

Summary
Signalling through Toll‐like receptors (TLRs) may play a role in the pathogenesis of autoimmune diseases, such as multiple sclerosis (MS). In the present study, the expression of TLR‐2, ‐4 and ‐9 was significantly higher on CD4+ and CD8+ T‐cells from MS patients compared to healthy individuals. Following in‐vitro activation, the proportion of interleukin (IL)‐17+ and IL‐6+ CD4+ and CD8+ T‐cells was higher in the patients. In addition, the proportion of IFN‐γ‐secreting TLR+ CD8+ T‐cells was increased in MS patients. Among different IL‐17+ T‐cell phenotypes, the proportion of IL‐17+ TLR+ CD4+ and CD8+ T‐cells producing IFN‐γ or IL‐6 were positively associated with the number of active brain lesions and neurological disabilities. Interestingly, activation of purified CD4+ and CD8+ T‐cells with ligands for TLR‐2 (Pam3Csk4), TLR‐4 [lipopolysaccharide (LPS)] and TLR‐9 [oligodeoxynucleotide (ODN)] directly induced cytokine production in MS patients. Among the pathogen‐associated molecular patterns (PAMPs), Pam3Csk4 was more potent than other TLR ligands in inducing the production of all proinflammatory cytokines. Furthermore, IL‐6, IFN‐γ, IL‐17 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) levels produced by Pam3Csk4‐activated CD4+ cells were directly associated with disease activity. A similar correlation was observed with regard to IL‐17 levels released by Pam3Csk4‐stimulated CD8+ T‐cells and clinical parameters. In conclusion, our data suggest that the expansion of different T helper type 17 (Th17) phenotypes expressing TLR‐2, ‐4 and ‐9 is associated with MS disease activity, and reveals a preferential ability of TLR‐2 ligand in directly inducing the production of cytokines related to brains lesions and neurological disabilities.
Keywords: Multiple sclerosis; PAMP, Th17/Tc‐17 cell subsets; TLR
Introduction
Infectious diseases are implicated in the development and exacerbation of autoimmune diseases (AID), such as multiple sclerosis (MS), a T‐cell‐mediated demyelinated and neurodegenerative disorder of the central nervous system (CNS).1, 2, 3, 4 Infections from influenza and herpesvirus families have been suggested as contributing agents to MS. Although infectious mononucleosis by Epstein–Barr virus (EBV) has been widely associated with MS development, this virus also appears to trigger other AID, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and Sjögren's syndrome.5, 6, 7 These observations suggest that in addition to molecular mimicry, bystander activation of the immune cells by microbial antigens may contribute to autoimmunity via breakdown of immunological tolerance in genetically predisposed individuals. In this context, some pathogen‐associated molecular patterns (PAMPs), by playing an adjuvant role, have been implicated in MS pathogenesis.8, 9
PAMPs mediate their effects through ligation to pattern recognition receptors (PRRs), such as Toll‐like receptors (TLRs), mainly expressed on innate immune cells.10 In humans, 10 different TLRs have been described, named 1–10, with different cellular distributions. While TLR‐1, ‐2, ‐4, ‐6 and ‐10 are expressed on the cell surface, TLR‐3, ‐7, ‐8 and ‐9 are present intracellularly (e.g. endosomes).11, 12 TLR‐1, ‐2, ‐4 and ‐6 recognize bacterial and fungal cell wall components, such as lipopolysaccharide (LPS) and lipopeptides, and TLR‐5 binds flagellin.10 In contrast, TLR‐3, ‐7, ‐8 and ‐9 recognize nucleic acid structures, which serve as molecular signatures for viral and bacterial infections.12 Upon PAMP engagement, the myeloid differentiation primary response protein 88 (MyD88), an adaptor protein associated with the majority of TLR, except TLR‐3, activates nuclear factor (NF)‐κβ, which in turn trigger signalling cascades via phosphatidylinositol‐4,5‐bisphosphate 3‐kinase/protein kinase B (PI3K/AKT) and Ras/mitogen‐activated protein kinase (MAPK). These events promote cell survival and proliferation, as well as production of cytokines.13 .
The reconnaissance of the involvement of PAMPs in MS comes from findings in the experimental model of multiple sclerosis, the experimental autoimmune encephalomyelitis (EAE).14 The classical way of inducing EAE in mice is through the administration of myelin sheath antigens, such as myelin oligodendrocyte glycoprotein (MOG), emulsified in complete Freund's adjuvant (CFA),14, 15 consisting of heat‐inactivated Mycobacterium tuberculosis containing ligands for TLR‐2, TLR‐4 and TLR‐9.16 Additionally, the disease may also be induced by different PAMPs.17 Prinz et al.18 demonstrated that TLR‐9‐deficient mice exhibited a significant delay in the onset of EAE and milder clinical symptoms. Furthermore, knock‐out mice for MyD88 are resistant to EAE induction.18, 19 Finally, LPS injection induces relapses of EAE mice via bystander activation of myelin‐specific CD4+ T‐cells.20 Altogether, these findings suggest that PAMP‐induced DC maturation may contribute to the differentiation and expansion of encephalitogenic T‐cells. TLRs might even influence on the conversion for progressive forms.21
With regard to immune phenotypes, myelin‐specific T helper type 17 (Th17) and Th1 cells have been implicated in the pathogenesis of MS.22 High levels of interleukin‐ (IL)‐17A mRNA are detected in the periphera|l blood and cerebrospinal fluid (CSF) of patients with relapsing–remitting MS during clinical relapses.23, 24 Further, the expression of IL‐17 has been detected in astrocytes and oligodendrocytes in areas of active MS lesions.24, 25 IL‐17‐producing CD8+ T‐cells (Tc‐17) also contribute to brain lesions.26 With regard to interferon IFN‐γ, an important cytokine produced by both Th1 cells and Tc‐1 cells, their levels are also elevated during clinical relapses, and co‐localize with apoptotic oligodendrocytes.27, 28 Although IL‐22 is produced by human Th17 cells, the frequency of a unique subset of IL‐22 producing CD4+ T‐cells (Th22), regardless of IL‐17 release, has been associated with apoptosis of oligodendrocytes, diminished expression of forkhead box protein 3 (FoxP3) and risk of new relapses and number of active brain lesions.29, 30, 31 Finally, an increased proportion of granulocyte–macrophage colony‐stimulating factor (GM‐CSF)‐secreting T CD4+ cells, some of these co‐expressing IL‐17 or IFN‐γ, were detected in the CNS of MS patients.32, 33 Therefore, the ability of some PAMPs to up‐regulate many co‐stimulatory pathways in dendritic cells (DCs) may impact the risk of developing and exacerbating MS.
Although PPRs are classically expressed on innate immune cells, activated human T‐cells also express detectable levels of TLRs,15, 34 which suggests an ability of PAMPs to directly modulate the behaviour of T lymphocytes.35 In SLE, elevated TLR‐3 and ‐9 expression on T‐cells is related to clinical parameters.36 In MS patients, Nyirenda et al.37 demonstrated an elevated TLR‐2 expression on CD4+ CD25hi FoxP3+ T‐cells. The addition of TLR‐2 agonist (Pam3Csk4) on regulatory T‐cells (Tregs) not only inhibited their in‐vitro suppressive actions, but also skews them towards a Th17 phenotype.37 However, these authors did not evaluate the expression of TLRs on different T‐cell phenotypes or correlate them with clinical parameters in MS patients, which are the objectives of the present study.
