

Summary
Aims
Metformin is a commonly prescribed anti‐hyperglycaemic pharmacological agent, and it remains a staple in the management of type II diabetes. In addition to metformin's glucose lowering effects, research has indicated that metformin inhibits glycation‐mediated and oxidative modification of lipoprotein residues. The purpose was to discuss the effects of metformin as it relates to high‐density lipoprotein (HDL) and low‐density lipoprotein (LDL) modification.
Materials and methods
The purpose was to conduct a narrative and pragmatic review on the effects of metformin as it pertains to HDL and LDL modification.
Results
High‐density lipoprotein (HDL) concentration is a quantitative measure and therefore does not provide insight into its function, which is a qualitative property. Dysfunctional HDLs are unable to carry out functions normally associated with HDL because they can be modified by glycating agents. Metformin may counteract HDL dysfunction by abating HDL modification. Reductions in HDL modification may improve reverse cholesterol transport ability and thus possibly diminish cardiovascular risk. Similarly, metformin‐mediated attenuations in LDL modification may reduce their atherogenic potency.
Conclusion
Metformin may partially ameliorate HDL dysfunction and reduce LDL modification by inhibiting alpha‐dicarbonyl‐mediated modification of apolipoprotein residues; consequently, the results are salient because cardiovascular disease incidence may be reduced given that reverse cholesterol transport activity predicts risk, and modified LDL are proatherogenic.
Keywords: Gycation, Lipoproteins, Metformin, Type II diabetes
Introduction
The disease type II diabetes (T2D) poses a significant burden to clinicians. Even though it has genetic underpinnings, it is clear that environmental, socio‐economic and behavioural factors contribute to the manifestation of T2D 1. Due to T2D diffuse effects, it is difficult to manage. Indeed, T2D poses challenges because it can affect a myriad of organs. For instance, the ocular, renal, cardiovascular and the skeletal systems can be negatively affected 1. The latter being denoted as diabetic osteopenia, which has recently garnered attention 2. Because of its widespread effects, the most popular way to manage T2D is via behavioural and pharmacological therapy; more recently, metabolic surgery has been added to the armamentarium as well. However, the pharmacological management of T2D is common, as the majority of individuals with T2D are prescribed with some class of anti‐hyperglycaemic agent.
To ameliorate T2D‐related complications, metformin is the first‐line form of pharmacotherapy 3. While its effects on glycaemia are well known, less is understood on how it can abate high‐density lipoprotein (HDL) dysfunction. As the name suggests, HDL dysfunction is a state in which HDLs are dysfunctional. Simply stated, the ability of HDL to facilitate reverse cholesterol transport and exert antioxidative functions is impaired. Due to oxidative stress, dyslipidemia and hyperglycaemia, HDLs become prone to modification, and as a result, their function becomes perturbed. While there will be mention of how dyslipidemia contributes to HDL dysfunction, the emphasis will be on the deleterious effects of oxidative stress and hyperglycaemia. Namely, the discussion will centre on reactive species (e.g. alpha‐dicarbonyl compounds) and their end products: advanced glycation end products (AGE).
The venerable anti‐hyperglycaemic agent metformin can counter the effects of reducing sugars and, consequently, AGE on HDL function. To date, there has been some preclinical research on the efficacy of metformin in abating HDL dysfunction, but the research has not been consolidated in a review. Thus, the purpose is to characterize the effects of metformin on HDL function and low‐density lipoprotein (LDL) modification, and the intention is to offer a pragmatic explanation of the topic at hand. To their knowledge, the authors' partake in providing the first review on the effects of metformin as it pertains to lipoprotein modification.
Pathophysiology
As is common in the western societies, people often ingest foods that are processed and sugar enriched. From a chronic perspective, the ramifications of this behaviour are dire because there is limited self‐restraint until metabolic diseases manifest; the relatively short‐lived hyperglycaemic spurts that occur postprandially give rise to oxidative stress and inflammation 4, 5, 6. Coupled with physical inactivity and genetic predispositions, the repetitive bursts of oxidative stress and inflammation ultimately induce disease states marked by metabolic derangements. Congruently, endothelial dysfunction is propagated partly because nitric oxide bioavailability is decreased (Figure 1) 7. More recently, evidence has indicated that glucose excursions, as manifested after meal intake, also give rise to alpha‐dicarbonyl species 8. These compounds can further induce oxidative stress and inflammation and react negatively with protein constituents.
Figure 1.
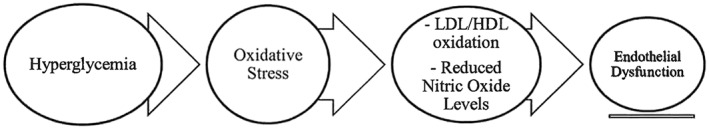
Effects of hyperglycaemia. HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
Traditionally, researchers believed that glycating species predominately reacted with amine groups on proteins with protracted half‐lives (e.g. collagen). However, that conclusion has been invalidated by more recent findings indicating that short‐lived apolipoproteins, as encapsulated in HDL, can indeed be modified 9. The Maillard reaction provides a conceptualization of the process. With the hyperglycaemic and prooxidant environment acting as an initial spark, reducing sugars such as glucose or ribose react with free amino acids, a process denoted as glycation 10. When the two entities react (sugar + free amino acid), they form one compound: a reversible sugar–protein adduct. Afterwards, the adduct gives rise to a more rigid covalently bonded compound: Amadori product 10. As an example, one of the most ubiquitous clinical measures is the assessment of haemoglobin A1c (HgbA1c), which is an Amadori product. After some additional incremental steps, the Amadori product can undergo further modification (e.g. oxidative modification) which gives rise to the formation of AGE at the endogenous level (Figure 2). AGE can also be ingested, and the concentration is influenced by food preparatory methods (e.g. heat application methods [broiling vs. boiling, etc.]) 11. Sometimes dicarbonyl compounds form and can be by‐products of the Maillard reaction, glycolytic pathway or lipid peroxidation 12. For the latter pathway, the terminal products are known as advanced lipoxidation end products (ALE). It is important to note that not all sugars and dicarbonyl species are equivalent in terms of their ability to hasten AGE or ALE formation. For instance, in relation to glucose, alpha‐dicarbonyl species are exponentially more potent in relation to glycating agents because they can induce AGE formation within hours 10, 13.
