

Abstract
Oxaliplatin is important for the clinical treatment of colorectal cancer and other gastrointestinal malignancies, but tumour resistance is limiting. Several oxaliplatin transporters were previously identified but their relative contributions to determining oxaliplatin tumour responses and gastrointestinal tumour cell sensitivity to oxaliplatin remains unclear. We studied clinical associations between tumour expression of oxaliplatin transporter candidate genes and patient response to oxaliplatin, then experimentally verified associations found with MRP2 in models of human gastrointestinal cancer. Among 18 oxaliplatin transporter candidate genes, MRP2 was the only one to be differentially expressed in the tumours of colorectal cancer patients who did or did not respond to FOLFOX chemotherapy. Over-expression of MRP2 (endogenously in HepG2 and PANC-1 cells, or induced by stable transfection of HEK293 cells) decreased oxaliplatin accumulation and cytotoxicity but those deficits were reversed by inhibition of MRP2 with myricetin or siRNA knockdown. Mice bearing subcutaneous HepG2 tumour xenografts were sensitised to oxaliplatin antitumour activity by concurrent myricetin treatment with little or no increase in toxicity. In conclusion, MRP2 limits oxaliplatin accumulation and response in human gastrointestinal cancer. Screening tumour MRP2 expression levels, to select patients for treatment with oxaliplatin-based chemotherapy alone or in combination with a MRP2 inhibitor, could improve treatment outcomes.
Introduction
Chemotherapy with the platinum-based drug oxaliplatin is of major importance for the clinical treatment of colorectal cancer and other gastrointestinal malignancies. Colorectal cancer and the other gastrointestinal malignancies treatable by oxaliplatin-based chemotherapy are among the most common cancer types and causes of cancer death in the world today1. Robust clinical evidence of the efficacy of oxaliplatin-based chemotherapy from well-designed randomised controlled trials have shown improved patient outcomes in colorectal cancer, both in the adjuvant2 and metastatic settings3,4, and in pancreatic5,6, oesophagogastric7,8 and hepatocellular9 cancer. Although oxaliplatin-based chemotherapy has been widely adopted as the standard and preferred chemotherapy regimen for treating many types of gastrointestinal cancer10,11, its toxicity and resistance are major clinical limitations.
Oxaliplatin must cross cell membranes before causing cytotoxicity in tumour cells by reacting with DNA and forming DNA–platinum adducts that induce cell cycle arrest and cell death12. Oxaliplatin’s inherent capacity for crossing cell membranes by passive diffusion may be limited by its hydrophilicity13,14 and chemical transformation into charged intermediates in biological fluids15. Over the last decade, evidence has accumulated for membrane transporter proteins controlling the movement of oxaliplatin into and out of cells16. Several membrane transporter proteins from the ATP binding cassette (ABC), solute carrier (SLC) and ATPase membrane protein superfamilies have been reported to transport oxaliplatin in vitro, in tumour lines selected for oxaliplatin resistance or in cells genetically modified to alter expression of individual membrane transporter genes (Table 1). However, the relative contributions of specific membrane transporters to determining gastrointestinal tumour clinical responses to oxaliplatin-based chemotherapy remains unclear.
Table 1.
Oxaliplatin transporter candidate genes. Adapted and updated from16. References are the original reports of oxaliplatin transport by each transporter.
Multidrug resistance-associated protein 2 (MRP2) is a 190 kDa glycoprotein encoded by the ABCC2 gene, which functions to transport a range of substrates across cell membranes using energy derived from ATP hydrolysis17. MRP2 is highly expressed in the normal gastrointestinal system, for example, on the apical membranes of colonic enterocytes and biliary canalicular membranes of hepatocytes, where it functions in the excretion of substances into the gut lumen and bile17. Some tumour cells also express MRP2, including colorectal, hepatocellular and other gastrointestinal cancer cells, in which MRP2 can confer multidrug resistance by virtue of its function as a poly-specific drug efflux pump17. Earlier work established MRP2 as an efflux transporter of cisplatin and mediator of cisplatin resistance18–22. However, there have been few studies of the influence of MRP2 in oxaliplatin therapy of gastrointestinal cancer23–26 despite its major therapeutic role in this clinical setting.
With this background, we carried out the study described here with the aim of identifying membrane transporter proteins that determine clinical sensitivity of human gastrointestinal cancer to oxaliplatin. First, we examined clinical associations between the tumour expression of oxaliplatin transporter candidate genes and patient response to oxaliplatin-based chemotherapy. Then, we experimentally verified the major clinical association found with MRP2 in models of human gastrointestinal cancer. In these in vitro and in vivo experimental systems, the expression and activity of MRP2 was manipulated by siRNA gene knockdown and pharmacological inhibition with a model compound (myricetin)27,28 that had low potential for reaction with platinum compounds.
Results
Clinical association
MRP2 was significantly overexpressed in the colorectal tumours of patients who did not respond to oxaliplatin chemotherapy. We searched the Oncomine transcriptome database for datasets of patients treated with oxaliplatin, who had tumour microarray gene expression profiling undertaken before treatment and annotation of their subsequent tumour response. Only one dataset was found, the Tsuji Colorectal dataset29 (GDS4393 and GDS4396) comprising of 83 patients with metastatic colorectal cancer who had tumour microarray gene expression profiling before treatment with FOLFOX. Patients were stratified into FOLFOX responders (n = 42) or non-responders (n = 41). Differences between the two groups in the expression of reporters of each oxaliplatin transporter candidate gene (Table 1) were calculated. Only one of 18 oxaliplatin transporter candidate genes showed significantly different expression. MRP2 (ABCC2) was overexpressed (1.5-fold) in the tumours of patients who did not respond (P < 0.0001 Bonferroni post-test following Two-Way ANOVA; Two-way ANOVA Factors: Tumour response, P = 0.0025; Gene, P < 0.0001; Interaction, P = 0014) (Fig. 1). The tumour expression of the other 17 oxaliplatin transporter candidate genes showed no significant difference (<−0.4- to 0.8-fold) between responding and non-responding patients.
Figure 1.
