

Abstract
Clinical studies indicate that psychosocial stress contributes to adverse chronic pain outcomes in patients, but it is unclear how this is initiated or amplified by stress. Repeated social defeat (RSD) is a mouse model of psychosocial stress that activates microglia, increases neuroinflammatory signaling, and augments pain and anxiety-like behaviors. We hypothesized that activated microglia within the spinal cord facilitate increased pain sensitivity following RSD. Here we show that mechanical allodynia in male mice was increased with exposure to RSD. This stress-induced behavior corresponded with increased mRNA expression of several inflammatory genes, including IL-1β, TNF-α, CCL2, and TLR4 in the lumbar spinal cord. While there were several adhesion and chemokine-related genes increased in the lumbar spinal cord after RSD, there was no accumulation of monocytes or neutrophils. Notably, there was evidence of microglial activation selectively within the nociceptive neurocircuitry of the dorsal horn of the lumbar cord. Elimination of microglia using the colony stimulating factor 1 receptor antagonist PLX5622 from the brain and spinal cord prevented the development of mechanical allodynia in RSD-exposed mice. Microglial elimination also attenuated RSD-induced IL-1β, CCR2, and TLR4 mRNA expression in the lumbar spinal cord. Together, RSD-induced allodynia was associated with microglia-mediated inflammation within the dorsal horn of the lumbar spinal cord.
SIGNIFICANCE STATEMENT Mounting evidence indicates that psychological stress contributes to the onset and progression of adverse nociceptive conditions. We show here that repeated social defeat stress causes increased pain sensitivity due to inflammatory signaling within the nociceptive circuits of the spinal cord. Studies here mechanistically tested the role of microglia in the development of pain by stress. Pharmacological ablation of microglia prevented stress-induced pain sensitivity. These findings demonstrate that microglia are critical mediators in the induction of pain conditions by stress. Moreover, these studies provide a proof of principle that microglia can be targeted as a therapeutic strategy to mitigate adverse pain conditions.
Keywords: allodynia, cytokines, microglia, repeated social defeat, stress
Introduction
Chronic pain is the most common cause of long-term disability, affecting >1.5 billion people (Global Industry Analysts, 2011). Despite available therapies, chronic pain remains inadequately treated and leads to personal suffering, reduced productivity, and significant health care costs (Alexander et al., 2009). Chronic pain is often associated with increased anxiety and depression that leads to reduced quality of life (Katz and Barkin, 2010), and psychological stress promotes the onset and progression of neuropathic pain (DeLeo and Yezierski, 2001; Greco et al., 2004). Psychological stress before surgery prolongs pain symptoms during postoperative recovery (Mathews and Ridgeway, 1981; Kiecolt-Glaser et al., 1998). Furthermore, chronically stressed individuals exhibit lower nociceptive thresholds in tests of tactile sensitivity (Ashkinazi and Vershinina, 1999) and pressure pain (Persson et al., 2000). Thus, stress-induced pain sensitivity and allodynia likely contribute to the pathophysiology of chronic pain conditions. Collectively, regulation of nociception by psychological stress has negative clinical outcomes that are not yet well understood.
It is possible that stress regulates nociception via neuroinflammatory signaling within the spinal cord. For instance, mouse models of chronic stress promote increased neuroinflammatory signaling within the spinal cord that may contribute to increased nociception and allodynia (Alexander et al., 2009, 2012; Sawicki et al., 2018). Moreover, clinical and preclinical data show that heightened neuroinflammatory signaling in the spinal cord contributes to exaggerated nociception (Maier and Watkins, 2003; Tsuda et al., 2005). For instance, microglial production of cytokines IL-1β and TNF-α promotes the development of allodynia in both peripheral nerve injury (PNI) (Raghavendra et al., 2003) and spinal cord injury (Detloff et al., 2008) After PNI, microglia are essential to the development of mechanical pain hypersensitivity in male mice (Sorge et al., 2015). Pharmacological interventions targeting microglial activation (Raghavendra et al., 2003; Ledeboer et al., 2005; Padi and Kulkarni, 2008) or preventing microglial proliferation (Gu et al., 2016) prevented neuropathic pain associated with PNI. Therefore, understanding the mechanism that mediates inflammatory interactions and the onset of nociceptive symptoms in the context of psychological stress may lead to novel therapeutic strategies for the management of chronic pain states.
Using a murine model of psychosocial stress, repeated social defeat (RSD), we showed that stress caused mechanical allodynia that persisted 1 week after stress termination (Sawicki et al., 2018). Similarly, restraint stress potentiated both allodynia and microglial activation following PNI (Alexander et al., 2009). Moreover, unpredictable sound stress in rats induced both hyperalgesia and neuroinflammatory signaling (Khasar et al., 2008). Therefore, microglial activation and the release of proinflammatory cytokines in the spinal cord likely contribute to exaggerated pain states associated with psychological stress. However, the interaction between microglia and pain, particularly in the presence or absence of concurrent stress, remains unclear.
The primary objective of this study was to determine the mechanism by which RSD induces mechanical allodynia in the absence of injury. Much of our previous work has focused on microglia in the brain and their regulation of complex behavioral responses to stress (Reader et al., 2015; Wohleb et al., 2015). The present study showed that RSD promoted microglial activation exclusively within the spinal cord dorsal horn, the region involved with nociceptive signaling, independent of peripheral monocyte recruitment. Corresponding to microglial activation, RSD increased the neuroinflammatory environment within the spinal cord, marked by increased gene expression of pronociceptive mediators. Pharmacological depletion of microglia prevented RSD-induced mechanical allodynia and attenuated the inflammatory environment within the lumbar spinal cord. These definitive findings show that microglia are necessary for the development of heightened pain states following stress exposure.
Materials and Methods
Mice.
Male C57BL/6 (6–8 weeks old) and male CD-1 (12 months, retired breeders) mice were purchased from Charles River Breeding Laboratories and allowed to acclimate to their surroundings for 7–10 d before experiments. Resident C57BL/6 mice were housed in cohorts of 3, and aggressor CD-1 mice were individually housed. All mice were housed in 11.5 inch × 7.5 inch × 6 inch polypropylene cages. Rooms were maintained at 21°C under a 12 h light-dark cycle (lights on at 0600) with ad libitum access to water and rodent chow. All procedures were in accordance with the National Institutes of Health Guidelines and were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee.
RSD.