Figure 2.
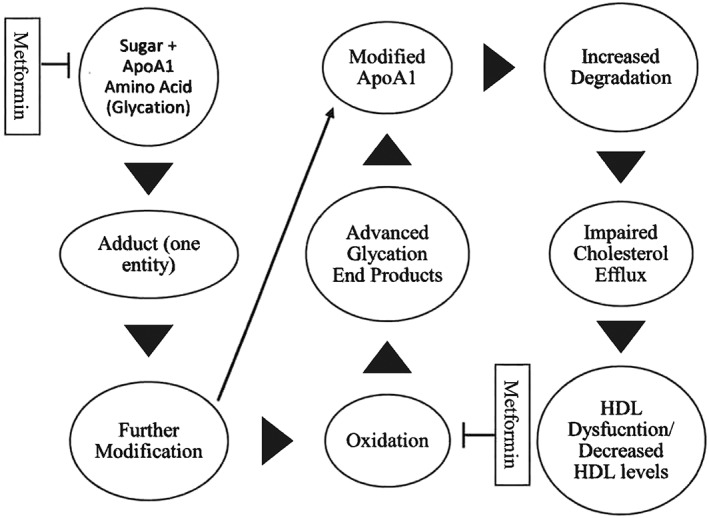
High‐density lipoprotein glycation and metformin. Simplified view of the effects of glycation. Metformin inhibits glycation by reducing blood glucose levels and glycating species (carbonyl compounds) and oxidative stress. ApoA‐1, Apolipoprotein A1; HDL, high‐density lipoprotein.
Whether derived from sugar–protein adducts or are by‐products of the glycolytic pathway, dicarbonyl species hastily react with amino acid residues 14; ultimately, the process may result in the formation of AGE or ALE depending on their origin. Afterwards, the tandem of AGE and the receptor for AGE (RAGE), among a few, increase oxidative stress and inflammation. Further, AGE cause physical alterations to proteins by making them more rigid and less labile. AGE and dicarbonyl levels are increased in patients with T2D and are implicated in microvascular and macrovascular complications. In addition to reducing blood glucose levels, inhibiting the formation of AGE can be achieved by (a) inhibiting the formation of sugar–protein adducts, (b) reducing oxidative stress, (c) scavenging dicarbonyl compounds (c) and potentiating dicarbonyl metabolism 15. Apart from reducing AGE‐related receptor expression and its glycaemic effects 16, metformin is known to be therapeutic by acting upon the latter three mechanisms.
High‐density lipoproteins
High‐density lipoproteins (HDLs) are heterogeneous because they can be classified into groups based upon their density, lipid content, size and antioxidant abilities to name a few. Although many subclasses exist, HDL2 and HDL3 define the more mature HDL subspecies 17. The mature classes manifest from nascent HDL and from its precursors: apolipoprotein A1 (ApoA‐1) and then pre‐B HDL. Other apolipoproteins associate with lipoprotein A1 (e.g. apolipoprotein A2), which, in contrast to ApoA‐1, is lapidated prior to secretion from the liver 18. As HDL becomes more saturated with cholesterol, phospholipids and triglycerides, it gradually becomes lipidated; under normal circumstances, maturing HDLs gain additional cardioprotective abilities when accessory proteins associate with them 17. The constituent proteins that comprise the subdivisions of HDL can be disparate and wide ranging; some of the aforesaid proteins or enzymes exert antioxidative and, therefore, anti‐atherogenic functions by inhibiting one of the primary propagators of endothelial dysfunction: oxidized LDL 17. In addition, ApoA‐1 initiates reverse cholesterol transport and serves as an anchor for other enzymes necessary for HDL maturation. There is also evidence that ApoA‐1 aids in antioxidative activity by removing or inhibiting the oxidation products of unsaturated fatty acids 19.
High‐density lipoprotein dysfunction
Within the clinical sector, there can be an oversimplified view of reverse cholesterol transport as it relates to HDL: the misconception that more is better with respect to HDL levels. To a certain threshold (about 60 mg/dL), while higher HDL concentrations are beneficial in healthy patients 20, elevated HDL levels do not always translate into improved outcomes in the clinical populations. Indeed, there were no benefits conferred by increasing HDL concentration in a long‐term randomized (36 months) study 21. Focusing strictly on HDL concentration neglects other facets or mediators of reverse cholesterol transport. HDL maturation is reliant upon several receptors and ancillary enzymes that collectively mediate its function. For example, under metabolic departures from normality, as observed in T2D, HDLs become enriched with triglycerides; as a result, their function becomes compromised due to increased enzyme activity 22. Indeed, there is one prime reason for the observed discrepancy in anti‐atherogenic, antioxidative and anti‐aptotic capacity of HDL in patients without T2D versus those with T2D: HDL dysfunction, which is precipitated by metabolic dysfunction (Figure 3).
Figure 3.