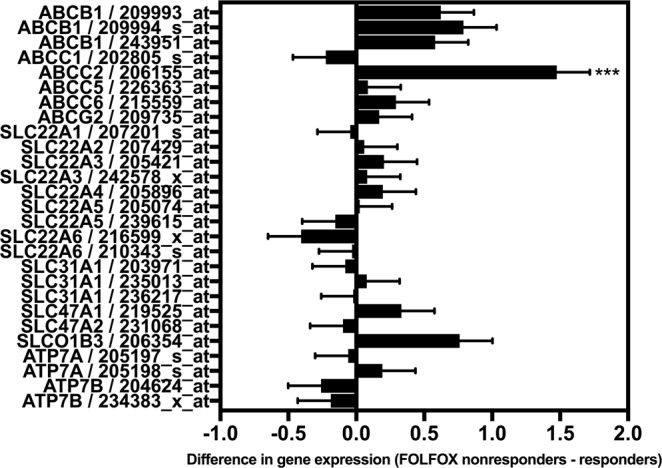
Differential expression of oxaliplatin transporter candidate genes in colorectal tumours of patients who responded or did not respond to FOLFOX chemotherapy. Data from the Tsuji Colorectal dataset29 from Oncomine included 83 colorectal cancer patients who had tumour microarray gene expression analysis prior to FOLFOX chemotherapy then stratification by tumour response. Bars and error bars are means and standard errors of differences in the expression of individual oxaliplatin transporter candidate genes between responding and non-responding patients. Positive differences indicate higher expression in non-responders whereas negative differences indicate higher expression in responders. The asterisk is a P value (***P < 0.0001) from a Bonferroni post-test that followed a Two-way ANOVA (Two-way ANOVA Factors: Tumour response, P = 0.0025; Gene, P < 0.0001; Interaction, P = 0014). ABCC2/MRP2 was over-expressed in colorectal tumours from patients who did not respond to FOLFOX chemotherapy compared to responders. Other oxaliplatin transporter candidate genes were expressed similarly in both responding and non-responding patients.
In vitro studies
In an isogenic pair of HEK293 cell lines, stable overexpression of MRP2 (HEK-MRP2 cells) decreased oxaliplatin accumulation and cytotoxicity but those deficits were reversed by inhibition of MRP2 with myricetin. Immunofluorescence confocal microscopy detected MRP2 protein localised to the plasma membranes of HEK-MRP2 cells but negligible immunoreactivity in parental HEK cells (HEK-P cells) (Fig. 2A). Compared to HEK-P cells, HEK-MRP2 cells accumulated up to 3-fold less platinum during exposure to oxaliplatin for up to 2 hours (Fig. 2C,D). Concurrent exposure to oxaliplatin with myricetin increased the accumulation of platinum in HEK-MRP2 cells by about 3-fold (P < 0.001) but did not change the accumulation of platinum in HEK-P cells (Fig. 2E). IC50 values for oxaliplatin-induced growth inhibition were increased in HEK-MRP2 cells by about 2-fold compared to HEK-P cells (Fig. 2B,F). Concurrent exposure to oxaliplatin with myricetin increased the sensitivity of HEK-MRP2 cells to oxaliplatin-induced growth inhibition but had no effect on HEK-P cells (Fig. 2F).
Figure 2.

Oxaliplatin accumulation and cytotoxicity in HEK293 cells stably transfected to overexpress MRP2 (HEK-MRP2 cells) compared to control cells (HEK-P cells). (A) MRP2 was localised to the plasma membranes of HEK-MRP2 cells as demonstrated by MRP2-immunofluorescence confocal microscopy. Red staining, MRP2; Blue staining, DAPI. (B) Representative inhibition of growth of HEK-MRP2 and HEK-P cells after 2-hour exposure to oxaliplatin. Symbols are means and standard errors of the mean [n = 4]. Solid lines are non-linear regression fits (Y = Bottom + (Top − Bottom)/(1 + 10((LogIC50 − X)*HillSlope))) to the data (r2 > 0.97). (C) Time-course of platinum accumulation during exposure to oxaliplatin for up to two hours showing lower accumulation of platinum in HEK-MRP2 cells. Asterisks are P values (***P < 0.001; ****P < 0.0001) for differences at each time point from Bonferroni post-tests that followed a Two-way ANOVA [n = 6]. (D) Kinetics of platinum accumulation during exposure to oxaliplatin for up to two hours showing a plateau in levels after one-hour exposure. Solid lines represent non-linear regression fits (y = ymax(1− e−kx) to the data (r2 > 0.92). (E,F) Effect of myricetin (60 μM) on oxaliplatin accumulation [n = 3] (E) and cytotoxicity [n = 3–4] (F). Asterisks are P values (***P < 0.001; ****P < 0.0001) for differences with each of the other three groups from Bonferroni post-tests that followed a Two-way ANOVA. Bars represent means and standard errors of the mean. HEK-MRP2 cells accumulated less platinum and were less sensitive to oxaliplatin-induced growth inhibition than control HEK-P cells. Myricetin increased the accumulation and cytotoxicity of oxaliplatin in HEK-MRP2 cells but not in HEK-P cells.
To identify clinically relevant experimental models, two human gastrointestinal tumour lines (HepG2 and PANC-1) were identified as having MRP2-mediated deficits in oxaliplatin accumulation reversed by inhibition of MRP2 with myricetin or siRNA ABCC2 gene knockdown. Like HEK-MRP2 cells, HepG2 and PANC1 cells accumulated less platinum than HEK-P cells during in vitro exposure to oxaliplatin (50 μM) for two hours (Fig. 3A). In contrast, other human gastrointestinal cancer lines (HCT116, MiaPACA-2, WiDr, SW620, and HT29) accumulated similar levels of platinum as HEK-P cells but more than by HEK-MRP2, HepG2 and PANC-1 cells (Fig. 3A). HepG2 cells accumulated less model MRP2 substrate (5(6)-carboxy-2,′7′-dichlorofluorescein (CDCF)) than HEK-P cells, as did the HEK-MRP2 cells (Fig. 3B). Real-time PCR array analysis of transporter gene expression revealed higher expression of MRP2 and lower expression of OCT2 (SLC22A2) in tumour lines with reduced accumulation of oxaliplatin (HepG2, PANC-1, HEK-MRP2) (Fig. 3C). Concurrent exposure to oxaliplatin with myricetin increased the accumulation and cytotoxicity of oxaliplatin, by 1.9 to 5.4-fold and 3.4 to 4.0-fold, respectively, in both HepG2 and PANC-1 cells (Fig. 3D,E).
Figure 3.

Identification of human gastrointestinal tumour lines with MRP2-mediated deficits in oxaliplatin accumulation. (A) Platinum accumulation after exposure to oxaliplatin (50 μM) for 2 h showing lower platinum accumulation in HepG2 and PANC-1 cells (open bars) compared to other tumour lines (closed bars) [n = 4]. (B) CDCF accumulation in HEK-P, HEK-MRP2 and HepG2 cells. Asterisks are P values (*P < 0.05; **P < 0.01) from Bonferroni post-tests that followed One-way ANOVA for comparisons to HEK-P cells [n = 3]; (C) Differential expression of oxaliplatin transporter candidate genes between tumour lines with high (6 cell lines) versus low (3 cell lines) oxaliplatin accumulation (closed versus open bars from panel (A)) determined by transporter real-time PCR array analysis. Positive differences indicate higher expression in lines with lower oxaliplatin accumulation whereas negative differences indicate higher expression in lines with higher oxaliplatin accumulation. Asterisks are P values (*P < 0.05; **P < 0.01) from Bonferroni post-tests that followed Two-way ANOVA (Two-way ANOVA factors: Gene P < 0.0001; Platinum accumulation low versus high, NS; Interaction, NS) [n = 3 replicates per cell line]. (D,E) Effect of myricetin (60 μM) on the accumulation (D) [n = 4–5] and cytotoxicity (E) [n = 3] of oxaliplatin in HepG2 and PANC1 cells. Asterisks are P values (**P < 0.01; ***P < 0.001; ****P < 0.0001) from Bonferroni post-tests that followed Two-way ANOVA. HepG2 and PANC-2 cells accumulated less oxaliplatin compared to other GI tumour lines. Inhibition of MRP2 by myricetin increased the accumulation and cytotoxicity of oxaliplatin in HepG2 and PANC-1 cells. Bars are the means and standard errors of the mean.
