95 °C at a rate of 0.2 °C/min. As the solution was heated, the fluorescence emission was monitored at (623 ± 14) nm after excitation at (580 ± 10) nm. Data were summarized by the value of Tm, which is the temperature at the midpoint of the thermal transition between native and denatured states.
Assays of Proteolytic Stability.
An RNase 1 glycoform (1 mg/mL) was incubated with sequencing-grade modified trypsin (10, 1, or 0.1 µg/mL) in 50 mM Tris–HCl buffer, pH 8.0, containing CaCl2 (1 mM) at 37 °C for 1 h. To terminate the reaction, phenylmethylsulfonyl fluoride (PMSF) was added to a final concentration of 1 mM. The reaction products were analyzed by SDS–PAGE.
RESULTS
Production, Purification, and Analysis of RNase 1 Glycoforms.
The three glycosylation sites within RNase 1 give rise to eight potential macroscopic glycoforms, specifically, one triglycosylated, three diglycosylated, three monoglycosylated, and one aglycosylated variant. We produced each macroscopic glycoform of RNase 1 by strategically installing Gln-Xaa-Ser/Thr sequences, which do not serve as substrates for oligosaccharyltransferase (Figure S3). Soluble RNase 1 glycoforms were produced by P. pastoris cells and secreted into the medium at ~3 mg per liter of culture.
We employed tangential-flow concentration, ion-exchange chromatography, and lectin-affinity chromatography to isolate each RNase 1 glycoform from the protein-rich culture medium. Purified RNase 1 glycoforms displayed a higher molecular mass than did aglycosylated controls, with shifts in electrophoretic mobility corresponding to the additional number of Man5GlcNAc2 glycans (Figures 3 and S4). Treatment of purified RNase 1 glycoforms with PNGase F, an amidase that hydrolyzes the bond between a GlcNAc residue and asparagine side chain, produced a single species that migrated similar to aglycosylated RNase 1, consistent with N-linked glycan attachment to the RNase 1 core (Figure 3).
Figure 3.

SDS–PAGE gels showing purified RNase 1 glycoforms. The enzymes were subjected to electrophoresis in a 15% w/v gel and visualized by staining with Coomassie blue. (A) Purified proteins. (B) Purified proteins after treatment with PNGase F (35.5 kDa).
The molecular mass of each purified RNase 1 glycoform was assessed by both MALDI–TOF and ESI mass spectrometry. The MALDI–TOF spectra indicated the presence of additional mass in the mono-, di-, and triglycosylated RNase 1, but was unable to resolve individual species (Figure S5). The ESI spectra indicated the appendage of the expected number of Man5GlcNAc2 units in each glycoform (Table 1, Figure S6). Some additional mannose monomers (i.e., microheterogeneity) were apparent in the RNase 1 glycoforms produced by the P. pastoris system, as is observed in bovine RNase B.44 Because these monomeric units are distal from the protein, they are likely to have less influence on RNase 1 than do the proximal Man5GlcNAc2 units.
Table 1.
Attributes of RNase 1 Glycoforms
|
m/z (Da) |
kcat/KMc |
||||||
|---|---|---|---|---|---|---|---|
| Source | RNase 1a | Glycans | expected | observed | Tm (°C)b | 106 M–1s–1 | % |
| E. coli | NNN | 0 | 14705.52d | 14698.35 | 55.9 ± 0.2 | 1.9 ± 0.2 | 100 |
| E. coli | QQQ | 0 | 14747.60d | 14740.03 | 56.6 ± 0.3 | 1.9 ± 0.1 | 100 |
| P. pastoris | QQQ | 0 | 14616.41 | 14608.88 | 56.4 ± 0.1 | 1.9 ± 0.1 | 100 |
| P. pastoris | NQQ | 1 | 15818.38 | 15811.70 | 58.6 ± 0.3 | 1.5 ± 0.1 | 79 |
| P. pastoris | QNQ | 1 | 15818.38 | 15811.91 | 56.8 ± 0.1 | 0.74 ± 0.07 | 39 |
| P. pastoris | QQN | 1 | 15818.38 | 15811.85 | 57.2 ± 0.2 | 0.30 ± 0.04 | 16 |
| P. pastoris | NNQ | 2 | 17020.36 | 17015.05 | 59.1 ± 0.2 | 0.72 ± 0.04 | 38 |
| P. pastoris | NQN | 2 | 17020.36 | 17014.49 | 58.7 ± 0.2 | 0.63 ± 0.03 | 33 |
| P. pastoris | QNN | 2 | 17020.36 | 17015.57 | 57.3 ± 0.1 | 0.21 ± 0.03 | 11 |
| P. pastoris | NNN | 3 | 18222.33 | 18218.39 | 59.4 ± 0.3 | 0.19 ± 0.09 | 10 |
Residues at positions 34, 76, and 88 (Figure 1D).