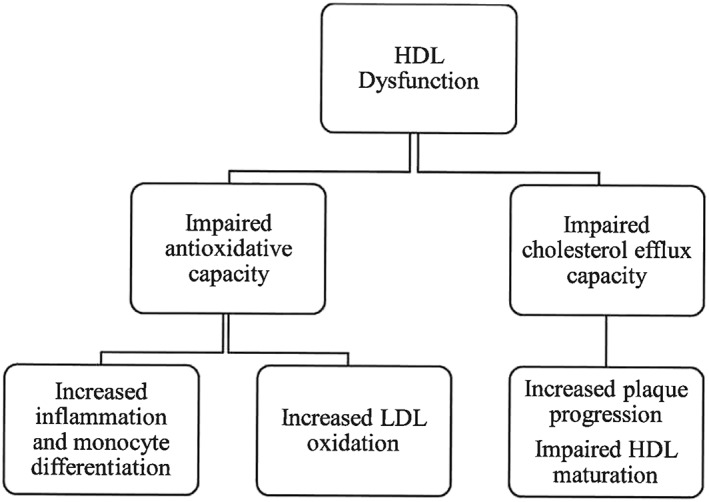
High‐density lipoprotein dysfunction: clinical repercussions. HDL, high‐density lipoprotein; LDL, low‐density lipoprotein.
HDLs are labelled as being chameleon‐like because their function is influenced by the physiologic environment (e.g. metabolic milieu). Upon exposure to virulent lipopolysaccharides (endotoxin) during the acute phase response, which induce a pro‐inflammatory effect, HDL in a likewise manner take on pro‐inflammatory properties; thus, their role becomes antithetical (opposite) to that under control conditions where anti‐inflammatory effects predominate 23. Similarly, in disease states marked by hyperglycaemia, dysfunctional HDLs are often pro‐inflammatory because they, as one reason, become glycated by reducing sugars (e.g. glucose or ribose) or modified by dicarbonyl compounds (e.g. methylglyoxal).
One of the most well‐known functions of HDL is its ability to siphon cholesterol from the periphery (e.g. from macrophages) and deliver it to the liver where it is ultimately excreted directly or as bile 24. Cholesterol efflux is an integral function of HDL because there is a negative relationship between cholesterol efflux capacity and coronary artery disease incidence; the incidence lessens as cholesterol efflux capacity improves 25.
While the majority of patients who are healthy have normal cholesterol efflux capacity, segments of the clinical population do not 26. Certainly, there are impediments to cholesterol efflux in the patient population consisting of people with T2D. Even in patients with T2D who are perceived to be optimally controlled (HgbA1c = 6.3%), cholesterol efflux is impaired 26. Therefore, while the concept or notion of being well controlled is traditionally associated with good glycaemic control, it may paint an inaccurate portrayal of the patient's current state because reverse cholesterol transport may still be hindered; indeed, relative to poorly controlled diabetes being well controlled confers benefit, but it should not thwart the patient or the overriding physician from striving for continued improvement. When dealing with an insidious and chronic disease such as T2D, adopting a static mentality does not behoove the patient because there will be continued deterioration in function as time elapses.
Similarly, impaired function manifests in well‐controlled patients with T2D (HgbA1c <7%) who are also non‐obese 27. In the aforementioned cohort and compared to a control group, HDL anti‐inflammatory and antioxidative functions were attenuated 27. Anti‐inflammatory activity can be construed as the ability to inhibit endothelial adhesion molecule expression and pro‐inflammatory cytokine release. In addition, diet controlled patients with T2D who exhibited mild hyperglycaemia (HgbA1 = 6.3%) were noted to have defects in antioxidative capacity 26. As one might expect, poorly controlled patients with T2D also had defective antioxidative and anti‐inflammatory activity 28. Therefore, HDL dysfunction manifests across the population continuum; as such, HDLs are unable to prevent LDL oxidation and monocyte infiltration into atherosclerotic plaques. As a consequence and over a long period of time, vulnerable plaques may be destabilized and ruptured, which may lead to an acute coronary syndrome.
Relative to HDL2, the type II diabetic state preferentially augments levels of the small dense subfraction of HDL: HDL3 29. Paradoxically, HDL3 may possess more potent antioxidative function 30. Also, HDL3 is able to maintain cell viability which may be mediated by sphingosine‐1‐phosphate 31. Improved cell viability translates into, for instance, a reduced infract area after cardiomyocytes are subjected to glucose deprivation and ischaemia 32 or a decreased risk of atherosclerotic plaque rupture due to reduced smooth muscle apoptosis. In a capacity, smooth muscle cells comprise and regulate the fibrous cap that overlies the plaque‐ridden area of macro vessels 33. Although the aforementioned properties may be predominately mediated by HDL3, the ability of HDL3 to carry out the pro‐survival functions is impaired by hyperglycaemia because of a decrease in sphingosine‐1‐phosphate association with HDL 34. Therefore, glycation or AGE accumulation may confound the relationship between HDL3 and cardiovascular risk.
HDL metabolism is hindered by two mechanisms: increased degradation and impaired maturation. Due to the hyperglycaemic and prooxidant milieu, HDL‐related proteins (e.g. ApoA‐1) become glycated. As a result, HDL behaviour becomes aberrant and more prone to degradation. Not only non‐enzymatic glycation leads to impaired function but also glycation induces a less stable particle, which is reflected by a decrease in particle size; HDL becomes more prone to degradation and may lead to the detachment of constituent proteins or enzymes that favourably modify cardiovascular risk 35. By using a novel isotope labelling technique to measure ApoA‐1 turnover in patients with diet controlled T2D, the researchers noted that in vivo glycated ApoA‐1 clearance rate was higher and associated with impaired HDL functions 26. The rapid degradation of both HDL and ApoA‐1 was closely associated with the severity of hyperglycaemia and resulted in reduced receptor‐dependent cholesterol efflux 26. As defined by traditional clinical criteria, the data highlight that normal or even elevated HDL levels in T2D does not equate to normal functionality. Rather, glycated ApoA‐1 is a marker of hyperglycaemia‐induced HDL dysfunction; the end result is that anti‐atherogenic and other protective functions are blunted.