MRP2 gene knockdown significantly increased the cellular accumulation of platinum and enhanced the sensitivity to oxaliplatin in HepG2 cells. HepG2 cells were transfected with three independent MRP2-siRNAs and negative control siRNA using Lipofectamine RNAiMAX. The expression of MRP2 mRNA was significantly decreased in MRP2-siRNA transfected HepG2 cells compared with controls (Fig. 4A). Silencing MRP2 led to increase of cellular accumulation of an MRP2 substrate, CDCF by 160% to 170% (Fig. 4B). The cellular platinum accumulation after 2 h exposure to 25 µM oxaliplatin in control HepG2 cells was 40.9 ± 5.9 pmol per mg protein. Compared with the controls, platinum accumulation in siRNA treated HepG2 cells was significantly higher (117 ± 5.3 (P < 0.001), 62.4 ± 1.0 (P < 0.05), 104.7 ± 2.9 (P < 0.001) pmol pt/mg protein in siRNA-1, siRNA-2 and siRNA-3; respectively) (Fig. 4C). IC50 values against oxaliplatin were 2- to 5-fold more potent in HepG2 cells transfected with control siRNA compared with those transfected with MRP2-siRNAs (Fig. 4D).
Figure 4.

Effect of ABCC2 siRNA gene knockdown on MRP2 mRNA expression (A), CDCF accumulation (B), oxaliplatin accumulation (C) and oxaliplatin-induced growth inhibition (D) in HepG2 cells. Asterisks are P values (*P < 0.05; **P < 0.01; **P < 0.001) from Bonferroni post-tests that followed one-way ANOVA. Inhibition of MRP2 by siRNA gene knockdown increased the accumulation and cytotoxicity of oxaliplatin in HepG2 cells. Bars are the means and standard errors of the mean (n = 3).
In vivo study
Mice bearing subcutaneous HepG2 tumour xenografts were sensitised to the in vivo antitumour activity of oxaliplatin by concurrent treatment with myricetin with little or no increase in toxicity. Nude mice (NIH-III) were implanted subcutaneously with HepG2 cells, which were demonstrated to have MRP2-mediated deficits in oxaliplatin accumulation in vitro. Tumour bearing mice were treated with oxaliplatin (3 mg/kg ip), myricetin (25 mg/kg iv) or their respective vehicles alone or in combination (n = 8/group), once weekly for five weeks, starting from when tumours measured about 200 mm3 (range, 161 to 237 mm3). The body weight of mice was reduced by oxaliplatin treatment when given alone or in combination with myricetin, compared to drug vehicle or myricetin alone (Repeated Measures Two-Way ANOVA Factors: Time, NS; Treatment group, P < 0.0001; Interaction, NS; Subjects, P < 0.0001) (Fig. 5A and Supplementary Table 1). Growth of HepG2 tumour xenografts was slowed by treatment with oxaliplatin combined with myricetin compared to treatment with drug vehicle alone, oxaliplatin alone or myricetin alone (Repeated Measures Two-Way ANOVA Factors: Time, P < 0.0001; Treatment group, P = 0.002; Interaction, P < 0.0001; Subjects, P < 0.0001)(Fig. 5B and Supplementary Table 2). The time taken for HepG2 tumours to quadruple in size was prolonged by treatment with oxaliplatin combined with myricetin (median, 32 days) compared to drug vehicle alone (median, 22.5 days; P = 0.0454), oxaliplatin alone (median, 17 days; P = 0.0016) or myricetin alone (median, 17 days; P = 0.015)(P value for comparison of all groups = 0.0112) (Fig. 5C and Supplementary Table 3). The time taken to reach the endpoint of euthanasia was prolonged by treatment with oxaliplatin combined with myricetin (median, 47 days) compared to drug vehicle alone (median, 33.5 days; P = 0.0968), oxaliplatin alone (median, 28 days; P = 0.0003) or myricetin alone (median, 17 days; P = 0.0181)(P value for comparison of all groups = 0.0032) (Fig. 5D and Supplementary Table 3).
Figure 5.

In vivo sensitisation to oxaliplatin antitumour activity by inhibition of MRP2 with myricetin. Nude mice (NIH-III) were implanted subcutaneously with HepG2 cells, which showed MRP2-mediated deficits in oxaliplatin accumulation in vitro. Tumour bearing mice (8 per group) were then treated with oxaliplatin (3 mg/kg ip), myricetin (25 mg/kg iv) or matching drug vehicle, alone or in combination once weekly for five weeks starting when tumours measured approximately 200 mm3. Arrowheads are the time of treatment. Experimental endpoints measured during and after treatment included body weight (A), tumour volume (B), time for tumours to quadruple in volume from baseline (C), and time to euthanasia from the start of treatment (D). Asterisks shown in panel B are P values (***P < 0.002; ****P < 0.0001) for differences between the oxaliplatin alone and oxaliplatin plus myricetin treatment groups at each time point from Bonferroni post-tests that followed Two-way Repeated Measures ANOVA. P values shown in panels C and D are for comparisons of all treatment groups. Oxaliplatin reduced body weight but had no antitumour activity when given alone. When combined with myricetin, oxaliplatin slowed tumour growth and extended the survival of animals with little or no increase in toxicity.
Discussion
Oxaliplatin and its combination regimens are of major importance in the clinical treatment of colorectal cancer and other gastrointestinal malignancies, but some patients fail to respond to treatment. National and international clinical practice guidelines10,11 now recommend oxaliplatin-based chemotherapy as a preferred regimen for the treatment of colon, gastric, oesophageal, pancreatic, rectal and other types of gastrointestinal cancer based on robust evidence from multiple randomised controlled trials2–9. However, in some patients the best tumour response is progressive disease and in others tumour responses are short-lived. Currently it is not possible to prospectively identify individual patients who are destined to fail oxaliplatin therapy prior to their exposure to treatment and observation of their unsatisfactory clinical outcomes. Moreover, there is an urgent need to identify alternative treatments for patients who will have such poor outcomes from standard oxaliplatin-based chemotherapy.