Values were determined by DSF at pH 7.4.
Values were determined by fluorescence spectroscopy for the cleavage of 6-FAM–dArUdAdA–6-TAMRA at pH 7.5.
The proteins produced in E. coli have an N-terminal methionine residue.
High-resolution analysis of the proteins from the P. pastoris system revealed additional peaks at ~80 Da and ~160 Da above that expected for each sample. The presence of these additional peaks, including with the aglycosylated control (QQQ), was indicative of further modification of the protein core by P. pastoris cells. Because phosphoryl groups have a mass of ~80 Da, we analyzed proteins from each expression system with a phosphoprotein-specific stain. The results confirmed the phosphorylation of proteins produced with the yeast system (Figure S7). Importantly, this additional modification (which is not added by E. coli cells) did not have a discernable impact on the attributes RNase 1 (vide infra), other than its mass. Protein phosphorylation, carried out by various kinases, is a common modification in eukaryotic expression systems.45 Secretory pathway-associated kinases have garnered attention for their role in phosphorylating extracellular proteins in human cells46 and could be at work in the P. pastoris system.
Ribonucleolytic Activity of RNase 1 Glycoforms.
Human RNase 1 catalyzes the cleavage of the P–O5′ bond on the 3′ side of pyrimidine ribonucleotides. The principal catalytic residues, His12, Lys41, and His 119 (Figure 1A), are conserved in RNase 1 homologs, suggestive of a similar enzymatic reaction mechanism.9 We assessed whether N-glycosylation impacts the ribonucleolytic activity of human RNase 1. We determined the ribonucleolytic activity of each RNase 1 glycoform with a fluorogenic substrate.42 A similar level of catalytic activity was observed for all aglycosylated variants, regardless of the expression system used in their production (Table 1, Figure 4A). Apparently, neither three extra methylene groups (QQQ versus NNN) nor an N-terminal methionine residue (NNN from E. coli) have a measurable effect on enzymatic catalysis by RNase 1. In contrast, all RNase 1 glycoforms exhibited lower ribonucleolytic activity. The monoglycosylated NQQ variant maintained the highest activity of all of the glycoforms.
Figure 4.

(A) Ribonucleolytic activity of RNase 1 and its glycoforms. Values of kcat/KM were determined for the cleavage of 6-FAM–dArU(dA)2–6-TAMRA at pH 7.5 and 25 °C. Each data point represents an individual measurement. (B) Thermostability of RNase 1 and its glycoforms. Values of Tm were determined were determined by using DSF in PBS containing SYPRO Orange (50×). Each data point represents an individual measurement.
RNase 1 and its homologs can catalyze the degradation of dsRNA substrates.33,40,47 We examined the ability of RNase 1 glycoforms to do the same. We observed that each RNase 1 glycoform, like the unglycosylated enzyme, can catalyze the degradation of a dsRNA (Figure S8).
Thermostability of RNase 1 Glycoforms.
N-Glycosylation has been reported to enhance the thermostability of proteins, including other ptRNases.48,49 We assessed the thermostability of each RNase 1 glycoform using DSF. We found that the Tm value of all aglycosylated variants, regardless of the expression system used in their production, was ~56 °C (Table 1, Figure 4B). As with enzymatic catalysis, neither three extra methylene groups (QQQ versus NNN) nor an N-terminal methionine residue (NNN from E. coli) has a measurable effect on the thermostability of RNase 1. In contrast, the addition of a single glycan at any position in RNase 1 increased its thermostability. The most thermostable of the monoglycosylated proteins had N-glycosylation at Asn34 (NQQ) and increased the Tm value by >2 °C to (58.6 ± 0.3) °C. N-Glycosylation at any two sequons resulted in minimal change in thermostability from the corresponding monoglycosylated species. Preventing N-glycosylation at Asn34 (QNN) generated the least stable diglycosylated protein—one having a Tm value (57.3 ± 0.1) °C, which is between the values for the related QNQ and QQN glycoforms. The triglycosylated RNase 1 had the highest thermostability with a Tm value of (59.4 ± 0.3) °C.