In one manner, the progression from ApoA‐1 to nascent HDL and then to HDL3 may be impeded by physiologically relevant concentrations of AGE; the aforesaid statement implies that the early and later stages of maturation are affected 36. Therefore, given that cholesterol efflux is impaired, it begets (gives rise to) impaired particle maturation. Secondly, hindered metabolism is due to an inability to activate an enzyme that is critical for maturation: lecithin cholesterol acyltransferase (LCAT). Indeed, HDL maturation may be impaired due to reduced coupling with LCAT 37, which facilitates the conversion of free cholesterol to cholesterol ester. The purpose of LCAT is to maintain an appropriate gradient for passive cholesterol efflux from cells and into HDL 24. An interesting discovery is the possibility that glycated HDLs associate more readily with hepatic lipase, which may explain why the denser and less mature HDL3 variant is more readily expressed in the T2D state 38. HDL metabolism may be further affected by the enzyme‐mediated rise in the exchange of triglycerides from LDL to HDL 39. As a consequence, HDL function is also negatively influenced. In a way, metabolism and HDL function reciprocate one another.
The clinical repercussions are salient for two reasons: coronary artery plaque progression is exacerbated, and as a result, cardiovascular disease risk is increased because antioxidative function, anti‐inflammatory function and reverse cholesterol transport are impaired 25, 40. Interestingly, across the spectrum of one, two and three vessel coronary artery disease among patients with T2D, HDL‐related antioxidative function decreased, while ApoA‐1 glycation increased 41.
Mechanisms of the metformin effect
Independent of its anti‐hyperglycaemic and insulin sensitizing actions, metformin moderates AGE accumulation by directly sequestering dicarbonyl compounds. The scavenging‐like characteristic is a unique characteristic of the guanidine class of medications. Specifically, metformin's molecular structure facilitates the reaction with dicarbonyl compounds (e.g. methylglyoxal and glyoxal) and, as a result, quells AGE formation 42. The product of the reaction was initially believed to be the formation of an innocuous seven‐ringed compound: triazepinone 42. More recently, researchers noted a condensation product with a similar molecular weight as in the antecedent study but classified it into a different category because of less carbon and nitrogen bonding within the ring of the molecule 43. Concurrently, the investigators noted that metformin treated patients with T2D excreted the product in their urine at clinically relevant concentrations (18.8 nM to 4.3 μM) given that it paralleled plasma levels of methylglyoxal 43. There was a direct positive relationship (r 2 = 0.5463) between urinary metformin concentration and levels of the inert by‐product, which further provides credence to the notion that metformin mediates dicarbonyl inactivation. It can be inferred from the aforesaid study that metformin may have varying interindividual effects given that genetic polymorphisms in organic cation transporters exist; organic cation transporters influence metformin's pharmacokinetics 44. Given that multiple products of the metformin‐methylglyoxal reaction exist, additional research is required to delineate which product is most stable and therefore that predominates.
Through the innate glyoxalase pathway, metformin may increase the degradation of methylglyoxal which results in the formation of lactic acid 45; the prior conclusion requires additional corroborating evidence. However, as the metformin dose was gradually titrated above 1 g/d, there was a reduction in plasma‐derived levels of methylglyoxal, while 3‐deoxyglucosone levels remained unscathed. Importantly, subjects on insulin or sulfonylurea therapy did not procure the benefits even though the level of glycaemic control was equitable between metformin and non‐metformin treated subjects (HbA1c ≈ 8.0%) 45. Subsequently, glycaemia is not the only or principal mediator of dicarbonyl levels. One drawback, however, was that there was some overlap between the medication regimens (i.e. some subjects who were taking metformin were also on sulfonylurea or insulin). Other trials have attained similar conclusions, as treatment with metformin (2 g/d) was shown to reduce 3‐deoxyglucosone levels 46. Further, an in vitro study has substantiated the fact that metformin mitigates AGE formation by noting reduced vascular and plasma‐derived AGE 47; the effects are diffuse as AGE accumulation has also been shown to be attenuated by metformin in renal and osteoblast‐like cell cultures 48, 49. Consequently, by acting as a scavenger, reducing agents such as dicarbonyl compounds are unable to modify proteins and exert their deleterious effects.
Another feature of metformin is its ability to dampen the hyperglycaemic‐induced generation of reactive oxygen species at the endothelial and systemic level 47. As a result, LDL and HDL become less prone to oxidative modification; also, there would be reduced monocyte differentiation and neutrophil recruitment. The latter (neutrophils) perpetuate redox reactions through the secretion of myeloperoxidase 50. Further, the production of AGE was diminished because a prooxidant milieu does play a role in their formation. Whether it is a by‐product of reductions in the feed‐forward effects of glycation species, RAGE expression was also diminished 49. By blocking the AGE‐RAGE axis, there is reduced oxidative stress, inflammation and programmed cell death 48, 49. Indeed, one reason for plaque rupture is smooth muscle death which is propagated by inflammatory mediators. Ultimately, although it remains to be definitively verified, cardiovascular disease risk may become lessened through the partial restoration in endothelial function 47.
Metformin and lipoprotein modification
Through the mechanisms that were elaborated on in the preceding two paragraphs, metformin is able to inhibit HDL dysfunction. With the glycation of specific HDL residues (e.g. lysine) and the subsequent formation of AGE, modified HDL fails to facilitate cholesterol efflux; the results are strikingly disparate from that of unmodified or control HDL. Moreover, initial glycation followed by AGE accumulation attenuates antioxidant activity as exemplified by reduced PON1 activity 51. Metformin is able to partially revert the negative effect of glycation and subsequent AGE formation.