In the current study, we demonstrated a significant clinical association between tumour expression of the oxaliplatin transporter MRP2 and lack of responsiveness of colorectal cancer patients to standard oxaliplatin-based chemotherapy (FOLFOX). In contrast, tumour expression of 17 other oxaliplatin transporter candidate genes showed no significant association with clinical response to oxaliplatin in this dataset. Unfortunately, only one dataset was available in Oncomine of gastrointestinal cancer patients with tumour gene expression profiling undertaken before treatment with oxaliplatin together with annotation of their subsequent tumour response. Moreover, in this dataset, only steady state MRP2 mRNA levels were measured, not MRP2 protein level or function, which may be altered by posttranslational modification, such as by glycosylation30,31. However, these data we now report add to an increasing number of independent studies reporting clinical associations between gastrointestinal tumour expression of MRP2 and non-responsiveness to chemotherapy with oxaliplatin or cisplatin23,32–35. Taken together, these findings suggest that MRP2 may cause clinical resistance to oxaliplatin in gastrointestinal cancer patients, but ultimate proof of a causal association will require generation and interrogation of other tumour gene expression clinical datasets along with prospective clinical trials evaluating the inhibition of MRP2 in combination with oxaliplatin in patients whose tumours overexpress MRP2.
Our in vitro experimental studies reported here provided insights into mechanisms underlying poor responses to oxaliplatin in gastrointestinal cancer. The mechanism identified here involves MRP2 actively transporting oxaliplatin and/or other cytotoxic platinum species derived from oxaliplatin, resulting in their extrusion from tumour cells, net reduction in cellular platinum accumulation and decreased inhibitory effects of oxaliplatin on cell growth. Previously, we demonstrated that MRP2 actively transports oxaliplatin-derived platinum in MRP2-expressing membrane vesicles36. Intact oxaliplatin and/or its anionic monochloro oxalate ring-opened early degradation product were identified in these studies as likely substrates for MRP2. Moreover, the active transport of oxaliplatin was shown to be inhibited by myricetin36. In the current study, we extended these findings to show lower accumulation of platinum and sensitivity to oxaliplatin-induced growth inhibition in HEK293 cells stably transfected to overexpress MRP2, and in human gastrointestinal tumour lines (HepG2 and PANC-1) with endogenous overexpression of MRP2. Inhibition of MRP2 by myricetin or siRNA knockdown increased the accumulation of platinum and sensitivity to oxaliplatin-induced growth inhibition in these MRP2 overexpressing tumour lines. None of the four colorectal cancer cell lines we studied displayed low oxaliplatin accumulation due to MRP2 overexpression but a hepatocellular (HepG2)37 and a pancreatic carcinoma (PANC-1) line did. This created an incomplete match in the data presented here between the clinical-gene expression dataset of colorectal cancer patients (Fig. 1) and our experimental work that mainly focussed on the HepG237 and PANC-1 lines that displayed MRP2-mediated transport-resistance to oxaliplatin in vitro (Fig. 3). This incomplete match in the presented data was further compounded by no tumour gene expression clinical datasets of hepatocellular or pancreatic carcinoma patients with annotated tumour response to oxaliplatin being available in Oncomine. However, previous studies have found increased MRP2 expression and activity in oxaliplatin-resistant colorectal cancer lines in vitro, and that inhibition of MRP2 expression and activity by genetic knockdown, myricetin or other inhibitors, can reverse MRP2-mediated accumulation deficits and resistance to oxaliplatin in colorectal cancer24–26.
This study also provided in vivo experimental proof of the therapeutic concept that targeting MRP2 could sensitise MRP2-overexpressing human gastrointestinal tumours to the anti-tumour activity of oxaliplatin without increased toxicity. In mice bearing subcutaneous xenografts of the HepG2 tumour line, which had shown MRP2-mediated deficits in oxaliplatin accumulation in vitro, oxaliplatin treatment given alone reduced body weight but had no significant antitumour activity. In contrast, when combined with myricetin, oxaliplatin treatment slowed tumour growth and extended the survival of animals without significant increase in toxicity. However, toxicity was measured only by body weight and the major clinical toxicity of oxaliplatin, neurotoxicity could have been increased by inhibition of MRP2. Further studies will be required to determine if MRP2 inhibition or genetic knockout increases oxaliplatin neurotoxicity, for example, using histomorphometric endpoints (eg. dorsal root ganglion neuron cell body size) of oxaliplatin neurotoxicity38,39. While other mechanisms cannot be ruled out26,40–42, it is reasonable on the basis of the findings reported here to hypothesise that myricetin sensitises tumours to oxaliplatin antitumour activity by inhibiting oxaliplatin transport mediated by MRP2. In another recent study of an animal model of human gastrointestinal cancer, myricetin enhanced the in vivo antitumour activity of 5-fluorouracil43, which is often given with oxaliplatin in combination regimens such as FOLFOX. These findings now set the scene for further exploration of the therapeutic concept of targeting MRP2 with myricetin or other inhibitors to enhance the antitumour activity of oxaliplatin-based combination regimens, both experimentally and in the clinic.
In conclusion, MRP2 limits oxaliplatin accumulation and responses in gastrointestinal cancer. As tumour MRP2 expression is both readily measurable and potentially targetable in gastrointestinal cancer patients, screening MRP2 tumour expression to select patients for treatment with oxaliplatin alone or in combination with an MRP2 inhibitor could improve their outcomes.
Materials and Methods
Oncomine dataset and analysis
Log2 median-centred intensity values for the tumour expression of eighteen oxaliplatin transporter candidate genes from 83 patients in the Tsuji Colorectal dataset (GDS4393 and GDS4396)29 were exported from Oncomine. Patients were stratified according to their response to FOLFOX chemotherapy. Differences between the responder (n = 42) and non-responder (n = 41) groups were analysed by Two-way ANOVA (Two-way ANOVA factors: transporter gene expression and tumour response to FOLFOX) followed by Bonferroni post-tests for each candidate gene. Data were assessed visually in graphs of absolute differences and standard errors of differences in mean log2 median-centred intensity values for each gene between the responder and non-responder groups.
Drugs and Reagents
Oxaliplatin (Actavis, New Zealand) solution at 5 mg/mL was prepared by dissolving 100 mg powder into 20 mL MilliQ water or 5% glucose. The stock solutions were stored frozen at −20 °C. One month after preparation, the stock solutions were discarded. The cell culture media RPMI 1640, Opti-MEM and stealth RNAi siRNA were from Invitrogen, and Dulbecco’s Modified Eagle Medium (DMEM), and foetal bovine serum (FBS) were from Life Technologies (Auckland, NZ). Myricetin was from Cayman Chem (MI, USA). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell Lines and Tissue Culture
Two isogenic HEK293 (Human Embryonic Kidney-293) sublines, one parental line (HEK-P) and another transfected to stably over-express MRP2 (HEK-MRP2), were kindly provided by Professor Piet Borst (Division of Molecular Biology and Centre for Biomedical Genetics, the Netherlands Cancer Institute, Amsterdam, Netherlands). Apart from the WiDR line that was from Dr Martin Ford (Glaxo-Wellcome, Stevenage, UK), the cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were grown and maintained in Dulbecco’s Modified Essential Medium (DMEM), supplemented with 10% FBS and 100 units of penicillin/streptomycin per ml, at 37 °C under 5% CO2 with 95% humidified air, except for the HepG2 cell line that was grown in Roswell Pack Memorial Institute (RPMI)−1640 or Eagle’s Minimum Essential Medium (MEM) supplemented with 10% (v/v) foetal bovine serum, 2 mmol/L L-glutamine, 100 units/mL penicillin and 100 units/mL streptomycin, in a humidified atmosphere of 5% carbon dioxide at 37 °C. The authenticity of the cell lines was confirmed by short tandem repeat DNA profiling (DNA Diagnostics, Auckland, New Zealand).