Proteolytic Stability of RNase 1 Glycoforms.
The proteolytic susceptibility of each RNase 1 glycoform was assessed by analysis with SDS–PAGE (Figure 5) following treatment with trypsin. The aglycosylated forms of RNase 1 were especially susceptible to trypsin and were largely degraded even at low concentrations of trypsin. Digestion products were observed as lower molecular weight bands even in the presence of trypsin at only 1 µg/mL. Nonetheless, any glycosylation produced an increase in resistance to digestion by trypsin, even at high concentrations of protease. Digestion products were not observed when glycosylated proteins were treated with 1 µg/mL trypsin.
Figure 5.

SDS–PAGE gels showing the effect of trypsin on RNase 1 glycoforms. An RNase 1 glycoform (1 mg/mL) was incubated with trypsin (0.1, 1, or 10 µg/mL) at 37 °C for 1 h. The trypsin was inactivated with PMSF, and the products were subjected to electrophoresis in a 15% w/v gel and visualized by staining with Coomassie blue.
DISCUSSION
Glycosylation Attenuates Catalysis by RNase 1.
Glycosylation can either enhance or diminish the catalytic activity of an enzyme.50 Catalysis by RNase 1 relies on the favorable Coulombic interaction between cationic enzymic residues and the anionic phosphoryl groups of an RNA substrate.51–53 During their purification by cation-exchange chromatography, the glycosylated forms of RNase 1 eluted more readily than did aglycosylated RNase 1, indicating that a pendant heptasaccharide diminishes affinity for an anionic resin. Similarly, N-glycosylation appears to reduce affinity for the rate-limiting transition state during catalysis of RNA cleavage, leading to a lower kcat/KM value (Table 1, Figure 4A).
Although monoglycosylation diminished the ability of RNase 1 to catalyze the cleavage of RNA (Table 1, Figure 4A), the impact was not equivalent across the three N-glycosylation sites. The least active monoglycoform, with glycosylation only at Asn88 (QQN), retained only ~20% of the activity of the aglycosylated controls. Residue 88 is proximal to the cationic RNA-binding site of RNase 1 (Figure 1B). A glycan appended to Asn88 could hinder access of an RNA substrate to the active site. Glycosylation of Asn76 (QNQ) yielded an enzyme with ~40% of the activity of the aglycosylated protein. Residue 76 is distal from the enzymic active site and the basis for its effect on catalysis is not clear.
In contrast to positions 76 and 88, N-glycosylation at position 34 (NQQ) enabled RNase 1 to maintain ~80% of its ribonucleolytic activity. Asn34 is relatively proximal to the active site, but the orientation of its side chain might allow for the appendage of a larger glycan without hindering access of an RNA substrate to the active site. This orientation is consistent with the results of analyses of bovine RNase B, which also has a glycan attached to Asn34.54
N-Glycosylation of RNase 1 at multiple sites likewise attenuated catalytic activity. The di- and triglycosylated proteins maintained ~30% and ~10% of control activity, respectively. Steric interactions incurred upon the addition of multiple glycans could distort the enzymic active site. Again, though, Asn34 is the most tolerable position for the installation of a glycan. Notably, all of the glycoforms retained the ability to cleave double-stranded RNA (Figure S8).
Glycosylation Increases Thermostability.
Human RNase 1 has a compact structure with thermostability (Tm ~56 °C) deriving largely from four disulfide bonds that crosslink the protein.55 Previous studies have demonstrated that N-glycosylation enhances the conformational stability of an amphibian homolog of RNase 1.49 We find that every permutation of N-glycosylation increases the thermostability of RNase 1 itself (Table 1, Figure 4B).
The three monoglycoforms of RNase 1 exhibited Tm values between 57 °C and 59 °C. N-Glycosylation at Asn34 yields the most significant increase in conformational stability. Its ∆Tm value of slightly more than 2 °C is in accord with previous analyses of RNase B. In that homologous context, N-glycosylation of Asn34 site resulted in a ∆Tm value of 1.5 °C.56 In RNase B, residues near Asn34 are known to be less dynamic than in RNase A.57
In terms of thermostability, diglycosylated and triglycosylated RNase 1 followed suit. We found that maintaining glycosylation at Asn34 generated the most stable diglycoforms: NNQ and NQN. When N-glycosylation at residue 34 was deterred, as in the QNN variant, the value of Tm was diminished. Moreover, N-glycosylation of all three sites increased the thermostability by ~3 °C. This gain in stability upon addition of multiple glycans was unknown for ptRNases.