Two in vitro studies have demonstrated that reverse cholesterol transport is, at least, partially preserved with metformin treatment. Although one study did not specify which HDL subclass was investigated, the other used HDL3 52, 53. Both studies investigated cholesterol efflux potential from macrophages. In the first study, HDL3 cholesterol efflux capacity was repressed in the presence of AGE modified albumin; the results were replicated even after AGE modified albumin was extracted from the medium 53. The residual effect may have been due to macrophage‐mediated engulfment of modified albumin; through inflammatory mechanisms, receptors integral to active or passive cholesterol transportation may be downregulated and, therefore, account for the discrepancy in cholesterol efflux capacity 53. In addition, incubation of mice peritoneal macrophages with glycating agents attenuated HDL3 cholesterol efflux. Through increased HDL3 interaction with efflux receptors or reduced AGE formation, metformin was able to significantly attenuate the impairment in cholesterol efflux capacity in both circumstances 53. In parallel, the second study corroborated the findings, but investigated the direct effects of 3‐deoxyglucosone‐mediated modification of human HDL on cholesterol efflux capacity 52. In the absence of metformin, there was a precipitous rise in AGE formation as judged by carboxyl methyl lysine levels, metformin‐attenuated HDL susceptibility to glycation‐mediated AGE formation. As a consequence, cholesterol efflux capacity from differentiated THP‐1 cells was significantly improved. The findings are intriguing because metformin improved cholesterol efflux in a similar manner to aminoguanidine. The latter inhibits glycation more significantly than metformin 52, but both share similarities in their chemical structure.
AGE accumulation hinders HDL maturation by two mechanisms: attenuated cholesterol efflux capacity and reduced LCAT activity. Indeed, the conversion of free cholesterol to cholesterol ester, an index of LCAT activity, is reduced in the presence of dicarbonyl species 37, 54. LCAT is integral to reverse cholesterol transport because it helps to foster HDL3 formation from nascent HDL. On a broader scale, across the compendium of one, two and three vessel coronary artery disease, LCAT activity progressively deteriorated 40. Glycation and subsequent AGE formation alter enzyme or protein conformation, thereby affecting the ability of enzymes to associate with their corresponding substrates. When metformin and methylglyoxal were incubated with ApoA‐1 of discoidal HDL at equimolar concentrations, cholesterol ester formation was significantly improved 54. As such, the indication is that metformin is able to partially preserve the ability of LCAT to anchor with ApoA‐1 of discoidal HDL because methylglyoxal‐induced modification was lessened 54. As a consequence, HDL maturation may proceed in a less hindered manner because LCAT plays a major role in the formation of more mature HDL.
Similar to oxidized LDL, glycated LDL and even modified albumin may be engulfed by macrophages, which propagate foam cell formation 55. While mitigating the formation of oxidative products, one study did indeed find that glycated LDL formed cholesterol rich macrophages. Albeit carried out at supraphysiological doses (10 mmol/L), equimolar levels of metformin were able to attenuate apolipoprotein B modification by glycolaldehyde or methylglyoxal as noted by an index of protein modification: particle charge; however, larger than equimolar concentrations of metformin were needed to prevent cross‐link formation 55. Moreover, in another study, Rabbani et al. noted that methylglyoxal‐induced AGE modification of apolipoprotein B, the lipoprotein of LDL, was diminished with metformin therapy; also, oxidative modification of LDL residues was lessened 9. Again, subjects not receiving metformin (e.g. insulin, gliclazide and glimepiride) did obtain the benefits. The median intake of metformin was 1.5 g/d. Another study corroborated the findings in 35 subjects with T2D; principally, subjects receiving 500 mg/d of metformin at baseline exhibited reductions in oxidative‐induced LDL modification after receiving an additional 500 mg/d over a 6‐month time frame 56. In their analysis, metformin dose served as an independent predictor of reduced LDL modification 56. Not only does posttranslational modification of LDL residues induce atherosclerosis, but there is evidence that suggests that methylglyoxal‐mediated modification does the same by increasing LDL density and their affinity for arterial proteoglycans 57. Therefore, inhibiting LDL modification would decrease foam cell formation because of reduced internalization by macrophages; as a result, the induction of pro‐inflammatory stimuli would be partly abrogated 58. However, the studies did not examine the effects of metformin in those respects.
Clinical ramifications
Recall, to abate dicarbonyl accumulation, a medication may need to have multifaceted effects because dicarbonyl compounds have complex origins (e.g. lipid peroxidation and glycolytic pathway). Thus, from a pharmacological perspective, strictly managing hyperglycaemia may not portend significant reductions in dicarbonyl stress. Although attenuating glucose spikes reduces oxidative stress, reducing postprandial blood glucose peaks with pharmacotherapy will not fully offset the ingestion of a sugar‐laden meal because glucose will still be metabolized; therefore, the formation of some dicarbonyl compounds (e.g. methylglyoxal) will persist because they manifest from glucose degradation. This contention is strengthened by empirical data noting no effect of insulin therapy on methylglyoxal concentration 59. At the clinical level, reductions in glycaemia are not trivial, but at the same juncture, modification of LDL or HDL residues is likely principally mediated by dicarbonyl species; for instance, there was no difference in glucose‐mediated glycation when comparing patients with T2D versus patients allocated into the control group even though fasting glucose levels differed significantly (8.75 ± 2.51 mM vs. 5.14 ± 0.74 mM, respectively) 9. Yes, it is important to improve glycaemia, but it is not the only clinical parameter that should be targeted via therapy; indeed, additional benefit can be garnered from reductions in dicarbonyl species as well.
To reap the benefits of metformin, it appears that ≥1 g/d may be needed. What differentiates metformin from other anti‐diabetic medications is its direct dampening effect on dicarbonyl compounds, which is in addition to its adjuvant effects: anti‐hyperglycaemia and antioxidant capabilities. By trapping dicarbonyl species and attenuating oxidative stress, the effects are twofold: reduced HDL and LDL modification. Thus, by limiting oxidative and dicarbonyl‐mediated modification of LDL, the induction of atherogenesis is partly mitigated 57. Also, not only are HDLs less prone to degradation 26, but HDL maturation would be less affected because cholesterol efflux capacity is partly retained. Although purely speculative at this juncture, HDL‐related antioxidative and anti‐apoptotic capacity may be less amenable to the deleterious effects of dicarbonyl compounds. In the clinical realm, the positive outcomes would prove to be auspicious because reverse cholesterol transport capacity is negatively related with cardiovascular disease incidence 25. Further, impeding AGE or ALE formation alone may confer reductions in cardiovascular risk and microvascular complications 60. There have been trials that suggest metformin does indeed reduce cardiovascular disease risk 61, but it remains an ongoing debate 62. With metformin therapy, cardiovascular risk reduction may also be a by‐product of improvement in lipoprotein subclasses: reduced small dense LDL and HDL 63.