Mice were subjected to RSD stress as previously described (McKim et al., 2018). In brief, an aggressive male intruder CD-1 mouse was introduced into cages of established male cohorts (3 per cage) of C57BL/6 mice for 2 h (h) between 17:00 and 19:00 for six consecutive nights. During each cycle, submissive behavior (e.g., upright posture, fleeing, and crouching) was observed to ensure defeat of the resident mice. A new intruder was introduced if an attack on the resident mice was not initiated within the first 5–10 min, or if the intruder was defeated by any of the resident mice. At the end of the 2 h period, the intruder was removed and the residents were left undisturbed until the following day when the paradigm was repeated. To avoid habituation, different intruders were used on consecutive nights. As described previously in studies with RSD, intermale aggression observed during each cycle resulted in minor tissue damage inflicted by the intruder mouse (McKim et al., 2018). The mice were monitored at least twice daily for any indication of distress or illness. Mice that were injured or moribund were removed from the study. Consistent with previous studies using RSD (Sawicki et al., 2015; McKim et al., 2016), <5% of mice met the early removal criteria. Control mice were left undisturbed in their home cages. In all studies with RSD, food intake and body weight were monitored. Our previously published studies indicate that our stress paradigm does not cause body weight loss, suggesting that the stressor is not severe and the mice are able to maintain regular eating habits (Avitsur et al., 2001). All social behavior and biological measures were obtained 12 h after the final cycle. This time point was selected because sympathetic nervous system and hypothalamic-pituitary-adrenal axis activation returns to baseline by 12 h after the final cycle (Hanke et al., 2012).
Pain behavior.
Mechanical allodynia was determined as previously described (Sawicki et al., 2018). Tactile mechanical sensitivity was analyzed by measuring threshold responses to a calibrated von Frey rigid tip (IITC Life Science). Mice were placed on a mesh platform in a clear compartment (8 cm × 12 cm × 5.5 cm) that allows unrestrained exploration, locomotion, and grooming. Animals acclimated to the testing environment for 30 min before testing. Mechanical thresholds were tested by probing the mid-line of the plantar surface of the right hindpaw by a blinded investigator to determine the force that repeatedly elicits withdrawal of the hindpaw (L5 dermatome) from the calibrated rigid tip. Baseline measurements were performed 24 h before RSD exposure. The readout value represents the maximum force at which the hindpaw was withdrawn (Martinov et al., 2013). The baseline mechanical withdrawal threshold was determined by averaging the threshold for five consecutive mechanical stimuli applied at 1 min intervals. In RSD and control animals, the force needed to elicit a withdrawal of the hindpaw was recorded following three stimulus presentations at ∼1 min intervals, and the mean values of the three readings were used for analysis. A lower withdrawal threshold (in grams) is indicative of increased pain sensitivity or mechanical allodynia. Subsequent behavioral testing for mechanical allodynia was completed 12 h after the first, third, and final day of RSD (n = 6 per group, 2 replicates).
RNA isolation and RT-PCR.
For spinal cord analyses, the cervical, thoracic, and lumbar regions were dissected from the spinal cord and immediately flash frozen in liquid nitrogen. RNA was isolated using tri-reagent/isopropanol precipitation, and RNA concentration was determined by NanoPhotometry (Implen). RNA (1.2 μg) was reverse transcribed to cDNA using an RT-RETROscript kit (Ambion, Thermo Fisher Scientific). For Percoll-enriched microglia, the USB PrepEase kit (Affymetrix) was used to isolate RNA according to the manufacturer's instructions. qRT-PCR was performed using the Assay-on-Demand Gene Expression protocol (Applied Biosystems). Experimental cDNA was amplified by real-time PCR where a target cDNA and reference cDNA (GAPDH) were amplified simultaneously using an oligonucleotide probe with a 5′ fluorescent reporter dye (FAM) and a 3′ quencher dye (nonfluorescent quencher). Florescence was determined on an ABI PRISM 7300-sequence detection system (Applied Biosystems). A blinded investigator analyzed relative gene expression using the ΔΔCT method, and results were expressed as fold difference from GAPDH (n = 6 per group).
Oral administration.
Plexxikon 5622 (PLX5622) was provided by Plexxikon and formulated in standard AIN-76A rodent chow by Research Diets at a concentration of 1200 mg/kg. Control diet consisted of the standard AIN-76A rodent chow. Mice were provided ad libitum access to PLX5622 or control diet for 14 d to deplete microglia before exposure to RSD.
Spinal cord histology.
Naive or RSD mice (n = 6 per group) were killed via overdose using a mixture of ketamine and xylazine. Blood was collected from the right ventricle before transcardial perfusion with 0.1 m PBS, pH 7.4, followed by 4% PFA, pH 7.2. Spinal cord segments were removed from the lumbar cord (L4-L6). Segments were postfixed for 1 h in 4% PFA, rinsed in 0.2 m PB, pH 7.4 overnight, then cryoprotected in 30% sucrose before being embedded in Optimal Cutting Temperature Compound (Thermo Fisher Scientific). Coronal sectioning the lumbar spinal cord was performed at 20 μm on a Microm HM505E cryostat and collected in a series of equally spaced sections.
Immunofluorescence.
Fluorescent immunohistochemistry was performed to assess microglial reactivity and monocyte infiltration. Briefly, tissue sections were blocked for 1 h using 2.5% NGS, 5% NDS, 0.1% Triton X-100 in PBS. To examine microglia/monocytes, a 1:500 dilution of rabbit anti-p2y12 (AS-55043A, AnaSpec) was combined with a 1:1000 dilution of rat anti-mouse CD45 (MCA1388, Bio-Rad) in PBS. Incubation of the primary antibody occurred overnight at 4°C. A 1:1000 dilution of donkey anti-rabbit 550 (ab96892, Abcam) was prepared with a 1:500 dilution of goat anti-rat 488 (A110006, Thermo Fisher Scientific) in PBS to visualize p2y12 and CD45. Secondary antibodies were incubated for 2 h at room temperature before coverslipping with ProLong Diamond antifade mountant with DAPI (P36962, Thermo Fisher Scientific). Slides were allowed to cure in a dark box overnight at room temperature. Microglial reactivity was also assessed in select experiments with a 1:1000 dilution of rabbit anti-mouse Iba-1 (019-19741, Wako Chemicals). Primary incubations were completed overnight at 4°C. Sections were then washed in PBS and incubated with a fluorochrome-conjugated secondary antibody (AlexaFluor-488). Sections were mounted on slides, coverslipped with Fluoromount G (Beckman Coulter), and stored at −20°C.
Imaging and quantification.