Glycosylation of RNase 1 Enhances Proteolytic Stability.
Previous work had demonstrated that the glycosylation of RNase A56,58,59 and other proteins60–62 diminishes their susceptibility to degradation by proteases. We likewise found that all glycoforms of RNase 1 are resistant to proteolysis compared to the aglycosylated protein (Figure 5). Each pendant Man5GlcNAc2 heptasaccharide has a mass (1.2 kDa) that is nearly 10% that of RNase 1 (14.6 kDa), and is likely to be disordermobile. These glycans could shield the main chain from proteases.
Glycosylation in human homologs of RNase 1.
N-Glycosylation sequons exist in a majority of human ptRNases (Figure S9). The potential for extensive glycosylation is large in two homologs, RNase 2 (eosinophil-derived neurotoxin) and RNase 3 (eosinophil cationic protein), that play crucial roles in host-defense through antiviral and antimicrobial actions, respectively.1,2 Interestingly, the antimicrobial activity of RNase 3 is squelched by its N-glycosylation, which could thereby play a regulatory role.63 Amongst human ptRNases, however, the glycosylation sequons at Asn34, Asn76, and Asn88 are each unique features of RNase 1. These three sequons are conserved in chimpanzee, gibbon, gorilla, and presumably other hominids (Figure 2).
CONCLUSIONS
The N-glycosylation sequons of RNase 1 have been conserved across vertebrates, indicative of biochemical significance. We have reported on the first production and characterization of human RNase 1 glycoforms with all possible permutations of endogenous N-glycosylation. We find that the N-glycosylation of human RNase 1 improves its resistance to both thermal denaturation and proteolytic degradation but reduces its catalytic activity. Among the observed N-glycosylation sites, Asn34 has been conserved most widely across diverse groups of mammals and has displayed the highest level of glycan occupancy in a majority of biological samples. In accord, we find that only the N-glycosylation of Asn34 in human RNase 1 significantly improves overall protein stability while maintaining robust catalytic activity.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Robert G. Pressler, Jr. for early contributions to this work.
Funding
V.T.R. was supported by a William H. Peterson Fellowship in Biochemistry (Department of Biochemistry, University of Wisconsin–Madison). This work was supported by Grant R01 CA073808 (NIH).
ABBREVIATIONS
- DSF
differential scanning fluorimetry
- EDTA
ethylenediaminetetraacetic acid
- ESI
electrospray ionization
- 6-FAM
6-carboxyfluorescein
- GlcNAc
N-acetyl-d-glucosamine
- MALDI–TOF
matrix-assisted laser desorption-ionization–time-of-flight
- Man
d-mannose
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
- ptRNase
pancreatic-type ribonuclease
- RNase
ribonuclease
- 6-TAMRA
6-carboxytetramethylrhodamine
- SDS–PAGE
polyacrylamide gel electrophoresis performed in the presence of sodium dodecyl sulfate
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
Notes
The authors declare no competing financial interest.