Other treatment options
If the patient heeds the clinician's advice, arguably the most impactful form of therapy will invariably be lifestyle modification, which is denoted as dietary modification and exercise adherence; it is untenable to suggest that pharmacotherapy can mimic or be on par with the effects of lifestyle modification because the burgeoning incidence of T2D can be largely attributed to physical inactivity and poor eating habits. Further, the effects of medications can be isolated (e.g. anti‐hyperglycaemic or antihyperlipidemic), while lifestyle modification positively influences multiple organs and clinical parameters (e.g. bone density, overall fitness and glycaemia). For instance, a study noted that intensive medical therapy did not fully restore HDL function 64. In relation, by partitioning healthy meals throughout the day and engaging in physical activity, blood glucose deviations will be minimized and antioxidative capacity bolstered. Notwithstanding metformin, other forms of pharmacotherapy may have some positive effects on dicarbonyl accumulation.
While the research remains sparse, other anti‐diabetic agents likely have a twofold effect on dicarbonyl accumulation and AGE formation via glucose depressing and antioxidative actions. For instance, although not as widely prescribed, thiazolidinediones fulfil that criteria 65; also, there is evidence that thiazolidinediones reduce RAGE expression 66. A sulfonylurea was also shown to reduce 3‐deoxyglucosone levels 46. Although theoretical, renal sodium glucose inhibitors also may impede dicarbonyl accumulation because glycosuria is promoted; consequently, glucose metabolism is attenuated and the induction of dicarbonyl stress may be lessened. Indeed, what the disparity is between metformin and other classes of medication is subject to further investigation.
Conclusions
The prime message that is being conveyed is that there is evidence that indicates metformin impedes glycation and oxidative‐mediated modification of HDL and/or LDL; thus, LDL‐induced atherogenesis may be mitigated, and HDL may retain their cardioprotective functions (i.e. antioxidative, reverse cholesterol transport and anti‐aptotic functions). In turn, there may be reduced cardiovascular risk. The purported effects are in line with the documented effects of metformin on oxidative stress and glycating species.
While the study of the effects of pharmacotherapy on HDL function and LDL modification remains in its infancy, there is empirical data demonstrating that metformin at least partially reverses HDL dysfunction and LDL modification. Metformin acts across the physiological spectrum by reducing glycaemia, oxidative stress, dicrabonyl accumulation, AGE accumulation and RAGE expression. In doing so, metformin may be able to salvage the positive effects of HDL in disease states marked by metabolic perturbations. To recapitulate, metformin may restore cholesterol efflux capacity, inhibit HDL and LDL glycation and maintain LCAT activity in the midst of chronic hyperglycaemia. It is to be determined whether the preclinical results translate into the clinical setting, but they are nonetheless intriguing because cardiovascular risk may be reduced as a result.
For future considerations, the reputed effects of metformin (i.e. improvements in cholesterol efflux capacity) will need to be verified in clinical and in additional in vitro studies. It is important to understand the effects of metformin by installing a paradigm that studies the effects under physiologically relevant conditions (e.g. metformin dose). Whether metformin improves HDL function through attenuations in lipid levels requires further study. It is pertinent to segregate the effects of early glycation versus AGE formation on HDL function. Additionally, although metformin may improve HDL function, it does not fully restore its capacity, and it is yet to be elucidated how it would impact cardiovascular risk. Finally, delineating to what extent metformin can limit HDL posttranslational modification spurred by oxidative stress is subject to further investigation.
Conflict of Interest Statement
The authors report no conflict of interest.
Kheniser, K. G. , Kashyap, S. R. , and Kasumov, T. (2019) A systematic review: the appraisal of the effects of metformin on lipoprotein modification and function. Obesity Science & Practice, 5: 36–45. 10.1002/osp4.309.