Three tissue sections were imaged per spinal cord level, per animal using an Olympus FV1000 filter confocal microscope (Ohio State University Confocal Microscopy Imaging Facility). Spinal cord hemisections were further divided into dorsal (laminae 1, 2, 3, 4), intermediate (laminae 5, 6, 7), and ventral (laminae 8, 9) gray matter segments based on predefined anatomical maps (Allen Brain Atlas, http://mousespinal.brain-map.org/). As both thoracic hemisections are contained within a single image, the quantified hemisection was chosen at random using a random sequence generator. Morphological assessment of microglial reactivity from digital images was performed using ImageJ. Quantification occurred within a single box, placed within the intermediate laminae and two boxes in the dorsal (deep dorsal horn, cap of the dorsal horn) and ventral horns (lateral and medial) to accommodate the size and irregular shape of these regions. Thresholds for positive staining were determined by a blinded investigator and then processed for densitometric scanning of thresholded targets. The positive labeling in each region was expressed as percentage area and averaged across the three tissue sections. Within the same regions in dorsal, intermediate, and ventral laminae, CD45+ cells were counted and represented infiltration of peripheral immune cells. Cells in the tissue parenchyma, but not within blood vessels, were quantified. For select experiments with Iba-1 labeling, fluorescent sections were visualized using an epi-fluorescent DM5000B microscope (Leica Microsystems). Images were captured using a DFC300 FX camera (Leica Microsystems) and imaging software. For each image, a threshold for positive staining was determined that included all cell bodies while excluding background staining (ImageJ). All results are expressed as average percentage area in the positive threshold for all representative images.
Isolation of CD11b+ cells from spinal cord.
CD11b+ cells were isolated from whole spinal cord homogenates as previously described (Sawicki et al., 2018). In brief, spinal cords were passed through a 70 μm nylon cell strainer and centrifuged at 600 × g for 6 min. Supernatants were removed, and cell pellets were resuspended in 70% isotonic Percoll (GE Healthcare). A discontinuous Percoll density gradient was layered as follows: 50%, 35%, and 0% isotonic Percoll. The gradient was centrifuged for 20 min at 2000 × g, and cells were collected from the interphase between the 70% and 50% Percoll layers. These cells were referred to as enriched CD11b+ cells based on previous studies demonstrating that viable cells isolated by Percoll density gradient yields >90% CD11b+ cells (McKim et al., 2018) (n = 6 per group).
Flow cytometry.
Labeling of cell surface antigens was performed as previously described (McKim et al., 2018). In brief, Fc receptors were blocked with anti-CD16/CD32 antibody (553142, BD Biosciences). Cells were washed and then incubated with the appropriate antibodies (CD45, CD11b, Ly6C, BD Biosciences) for 1 h at 4°C. Cells were washed and then resuspended in FACS buffer for analysis. Cell numbers were estimated using counting beads (BD Biosciences). Nonspecific binding was assessed using isotype-matched antibodies. Antigen expression was determined using a FACSCalibur four-color cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star) by a blinded investigator, and positive labeling for each antibody was determined based on isotype stained controls (n = 6 per group).
GFP+ bone marrow (BM)-chimera.
To establish chimerism, recipient C57BL/6 male mice (6 weeks old) were injected intraperitoneally once daily for 2 consecutive days with busulfan in a 1:1 solution of DMSO and deionized water (30 mg/kg/100 μl). This dose of busulfan resulted in high myeloid ablation with limited complications (Wohleb et al., 2013). Donor BM-derived cells were isolated from the femur and passed through a 70 μm nylon cell strainer. Total number of cells was determined with a Particle Count and Size Analyzer (Beckman Coulter). Donor BM-derived cells were obtained from male C57BL/6-Tg(CAG-EGFP)131Osb/LeySopJ; strain #006567) mice. BM-derived cells (1 × 106) were transferred to recipient mice by tail vein injection (100 μl) 48 h after the second dose of busulfan. Mice were left undisturbed for 4 weeks to allow engraftment. Engraftment was verified by determining the percentage of chimerism in the BM and blood. All mice had >90% BM engraftment in the present study.
Isolation of cells from blood.
Tissues were collected immediately following CO2 asphyxiation. Whole blood was collected with EDTA-lined syringes by cardiac puncture, and red blood cells were lysed. Tissue samples were washed with HBSS, filtered through a 70 μm nylon cell strainer, and then the total number of cells was determined with a Particle Count and Size Analyzer (Beckman Coulter) (n = 6 per group).
Experimental design and statistical analysis.
The number of individual animals is described throughout Materials and Methods. Data are expressed as treatment mean ± SEM. To achieve the recommended level of statistical significance, previous power analyses for flow cytometric studies indicated that a sample size of n = 6 was needed for each experimental group during biochemical assays, and a sample size of n = 12 was required for each experimental group in behavioral assays (McKim et al., 2018). Individual data points >2 SDs above and below the mean were counted as outliers and were excluded in the subsequent analyses. To determine significant main effects and interactions between main factors, data were analyzed using one- or two-way ANOVA using GraphPad Prism Statistical Software. In the event of a main effect of experimental treatment, differences between group means were evaluated by an F-protected t test. Post hoc analyses are graphically presented in figures. Threshold for statistical significance was set at p < 0.05.
Results
RSD stress caused mechanical allodynia in an exposure-dependent manner
We have reported that mice exposed to RSD develop mechanical allodynia, and this increased pain sensitivity persisted for 1 week after stress cessation (Sawicki et al., 2018). To confirm this finding, mice were exposed to RSD and mechanical allodynia was assessed 12 h after the first, third, and sixth day of stress (Fig. 1). Before RSD, each group had similar withdrawal thresholds. Exposure to RSD induced mechanical allodynia throughout the testing period (stress × time interaction; F(3,40) = 52.88, p < 0.0001). Post hoc analysis revealed increased mechanical allodynia after 3 d of RSD that was further increased by 6 d (p < 0.05). Together, these results confirm previous findings (Sawicki et al., 2018) and demonstrate that 6 d of RSD promoted the development of mechanical allodynia.
Figure 1.
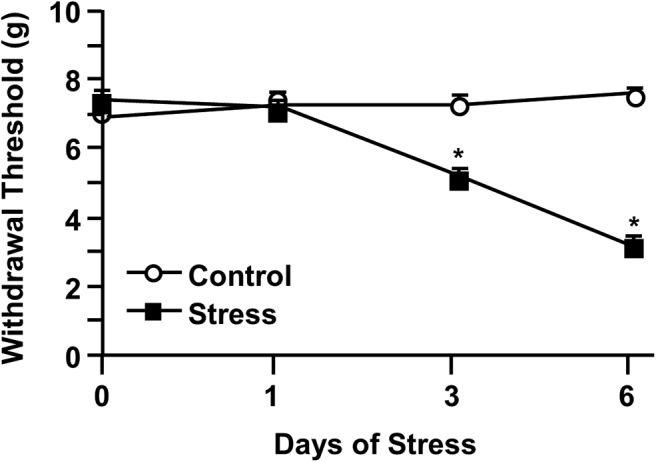
RSD caused mechanical allodynia in an exposure-dependent manner. Male C57BL/6 mice were subjected to 6 d of RSD (Stress) or left undisturbed as controls (Control). Mechanical allodynia was assessed before stress exposure and 12 h after the first, third, and sixth day of RSD. Withdrawal threshold of mechanical stimulation to the hindpaw using the von Frey behavior test was determined. Error bars indicate mean ± SEM. *Significantly different from control mice (p < 0.05).