MRP2 Fluorescence Immunocytochemistry
Fluorescent immunocytochemistry was undertaken to detect cellular MRP2 protein expression in the HEK-293 sublines using a primary antibody, anti-MRP2 (1:100; ab3373 from Abcam), and a secondary antibody, Alexa Fluor 594-labeled anti-mouse IgG (Invitrogen, Carlsbad, CA, USA). Cells were cultured in the chamber slides until they reached the confluence of 60–70%. The growth medium was then replaced with pre-warmed PBS which was left for 5 min followed by fixation of cells with 4% paraformaldehyde for 15 min at room temperature. Then, cells were permeabilised with 0.2% Triton X-100 in PBS for 15 min. Blocking of the non-specific protein binding was achieved by incubating cells with blocking solution (PBS containing 0.2% Triton X-100, 3% goat serum, and 2% bovine serum albumin) for 1 h at room temperature. After blocking, cells were incubated with the primary antibody, anti-MRP2 (ab3373 from Abcam) prepared in immunobuffer (PBS containing 0.2% Triton X-100 and 3% goat serum) with the dilution ratio of 1:100 for overnight at 4 °C. After that, the cells were washed for 3–4 times with PBS containing 0.2% Triton X-100 followed by incubation of cells with the secondary antibody (Alexa Fluor 594-labeled anti-mouse IgG, Invitrogen, Carlsbad, CA, USA) diluted in the immunobuffer (dilution ratio was 1:500) for 3 h at 4 °C with the cover to protect from light. Cells were then washed with PBS and cover-slipped with VECTASHIELD Mounting Medium with DAPI (6-diamidino-2-phenylindole) to stain nucleic acid (Vector Laboratories, Burlingame, CA, USA). Images were captured using an Olympus FV1000 confocal laser scanning microscope (Olympus Inc., Tokyo) attached to a Nikon digital camera and then analysed using Nikon Elipse Net (Nikon, Melville, NY, USA) and ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Platinum accumulation
Inductively coupled plasma mass spectrometry (ICP-MS) was used to measure the cellular content of platinum after exposure to oxaliplatin. For these experiments, cells were seeded at 250,000 cells per well (350,000 cells per well for HepG2 and PANC-1) in collagen-coated six-well plates, and grown in the normal growth medium until around 80% confluent. Cells were then incubated with oxaliplatin at designated concentrations and treatment durations. At the end of the treatment period, cells were washed three times with 1 mL of ice-cold PBS (phosphate buffered saline) then dried in room air for 30 min. Then 330 μL 70% nitric acid was added to each well and the plate gently shaken at room temperature for 2 hours to digest the cells. A 300 μL aliquot of the digest was then added to a 96-well plate to determine protein content using a modified tyrosine nitration assay44. After the protein assay, aliquots were transferred to 5 mL screwtop vials for further digestion in 70% nitric acid at room temperature overnight before heating at 95 °C for 2 hours. Digests were diluted in milliQ water containing thallium 50 ppb as an internal standard before ICP-MS analysis using a Varian 820MS ICP-MS (Agilent Technologies Inc., Santa Clara, CA, USA) at LabPLUS (Auckland, New Zealand). Optimisation of signal/noise was carried prior to each run using a tuning solution of platinum and thallium. Platinum isotopes (m/z 194 and 195) and thallium (m/z 205) were monitored during the run. Platinum counts for samples and standards were normalized for thallium internal standard counts. The platinum content of each experimental sample was calculated from the platinum to thallium count ratios and a calibration curve made from standard solutions at desired platinum concentrations made up in the same matrix as the samples. Quality control samples at three different concentrations were included for determing the accuracy, precision and reliability of each run. An ICP-MS analysis run was accepted if the standard curve was linear, and quality control and calibration standards were ±15% of their nominal value and replicate quality control samples had % coefficient of variation of ≤15%. The limit of detection and lower limit of quantification were 0.3 ppb and 1 ppb, respectively.
Cell growth inhibition
To evaluate the sensitivity of different cell lines to oxaliplatin-induced cell growth inhibition, cells were exposed to oxaliplatin at different concentrations for 2 h followed by measurement of viable cells with MTT assay45. Cells were seeded at 5000 cells/well in a collagen-coated 96-well plates then allowed to attach to the well surface over 24 h incubation in normal drug-free growth medium. After attachment, cells were exposed to oxaliplatin at different concentrations for the designated time at 37 °C with 5% CO2/95% air. When myricetin was used, cells were exposed to myricetin for 30 min immediately prior to and during the 2 h exposure to oxaliplatin. To terminate drug exposure, the medium containing drug was removed and replaced with the drug-free growth medium. Cells were then allowed to grow for 72 hours under usual incubation conditions before the MTT assay was performed. Percent cell viability at different drug concentrations was calculated from 540 nm absorbance values normalised to the mean absorbance of control untreated cells. IC-50 values for oxaliplatin-induced growth inhibition were determined from the data using nonlinear regression in GraphPad Prism version 6.
MRP2 function
Cellular 5(6)-carboxy-2,′7′-dichlorofluorescein (CDCF) accumulation in HEK-P, HEK-MRP2 and HepG2 cell lines was measured to evaluate the transporter activity of MRP246. Cells were seeded at the density of 50,000 cells per well (80,000 cells for HepG2) in the collagen coated 24-well plates. Once cells reached 80–90% confluence, they were incubated with 10 μM 5(6)-carboxy-2,′7′-dichlorofluorescein diacetate (CDCFDA) dissolved in 10 mM HEPES-HBSS medium for 90 min at 37 °C. Cells were then washed twice with 10 mM HEPES-HBSS medium, then treated with 0.1% Triton dissolved in 10 mM HEPES-HBSS medium for 10 min at room temperature. Cell lysate samples were then collected in black 96-well plates for the reading of fluorescence intensity using fluorimeter at 485/528 nm with 20 nm wavelength. Protein levels of samples were measured using a modified tyrosine nitration assay44 after the cells were digested with 70% nitric acid for 2 hr. The cellular accumulation of CDCF in fluorescence units (FU) per mg of protein was calculated using the fluorescence intensity of lysate measured as arbitrary fluorescence units and the amount of protein of cell lysate measured as mg of protein.