References
- 1. Wu Y, Ding Y, Tanaka Y, Zhang W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci 2014; 11: 1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallander M, Axelsson K, Nilsson A, Lundh D, Lorentzon M. Type 2 diabetes and risk of hip fractures and non‐skeletal fall injuries in the elderly: a study from the fractures and fall injuries in the elderly cohort (FRAILCO). J Bone Miner Res 2017; 32: 449–460. [DOI] [PubMed] [Google Scholar]
- 3. Association AD . Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes‐2018. Daibetes Care 2018; 41: S73–S85. [DOI] [PubMed] [Google Scholar]
- 4. Gallo A, Ceolotto G, Pinton P, et al. Metformin prevents glucose‐induced protein kinase C‐B2 activation in human umbilical vein endothelial cells through an antioxidant mechanism. Diabetes 2005; 54: 1123–1131. [DOI] [PubMed] [Google Scholar]
- 5. Ding H, Aljofan M, Triggle C. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J Cell Physiol 2007; 212: 682–689. [DOI] [PubMed] [Google Scholar]
- 6. Aljofan M, Ding H. High glucose increases expression of cyclooxygenase‐2, increases oxidative stress and decreases the generation of nitric oxide in mouse microvessel endothelial cells. J Cell Physiol 2010; 222: 669–675. [DOI] [PubMed] [Google Scholar]
- 7. Mahrouf‐Yorgov M, Marie N, Borderie D, et al. Metformin suppresses high glucose‐induced poly (adenosine diphosphate‐ribose) polymerase overactivation in aortic endothelial cells. Metabolism 2009; 58: 525–533. [DOI] [PubMed] [Google Scholar]
- 8. Beisswenger P, Howell S, O'Dell R, Wood M, Touchette A, Szwergold B. alpha‐Dicarbonyls increase in the postprandial period and reflect the degree of hyperglycemia. Diabetes Care 2001; 24: 726–732. [DOI] [PubMed] [Google Scholar]
- 9. Rabbani N, Chittari M, Bodmer C, Zehnder D, Ceriello A, Thornalley P. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes 2010; 59: 1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thornalley P, Langborg A, Minhas H. Formation of glyoxal, methylglyoxal and 3‐deoxyglucosone in the glycation of proteins by glucose. Biochem J 1999; 344: 109–116. [PMC free article] [PubMed] [Google Scholar]
- 11. Goldberg T, Cai W, Peppa M, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc 2004; 104: 1287–1291. [DOI] [PubMed] [Google Scholar]
- 12. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol 2014; 9: 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thornalley P. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification – a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol 1996; 27: 565–573. [DOI] [PubMed] [Google Scholar]
- 14. Golizeh M, Lee K, Ilchenko S, et al. Increased serotransferrin and ceruloplasmin turnover in diet‐controlled patients with type 2 diabetes. Free Radic Biol Med 2017; 113: 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khalifah R, Baynes J, Hudson B. Amadorins: novel post‐Amadori inhibitors of advanced glycation reactions. Biochem Biophys Res Commun 1999; 257: 251–258. [DOI] [PubMed] [Google Scholar]
- 16. Ouslimani N, Mahrouf M, Peynet J, et al. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin‐like oxidized receptor 1. Metabolism 2007; 56: 308–313. [DOI] [PubMed] [Google Scholar]
- 17. Kontush A, Chapman MJ. Functionally defective high‐density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev 2006; 58: 342–374. [DOI] [PubMed] [Google Scholar]
- 18. Pownall H, Gillard B, Gotto A. Setting the course for ApoAII: a port in sight? Clin Lipidol 2013; 8: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Navab M, Hama SY, Anantharamaiah GM, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res 2000; 41: 1495–1508. [PubMed] [Google Scholar]
- 20. Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009; 302: 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boden W, Probstfield J, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365: 2255–2267. [DOI] [PubMed] [Google Scholar]
- 22. Patel S, Puranik R, Nakhla S, et al. Acute hypertriglyceridaemia in humans increases the triglyceride content and decreases the anti‐inflammatory capacity of high density lipoproteins. Atherosclerosis 2009; 204: 424–428. [DOI] [PubMed] [Google Scholar]
- 23. Vaisar T, Tang C, Babenko I, et al. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J Lipid Res 2015; 56: 1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. März W, Kleber M, Scharnagl H, et al. HDL cholesterol: reappraisal of its clinical relevance. Clin Res Cardiol 2017; 106: 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saleheen D, Scott R, Javad S, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case‐control study. Lancet Diabetes Endocrinol 2015; 3: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kashyap S, Osme A, Ilchenko S, et al. Glycation reduces the stability of ApoAI and increases HDL dysfunction in diet‐controlled type 2 diabetes. J Clin Endocrinol Metab 2018; 103: 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ebtehaj S, Gruppen E, Parvizi M, Tietge U, Dullaart R. The anti‐inflammatory function of HDL is impaired in type 2 diabetes: role of hyperglycemia, paraoxonase‐1 and low grade inflammation. Cardiovasc Diabetol 2017; 16: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgantini C, Natali A, Boldrini B, et al. Anti‐inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011; 60: 2617–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krauss R. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care 2004; 27: 1496–1504. [DOI] [PubMed] [Google Scholar]
- 30. Kontush A, Chantepie S, Chapman M. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol 2003; 23: 1881–1888. [DOI] [PubMed] [Google Scholar]
- 31. Christoffersen C, Obinata H, Kumaraswamy S, et al. Endothelium‐protective sphingosine‐1‐phosphate provided by HDL‐associated apolipoprotein M. Proc Natl Acad Sci U S A, Early Ed 2011; 108: 9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Durham K, Chathely K, Trigatti B. High‐density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR‐B1, PI3K, and AKT1 and 2. Biom J 2018; 475: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clarke M, Bennett M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am J Nephrol 2006; 26: 531–535. [DOI] [PubMed] [Google Scholar]
- 34. Brinck J, Thomas A, Lauer E, et al. Diabetes mellitus is associated with reduced high‐density lipoprotein sphingosine‐1‐phosphate content and impaired high‐density lipoprotein cardiac cell protection. Arterioscler Thromb Vasc Biol 2016; 36: 817–824. [DOI] [PubMed] [Google Scholar]
- 35. Godfrey L, Yamada‐Fowler N, Smith J, Thornalley P, Rabbani N. Arginine‐directed glycation and decreased HDL plasma concentration and functionality. Nutr Diabetes 2014; 4: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoang A, Murphy A, Coughlan M, et al. Advanced glycation of apolipoprotein A‐I impairs its anti‐atherogenic properties. Diabetologia 2007; 50: 1770–1779. [DOI] [PubMed] [Google Scholar]