RSD caused region-specific microglial activation in the spinal cord
Previous studies demonstrate that RSD activates microglia in the brain, which promotes the development of prolonged anxiety (Wohleb et al., 2013; McKim et al., 2018). Our next objective was to determine whether there was microglial activation the spinal cord in response to RSD. Here mice were exposed to 6 d of RSD or left undisturbed as controls, and P2Y12 labeling of microglia was determined in three key areas of the lumbar cord. We are reporting the lumbar data because it reflects the behavioral assay (L5 dermatome and L4-L6 spinal level histology). These include the cap of the dorsal horn (receives input from small diameter pain afferents), the lateral ventral horn (contains motor neurons for lower extremity limb control), and the intermediate laminae (maintains central pattern generators). There was a significant increase in microglial restructuring (percentage P2Y12 area) in the cap of the dorsal horn in mice exposed to RSD (Fig. 2A,B; p < 0.05). In the lateral ventral horn (Fig. 2C,D) and the intermediate laminae (Fig. 2E,F), however, there was reduced expression of P2Y12 after RSD compared with controls. Collectively, these findings indicate that RSD activated microglia in a regionally dependent manner, with enhanced expression in the dorsal horn of the spinal cord, the first relay for pain transmission in the CNS (Zhuo et al., 2011).
Figure 2.

RSD caused region-specific microglial activation in the spinal cord. Male C57BL/6 mice were subjected to 6 d of RSD (Stress) or left undisturbed as controls (Con). Mice were perfused, and spinal cords were PFA fixed 14 h after the last day of stress. Microglial activation (P2Y12 expression) was assessed in the lumbar spinal cord. A, Representative images within the cap of the dorsal horn of P2Y12 labeling. B, P2Y12 proportional area was determined in the cap of the dorsal horn. C, Representative images within the lateral ventral horn of P2Y12 labeling. D, P2Y12 proportional area was determined in the lateral ventral horn. E, Representative images within the intermediate laminae of P2Y12 labeling. F, P2Y12 proportional area was determined in the intermediate laminae. Boxed insets represent location where proportional area was measured. Error bars indicate mean ± SEM. *Significantly different from control mice (p < 0.05; F-protected after analysis).
mRNA expression of immune and inflammatory mediators increased in the lumbar spinal cord after RSD
Previous studies indicate that RSD is associated with increased gene expression of inflammatory cytokines, chemokines, and adhesion molecules in the brain (Sawicki et al., 2015; Wohleb et al., 2015; McKim et al., 2018). Inflammation within the spinal cord may also contribute to increased pain sensitivity with stress (Alexander et al., 2009). Because we observed increased P2Y12 labeling in the lumbar spinal cord, mRNA levels of several cytokines, chemokines, and regulatory/adhesion molecules were determined after RSD exposure in this region (Table 1). Mice exposed to RSD had increased mRNA levels of proinflammatory cytokines IL-1β and TNF-α in the lumbar region of the spinal cord (Table 1; p < 0.05 for each). Consistent with our previous studies in the brain (Weber et al., 2017), there was a significant increase in the mRNA expression of CCL2 and CCR2 in the lumbar spinal cord of mice exposed to RSD (Table 1; p < 0.05 for each). Levels of CXCR2, E-Selectin, and TLR4 mRNA were also increased in the lumbar spinal cord after RSD (Table 1; p < 0.05 for each). Together, these findings indicate that RSD induced an inflammatory gene profile in the lumbar spinal cord characterized by enhanced proinflammatory cytokine expression, chemokine ligand/receptor interactions, adhesion molecules, and immunoregulatory markers.
Table 1.
Lumbar cord mRNA levels (fold Δ)a
| Gene | Control | Stress | |
|---|---|---|---|
| Cytokines | IL-1β | 1.08 ± 0.19 | 6.33 ± 2.45* |
| TNFα | 1.03 ± 0.09 | 1.69 ± 0.17* | |
| IL-6 | 1.01 ± 0.04 | 0.82 ± 0.08* | |
| MIF | 1.02 ± 0.08 | 1.10 ± 0.06 | |
| Chemokines | CXCR2 | 1.11 ± 0.16 | 5.77 ± 1.25* |
| CCR2 | 1.13 ± 0.23 | 2.04 ± 0.23* | |
| CX3CR1 | 1.01 ± 0.05 | 1.12 ± 0.10 | |
| CCL2 | 1.06 ± 0.15 | 2.88 ± 0.43* | |
| CXCL2 | 1.34 ± 0.47 | 2.02 ± 0.63 | |
| CCL7 | 1.15 ± 0.22 | 2.43 ± 0.63# | |
| CXCL2 | 1.04 ± 0.12 | 1.17 ± 0.17 | |
| Regulatory/adhesion | GFAP | 1.02 ± 0.08 | 1.07 ± 0.05 |
| Arginase | 1.72 ± 0.99 | 0.87 ± 0.22 | |
| IL-4Ra | 1.04 ± 0.12 | 1.35 ± 0.28 | |
| CD206 | 1.34 ± 0.43 | 1.43 ± 0.21 | |
| TGF-β | 1.00 ± 0.03 | 1.77 ± 0.17* | |
| E-Selectin | 1.03 ± 0.20 | 5.13 ± 1.48* | |
| ICAM-1 | 1.12 ± 0.25 | 1.19 ± 0.36 | |
| Miscellaneous | GLAST | 1.16 ± 0.27 | 1.46 ± 0.24 |
| EAAT3 | 1.12 ± 0.20 | 1.38 ± 0.38 | |
| TLR4 | 1.05 ± 0.14 | 1.88 ± 0.20* |
amRNA expression of immune and inflammatory mediators increased in the lumbar spinal cord after RSD. Male C57BL/6 mice were subjected to 6 d of RSD (Stress) or left undisturbed as controls. Fourteen hours after the last day of stress, spinal cords were collected and dissected for mRNA analyses. mRNA levels of inflammatory cytokines, chemokines, regulatory/adhesion molecules, and immune mediators were determined in the lumbar spinal cord.
*p < 0.05 (F-protected after analysis).
#p < 0.1 (F-protected after analysis).