Material and methods
Patients
Thirty patients with a definite RRMS diagnosis, according to criteria (2010)38 during the clinical remission phase, were recruited from Lagoa Hospital and Gaffrée Guinle University Hospital/UNIRIO (Rio de Janeiro, Brazil). Demographic data such as gender and age at disease onset were obtained from medical records (Table 1). All patients were naive for disease‐modifying therapies (DMT) and corticoid therapy for at least 2 months. The occurrence of infectious and other autoimmune diseases was excluded by clinical and serological tests. The neurological disability status of the patients was evaluated by authors (A.C.W. and C.C.V), and was determined according to the Expanded Disability Status Scale (EDSS).39 To quantify the number of active brain lesions, some of the MS patients underwent brain magnetic resonance imaging (MRI) at the time of blood sampling and clinical evaluation. Imaging was performed using the Siemens Trio 3 Tesla machine. The sequences obtained were T1 GRE 3D (ECHO gradient) on the sagittal plane, with multiplanar reformatting before and after intravenous contrast, weighted sequences in T2 and proton density (PD), fluid attenuation inversion recovery (FLAIR) sequence and T1 magnetization transfer and dissemination with apparent diffusion coefficient (ADC) mapping in the axial plane. Images were analysed by a single neuroradiologist (F.R.), a specialist in demyelinating diseases and blind to the degree of the patient's disability. As a control group, 20 healthy subjects matched by age, gender and ethnic background were recruited. The study was approved by the Ethics Committee for Research on Human Subjects at the Federal University of the State of Rio de Janeiro (UNIRIO) and blood was collected only after written informed consent was obtained from each individual.
Table 1.
Demographic and clinical features of the multiple sclerosis (MS) patients and controls
| Control1 | MS2 | |
|---|---|---|
| No. of subjects (n) | 20 | 30 |
| Gender, female/male (n) | 15/05 | 21/09 |
| Age in years (mean ± SD) | 29·1 ± 7·3 | 27·7 ± 6·8 |
| Disease duration in years [mean (range)] | NA5 | 5·2 (1–13) |
| EDSS [mean (range)]3 | NA | 2·1 (0–5) |
| No. of active brain lesions [mean (range)]4 | NA | 3·59 (0–25) |
Data from 1healthy individuals, 2relapsing–remitting multiple sclerosis (RRMS) in remission phase and at disease onset. Age (years) refers to age at point in time that the blood samples were collected. Disease duration refers to the years from the definitive diagnosis of MS. 3EDSS, Expanded Disability Status Scale and 4the number of active brain lesions by magnetic resonance imaging (MRI) scan were determined at the time‐point that the blood was collected to perform the immune assays. 5Not applicable. SD, standard deviation.
Cultures
Peripheral blood mononuclear cells (PBMC) were separated by a Ficoll‐Paque gradient. These cells were then washed three times in Hanks's balanced salt solution (HBSS) and suspended in RPMI‐1640 medium supplemented with 2 μm of L‐glutamine (Gibco, Carlsbad, CA), 10% of fetal calf serum, 20 U/ml of penicillin, 20 μg/ml of streptomycin and 20 mm of HEPES buffer. One × 106 cells/ml of viable PBMC were cultivated in 24‐well plates with 2 ml of complete medium in the presence or absence of anti‐CD3/anti‐CD28 beads (10 μl/ml) for 3 days at 37° and 5% CO2. In some experiments, enriched CD4+ and CD8+ T‐cells were obtained via negative selection using magnetic columns, according to the manufacturer's instructions (EasySep™; StemCell Technology, Vancouver, Canada). Briefly, 50 μl of the isolation cocktail were added to a cell suspension of 1 × 107 cells in 1 ml of HBSS in a 14‐ml tube. After rapidly mixing, the suspension was incubated for 10 min at room temperature. Then, 100 μl for CD4+ and 150 μl for CD8+ of the RapidSpheres suspension, already homogenated, were added to the cell suspension. After rapidly mixing, the cell suspension was incubated at room temperature for 5 min. Finally, 4 ml of HBSS was added to the cell suspension and, after pipetting, the tube was then placed at the magnet for 5 min. Finally, the supernatants were recovered. The purity of CD4+ T‐cells and CD8+ T‐cells was > 98%, as measured by flow cytometry (data not shown). Enriched CD4+ and CD8+ T‐cell cultures were maintained for 48 hr in the absence or presence of TLR‐4 agonist lipopolysacharide (LPS; 100 ng/ml) from Escherichia coli (Sigma‐Aldrich, St Louis, MO)], TLR‐2 agonist synthetic triacylated lipopeptide (Pam3Csk4, 1 μg/ml) from InvivoGen, San Diego, CA)] or TLR‐9 agonist cytosine–phosphate–guanosine (CpG) oligodeoxynucleotides (ODN M362 1 μm/ml, from InvivoGen). These concentrations were chosen from a previous study conducted by Voo et al.40 All cell cultures were kept for 48 hr at 37° and 5% CO2.
Immunofluorescence labelling and flow cytometry
The mouse anti‐human monoclonal antibodies (mAbs) to CD3‐phycoerythrin‐cyanin 5 (PE‐Cy5), CD8‐fluorescein isothiocyanate (FITC), CD4‐FITC, TLR‐2‐PE, TLR‐4‐PE, TLR‐9‐PE, IL‐17A‐PE‐Cγ7, IFN‐γ ‐allophycocyanin (APC), IL‐6‐APC, IL‐10‐APC and all isotype‐control antibodies were purchased from BD Bioscience (San Diego, CA), and were used to quantify the percentage of different T‐cell subsets. Briefly, various combinations of mAbs directed for surface markers were added to PBMC (2 × 105/tube) and incubated for 30 min at room temperature in the dark. The cells were washed with phosphate‐buffered saline (PBS), then permeabilized by incubating cells with Cytofix/Cytoperm (BD Pharmingen, San Diego, CA) at 4° for 20 min. After washing, the antibodies for intracellular staining (anti‐IL‐17, anti‐IFN‐γ, anti‐IL‐10, anti‐IL‐6) or the corresponding isotype control anti‐immunoglobulin (Ig)G1 were added in various combinations and incubated for 30 min at 4°. The cells were analysed in the Accuri C6 (Accuri™; Ann Arbor, MI) and CFlow software (Accuri™; Ann Arbor). Isotype control antibodies and single‐stained samples were used to periodically check the settings and gates on the flow cytometer. After acquisition of 200 000 events, lymphocytes were gated based on forward‐ and side‐scatter properties after the exclusion of dead cells by using propidium iodide and doublets. Further, the gated cells were negative for CD14 marker.