- 37. Nobecourt E, Davies M, Brown B, et al. The impact of glycation on apolipoprotein A‐I structure and its ability to activate lecithin: cholesterol acyltransferase. Diabetologia 2007; 50: 643–653. [DOI] [PubMed] [Google Scholar]
- 38. Hedrick C, Thorpe S, Fu M, et al. Glycation impairs high‐density lipoprotein function. Diabetologia 2000; 43: 312–320. [DOI] [PubMed] [Google Scholar]
- 39. Dallinga‐Thie G, Dullaart R, van Tol A. Concerted actions of cholesteryl ester transfer protein and phospholipid transfer protein in type 2 diabetes: effects of apolipoproteins. Curr Opin Lipidol 2007; 18: 251–257. [DOI] [PubMed] [Google Scholar]
- 40. Pu L, Lu L, Zhang R, et al. Glycation of apoprotein A‐I is associated with coronary artery plaque progression in type 2 diabetic patients. Diabetes Care 2013; 36: 1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen Y, Ding F, Sun J, et al. Association of elevated ApoA‐I glycation and reduced HDL‐associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc Diabetol 2015; 14: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruggiero‐Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol 1999; 58: 1765–1773. [DOI] [PubMed] [Google Scholar]
- 43. Kinsky O, Hargraves T, Anumol T, et al. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem Res Toxicol 2016; 29: 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tzvetkov M, Vormfelde S, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther 2009; 86: 299–306. [DOI] [PubMed] [Google Scholar]
- 45. Beisswenger P, Howell S, Touchette A, Lal S, Szwergold B. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999; 48: 198–202. [DOI] [PubMed] [Google Scholar]
- 46. Engelen L, Lund S, Ferreira I, et al. Improved glycemic control induced by both metformin and repaglinide is associated with a reduction in blood levels of 3‐deoxyglucosone in nonobese patients with type 2 diabetes. Eur J Endocrinol 2011; 164: 371–379. [DOI] [PubMed] [Google Scholar]
- 47. Sena C, Matafome P, Louro T, Nunes E, Fernandes R, Seiça R. Metformin restores endothelial function in aorta of diabetic rats. Br J Pharmacol 2011; 163: 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schurman L, McCarthy A, Sedlinsky C, et al. Metformin reverts deleterious effects of advanced glycation end‐products (AGEs) on osteoblastic cells. Exp Clin Endocrinol Diabetes 2008; 116: 333–340. [DOI] [PubMed] [Google Scholar]
- 49. Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Metformin inhibits advanced glycation end products (AGEs)‐induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm Metab Res 2012; 44: 891–895. [DOI] [PubMed] [Google Scholar]
- 50. Mayyas F, Al‐jarrah M, Ibrahim K, Alzoubi K. Level and significance of plasma myeloperoxidase and the neutrophil to lymphocyte ratio in patients with coronary artery disease. Exp Ther Med 2014; 8: 1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bacchetti T, Masciangelo S, Armeni T, Bicchiega V, Ferretti G. Glycation of human high density lipoprotein by methylglyoxal: effect on HDL‐paraoxonase activity. Metabolism 2014; 63: 307–311. [DOI] [PubMed] [Google Scholar]
- 52. Matsuki K, Tamasawa N, Yamashita M, et al. Metformin restores impaired HDL‐mediated cholesterol efflux due to glycation. Atherosclerosis 2009; 206: 434–438. [DOI] [PubMed] [Google Scholar]
- 53. Machado A, Pinto R, Moysés Z, Nakandakare E, Quintão E, Passarelli M. Aminoguanidine and metformin prevent the reduced rate of HDL‐mediated cell cholesterol efflux induced by formation of advanced glycation end products. Int J Biochem Cell Biol 2006; 38: 392–403. [DOI] [PubMed] [Google Scholar]
- 54. Nobécourt E, Zeng J, Davies MJ, et al. Effects of cross‐link breakers, glycation inhibitors and insulin sensitisers on HDL function and the non‐enzymatic glycation of apolipoprotein A‐I. Diabetologia 2008; 51: 1008–1017. [DOI] [PubMed] [Google Scholar]
- 55. Brown B, Mahroof F, Cook N, van Reyk D, Davies M. Hydrazine compounds inhibit glycation of low‐density lipoproteins and prevent the in vitro formation of model foam cells from glycolaldehyde‐modified low‐density lipoproteins. Diabetologia 2006; 49: 775–783. [DOI] [PubMed] [Google Scholar]
- 56. Ohira M, Yamaguchi T, Saiki A, et al. Metformin reduces circulating malondialdehyde‐modified low‐density lipoprotein in type 2 diabetes mellitus. Clin Invest Med 2014; 37: E243–E251. [DOI] [PubMed] [Google Scholar]
- 57. Rabbani N, Godfrey L, Xue M, et al. Glycation of LDL by methylglyoxal increases arterial atherogenicity: a possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011; 60: 1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hodgkinson C, Laxton R, Patel K, Ye S. Advanced glycation end‐product of low density lipoprotein activates the toll‐like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol 2008; 28: 2275–2281. [DOI] [PubMed] [Google Scholar]
- 59. Sakharova O, Lleva R, Dziura J, et al. Effects on post‐prandial glucose and AGE precursors from two initial insulin strategies in patients with type 2 diabetes uncontrolled by oral agents. J Diabetes Complications 2012; 26: 333–338. [DOI] [PubMed] [Google Scholar]
- 60. Koska J, Saremi A, Howell S, et al. Advanced glycation end products, oxidation products, and incident cardiovascular events in patients with type 2 diabetes. Diabetes Care 2018; 41: 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Group UPDSU . Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352: 854–865. [PubMed] [Google Scholar]
- 62. Griffin S, Leaver J, Irving G. Impact of metformin on cardiovascular disease: a meta‐analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017; 60: 1620–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goldberg R, Temprosa M, Otvos J, et al. Lifestyle and metformin treatment favorably influence lipoprotein subfraction distribution in the diabetes prevention program. J Clin Endocrinol Metabol 2013; 98: 3989–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kashyap S, Kheniser K, Ling L, Bena J, Kasumov T. The therapeutic efficacy of intensive medical therapy in ameliorating high‐density lipoprotein dysfunction in subjects with type two diabetes. Lipids Health Dis 2016; 15: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chung S, Kim M, Lee J, et al. Mechanism for antioxidative effects of thiazolidinediones in pancreatic β‐cells. Am J Physiol Endocrinol Metab 2011; 301: E912–E921. [DOI] [PubMed] [Google Scholar]
- 66. Marx N, Walcher D, Ivanova N, et al. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 2004; 53: 2662–2668. [DOI] [PubMed] [Google Scholar]