Mechanical allodynia during RSD stress occurred independent of peripheral monocyte recruitment to the spinal cord
We have reported that monocytes are actively recruited to the brain by microglia and function to augment anxiety-like behavior (Wohleb et al., 2013; McKim et al., 2018). The mRNA profile within the spinal cord of increased adhesion molecules, cytokines, and chemokines suggests that monocytes may also be recruited to the spinal cord. Therefore, monocyte accumulation in the lumbar region of the spinal cord was assessed after RSD (Fig. 3). Representative bivariate dot plots of CD11b and CD45 labeling for spinal cord macrophages (CD11b+/CD45high) for control and stressed mice are shown (Fig. 3A). There was no difference in the number of macrophages in the spinal cord between mice exposed to RSD compared with controls (Fig. 3B). In a separate experiment, the number of CD45+ cells was assessed in the lumbar cord (Fig. 3C,D). Again, there was no difference in the number of CD45+ cells in the spinal cord between control and stressed mice. To further confirm these results, GFP+ BM chimeras were generated with BM-derived donor cells that ubiquitously express GFP (Fig. 3E). Consistent with the CD45 data, there was no difference in the number of GFP+ cells in the lumbar spinal cord of control versus stressed mice. Collectively, these findings demonstrate that monocytes were not recruited to the spinal cord with RSD.
Figure 3.

Mechanical allodynia during RSD occurred independent of peripheral monocyte recruitment to the spinal cord. Male C57BL/6 mice were subjected to 6 d of RSD (Stress) or left undisturbed as controls (Con). Fourteen hours after the last day of stress, spinal cords were collected and Percoll gradient-enriched. A, Representative flow bivariate dot plots of CD11b and CD45 labeling of Percoll gradient-enriched cells from the spinal cord. B, Number of CD11b+/CD45hi monocytes/macrophages in the spinal cord. In a separate experiment, mice were perfused, and spinal cords were PFA fixed 14 h after the last day of stress. Presence of monocytes (CD45+) was assessed in the lumbar spinal cord parenchyma. C, Representative images within the lumbar spinal cord parenchyma of CD45 labeling. D, The number of CD45+ cells was quantified in the lumbar spinal cord parenchyma. E, In another experiment, GFP+ BM-chimeric mice were generated. GFP+ BM-chimera mice were generated with BM-derived donor cells that ubiquitously express GFP. In this model, resident (GFP−) and BM-derived cells (GFP+) can be distinguished based on GFP expression. Four weeks after BM reconstitution, GFP+ BM-chimeric mice were exposed to RSD or left undisturbed as controls. F, Mice were perfused, and spinal cords were PFA fixed 14 h after the last day of stress. Presence of peripheral monocytes (GFP+) was assessed in the lumbar spinal cord parenchyma. G, The number of GFP+ cells was quantified in the lumbar spinal cord parenchyma. Error bars indicate mean ± SEM.
Colony stimulating factor 1 receptor (CSF1R) antagonist PLX5622 depleted microglia in the spinal cord
Given the absence of increased monocytes in the spinal cord after RSD, we sought to further define the role of microglia in RSD-associated pain responses using a series of experiments using the specific CSF1R antagonist PLX5622 (Dagher et al., 2015; McKim et al., 2018). Following 14 d of treatment with PLX5622, microglia were eliminated from the spinal cord, based on Iba-1 labeling (Fig. 4A,B; p < 0.05). In addition, microglia-related CX3CR1 mRNA was determined in the spinal cord. The mRNA expression of CX3CR1 was reduced by PLX5622 in the cervical (Fig. 4C; p < 0.05), thoracic (Fig. 4D; p < 0.05), and lumbar (Fig. 4E; p < 0.05) spinal cord. Together, PLX5622 eliminated microglia from the spinal cord.
Figure 4.

CSF1R antagonist PLX5622 depleted microglia in the spinal cord. Male C57BL/6 mice were provided ad libitum diets of PLX5622 (PLX, 1200 ppm chow) or vehicle (Veh) chow for 14 d. Mice were perfused, and spinal cords were PFA fixed to determine microglial ablation. Microglial activation (Iba-1) was assessed in the lumbar spinal cord parenchyma. A, Representative images within the lumbar spinal cord parenchyma of Iba-1 labeling. B, Iba-1 proportional area was determined in the lumbar spinal cord parenchyma. A separate set of mice provided with PLX5622 or vehicle chow for 14 d were then exposed to RSD (Stress) or left undisturbed as controls. Fourteen hours after the last day of stress, spinal cords were collected and dissected for mRNA analyses. CX3CR1 mRNA levels were determined in the (C) cervical, (D) thoracic, and (E) lumbar spinal cord. Error bars indicate mean ± SEM. *Significantly different from control mice (p < 0.05; F-protected after analysis).
Microglial depletion with CSF1R antagonist prevented mechanical allodynia during RSD
In the next experiment, microglia were eliminated, and then mice were exposed to 6 d of RSD or left undisturbed as controls. Microglial depletion with PLX5622 had no effect on RSD-induced splenomegaly (Fig. 5A) or the peripheral production and release of Ly6Chi monocytes (Fig. 5B). These results are consistent with our previous findings with RSD and microglial elimination (McKim et al., 2018).
Figure 5.

Microglial depletion with CSF1R antagonist prevented mechanical allodynia during RSD. Male C57BL/6 mice were provided ad libitum diets of PLX5622 (PLX, 1200 ppm chow) or vehicle (Veh) chow for 14 d and then exposed to RSD (Stress) or left undisturbed as controls (Con). Fourteen hours after the last day of stress, samples (blood, spleen, spinal cord) were collected for cell or mRNA analyses. A, Spleen weight was determined. B, Percentage of Ly6Chi monocytes in circulation. In a separate experiment, mRNA levels of (C) CXCR2, (D) TNF-α, (E) IL-1β, (F) CCR2, and (G) TLR4 were determined in the lumbar spinal cord. Mice were tested for mechanical allodynia before RSD exposure and 12 h after the first, third, and sixth day of stress. H, Withdrawal threshold of mechanical stimulation to the hindpaw using the von Frey behavior test. Error bars indicate mean ± SEM. *Significantly different from control mice (p < 0.05; F-protected after analysis). Significantly different from control mice (p < 0.1; F-protected after analysis).
Next, mRNA levels of several cytokines, chemokines, and regulatory/adhesion molecules (Fig. 5C–G) were determined in the lumbar cord after PLX5622 treatment and RSD. IL-1β (Fig. 5C; stress × intervention interaction; F(1,31) = 6.743, p = 0.0143), CCR2 (Fig. 5D; stress × intervention interaction; F(1,12) = 6.152, p = 0.0289), and TLR4 (Fig. 5E; stress × intervention interaction; F(1,23) = 4.341, p = 0.0485) were increased with RSD, and induction of these genes was attenuated by microglial elimination. RSD also increased CXCR2 (Fig. 5F; main effect of stress; F(1,13) = 10.35, p = 0.0067) and TNF-α (Fig. 5G; main effect of stress; F(1,10) = 9.503, p = 0.0116) mRNA levels. These increases, however, were unaffected by microglial elimination.