ELISA technique
After 48 hr, the supernatant from PAMP‐activated CD4+ and CD8+ T‐cells were collected and the cytokines were quantified by enzyme‐linked immunosorbent assay (ELISA) using OptEIA ELISA kits (BD Pharmigen, San Diego, CA), according to the manufacturer's instructions. Briefly, each ELISA was performed using pairs of antibodies against IL‐1β, IL‐6, TNF‐α, IL‐17A (IL‐17), IL‐22, GM‐CSF and IL‐10. The reaction was revealed with streptavidin–horseradish peroxidase, using 3,3′,5,5′‐tetramethylbenzidine (TMB) as substrate. Recombinant human cytokines, at concentrations ranging from 3·5 to 500 pg/ml, were used to construct standard curves.
Statistical analysis
Statistical analysis was performed using prism version 5·0 software (GraphPad Software). All immunological evaluations were performed in triplicate or quadruplicate in each individual and the intra‐assay variability ranged from 8·7 to 15·1% (median value of 10·1%), as calculated by the software. The non‐parametric Mann–Whitney U‐test and Student's t‐test were applied to determine whether the two groups were statistically different for non‐parametric and parametric variables, respectively. Correlations between variables were investigated using Pearson's correlation. The significance in all experiments was defined as P < 0·05.
Results
Clinical and demographic data
Our study was performed with 30 MS patients, nine males (30%) and 21 females (70%), all in remission phase (Table 1). The mean age at disease onset was 27·7 years (range 17–38 years). Mean time between MS diagnosis and blood sampling was approximately 5·2 years (range 1–13 years). All 30 patients were naive for disease‐modifying therapies (DMT); 70% of patients had been previously treated with oral or intravenous corticosteroids to control the acute neurological bouts. However, at the time of the study none of these patients were on corticosteroids for 60 days. Despite all patients being clinically asymptomatic, at the moment of evaluation the data from the brain MRI scan revealed that 18 patients (60%) had active lesions (Table 1).
TLR expression on T‐cells from MS patients and the cytokine profile
Following the gating strategy shown in Fig. 1a,b, we observed that TLR‐2, ‐4 and ‐9 expression was significantly higher at the basal level in both CD4+ and CD8+ T‐cells from MS patients than in healthy controls (Fig. 1c,d). Moreover, as shown in Supporting information, Fig. S1, in MS patients the expression levels of TLR‐2, TLR‐4 and TLR‐9 were significantly higher in CD8+ T‐cells. Among TLRs (‐2, ‐4 and ‐9)+ cells, IL‐17A (IL‐17) was consistently detected in unstimulated CD4+ (Fig. 2a and Supporting information, S2a–d) and CD8+ (Fig. 2c and Supporting information, S3a–d) T‐cells from MS patients, but not in the control group. Of note, no difference was observed regarding both percentage and mean fluorescence intensity of MS‐derived CD4+ or CD8+ T‐cells positive for IL‐17 cytokine (data not shown). Following in‐vitro T‐cell activation with anti‐CD3‐anti‐CD28 beads, the proportion of those IL‐17+ cells increased in both experimental groups, but remained higher among TLR‐expressing CD4+ (Fig. 2a and Supporting information, S2a–d) and CD8+ (Fig. 2b and Supporting information, S3a–d) T‐cells from MS patients. Moreover, with regard to other cytokines, the proportion of IL‐6‐producing CD4+ and CD8+ T‐cells positive for TLR‐2, ‐4 and ‐9 was also higher in MS samples when compared with control (Fig. 2a,b). No significance was observed in the percentage of TLR+ (‐2, ‐4 and ‐9) CD4+ T‐cells capable of producing IFN‐γ or IL‐10 between MS patients and controls (Fig. 2a). By contrast, a higher proportion of IFN‐γ + TLR+ CD8+ T‐cells were observed in patients with MS (Fig. 2b). The proportions of TLR+ CD8+ T‐cells positive for IL‐10 were almost undetectable in majority of patents and controls subjects (Fig. 2b). Among T‐cells negative for TLR‐2, ‐4 and ‐9, detectable cytokine production was seen only after cell activation (Fig. 2b and 2b). While the percentage of activated peripheral IL‐6‐secreting TLR− (CD4 and CD8) T‐cells were significantly higher in patients, a significantly higher proportion of activated IL‐17+ TLR− T‐cells was observed only in the MS‐derived CD8+ compartment (Fig. 2b). In contrast to TLR‐positive subsets, the percentage of TLR− (CD4 and CD8) T‐cells able to produce IFN‐γ and IL‐10 was significantly lower in the patient sample (Fig. 2a,b).
Figure 1.

Comparison of the proportion of circulating CD4+ and CD8+ T‐cells expressing Toll‐like receptor (TLR)‐2, ‐4 and ‐9 from multiple sclerosis (MS) patients and healthy subjects. Panels (c) and (d) show the proportion [mean ± standard deviation (SD)] of TLRs‐expressing CD4+ and CD8+ T‐cells, respectively. The percentage of T‐cells able to express TLRs (‐2, ‐4 and ‐9) obtained from healthy subjects (n = 20) or MS patients (n = 30) was evaluated in non‐activated or stimulated cell cultures with anti‐CD3/anti‐CD28 beads for 3 days. P‐values were obtained by the Mann–Whitney U‐test. Representative dot‐plots showing identification of TLR‐secreting CD4+ (a) and CD8+ (b) T‐cells from control and MS patients, after acquisition of 200 000 events, are indicated. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
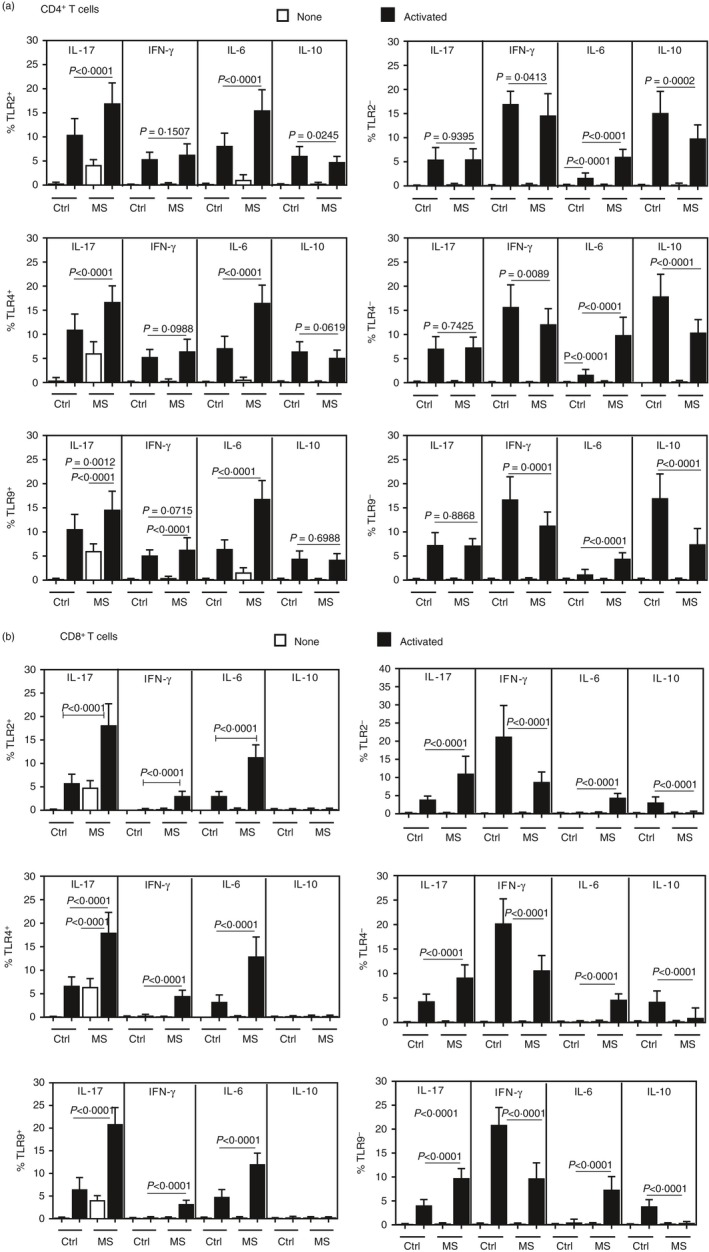
The cytokine profile of Toll‐like receptor (TLR)+ T‐cells in MS patients. The percentage [± standard deviation (SD)] of CD4+ (a) and CD8+ (b) T‐cells positive for TLR‐2, ‐4 and ‐9 able to produce interleukin (IL)‐17, interferon (IFN)‐γ, IL‐6 or IL‐10 was determined before and after activating T‐cells (1 × 106/ml) from control individuals (n = 20) and multiple sclerosis (MS) patients (n = 30). The cells were activated with anti‐CD3/anti‐CD28 (5 μl/ml), for 3 days. The P‐values were obtained by the Mann–Whitney U‐test. Representative histograms showing identification of IL‐17, IFN‐γ, IL‐6 or IL‐10 production by TLR+ CD4+ (Supporting information, Fig. S1) and TLR+ CD8+ (Supporting information, Fig. S2) T‐cells from both groups, after acquisition of 200 000 events, are shown in the supplementary figures.