Real-time Transporter PCR array analysis
Total RNA from cultured cells was extracted using an RNeasy Mini Kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s instructions. Total RNA content was determined by measuring the absorbance at 260 nm with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesised from 400 ng RNA using an RT2 First Strand Kit (QIAGEN) according to the manufacturer’s instructions, and then mixed with RT2 SYBR Green qPCR Mastermix (QIAGEN). The mixtures were loaded into a commercially-available pre-validated RT2 Profiler PCR Array 96-well plate containing optimized primer assays for a set of five reference genes (GUSB, HPRT, HSP90AB1, GAPDH and ACTB) and 86 ABC and SLC drug transporter genes (QIAGEN). Real-time PCR reactions were performed according to the array manufacturer’s instructions using Applied Biosystems 7900HT Fast Real-Time PCR System and SDS 2.3 software (Life Technologies) repeated three times for each cell line. The temperature profile was as follows: 95 °C for 10 min, and then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The threshold cycle (Ct) values were obtained for each sample. As previously47,48, the expression of the five housekeeping genes varied widely between the different cell lines and were unsuitable for use as controls, and expression level Ct values were normalised by the mean expression of all genes of each cell line and PCR run, then multiplied by −1 so that positive values indicated higher expression. Cell lines were stratified into two groups according to whether they had high or low accumulation of platinum after exposure to oxaliplatin. Differences in transporter gene expression between the groups of cell lines were analysed by Two-way ANOVA (Two-way ANOVA factors: transporter gene expression and platinum accumulation) followed by Bonferroni post-tests for each candidate gene. Data were assessed visually in graphs of absolute differences and standard errors of differences in the expression of each gene between the high and low platinum accumulation groups of cell lines.
Transfections of siRNA
siRNA targeting the ABCC2 gene was transfected into HepG2 cells using Invitrogen transfection reagent. A non-targeting negative stealth siRNA (scrambled) was used as a negative control. The cells were seeded at a density of 1.5 × 105 cells per well in 12-well plates. Cells were transfected with different ABCC2 siRNA subtypes (siRNA-1, siRNA-2 and siRNA-3) at 60 pmol using lipofectamine RNAiMAX (Invitrogen) in Opti-MEM I Reduced Serum Medium (Invitrogen). At 24–96 hr after transfection, experiments were done to assess effects on MRP2 expression and function, platinum accumulation and oxaliplatin-induced growth inhibition. Quantitative real-time PCR was performed to confirm the specific gene silencing by using LightCycler-FastStart DNA Master SYBR Green 1 Master Mix (Roche Applied Science) and gene-specific primers. Total RNA was isolated from cultured cells using RNeasy Mini kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. The quantity of total RNA was measured using Qubit® 2.0 Fluorometer (Invitrogen). Thereafter, cDNA was synthesised from total RNA using Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science) according to the kit protocol. Primer sequences (MRP2 forward, 5′-AATCAGAGTCAAAGCCAAGATGCC-3′ and reverse, 5′- TAGCTTCAGTAGGAATGATTTCAGGAGCAC-3′; GAPDH forward, 5′- GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′- GCCTTCTCCATGGTGGTGAA-3′) used to detect the expression of targeted genes were purchased from IDT (Integrated DNA Technology, Singapore). Quantitative real-time PCR was performed with a LightCycler 480 Instrument II (Roche Diagnostics, New Zealand) using LightCycler-FastStart DNA Master SYBR Green 1 Master Mix (Roche Diagnostics, New Zealand) and gene-specific primers at 180 nM. The reaction conditions were as follows: 95 °C for 10 min, followed by 45 cycles at 95 °C for 15 s, at 58 °C for 30 s, and 72 °C for 30 s. The results were analysed using comparative threshold cycle method. Accumulation of an MRP2 specific substrate, 5(6)-carboxy-2′,7′-dichlorofluorescein (CDCF) by transfected cells were performed by incubating cells of density 800,000 cells/mL with 5 µM of CDCFDA for 20 min at 37 °C. After the incubation, cells were washed twice, re-suspended in ice-cold PBS and kept at 4 °C until analysis by flow cytometry (Beckman Coulter MoFlow XDP) with standard laser for excitation at 488 nm and band pass filter at 525 nm. Cells were gated to exclude dead cells, cellular debris and cell doublets. The mean fluorescence intensity was measured using Kaluza software (Beckman Coulter). Effects of siRNA transfection on cellular accumulation of platinum and oxaliplatin-induced growth inhibition was determined as described above.
Animal experiments
Animals were housed in a temperature-controlled environment with access to food and water ad libitum, and were acclimatized to handling prior to the experiments. Pilot studies were undertaken to establish the treatment protocol prior to the definitive experiment. The pilot studies showed similar trends as shown in the final experiment. For the definitive experiment, 32 NIH-III nude mice weighing 18 to 21 g were injected subcutaneously into the right flank with 107 of HepG2 cells in three batches of 10 or 11 animals each. When tumours measured approximately 200 mm3 (range, 161 to 237 mm3), animals were randomly allocated to one of four treatment groups (control, oxaliplatin alone, myricetin alone or oxaliplatin plus myricetin). Accordingly, animals were treated with oxaliplatin (3 mg/kg ip), myricetin (25 mg/kg iv) or matching drug vehicle, alone or in combination once weekly for up to five weeks. Oxaliplatin was given 30 mins after myricetin or its vehicle. Animals were monitored during the treatment period twice each week by general inspection, and measurement of body weight and perpendicular tumour dimensions using calipers. Animals were euthanased if they had poor general condition, greater than 15% body weight reduction or had tumours greater than 20 mm in average diameter or 10% of body weight, with ulceration or restricting movement. Body weight was expressed as percentage of baseline. Tumour volumes were calculated using the formula π/6*w*l2, where w = the longest diameter and l = the shortest diameter. Change in body weight and tumour volume from baseline was assessed visually by plotting mean and standard error of the mean versus time, and analysed by Two-way Repeated Measures ANOVA with Bonferroni post-tests. Time to event endpoints (quadrupling of tumour volume and euthanasia) were assessed by Kaplan-Meier plots and analysis by Gehan-Breslow-Wilcoxon test without adjustment for multiple testing. All animal procedures were approved by and performed in compliance with ethical guidelines of the Animal Ethics Committee of the University of Auckland.
Data analysis
Data were analysed using GraphPad Prism 6.2 software (San Diego, CA, USA). In vitro experiments were replicated at least three times. The significance of differences between means was assessed by ANOVA and post-tests with correction for repeated testing, as appropriate for the data. For repeated measures ANOVA, missing values (3 out of a total of 64 values (<5%)) were replaced by imputation. Differences in time to event (survival) curves were analysed using the Gehan-Breslow-Wilcoxon test without adjustment for multiple testing. A P < 0.05 was regarded as statistically significant for all statistical tests.