UniProt Accession ID
Human RNase 1 P07998
ASSOCIATED CONTENT
Supporting Information
Figures S1–S9. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Sorrentino S (2010) The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins, FEBS Lett 584, 2194–2200. [DOI] [PubMed] [Google Scholar]
- (2).Lu L, Li J, Moussaoui M, and Boix E (2018) Immune modulation by human secreted RNases at the extracellular space, Front. Immunol 9, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kunitz M (1936) Isolation from beef pancreas of a crystalline protein possessing ribonuclease activity, Science 90, 112–113. [DOI] [PubMed] [Google Scholar]
- (4).Richards FM (1972) The 1972 Nobel prize for chemistry, Science 178, 492–493. [DOI] [PubMed] [Google Scholar]
- (5).Richards FM (1992) Linderstrøm-Lang and the Carlsberg Laboratory: The view of a postdoctoral fellow in 1954, Protein Sci 1, 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Raines RT (1998) Ribonuclease A, Chem. Rev 98, 1045–1066. [DOI] [PubMed] [Google Scholar]
- (7).Kresge N, Simoni RD, and Hill RL (2006) The thermodynamic hypothesis of protein folding: The work of Christian Anfinsen, J. Biol. Chem 281, e11. [Google Scholar]
- (8).Marshall GR, Feng JA, and Kuster DJ (2008) Back to the future: Ribonuclease A, Biopolymers 90, 259–277. [DOI] [PubMed] [Google Scholar]
- (9).Cuchillo CM, Nogués MV, and Raines RT (2011) Bovine pancreatic ribonuclease: Fifty years of the first enzymatic reaction mechanism, Biochemistry 50, 7835–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martin AJP, and Porter RR (1951) The chromatographic fractionation of ribonuclease, Biochem. J 49, 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hirs CHW, Moore S, and Stein WH (1952) A chromatographic investigation of pancreatic ribonuclease, J. Biol. Chem 200, 493–506. [PubMed] [Google Scholar]
- (12).Plummer TH Jr., and Hirs CHW (1963) The isolation of ribonuclease B, a glycoprotein, from bovine pancreatic juice, J. Biol. Chem 238, 1396–1401. [PubMed] [Google Scholar]
- (13).Plummer TH Jr. (1968) Glycoproteins of bovine pancreatic juice, J. Biol. Chem 243, 5961–5966. [PubMed] [Google Scholar]
- (14).Marshall RD (1974) The nature and metabolism of the carbohydrate-peptide linkage of glycoproteins, Biochem. Soc. Symp, 17–26. [PubMed]
- (15).Imperiali B, and Hendrickson TL (1995) Asparagine-linked glycosylation: Specificity and function of oligosaccharyl transferase, Bioorg. Med. Chem 3, 1565–1578. [DOI] [PubMed] [Google Scholar]
- (16).Aebi M, Bernasconi R, Clerc S, and Molinari M (2009) N-Glycan structures: Recognition and processing in the ER, Trends Biochem. Sci 35, 74–82. [DOI] [PubMed] [Google Scholar]
- (17).Levine ZG, and Walker S (2016) The biochemistry of O-GlcNAc transferase: Which functions make it essential in mammalian cells?, Annu. Rev. Biochem 85, 632–657. [DOI] [PubMed] [Google Scholar]
- (18).Bause E, and Hettkamp H (1979) Primary structural requirements for N-glycosylation of peptides in rat liver, FEBS Lett 108, 341–344. [DOI] [PubMed] [Google Scholar]
- (19).Morita T, Niwata Y, Ohgi K, Ogawa M, and Irie M (1986) Distribution of two urinary ribonuclease-like enzymes in human organs and body fluids, J. Biochem 99, 17–25. [DOI] [PubMed] [Google Scholar]
- (20).Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, and Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes, Proc. Natl. Acad. Sci. USA 101, 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Weickmann JL, Olson EM, and Glitz DG (1984) Immunological assay of pancreatic ribonuclease in serum as an indicator of pancreatic cancer, Cancer Res 44, 1682–1687. [PubMed] [Google Scholar]
- (22).Johnson RJ, McCoy JG, Bingman CA, Phillips GN Jr., and Raines RT (2007) Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein, J. Mol. Biol 367, 434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ribó M, Beintema JJ, Osset M, Fernández E, Bravo J, de Llorens R, and Cuchillo CM (1994) Heterogeneity in the glycosylation pattern of human pancreatic ribonuclease, Biol. Chem. Hoppe-Seyler 375, 357–363. [PubMed] [Google Scholar]
- (24).Beintema JJ, Blank A, Schieven GL, Dekker CA, Sorrentino S, and Libonati M (1988) Differences in glycosylation pattern of human secretory ribonucleases, Biochem. J 255, 501–505. [PMC free article] [PubMed] [Google Scholar]