The proportion of IL‐6‐ and IFN‐γ‐producing TLR+ IL‐17+ T‐cells was associated with clinical parameters in MS patients
Although IL‐17 is the signature cytokine of the Th17 phenotype, these CD4+ T‐cells present a functional heterogeneous lineage, which may perform antagonistic functions.41, 42, 43, 44 The percentage of Th17‐like subsets able to produce IL‐17 along with IFN‐γ or with IL‐6 was significantly higher among CD4+ and CD8+ T‐cells positive for TLR‐2, ‐4 and ‐9 in MS patients than in the control group (Fig. 3b). No significant difference was observed in terms of dual IL‐10‐ and IL‐17‐secreting T‐cells. Moreover, in MS patients, the proportion of IL‐6‐secreting IL‐17+ TLR+ (‐2, ‐4 and ‐9) T‐cells, either CD4 or CD8 cells, was significantly higher than those able to co‐produce IFN‐γ or IL‐10 (Fig. 3a,b). In our previous study, IFN‐γ + IL‐17+ CD4+ or CD8+ T‐cell frequencies were positively associated with neurological disabilities, as determined by Expanded Disability Status Scale (EDSS) score.44 The percentage of IL‐6+ IL‐17+ TLR+ and IFN‐γ + IL‐17+ TLR+ subsets, for both activated CD4+ and CD8+ T‐cells, were positively correlated with radiological activity of the disease and neurological disabilities (Table 2). Nevertheless, taking into consideration the P‐values, in general a stronger correlation was observed between the frequency of IFN‐γ + IL‐17+ TLR+ T‐cell subsets and the number of active brain lesions, mainly those positive for TLR‐2. No correlation was observed between clinical parameters and dual IL‐17 and IL‐6‐producing (CD4+ and CD8+) T‐cells negative for TLR‐2, ‐4 and ‐9 (data not shown). Furthermore, no correlation was seen between these signs of clinical activity of the disease and the proportion of all IL‐17‐secreting T‐cell subsets when maintained with medium alone (data not shown).
Figure 3.
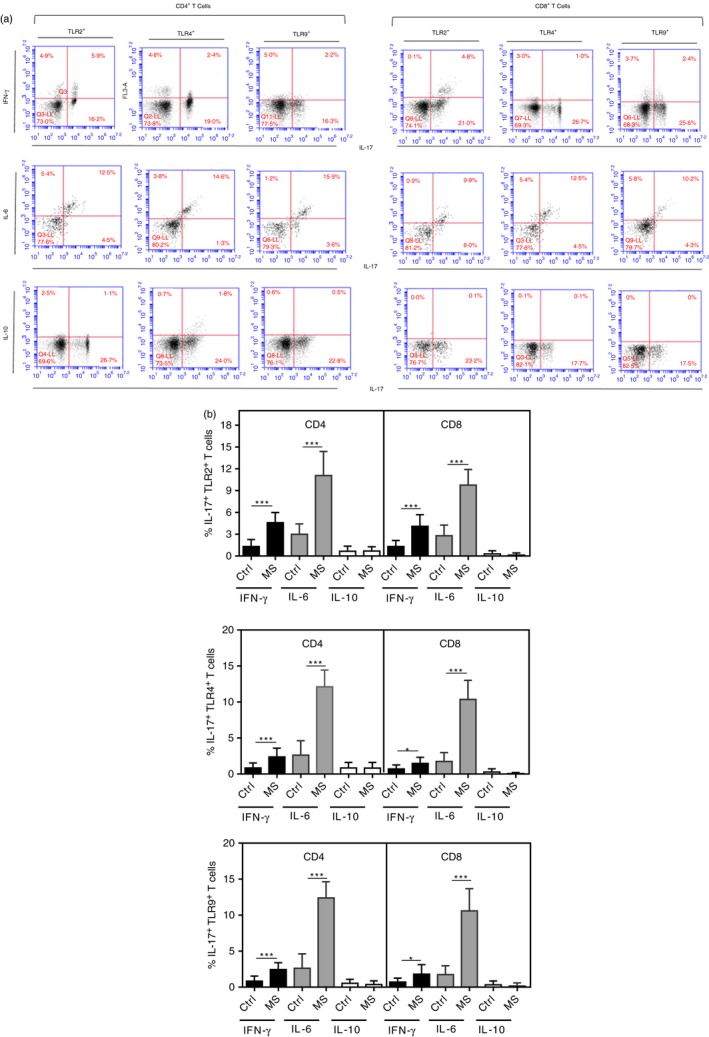
The proportion of different interleukin (IL)‐17‐secreting CD4+ and CD8+ T‐cells positives for Toll‐like receptors (TLR) in MS patients. Peripheral blood mononuclear cells (PBMC) (1 × 106/ml), obtained from multiple sclerosis (MS) patients (n = 30), were stimulated with anti‐CD3/anti‐CD28 (5 μl/ml), for 3 days. In (b), the proportion [mean ± standard deviation (SD)] of dual IL‐17/IL‐10‐, IL‐17/interferon (IFN)‐γ‐ or IL‐17/IL‐6 by CD4+ or CD8+ T‐cells able to express TLR‐2, TLR‐4 and TLR‐9 are shown. In (a), representative dot‐plots showing identification of different IL‐17‐producing TLR+ T‐cell subsets, after acquisition of 200 000 events, are indicated. The P‐values were obtained by the Mann–Whitney U‐test. *P < 0.05; ***P < 0.0001. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 2.