Supplementary information
Acknowledgements
Funding for this project was from a New Zealand Aid Programme Scholarship (KM) and grants from the Cancer Society of New Zealand (Project Grants 10/23 and 15/21) (MM) and Health Research Council of New Zealand (Project Grant 12/254) (MM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful for the contributions to this work from Associate Professor James Paxton and Angela Ding.
Author Contributions
Y.L., C.H., C.S., T.N., I.T. and M.M. were responsible for the study concept and design, and obtained funding. K.M., R.B. and N.J. undertook the in vitro experimental work under the supervision of Y.L., J.Liu, F.M., J.Lu, T.N., I.T. and M.M. S.J. and M.M. extracted and analysed the Oncomine dataset, and designed, oversaw and analysed data from the in vivo experiment. K.M., Y.L., S.J. and M.M. wrote the first draft of the report that was reviewed and then the final version approved by all authors.
Data Availability
The datasets generated during and analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-38667-8.
References
- 1.Torre LA, et al. Global Cancer Statistics, 2012. CA-Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Andre T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004;350:2343–2351. doi: 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- 3.Cassidy J, et al. XELOX (capecitabine plus oxaliplatin): active first-line therapy for patients with metastatic colorectal cancer. J. Clin. Oncol. 2004;22:2084–2091. doi: 10.1200/JCO.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 4.de Gramont A, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000;18:2938–2947. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 5.Conroy T, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 6.Oettle H, et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: outcomes from the CONKO-003 trial. J. Clin. Oncol. 2014;32:2423–2429. doi: 10.1200/JCO.2013.53.6995. [DOI] [PubMed] [Google Scholar]
- 7.Al-Batran SE, et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: A study of the arbeitsgemeinschaft internistische onkologie. J. Clin. Oncol. 2008;26:1435–1442. doi: 10.1200/JCO.2007.13.9378. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham D, et al. Capecitabine and Oxaliplatin for Advanced Esophagogastric Cancer. N. Engl. J. Med. 2008;358:36–46. doi: 10.1056/NEJMoa073149. [DOI] [PubMed] [Google Scholar]
- 9.Qin S, et al. Randomized, Multicenter, Open-Label Study of Oxaliplatin Plus Fluorouracil/Leucovorin Versus Doxorubicin As Palliative Chemotherapy in Patients With Advanced Hepatocellular Carcinoma From Asia. J. Clin. Oncol. 2013;31:3501–3508. doi: 10.1200/JCO.2012.44.5643. [DOI] [PubMed] [Google Scholar]
- 10.Anonymous. National Comprehensive Cancer Network Guidelines for Treatment of Cancer by Site, www.nccn.org/professionals/physician_gls/default.aspx#site (2017).
- 11.Anonymous. ESMO Clinical Practice Guidelines: Gastrointestinal Cancers, www.esmo.org/Guidelines/Gastrointestinal-Cancers (2017).
- 12.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 13.Platts JA, Hibbs DE, Hambley TW, Hall MD. Calculation of the Hydrophobicity of Platinum Drugs. J. Med. Chem. 2000;44:472–474. doi: 10.1021/jm001080k. [DOI] [PubMed] [Google Scholar]
- 14.Screnci D, et al. Relationships between hydrophobicity, reactivity, accumulation and peripheral nerve toxicity of a series of platinum drugs. Br. J. Cancer. 2000;82:966–972. doi: 10.1054/bjoc.1999.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jerremalm E, Hedeland M, Wallin I, Bondesson U, Ehrsson H. Oxaliplatin degradation in the presence of chloride: Identification and cytotoxicity of the monochloro monooxalato complex. Pharm. Res. 2004;21:891–894. doi: 10.1023/B:PHAM.0000026444.67883.83. [DOI] [PubMed] [Google Scholar]
- 16.Liu JJ, Lu J, McKeage MJ. Membrane transporters as determinants of the pharmacology of platinum anticancer drugs. Curr. Cancer Drug Targets. 2012;12:962–986. doi: 10.2174/156800912803251199. [DOI] [PubMed] [Google Scholar]
- 17.Jemnitz K, et al. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab. Rev. 2010;42:402–436. doi: 10.3109/03602530903491741. [DOI] [PubMed] [Google Scholar]
- 18.Koike K, et al. A canalicular multispecific organic anion transporter (cMOAT) antisense cDNA enhances drug sensitivity in human hepatic cancer cells. Cancer Res. 1997;57:5475–5479. [PubMed] [Google Scholar]
- 19.Taniguchi K, et al. A human canalicular multispecific organic anion transporter (cMOAT) gene is overexpressed in cisplatin-resistant human cancer cell lines with decreased drug accumulation. Cancer Res. 1996;56:4124–4129. [PubMed] [Google Scholar]
- 20.Kawabe T, et al. Enhanced transport of anticancer agents and leukotriene C4 by the human canalicular multispecific organic anion transporter (cMOAT/MRP2) FEBS Lett. 1999;456:327–331. doi: 10.1016/S0014-5793(99)00979-5. [DOI] [PubMed] [Google Scholar]
- 21.Cui YH, et al. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- 22.Tamai M, et al. Conjugated bilirubin induces multidrug resistance-associated protein 2 mRNA expression and in vivo cisplatin resistance in rat hepatoma AH66 cells. Anticancer Res. 2003;23:4781–4787. [PubMed] [Google Scholar]
- 23.Li ZY, et al. ABCC2-24C > T polymorphism is associated with the response to platinum/5-Fu-based neoadjuvant chemotherapy and better clinical outcomes in advanced gastric cancer patients. Oncotarget. 2016;7:55449–55457. doi: 10.18632/oncotarget.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohn C, et al. Contribution of glutathione and MRP-mediated efflux to intracellular oxaliplatin accumulation. Int. J. Clin. Pharmacol. Ther. 2010;48:445–447. doi: 10.5414/CPP48445. [DOI] [PubMed] [Google Scholar]
- 25.Shen K, et al. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J. Cell. Biochem. 2012;113:2086–2097. doi: 10.1002/jcb.24080. [DOI] [PubMed] [Google Scholar]
- 26.Wang ZY, et al. Dihydromyricetin reverses MRP2-mediated MDR and enhances anticancer activity induced by oxaliplatin in colorectal cancer cells. Anti-Cancer Drugs. 2017;28:281–288. doi: 10.1097/CAD.0000000000000459. [DOI] [PubMed] [Google Scholar]
- 27.van Zanden JJ, et al. Reversal of in vitro cellular MRP1 and MRP2 mediated vincristine resistance by the flavonoid myricetin. Biochem. Pharmacol. 2005;69:1657–1665. doi: 10.1016/j.bcp.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 28.van Zanden JJ, et al. Quantitative structure activity relationship studies on the flavonoid mediated inhibition of multidrug resistance proteins 1 and 2. Biochem. Pharmacol. 2005;69:699–708. doi: 10.1016/j.bcp.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Tsuji S, et al. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer. 2012;106:126–132. doi: 10.1038/bjc.2011.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beretta GL, et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem. Pharmacol. 2010;79:1108–1117. doi: 10.1016/j.bcp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Ferreira JA, et al. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug Resist. Updat. 2016;24:34–54. doi: 10.1016/j.drup.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Hinoshita E, et al. Increased expression of an ATP-binding cassette superfamily transporter, multidrug resistance protein 2, in human colorectal carcinomas. Clin. Cancer Res. 2000;6:2401–2407. [PubMed] [Google Scholar]