Next, mechanical allodynia was determined and analyzed by two-way ANOVA at baseline and 12 h after the first, third, and final day of RSD (Fig. 5H). Before stress, each of the four treatment groups had comparable baseline withdrawal thresholds. Microglial depletion by PLX5622 prevented RSD-induced allodynia after 3 (stress × drug interaction; F(1,36) = 67.15, p < 0.0001) and 6 d of RSD (stress × drug interaction; F(1,36) = 235.5, p < 0.0001). Post hoc analysis confirmed that the Stress-vehicle group had the lowest withdrawal threshold at 3 and 6 d compared with all other experimental groups (p < 0.01 for each). These findings suggest that CNS microglia are critical for the promotion of increased mechanical allodynia in mice exposed to RSD.
Discussion
Here we reveal a novel mechanism by which stress promotes allodynia in the absence of injury. Stress promoted a heightened neuroinflammatory environment in the spinal cord marked by increased expression of inflammatory and immune mediators. Moreover, stress induced microglial activation in the spinal cord in regions specifically associated with pain signaling, independent of peripheral monocyte recruitment. Depletion of microglia with a CSF1R antagonist prevented the development of mechanical allodynia and reduced the mRNA expression of critical inflammatory markers involved in nociceptive signaling. These results indicate that microglia were involved in the development of pain during stress through an inflammatory-driven mechanism. These findings suggest that disruption in microglial functioning likely influences the neurocircuitry that underlies the development of pain associated with stress, and provide a novel cellular perspective on the mechanism of stress-induced pain.
A key finding was that RSD stress promotes region-specific microglial activation in the spinal cord. Activated microglia in the spinal cord have been directly implicated in the development of abnormal and exaggerated pain states (Ledeboer et al., 2005). The spinal cord dorsal horn is the first relay for pain transmission in the CNS (Zhuo et al., 2011). Nociceptive signaling initiated in peripheral sensory neurons enters the dorsal horn of the spinal cord and is transmitted to supraspinal structures, including the thalamus (Zhuo, 2007). Here we report microglial activation, marked by increased protein expression of microglia-specific marker P2Y12, specifically in the cap of the dorsal horn of the spinal cord. This effect was not seen in the lateral ventral horn or intermediate laminae, regions related to motor and locomotion processing. These same findings were corroborated using Iba-1 labeling for microglial activation in all three regions of the lumbar spinal cord (data not shown). These results are consistent with previous studies of microglial activation in the spinal cord dorsal horn associated with stress-induced hyperalgesia (Qi et al., 2016). Previous studies suggest that nerve injury-induced microglial activation, specifically within the spinal cord dorsal horn, contributes to the development of abnormal pain sensations (Yamamoto et al., 2015). We speculate that microglial activation during RSD induces a reorganization of the circuitry within the spinal cord dorsal horn that mediates the development of mechanical allodynia.
A notable finding was that stress promoted mechanical allodynia independent of peripheral monocyte recruitment. RSD corresponds with the recruitment of peripherally derived Ly6Chi monocytes to stress-responsive brain regions, where they differentiate into macrophages and propagate neuroinflammatory signaling to promote anxiety-like behavior (Wohleb et al., 2013; Weber et al., 2017; McKim et al., 2018). Despite increased inflammatory signaling in the spinal cord, mice exposed to RSD in the current study did not exhibit increased macrophage accumulation in the spinal cord by either flow cytometry (CD11b and CD45) or immunohistochemistry (CD45 or GFP). This spinal cord environment mimics what is seen after peripheral nerve injury in which there is increased immunoreactivity in the dorsal horn of the spinal cord but no monocyte recruitment (Alexander et al., 2009; Gu et al., 2016). Thus, we conclude that mechanical allodynia during RSD occurs independent of peripheral monocyte recruitment to the spinal cord.
The principal finding of this study was that stress promotes allodynia in a microglia-dependent manner. Here we show that microglial depletion with CSF1R antagonist PLX5622 prevented the development of mechanical allodynia after RSD. Macrophage-colony stimulating factor, which binds to CSF1R, is integral in initiating microglial activation in the dorsal horn of the lumbar spinal cord after peripheral nerve injury (Guan et al., 2016). Recent studies using PLX5622 show that microglia in the spinal cord are necessary for the generation and maintenance of neuropathic pain associated with peripheral nerve injury (Lee et al., 2018). Notably, microglial depletion in the absence of injury (i.e., during stress exposure) leads to similar behavioral outcomes observed after peripheral nerve damage (Lee et al., 2018). It is important to highlight that systemic administration of PLX5622 depletes microglia not only in the spinal cord, but also the brain and myeloid cells in other tissues (Dagher et al., 2015). Despite this, the results presented here provide evidence for the critical role of microglia in stress-induced pain, and implicate microglia as a potential cellular target for the alleviation of pain symptoms associated with stress. These findings provide further support for the role of microglia in mediating behavioral outcomes during stress (McKim et al., 2018).
A relevant finding was that stress promoted microglial activation through signaling pathways involving CCR2 and TLR4. RSD is associated with the production and release of Ly6Chi monocytes that express a chemokine receptor profile that allows for their trafficking to inflamed tissue (Weber et al., 2017). Inflammation and chemokine-dependent recruitment of monocytes to the spinal cord often coincide with chronic pain symptoms (Milligan and Watkins, 2009). For example, following nerve injury, CCR2-deficient mice did not show increased monocytes in the spinal cord and did not develop mechanical allodynia (Zhang and An, 2007; Zhang et al., 2007). Furthermore, mice lacking CCR2 showed a marked attenuation of monocyte recruitment and mechanical allodynia in response to inflammatory stimuli (Abbadie et al., 2003). Although we did not detect increased monocyte recruitment to the spinal cord with stress, microglia-depleted mice exhibited a significant reduction in CCR2 mRNA levels after RSD. CCR2 is expressed by sensory neurons and, when ligated by CCL2, can directly excite nociceptive neurons to promote pain behavior (Miotla Zarebska et al., 2017). Therefore, it is plausible that CCL2/CCR2 signaling pathways influence RSD-induced mechanical allodynia independent of monocyte recruitment. Microglial depletion was also associated with a significant reduction in TLR4 mRNA levels in the spinal cord after RSD. Evidence suggests that TLR4 is a critical microglial receptor in the onset and maintenance of pain (Scholz and Woolf, 2007). Microglial TLR4 is required for the induction of behavioral hypersensitivity in rodent models of neuropathy (Tanga et al., 2005), and blockade of TLR4 attenuated neuropathic pain symptoms in rats following chronic constriction injury (Jurga et al., 2016). TLR4 is also involved in the formation and activation of the NLRP3 inflammasome, which prolongs opioid-induced mechanical allodynia (Grace et al., 2016). Our findings here suggest that TLR4 signaling pathways may activate spinal microglia and potentiate pain transmission by neurons during stress exposure. We conclude that microglial activation of TLR4 and CCR2 signaling pathways contribute to mechanical allodynia during RSD.