Correlation coefficient (r) between the proportion of different interleukin (IL)‐17‐secreting CD4+ and CD8+ T‐cell subsets expressing Toll‐like receptors (TLRs) and clinical parameters in multiple sclerosis (MS) patients
| No. active brain lesions | EDSS | |||
|---|---|---|---|---|
| r | P | r | P | |
| % TLR‐2+ CD4+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·9537 | <0·0001 | 0·5556 | 0·0110 |
| IL‐17+ IL‐6+ | 0·4798 | 0·0323 | 0·4553 | 0·0437 |
| IL‐17+ IL‐10+ | −0·035 | 0·8807 | −0·024 | 0·92 |
| % TLR‐4+ CD4+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·6420 | 0·0023 | 0·5013 | 0·0312 |
| IL‐17+ IL‐6+ | 0·4909 | 0·0280 | 0·4707 | 0·0362 |
| IL‐17+ IL‐10+ | −0·1147 | 0·6303 | −0·1111 | 0·6411 |
| % TLR‐9+ CD4+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·5565 | 0·0018 | 0·5136 | 0·0396 |
| IL‐17+ IL‐6+ | 0·6359 | 0·0026 | 0·4511 | 0·0471 |
| IL‐17+ IL‐10+ | −0·3699 | 0·1085 | −0·3193 | 0·1699 |
| % TLR‐2+ CD8+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·7478 | 0·0002 | 0·5014 | 0·0251 |
| IL‐17+ IL‐6+ | 0·4739 | 0·0348 | 0·4509 | 0·0460 |
| IL‐17+ IL‐10+ | 0·30 | 0·7974 | 0·3098 | 0·1837 |
| % TLR‐4+ CD8+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·6137 | 0·0040 | 0·4924 | 0·0274 |
| IL‐17+ IL‐6+ | 0·5435 | 0·0133 | 0·4115 | 0·0373 |
| IL‐17+ IL‐10+ | 0·1234 | 0·6043 | 0·1346 | 0·6043 |
| % TLR‐9+ CD8+ T‐cells | ||||
| IL‐17+ IFN‐γ + | 0·5649 | 0·0151 | 0·437 | 0·0412 |
| IL‐17+ IL‐6+ | 0·5449 | 0·0130 | 0·478 | 0·0406 |
| IL‐17+ IL‐10+ | 0·3301 | 0·1552 | 0·3057 | 0·190 |
The proportion of different T helper type 17 (Th17)‐like subsets from MS patients was compared with radiological activity of the disease (n = 18) and neurological disabilities (n = 30), determined by the Expanded Disability Status Scale (EDSS) score. IFN, interferon; IL, interleukin; TNF, tumour necrosis factor. Significant P values are highlighted in bold.
The levels of IL‐6, IL‐17, IFN‐γ and GM‐CSF released by purified T‐cells were correlated with disease activity
The ability of T‐cells to express TLR‐2, ‐4 and ‐9 suggests that those cells may directly respond to agonists for these PRRs. As observed in Fig. 4, the Pam3Csk4 (TLR‐2L), LPS (TLR‐4L) and ODN (TLR‐9L) were able to directly induce cytokine production by purified CD4+ and CD8+ T‐cells from MS patients. Among the PAMPs, Pam3Csk4 was more potent at inducing the production of all proinflammatory cytokines by CD4+ T‐cell cultures (Fig. 4a). Moreover, the production of IL‐1β, IL‐6 and IL‐17 by Pam3Csk4‐activated CD8+ T‐cells was higher when compared with LPS and ODN. In terms of IL‐10, its production was elevated in CD4+ and CD8+ T‐cells after addition of LPS. More importantly, the number of active brain lesions and EDSS scores were positively associated with IL‐6 levels produced by CD4+ and CD8+ T‐cells in response to LPS and Pam3Csk4 (Table 3). Further, the release of IFN‐γ and IL‐17 by CD4+ T‐cells, as well as IL‐17 from CD8+ T‐cells, in response to Pam3Csk4 was directly correlated with clinical parameters (Table 3). Finally, CD4+ T‐cells from patients with the highest number of brain lesions responded in vitro to Pam3Csk4 stimulation, releasing higher GM‐CSF levels (Table 3). No correlation was observed between clinical parameters and cytokine content by ODN‐activated CD4+ and CD8+ T‐cells (data not shown).
Figure 4.
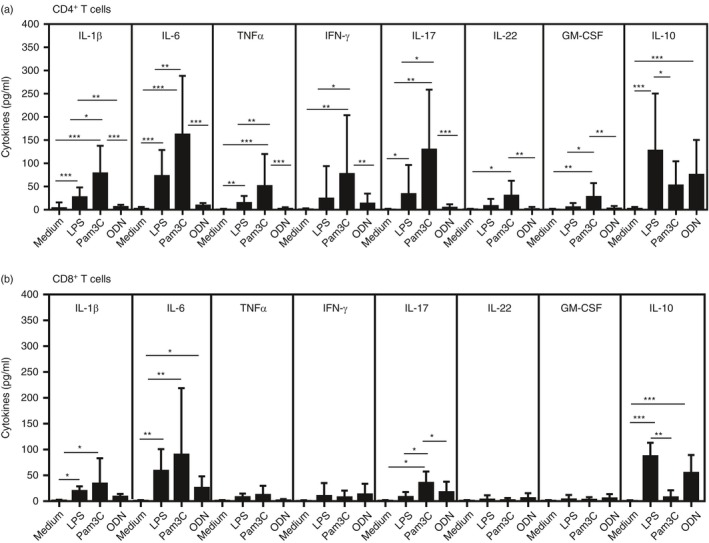
Cytokine production by T‐cells from multiple sclerosis (MS) patients in response to pathogen‐associated molecular patterns (PAMPs). The cytokine content in the supernatants collected from purified CD4+ (a) and CD8+ T‐cell (b) cultures maintained for 2 days in the absence or presence of lipopolysaccharide (LPS) (100 ηg/ml), Pam3Csk4 (1 μg/ml) and oligodeoxynucleotide (ODN) (1 μm/ml) was determined using enzyme‐linked immunosorbent assay (ELISA). In the figure, (*), (**) and (***) indicate P‐values < 0·05, < 0·001 and < 0·0001.
Table 3.