- 33.Korita PV, et al. Multidrug resistance-associated protein 2 determines the efficacy of cisplatin in patients with hepatocellular carcinoma. Oncol. Rep. 2010;23:965–972. doi: 10.3892/or_00000721. [DOI] [PubMed] [Google Scholar]
- 34.Yamasaki M, et al. Role of multidrug resistance protein 2 (MRP2) in chemoresistance and clinical outcome in oesophageal squamous cell carcinoma. Br. J. Cancer. 2011;104:707–713. doi: 10.1038/sj.bjc.6606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rumiato E, et al. A germline predictive signature of response to platinum chemotherapy in esophageal cancer. Transl. Res. 2016;171:29–37. doi: 10.1016/j.trsl.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Myint K, Li Y, Paxton J, McKeage M. Multidrug Resistance-Associated Protein 2 (MRP2) Mediated Transport of Oxaliplatin-Derived Platinum in Membrane Vesicles. PLoS One. 2015;10:e0130727. doi: 10.1371/journal.pone.0130727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human-liver carcinoma-derived cell-line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 38.Jamieson SM, et al. Oxaliplatin-induced loss of phosphorylated heavy neurofilament subunit neuronal immunoreactivity in rat DRG tissue. Mol. Pain. 2009;5:66. doi: 10.1186/1744-8069-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jamieson SM, Liu J, Connor B, McKeage MJ. Oxaliplatin causes selective atrophy of a subpopulation of dorsal root ganglion neurons without inducing cell loss. Cancer Chemother. Pharmacol. 2005;56:391–399. doi: 10.1007/s00280-004-0953-4. [DOI] [PubMed] [Google Scholar]
- 40.Zhang S, Wang L, Liu H, Zhao G, Ming L. Enhancement of recombinant myricetin on the radiosensitivity of lung cancer A549 and H1299 cells. Diagn. Pathol. 2014;9:68. doi: 10.1186/1746-1596-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang XH, Zou ZQ, Xu CW, Shen YZ, Li D. Myricetin induces G2/M phase arrest in HepG2 cells by inhibiting the activity of the cyclin B/Cdc2 complex. Mol. Med. Rep. 2011;4:273–277. doi: 10.3892/mmr.2011.417. [DOI] [PubMed] [Google Scholar]
- 42.Phillips PA, et al. Myricetin induces pancreatic cancer cell death via the induction of apoptosis and inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Cancer Lett. 2011;308:181–188. doi: 10.1016/j.canlet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, et al. Myricetin enhance chemosensitivity of 5-fluorouracil on esophageal carcinoma in vitro and in vivo. Cancer Cell Int. 2014;14:71. doi: 10.1186/s12935-014-0071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bible KC, Boerner SA, Kaufmann SH. A one-step method for protein estimation in biological samples: Nitration of tyrosine in nitric acid. Anal. Biochem. 1999;267:217–221. doi: 10.1006/abio.1998.2980. [DOI] [PubMed] [Google Scholar]
- 45.Mosmann T. Rapid colorimetric assay for cellular growth and survival - application to proliferation and cyto-toxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 46.Munic V, Hlevnjak M, Haber VE. Characterization of rhodamine-123, calcein and 5(6)-carboxy-2′,7 ′-dichlorofluorescein (CDCF) export via MRP2 (ABCC2) in MES-SA and A549 cells. Eur. J. Pharm. Sci. 2011;43:359–369. doi: 10.1016/j.ejps.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Okabe M, et al. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008;7:3081–3091. doi: 10.1158/1535-7163.MCT-08-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szakács G, et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell. 2004;6:129–137. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 49.Lancaster CS, et al. Modulation of OATP1B-Type Transporter Function Alters Cellular Uptake and Disposition of Platinum Chemotherapeutics. Mol. Cancer Ther. 2013;12:1537–1544. doi: 10.1158/1535-7163.MCT-12-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li S, et al. Role of Organic Cation Transporter 1, OCT1 in the Pharmacokinetics and Toxicity of cis-Diammine(pyridine)chloroplatinum(II) and Oxaliplatin in Mice. Pharm. Res. 2011;28:610–625. doi: 10.1007/s11095-010-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang SZ, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lovejoy KS, et al. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proc. Natl. Acad. Sci USA. 2008;105:8902–8907. doi: 10.1073/pnas.0803441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burger H, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br. J. Pharmacol. 2010;159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J. Pharmacol. Exp. Ther. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 55.Yokoo S, et al. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007;74:477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 56.Yokoo S, et al. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab. Dispos. 2008;36:2299–2306. doi: 10.1124/dmd.108.023168. [DOI] [PubMed] [Google Scholar]
- 57.Jong NN, Nakanishi T, Liu JJ, Tamai I, McKeage MJ. Oxaliplatin transport mediated by OCTN1 and OCTN2 in overexpressing HEK293 cells and rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2011;338:537–547. doi: 10.1124/jpet.111.181297. [DOI] [PubMed] [Google Scholar]
- 58.Kunii E, et al. Organic cation transporter OCT6 mediates cisplatin uptake and resistance to cisplatin in lung cancer. Cancer Chemother. Pharmacol. 2015;75:985–991. doi: 10.1007/s00280-015-2723-x. [DOI] [PubMed] [Google Scholar]
- 59.Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 60.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol. Pharmacol. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin X, Okuda T, Holzer A, Howell SB. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002;62:1154–1159. doi: 10.1124/mol.62.5.1154. [DOI] [PubMed] [Google Scholar]
- 62.Song IS, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol. Cancer. Ther. 2004;3:1543–1549. [PubMed] [Google Scholar]
- 63.Chen CC, et al. Combined modalities of resistance in an oxaliplatin-resistant human gastric cancer cell line with enhanced sensitivity to 5-fluorouracil. Br. J. Cancer. 2007;97:334–344. doi: 10.1038/sj.bjc.6603866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katano K, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–6565. [PubMed] [Google Scholar]
- 65.Samimi G, Katano K, Holzer AK, Safaei R, Howell SB. Modulation of the cellular pharmacology of cisplatin and its analogs by the copper exporters ATP7A and ATP7B. Mol. Pharmacol. 2004;66:25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 66.Samimi G, et al. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004;10:4661–4669. doi: 10.1158/1078-0432.CCR-04-0137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and analysed during the current study are available from the corresponding author on reasonable request.