- (25).Peracaula R, Royle L, Tabarés G, Mallorquí-Fernández G, Barrabés S, Harvey DJ, Dwek RA, Rudd PM, and de Llorens R (2003) Glycosylation of human pancreatic ribonuclease: Differences between normal and tumor states, Glycobiology 13, 227–244. [DOI] [PubMed] [Google Scholar]
- (26).Barrabés S, Pagès-Pons L, Radcliffe CM, Tabarés G, Fort E, Royle L, Harvey DJ, Moenner M, Dwek RA, Rudd PM, De Llorens R, and Peracaula R (2007) Glycosylation of serum ribonuclease 1 indicates a major endothelial origin and reveals an increase in core fucosylation in pancreatic cancer, Glycobiology 17, 388–400. [DOI] [PubMed] [Google Scholar]
- (27).Bravo J, Fernández E, Ribó M, de Llorens R, and Cuchillo CM (1994) A versatile negative-staining ribonuclease zymogram, Anal. Biochem 219, 82–86. [DOI] [PubMed] [Google Scholar]
- (28).Petrescu AJ, Milac A-L, Petrescu SM, Dwek RA, and Wormald MR (2004) Statistical analysis of the protein environment of N-glycosylation sites: Implications for occupancy, structure, and folding, Glycobiology 14, 103–114. [DOI] [PubMed] [Google Scholar]
- (29).Beintema JJ (1987) Structure, properties and molecular evolution of pancreatic-type ribonucleases, Life Chem. Rep 4, 333–389. [Google Scholar]
- (30).Lomax JE, Eller CH, and Raines RT (2017) Comparative functional analysis of ribonuclease 1 homologs: Molecular insights into evolving vertebrate physiology, Biochem. J 474, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Beintema JJ, and Lenstra JA (1982) Evolution of mammalian pancreatic ribonucleases, In Macromolecular Sequences in Systematic and Evolutionary Biology (Goodman M, Ed.), pp 43–73, Plenum, New York. [Google Scholar]
- (32).Beintema JJ, Breukelman HJ, Carsana A, and Furia A (1997) Evolution of vertebrate ribonucleases: Ribonuclease A superfamily, In Ribonucleases: Structures and Functions (D’Alessio G, and Riordan JF, Eds.), pp 245–269, Academic Press, New York. [Google Scholar]
- (33).Eller CH, Lomax JE, and Raines RT (2014) Bovine brain ribonuclease is the functional homolog of human ribonuclease 1, J. Biol. Chem 289, 25996–26006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Russo N, de Nigris M, Ciardiello A, Di Donato A, and D’Alessio G (1993) Expression in mammalian cells, purification and characterization of recombinant human pancreatic ribonuclease, FEBS Lett 333, 233–237. [DOI] [PubMed] [Google Scholar]
- (35).Ribó M, delCardayré SB, Raines RT, de Llorens R, and Cuchillo CM (1996) Production of human pancreatic ribonuclease in Saccharomyces cerevisiae and Escherichia coli, Protein Express. Purific 7, 253–261. [DOI] [PubMed] [Google Scholar]
- (36).Brooks SA (2009) Strategies for analysis of the glycosylation of proteins: Current status and future perspectives, Mol. Biotechnol 43, 76–88. [DOI] [PubMed] [Google Scholar]
- (37).Vervecken W, Callewaert N, Kaigorodov V, Geysens S, and Contreras R (2007) Modification of the N-glycosylation pathway to produce homogeneous, human-like glycans using GlycoSwitch plasmids, In Pichia Protocols (Cregg JM, Ed.), pp 119–138, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- (38).Jacobs PP, Geysens S, Vervecken W, Contreras R, and Callewaert N (2009) Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology, Nat. Protoc 4, 58–70. [DOI] [PubMed] [Google Scholar]
- (39).Niesen FH, Berglund H, and Vadadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability, Nat. Protoc 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (40).Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases, Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (41).Thomas SP, Kim E, Kim J-S, and Raines RT (2016) Knockout of the ribonuclease inhibitor gene leaves human cells vulnerable to secretory ribonucleases, Biochemistry 55, 6359–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kelemen BR, Klink TA, Behlke MA, Eubanks SR, Leland PA, and Raines RT (1999) Hypersensitive substrate for ribonucleases, Nucleic Acids Res 27, 3696–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Park C, Kelemen BR, Klink TA, Sweeney RY, Behlke MA, Eubanks SR, and Raines RT (2001) Fast, facile, hypersensitive assays for ribonucleolytic activity, Methods Enzymol 341, 81–94. [DOI] [PubMed] [Google Scholar]