Correlation coefficient (r) between the in‐vitro cytokine production by CD4+ and CD8+ T‐cells from multiple sclerosis (MS) patients following different Toll like receptors (TLRs) agonists and clinical parameters
| No. active brain lesions | EDSS | |||
|---|---|---|---|---|
| r | P | r | P | |
| CD4+ T‐cells | ||||
| TLR‐4 agonist | ||||
| IL‐1β | 0·2801 | 0·2904 | 0·2332 | 0·3809 |
| IL‐6 | 0·6993 | 0·0034 | 0·6690 | 0·0058 |
| TNF‐α | 0·3085 | 0·2424 | 0·3684 | 0·1596 |
| IFN‐γ | 0·3608 | 0·1762 | 0·2785 | 0·3024 |
| IL‐17 | 0·1178 | 0·66 | 0·1386 | 0·6034 |
| IL‐22 | 0·2162 | 0·4637 | 0·1327 | 0·4680 |
| GM‐CSF | 0·2414 | 0·3668 | 0·1945 | 0·4680 |
| IL‐10 | 0·2445 | 0·3587 | 0·2272 | 0·3942 |
| TLR‐2 agonist | ||||
| IL‐1β | 0·5123 | 0·0912 | 0·4735 | 0·1218 |
| IL‐6 | 0·7754 | 0·0042 | 0·8022 | 0·0026 |
| TNF‐α | 0·1857 | 0·5577 | 0·2680 | 0·3939 |
| IFN‐γ | 0·5921 | 0·0464 | 0·5991 | 0·04321 |
| IL‐17 | 0·5986 | 0·0429 | 0·7713 | 0·0045 |
| IL‐22 | 0·4170 | 0·1773 | 0·3950 | 0·2030 |
| GM‐CSF | 0·6387 | 0·0290 | 0·5045 | 0·0960 |
| IL‐10 | −0·3189 | 0·2981 | −0·3292 | 0·2740 |
| CD8+ T‐cells | ||||
| TLR‐4 agonist | ||||
| IL‐1β | 0·4857 | 0·3556 | 0·2942 | 0·5667 |
| IL‐6 | 0·8697 | 0·0333 | 0·9553 | 0·0222 |
| TNF‐α | 0·4414 | 0·4333 | 0·5909 | 0·2444 |
| IFN‐γ | 0·3928 | 0·6667 | 0·4045 | 0·6661 |
| IL‐17 | 0·3816 | 0·6605 | 0·4112 | 0·6567 |
| IL‐22 | 0·3928 | 0·6645 | 0·4043 | 0·6661 |
| GM‐CSF | 0·3921 | 0·6660 | 0·4045 | 0·6667 |
| IL‐10 | 0·2310 | 0·65 | 0·1941 | 0·7722 |
| TLR‐2 agonist | ||||
| IL‐1β | 0·2571 | 0·6583 | 0·4414 | 0·4111 |
| IL‐6 | 0·8407 | 0·0444 | 0·8359 | 0·0667 |
| TNF‐α | 0·5161 | 0·3333 | 0·4690 | 0·3101 |
| IFN‐γ | 0·6761 | 0·2 | 0·6093 | 0·2667 |
| IL‐17 | 0·9429 | 0·0167 | 0·9122 | 0·022 |
| IL‐22 | 0·1309 | 0·6667 | 0·435 | 0·3339 |
| GM‐CSF | 0·4395 | 0·4 | 0·4553 | 0·2771 |
| IL‐10 | −0·3043 | 0·3333 | −0·3482 | 0·2665 |
The levels of different cytokines produced by CD4 and CD8 T‐cell cultures maintained for 48 hr in the presence of lipopolysaccharide (LPS) [Toll‐like receptor (TLR)‐4L], Pam3Csk4 (TLR‐2L) and oligodeoxynucleotide (ODN) (TLR‐9L) were correlated with the number of active brain lesions (n = 18) and neurological disabilities, determined (n = 20) using the Expanded Disability Status Scale (EDSS) score. IFN, interferon; IL, interleukin; TNF, tumour necrosis facot; GM‐CSF, granulocyte–macrophage colony‐stimulating factor. Significant P values are highlighted in bold.
Discussion
Pathogens and their PAMPs have been linked to autoimmune diseases, and are associated with the breakdown of immunological tolerance via activation of self‐reactive T‐cells.8, 15, 17, 18, 45 In the present study, we demonstrated a relationship between elevated TLR‐2, ‐4 and ‐9 expression on IL‐17‐secreting T‐cell subsets and disease severity in MS patients.
In MS, the TLR engagement by different PAMPs on DCs appears to favour both Th1 and Th17 phenotypes by releasing high levels of IL‐12 and IL‐23, respectively.45 Activated human T‐cells also express TLRs, and studies suggest that TLR ligands may result in a lower threshold for T‐cell reactivation via TCR.35, 46, 47, 48 In the present study, the frequency of CD4+ and CD8+ T‐cells expressing TLR‐2, ‐4 and ‐9 was higher in MS patients than healthy individuals, even in the absence of stimulus. Of note, a higher percentage of those TLR+ T‐cells in patients was also observed ex vivo, i.e. after immediate staining of blood sampling (data not shown). In the future, we hope to evaluate the expression of other types of TLRs.
In EAE, CD4+ T‐cells deficient in TLR‐2 expression were partially protected from experimental autoimmune encephalomyelitis.49, 50 In humans, TLRs are involved in the pathogenesis of others Th17‐mediated autoimmune diseases, such as SLE,51 rheumatoid arthritis52 and neuromyelitis optic spectrum disorder.53 A recent study by Zastepa et al.54 demonstrated sustained TLR‐2 and ‐4 expression on naive CD4+ T‐cells following in‐vitro polyclonal activation in MS patients who evolved rapidly to the progressive form of the disease. These findings suggest the involvement of TLR expression in T‐cells in MS pathogenesis.
In the present study, although elevated expression of TLR‐2, ‐4 and ‐9 on MS‐derived T‐cells was not correlated per se with clinical parameters (data not shown), IL‐17+ IL‐6+ and IL‐17+ IFN‐γ + among TLR+ CD4+ and CD8+ T‐cell subsets directly correlated with the number of active brain lesions and neurological disabilities, as determined by EDSS score. These data suggest an involvement of different Th17‐like phenotypes in MS.
It is well recognized that IL‐17‐secreting T‐cells present a functional heterogeneous lineage. Among CD4+ T‐cells, some pathogenic human Th17 cell subsets have been identified through co‐expression of CCR6 and CXCR3 and simultaneous production of cytokines IL‐17 and IFN‐γ.55 The differentiation of this T‐cell subset depends upon IL‐6, IL‐1β and IL‐23 associated with an absence of transforming growth factor (TGF)‐β.55, 56, 57, 58 Upon transfer, these IFN‐γ‐secreting Th17 cells induced more severe EAE than those that received the classical Th17 cells.55 Although these cells present a minor T‐cell phenotype among those that secrete IL‐17 or IFN‐γ, they were found in the blood and CSF from MS patients during clinical relapses.23 Moreover, in our previous study, a proportion of total IFN‐γ + IL‐17+ (CD4+ and CD8+) T‐cells was associated with EDSS score.44 These data suggest that circulating IL‐17+ IFN‐γ + T‐cells, regardless of expression of TLR‐2, ‐4 and ‐9, may present pathogenic phenotypes in MS. Furthermore, to the best of our knowledge, we report for the first time an expansion of IL‐17+ IL‐6+ TLR+ T‐cells that was associated with MS severity.
It is known that knock‐out mice for IL‐6 are resistant to EAE induction.59 Ferreira et al.60 also demonstrated that endogenous IL‐6 is important to maintain the production of IL‐17 by CD4+ T‐cells from MS patients, as well as its role in favouring Th17 phenotype differentiation. Further, excess of IL‐6 damaged Treg cells function through reduction of FoxP3 protein expression and release of IL‐10, a potent anti‐inflammatory cytokine.61, 62 In the present study, the percentage of IL‐10+ (CD4+ and CD8+) T‐cells negative for TLR‐2, ‐4 and ‐9 was significantly lower in patients; however, no relationship was observed with clinical parameters (data not shown). Nevertheless, there is a possibility that, in vivo, excessive production of IL‐6 by different T‐cell subsets co‐stimulated with different TLR ligands may overcome Treg‐mediated suppression of encephalitogenic T‐cells. Nyirenda et al.37 showed that TLR‐2 ligand Pam3Csk4 and TLR‐4 ligand LPS directly blocked anti‐CD3‐activated Treg cell function by reducing IL‐10 production, and by inducing resistance in effector T‐cells to Treg suppression. In general, these results suggest that PAMPs, via different mechanisms, may play a pathogenic role in autoimmunity.