Another relevant finding was that stress promoted an inflammatory gene profile in the spinal cord that corresponded with the development mechanical allodynia. A characteristic of the RSD model is minor cutaneous tissue damage (bite wounds) that is inflicted on the resident mice during stress. Because tissue injury may induce local inflammatory cytokine expression (Zhang and An, 2007), there is a possibility wounding might have an effect on pain. Activation of microglia in the spinal cord leads to the release of proinflammatory cytokines that may facilitate pain transmission (Ledeboer et al., 2005; Yamamoto et al., 2015). Among the proinflammatory cytokines, IL-1β and TNF-α showed the highest increases in mRNA expression after RSD in the lumbar spinal cord. Several studies show a link between inflammation and pain after IL-1β as a potent pronociceptive agent in the periphery and CNS (Ren and Torres, 2009). Genetic impairment of IL-1β signaling (Honore et al., 2006; Wolf et al., 2006) or intrathecal administration of IL-1 receptor antagonist (Sweitzer et al., 2001) reduces pain associated with nerve injury. Here microglia-depleted mice showed a significant reduction in the mRNA expression of IL-1β. Therefore, it is plausible that spinal microglia activated by RSD release IL-1β to enhance the transmission of pain information to the brain via ascending tracts originating in the spinal cord. TNF-α was also increased in the lumbar spinal cord during RSD, and several studies show a critical role for TNF-α in the modulation of nociceptive signaling (Leung and Cahill, 2010). Nonetheless, the RSD-associated increase in TNF-α was unaffected by PLX5622-mediated microglial depletion. Notably, the effects of cytokine and chemokine gene expression transcended the region-specific microglial activation pattern observed in the spinal cord. Other cell types may contribute to inflammatory gene expression and the maintenance of pain induced by inflammation, including astrocytes (Tanga et al., 2006). We conclude from the current findings that spinal IL-1β is a key inflammatory mediator in the transmission of pain to the brain in response to stress.
In conclusion, these findings reveal a critical role for microglia in the development of allodynia during RSD stress. The present study showed that mechanical allodynia during stress corresponded with enhanced inflammation within the lumbar spinal cord. Furthermore, microglia were activated selectively within the nociceptive neurocircuitry of the dorsal horn of the lumbar cord during stress independent of peripheral monocyte recruitment. Microglial depletion with CSF1R antagonist PLX5622 prevented the development of mechanical allodynia during stress that corresponded with a reduction in inflammatory markers associated with nociceptive signaling. These results indicate that microglia facilitate the transmission of pain to the brain during stress through an inflammatory-driven mechanism. Thus, microglia may serve as a therapeutic cellular target in the alleviation of pain associated with stress.
Footnotes
This work was supported by National Institute of Mental Health Grants R01-MH-093473 and R01-MH097243 to J.F.S. and a National Institute on Aging Grant R01-AG051902 to J.P.G. M.D.W. and C.M.S. were supported by National Institute of Dental and Craniofacial Research Training Grant T32-DE014320. D.B.M. was supported by National Institute of Mental Health Grant F31-MH109234. C.M.S. was supported by National Institute of Dental And Craniofacial Research Grant F30-DE026075. T.D.F. was supported by National Institute of Neurological Disorders and Stroke Grant F31-NS096921 and Foundation for Physical Therapy Promotion of Doctoral Studies Level II Scholarship. D.M.B. was supported by R01-NS074882. The Center for Brain and Spinal Cord Repair was supported by National Institute of Neurological Disorders and Stroke Core Grant P30-NS04587. We thank Plexxikon Inc. for the use of PLX5622; Rochelle Deibert for technical support; and Ohio State University Comprehensive Cancer Center's Analytical Cytometry and Nucleic Acid Shared Resources.
The authors declare no competing financial interests.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ (2003) Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A 100:7947–7952. 10.1073/pnas.1331358100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JK, DeVries AC, Kigerl KA, Dahlman JM, Popovich PG (2009) Stress exacerbates neuropathic pain via glucocorticoid and NMDA receptor activation. Brain Behav Immun 23:851–860. 10.1016/j.bbi.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JK, Cox GM, Tian JB, Zha AM, Wei P, Kigerl KA, Reddy MK, Dagia NM, Sielecki T, Zhu MX, Satoskar AR, McTigue DM, Whitacre CC, Popovich PG (2012) Macrophage migration inhibitory factor (MIF) is essential for inflammatory and neuropathic pain and enhances pain in response to stress. Exp Neurol 236:351–362. 10.1016/j.expneurol.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkinazi IY, Vershinina EA (1999) Pain sensitivity in chronic psychoemotional stress in humans. Neurosci Behav Physiol 29:333–337. 10.1007/BF02465346 [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Sheridan JF (2001) Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav 39:247–257. 10.1006/hbeh.2001.1653 [DOI] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL, Green KN (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation 12:139. 10.1186/s12974-015-0366-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP (2001) The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 90:1–6. 10.1016/S0304-3959(00)00490-5 [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM (2008) Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol 212:337–347. 10.1016/j.expneurol.2008.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Industry Analysts (2011) Global Pain Management Market to reach US $60 billion by 2015, according to a new report by Global Industry Analysts, Inc. PRWeb. Available at https://www.prweb.com/releases/2011/1/prweb8052240.htm.
- Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Reddy MK, Dagia NM, Sielecki T, Zhu MX, Satoskar AR, McTigue DM, Whitacre CC, Watkins LR (2016) Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A 113:E3441–E3450. 10.1073/pnas.1602070113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Rudy TE, Manzi S (2004) Effects of a stress-reduction program on psychological function, pain, and physical function of systemic lupus erythematosus patients: a randomized controlled trial. Arthritis Rheum 51:625–634. 10.1002/art.20533 [DOI] [PubMed] [Google Scholar]
- Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H, Wu LJ (2016) Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Rep 16:605–614. 10.1016/j.celrep.2016.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI (2016) Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat Neurosci 19:94–101. 10.1038/nn.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF (2012) Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26:1150–1159. 10.1016/j.bbi.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Wade CL, Zhong C, Harris RR, Wu C, Ghayur T, Iwakura Y, Decker MW, Faltynek C, Sullivan J, Jarvis MF (2006) Interleukin-1alphabeta gene-deficient mice show reduced nociceptive sensitivity in models of inflammatory and neuropathic pain but not post-operative pain. Behav Brain Res 167:355–364. 10.1016/j.bbr.2005.09.024 [DOI] [PubMed] [Google Scholar]
- Jurga AM, Rojewska E, Piotrowska A, Makuch W, Pilat D, Przewlocka B, Mika J (2016) Blockade of toll-like receptors (TLR2, TLR4) attenuates pain and potentiates buprenorphine analgesia in a rat neuropathic pain model. Neural Plast 2016:5238730. 10.1155/2016/5238730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz WA, Barkin RL (2010) Dilemmas in chronic/persistent pain management. Dis Mon 56:233–250. 10.1016/j.disamonth.2009.12.006 [DOI] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD (2008) Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci 28:5721–5730. 10.1523/JNEUROSCI.0256-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Page GG, Marucha PT, MacCallum RC, Glaser R (1998) Psychological influences on surgical recovery: perspectives from psychoneuroimmunology. Am Psychol 53:1209–1218. 10.1037/0003-066X.53.11.1209 [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR (2005) Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115:71–83. 10.1016/j.pain.2005.02.009 [DOI] [PubMed] [Google Scholar]
- Lee S, Shi XQ, Fan A, West B, Zhang J (2018) Targeting macrophage and microglia activation with colony stimulating factor 1 receptor inhibitor is an effective strategy to treat injury-triggered neuropathic pain. Mol Pain 14:1744806918764979. 10.1177/1744806918764979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L, Cahill CM (2010) TNF-α and neuropathic pain: a review. J Neuroinflammation 7:27. 10.1186/1742-2094-7-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF, Watkins LR (2003) Immune-to-central nervous system communication and its role in modulating pain and cognition: implications for cancer and cancer treatment. Brain Behav Immun 17 [Suppl 1]:S125–S131. 10.1016/S0889-1591(02)00079-X [DOI] [PubMed] [Google Scholar]
- Martinov T, Mack M, Sykes A, Chatterjea D (2013) Measuring changes in tactile sensitivity in the hind paw of mice using an electronic von Frey apparatus. J Vis Exp 82:e51212. 10.3791/51212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews A, Ridgeway V (1981) Personality and surgical recovery: a review. Br J Clin Psychol 20:243–260. 10.1111/j.2044-8260.1981.tb00525.x [DOI] [PubMed] [Google Scholar]
- McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP (2016) Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci 36:2590–2604. 10.1523/JNEUROSCI.2394-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, Ramirez-Chan K, Wang Y, Roeth RM, Sucaldito AD, Sobol CG, Quan N, Sheridan JF, Godbout JP (2018) Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry 23:1421–1431. 10.1038/mp.2017.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36. 10.1038/nrn2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, Stott B, Vincent TL (2017) CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage 25:406–412. 10.1016/j.joca.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padi SS, Kulkarni SK (2008) Minocycline prevents the development of neuropathic pain, but not acute pain: possible anti-inflammatory and antioxidant mechanisms. Eur J Pharmacol 601:79–87. 10.1016/j.ejphar.2008.10.018 [DOI] [PubMed] [Google Scholar]
- Persson AL, Hansson GA, Kalliomäki A, Moritz U, Sjölund BH (2000) Pressure pain thresholds and electromyographically defined muscular fatigue induced by a muscular endurance test in normal women. Clin J Pain 16:155–163. 10.1097/00002508-200006000-00009 [DOI] [PubMed] [Google Scholar]
- Qi J, Chen C, Meng QX, Wu Y, Wu H, Zhao TB (2016) Crosstalk between activated microglia and neurons in the spinal dorsal horn contributes to stress-induced hyperalgesia. Sci Rep 6:39442. 10.1038/srep39442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA (2003) Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 306:624–630. 10.1124/jpet.103.052407 [DOI] [PubMed] [Google Scholar]
- Reader BF, Jarrett BL, McKim DB, Wohleb ES, Godbout JP, Sheridan JF (2015) Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation, and anxiety. Neuroscience 289:429–442. 10.1016/j.neuroscience.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Torres R (2009) Role of interleukin-1beta during pain and inflammation. Brain Res Rev 60:57–64. 10.1016/j.brainresrev.2008.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, Godbout JP, Sheridan JF (2015) Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience 302:151–164. 10.1016/j.neuroscience.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki CM, Kim JK, Weber MD, Jarrett BL, Godbout JP, Sheridan JF, Humeidan M (2018) Ropivacaine and Bupivacaine prevent increased pain sensitivity without altering neuroimmune activation following repeated social defeat stress. Brain Behav Immun 69:113–123. 10.1016/j.bbi.2017.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10:1361–1368. 10.1038/nn1992 [DOI] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, et al. (2015) Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18:1081–1083. 10.1038/nn.4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA (2001) Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 103:529–539. 10.1016/S0306-4522(00)00574-1 [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 102:5856–5861. 10.1073/pnas.0501634102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, Deleo JA (2006) Role of astrocytic S100β in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience 140:1003–1010. 10.1016/j.neuroscience.2006.02.070 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW (2005) Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 28:101–107. 10.1016/j.tins.2004.12.002 [DOI] [PubMed] [Google Scholar]
- Weber MD, Godbout JP, Sheridan JF (2017) Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 42:46–61. 10.1038/npp.2016.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013) Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33:13820–13833. 10.1523/JNEUROSCI.1671-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2015) Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8:447. 10.3389/fnins.2014.00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y (2006) Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 120:315–324. 10.1016/j.pain.2005.11.011 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Terayama R, Kishimoto N, Maruhama K, Mizutani M, Iida S, Sugimoto T (2015) Activated microglia contribute to convergent nociceptive inputs to spinal dorsal horn neurons and the development of neuropathic pain. Neurochem Res 40:1000–1012. 10.1007/s11064-015-1555-8 [DOI] [PubMed] [Google Scholar]
- Zhang J, An J (2007) Cytokines, inflammation, and pain. Int Anesthesiol Clin 45:27–37. 10.1097/AIA.0b013e318034194e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S (2007) Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci 27:12396–12406. 10.1523/JNEUROSCI.3016-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. (2007) Neuronal mechanism for neuropathic pain. Mol Pain 3:14. 10.1186/1744-8069-3-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Wu G, Wu LJ (2011) Neuronal and microglial mechanisms of neuropathic pain. Mol Brain 4:31. 10.1186/1756-6606-4-31 [DOI] [PMC free article] [PubMed] [Google Scholar]