- (44).Bourgoin-Voillard S, Leymarie N, and Costello CE (2014) Top-down tandem mass spectrometry on RNase A and B using a Qh/FT-ICR hybrid mass spectrometer, Proteomics 14, 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kyriakis JM (2014) In the beginning, there was protein phosphorylation, J. Biol. Chem 289, 9460–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, Xiao J, Grishin NV, and Dixon JE (2012) Secreted kinase phosphorylates extracellular proteins that regulate biomineralization, Science 336, 1150–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Libonati M, and Sorrentino S (2001) Degradation of double-stranded RNA by mammalian pancreatic-type ribonucleases, Methods Enzymol 341, 234–248. [DOI] [PubMed] [Google Scholar]
- (48).Rudd PM, Joao HC, Coghill E, Fiten P, Saunders MR, Opdenakker G, and Dwek RA (1994) Glycoforms modify the dynamic stability and functional activity of an enzyme, Biochemistry 33, 17–22. [DOI] [PubMed] [Google Scholar]
- (49).Kim B-M, Kim H, Raines RT, and Lee Y (2004) Glycosylation of onconase increases its conformational stability and toxicity for cancer cells, Biochem. Biophys. Res. Commun 315, 976–983. [DOI] [PubMed] [Google Scholar]
- (50).Goettig P (2016) Effects of glycosylation on the enzymatic activity and mechanisms of proteases, Int. J. Mol. Sci 17, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Nogués MV, Moussaoui M, Boix E, Vilanova M, Ribó M, and Cuchillo CM (1998) The contribution of noncatalytic phosphate-binding subsites to the mechanism of bovine pancreatic ribonuclease A, Cell. Mol. Life Sci 54, 766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fisher BM, Ha J-H, and Raines RT (1998) Coulombic forces in protein-RNA interactions: Binding and cleavage by ribonuclease A and variants at Lys7, Arg10 and Lys66, Biochemistry 37, 12121–12132. [DOI] [PubMed] [Google Scholar]
- (53).Fisher BM, Grilley JE, and Raines RT (1998) A new remote subsite in ribonuclease A, J. Biol. Chem 273, 34134–34138. [DOI] [PubMed] [Google Scholar]
- (54).Joao HC, and Dwek RA (1993) Effects of glycosylation on protein structure and dynamics in ribonuclease B and some of its individual glycoforms, Eur. J. Biochem 218, 239–244. [DOI] [PubMed] [Google Scholar]
- (55).Klink TA, Woycechowsky KJ, Taylor KM, and Raines RT (2001) Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A, Eur. J. Biochem 267, 566–572. [DOI] [PubMed] [Google Scholar]
- (56).Arnold U, and Ulbrich-Hofmann R (1997) Kinetic and thermodynamic thermal stabilities of ribonuclease A and ribonuclease B, Biochemistry 36, 2166–2172. [DOI] [PubMed] [Google Scholar]
- (57).Joao HC, Scragg IG, and Dwek RA (1992) Effects of glycosylation on protein conformation and amide proton exchange in RNase B, FEBS Lett 307, 343–346. [DOI] [PubMed] [Google Scholar]
- (58).Arnold U, Schierhorn A, and Ulbrich-Hofmann R (1998) Influence of the carbohydrate moiety on the proteolytic cleavage sites in ribonuclease B, J. Protein Chem 17, 397–405. [DOI] [PubMed] [Google Scholar]
- (59).Arnold U, Schierhorn A, and Ulbrich-Hofmann R (1999) Modification of the unfolding region in bovine pancreatic ribonuclease and its influence on the thermal stability and proteolytic fragmentation, Eur. J. Biochem 259, 470–475. [DOI] [PubMed] [Google Scholar]
- (60).Mitra N, Sinha S, Ramya TN, and Surolia A (2006) N-Linked oligosaccharides as outfitters for glycoprotein folding, form and function, Trends Biochem. Sci 31, 156–163. [DOI] [PubMed] [Google Scholar]
- (61).Solá RJ, and Griebenow KAI (2009) Effects of glycosylation on the stability of protein pharmaceuticals, J. Pharm. Sci 98, 1223–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Russell D, Oldham NJ, and Davis BG (2009) Site-selective chemical protein glycosylation protects from autolysis and proteolytic degradation, Carbohydr. Res 344, 1508–1514. [DOI] [PubMed] [Google Scholar]
- (63).Rubin J, and Venge P (2013) Asparagine-linked glycans determine the cytotoxic capacity of eosinophil cationic protein (ECP), Mol. Immunol 55, 372–380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.