With regard to IFN‐γ, no difference was observed in the total IFN‐γ‐secreting CD4+ and CD8+ T‐cells between the two experimental groups (data not shown). Nevertheless, the IFN‐γ‐producing (CD4+ and CD8+) T‐cells negative for TLR‐2, ‐4 and ‐9 were significantly lower in MS patients than in healthy subjects, which may be a consequence of a trend of higher percentage of IFN‐γ‐secreting TLR+ T‐cell subsets.
Although PAMPs co‐stimulate T‐cell activation, in the present study we have demonstrated that TLR‐2, ‐4 and ‐9 can directly induce cytokine production by purified CD4+ T and CD8+ T‐cells from MS patients. In general, among PAMPs, Pam3Csk4, a TLR‐2 ligand (TLR‐2L), was more potent in inducing the production of all proinflammatory cytokines by CD4+ T‐cell cultures. Moreover, the production of IL‐1β, IL‐6 and IL‐17 by Pam3Csk4‐activated CD8+ T‐cells was higher when compared with LPS and ODN. In contrast, the Pam3Csk4 induced almost no IL‐10. These findings are in agreement with reported studies demonstrating the ability of Pam3Csk4 to promote production of proinflammatory cytokines by memory T‐cells from healthy individuals and MS patients, whereas it inhibits IL‐10 production by Treg cell subsets.37, 40, 46, 49 When we correlate the cytokine content and clinical parameters, IL‐6 levels produced by CD4+ and CD8+ T‐cells in response to LPS and Pam3Csk4 correlated positively with the number of active brain lesions and EDSS scores. The same relationship was seen between IFN‐γ, released by CD4+ T‐cells, and IL‐17, produced by both CD4+ and CD8+ T‐cells in response to TLR‐2L. In addition, CD4+ T‐cell cultures from patients with radiological active disease secreted higher GM‐CSF levels when activated with Pam3Csk4. No relationship was observed between the cytokine response of CD4+ and CD8+ T‐cells to ODN and clinical parameters, which could be explained by lower levels of all proinflammatory cytokines in those cultures. Nevertheless, ODN, like other PAMPs, could co‐stimulate TCR‐induced T‐effector function.47, 63 At present, we are dedicated to analysing the impact of different PAMPs on modulating cytokine production by T‐cells stimulated by myelin antigens. These data are in line with some studies showing the ability of PAMPs to preferentially activate Th1 and Th17 phenotypes, probably due to high TLR expression.15, 45, 50, 64
The presence of GM‐CSF+ T‐cell phenotypes have been associated with disease activity in both EAE and MS.32, 33 This haematopoietic factor should contribute to neuronal damage by inducing recruitment and activation of monocytes into the CNS.65 Here, the ability of TLR‐2L to induce the production of GM‐CSF by CD4+ T‐cells reinforced the adverse impact of infections in MS.
While TLR‐4 and ‐9 recognize LPS and DNA from bacteria and virus, respectively, TLR‐2 is required for recognition of diverse microbial molecules from various types of microorganisms, such as Gram‐positive bacteria rich in TLR‐2 ligands and Clostridium perfringens, recently associated with NMOSD, another CNS neurodegenerative autoimmune disease.66, 67 Interestingly, dysbiosis with overgrowth of commensal species belonging to the Clostridia cluster has been reported in MS patients.68 Excessive TLR‐2 ligands from these bacteria are known to induce colitis in animal models of inflammatory bowel disease.69 These findings suggest the adverse impact of microbial translocation in MS. In this context, a study published by our group70 demonstrated elevated plasma levels of LPS in MS patients, which were directly related to in‐vivo IL‐6 production. Finally, the presence of endogenous TLR ligands, called danger‐associated molecular patterns (DAMPs), could also contribute to MS. In MS, heat shock protein (HSP)70 is a DAMP highly expressed on brain lesions71 that binds to myelin basic protein, forming a immunogenic complex72 able to influence the induction of EAE.73 Although further studies are required, these findings suggest that infections or events that elevate intestinal translocation of bacteria should impact upon the progression of MS by favouring the activation of encephalitogenic memory TLR+ Th17 cell subsets, both directly and indirectly, through release of alarmins (DAMPs) from damaged cells during inflammatory reactions against pathogens and/or their PAMPs.
Conclusions
In conclusion, our data, although preliminary, suggest that the expansion of different Th17/Tc‐17‐like phenotypes expressing TLR‐2, ‐4 and ‐9 are associated with MS severity, and reveal a particular ability of the TLR‐2 ligand to directly induce cytokine production related to brain lesions and neurological disabilities. This study is significant, because we believe that our data highlight possible molecular mechanisms by which microorganisms negatively impact autoimmunity, which could help to design immunotherapeutic tools for MS patients.
Disclosure
All authors declare that there are no conflicts of interest.
Financial support
This work was supported by Fundação de Amparo à Pesquisa Carlos Chagas Filho (Grant number: 26/201.208/2014) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (Grant number: 302714/2013‐8)].
Supporting information
Figure S1. The levels of expression of Toll‐like receptor (TLR) on T‐cells from multiple sclerosis (MS) patients.
Figure S2. Representative flow cytometric histograms showing the percentage of peripheral cytokine‐producing CD4+ T‐cells able to express, or not, Toll‐like receptor (TLR)‐2, ‐4 and ‐9 in healthy and multiple sclerosis (MS) patients.
Figure S3. Representative flow cytometric histograms showing the percentage of peripheral cytokine‐producing CD8+ T‐cells able to express, or not, Toll‐like receptor (TLR)‐2, ‐4 and ‐9 in healthy and multiple sclerosis (MS) patients.
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372:1502–17. [DOI] [PubMed] [Google Scholar]
- 2. Chen Q, Liu Y, Lu A, Ni K, Xiang Z, Wen K et al Influenza virus infection exacerbates experimental autoimmune encephalomyelitis disease by promoting type I T‐cells infiltration into central nervous system. J Autoimmun 2017; 77:1–10. [DOI] [PubMed] [Google Scholar]
- 3. Panitch HS. Influence of infection on exacerbations of multiple sclerosis. Ann Neurol 1994; 36:25–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steelman AJ. Infection as an environmental trigger of multiple sclerosis disease exacerbation. Front Immunol 2015; 6:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lossius A, Johansen JN, Torkildsen Ø, Vartdal F, Holmoy T. Epstein–Barr virus in systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis‐association and causation. Viruses 2012; 4:3701–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Igoe A, Scofield RH. Autoimmunity and infection in Sjögren's syndrome. Curr Opin Rheumatol 2013; 25:480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiménez‐Dalmaroni MJ, Gerswhin ME, Adamopoulos IE. The critical role of toll‐like receptors – from microbial recognition to autoimmunity: a comprehensive review. Autoimmun Rev 2015;15:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hernández‐Pedro N, Magana‐Maldonado R, Ramiro AS, Pérez‐De la Cruz V, Rangel‐López E, Sotelo J et al PAMP‐DAMPs interactions mediates development and progression of multiple sclerosis. Front Biosci (Schol Ed). 2016; 1:13–28. [DOI] [PubMed] [Google Scholar]
- 9. Chen J‐Q, Szodoray P, Zeher M. Toll‐like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol 2015; 50:1–17. [DOI] [PubMed] [Google Scholar]
- 10. Takeda K, Akira S. Toll‐like receptors in innate immunity. Int Immunol 2005; 17:1–14. [DOI] [PubMed] [Google Scholar]
- 11. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783–801. [DOI] [PubMed] [Google Scholar]
- 12. Dunne A, O'Neill LAJ. Adaptor usage and Toll‐like receptor signaling specificity. FEBS Lett 2005; 579:3330–5. [DOI] [PubMed] [Google Scholar]
- 13. Takeda K, Akira S. TLR signaling pathways. Semin Immunol 2004; 16:3–9. [DOI] [PubMed] [Google Scholar]
- 14. Miranda‐Hernandez S, Baxter AG. Role of Toll‐like receptors in multiple sclerosis. Am J Clin Exp Immunol. 2013; 2:75–93. [PMC free article] [PubMed] [Google Scholar]
- 15. Mills KHG. TLR‐dependent T‐cell activation in autoimmunity. Nat Rev Immunol 2011; 11:807–22. [DOI] [PubMed] [Google Scholar]
- 16. Dowling JK, Mansell A. Toll‐like receptors: the Swiss army knife of immunity and vaccine development. Clin Transl Immunol 2016; 5:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hansen BS, Hussain RZ, Lovett‐Racke AE, Thomas JA, Racke MK. Multiple Toll‐like receptor agonists act as potent adjuvants in the induction of autoimmunity. J Neuroimmunol 2006; 172:94–103. [DOI] [PubMed] [Google Scholar]
- 18. Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K et al Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006; 116:456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marta M, Andersson Å, Isaksson M, Kämpe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol 2008; 38:565–75. [DOI] [PubMed] [Google Scholar]
- 20. Nogai A, Siffrin V, Bonhagen K, Pfueller CF, Hohnstein T, Volkmer‐Engert R et al Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J Immunol 2005; 175:959–66. [DOI] [PubMed] [Google Scholar]
- 21. Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll‐like receptor 2 and poly(ADP‐ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol 2009; 10:958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lovett‐Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T‐cell cytokines relevant in multiple sclerosis? Biochim Biophys Acta ‐ Mol Basis Dis 2011; 1812:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brucklacher‐Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009; 132:3329–41. [DOI] [PubMed] [Google Scholar]
- 24. Matusevicius D, Kivisäkk P, He B, Kostulas N, Özenci V, Fredrikson S et al Interleukin‐17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler J 1999; 5:101–4. [DOI] [PubMed] [Google Scholar]
- 25. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H et al Gene‐microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 2002; 8:500–8. [DOI] [PubMed] [Google Scholar]
- 26. Salehi Z, Doosti R, Beheshti M, Janzamin E, Sahraian MA, Izad M. Differential frequency of CD8+ T‐cell subsets in multiple sclerosis patients with various clinical patterns. PLOS ONE 2016; 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pouly S, Becher B, Blain M, Antel JP. Interferon‐gamma modulates human oligodendrocyte susceptibility to Fas‐mediated apoptosis. J Neuropathol Exp Neurol 2000; 59:280–6. [DOI] [PubMed] [Google Scholar]
- 28. Vartanian T, Li Y, Zhao M, Stefansson K. Interferon‐gamma‐induced oligodendrocyte cell death: implications for the pathogenesis of multiple sclerosis. Mol Med 1995; 1:732–43. [PMC free article] [PubMed] [Google Scholar]
- 29. Rolla S, Bardina V, De Mercanti S, Quaglino P, De Palma R, Gned D et al Th22 cells are expanded in multiple sclerosis and are resistant to IFN‐β. J Leukoc Biol 2014; 96:1–10. [DOI] [PubMed] [Google Scholar]
- 30. Wing AC, Ferreira TB, Kasahara M, Barros PO, Camargo S, Rueda F et al Interleukin‐17‐ and interleukin‐22‐secreting myelin‐specific CD4+ T‐cells resistant to corticoids are related with active brain lesions in multiple sclerosis patients. Immunology 2015; 147: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhen J, Yuan J, Fu Y, Zhu R, Wang M, Chang H et al IL‐22 promotes Fas expression in oligodendrocytes and inhibits FOXP3 expression in T‐cells by activating the NF‐κB pathway in multiple sclerosis. Mol Immunol 2017; 82:84–93. [DOI] [PubMed] [Google Scholar]
- 32. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K et al Expression of GM‐CSF in T‐cells is increased in multiple sclerosis and suppressed by IFN‐β therapy. J Immunol 2015; 194:5085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Restorick SM, Durant L, Kalra S, Hassan‐Smith G, Rathbone E, Douglas MR et al CCR6+ Th cells in the cerebrospinal fluid of persons with multiple sclerosis are dominated by pathogenic non‐classic Th1 cells and GM‐CSF‐only‐secreting Th cells. Brain Behav Immun 2017; 64:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Komai‐Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T‐cells as a costimulatory receptor. Proc Natl Acad Sci USA 2004; 101:3029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marsland BJ, Kopf M. Toll‐like receptors: paving the path to T‐cell‐driven autoimmunity? Curr Opin Immunol 2007; 19:611–4. [DOI] [PubMed] [Google Scholar]
- 36. Klonowska‐szymczyk A, Wolska A, Robak T, Cebula‐obrzut B, Smolewski P, Robak E. Expression of Toll‐like receptors 3, 7, and 9 in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Mediators Inflamm 2014; 2014:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nyirenda MH, Morandi E, Vinkemeier U, Constantin‐Teodosiu D, Drinkwater S, Mee M et al TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T‐cell function in multiple sclerosis. J Immunol 2015; 194:5761–74. [DOI] [PubMed] [Google Scholar]
- 38. Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M et al Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann Neurol 2011; 69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33:1444–52. [DOI] [PubMed] [Google Scholar]
- 40. Voo KS, Bover L, Harline ML, Weng J, Sugimoto N, Liu Y‐J. Targeting of Toll‐like receptors inhibits CD4+ regulatory T‐cell function and activates lymphocytes in human PBMCs. J Immunol 2014; 193:627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood 2013; 121:2402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bhaumik S, Basu R. Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front Immunol 2017; 8:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA et al Pro‐inflammatory human Th17 cells selectively express P‐glycoprotein and are refractory to glucocorticoids. J Exp Med 2014; 211:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. da Costa DSMM, Hygino J, Ferreira TB, Kasahara TM, Barros PO, Monteiro C et al Vitamin D modulates different IL‐17‐secreting T‐cell subsets in multiple sclerosis patients. J Neuroimmunol 2016; 299:8–18. [DOI] [PubMed] [Google Scholar]
- 45. Shi G, Vistica BP, Nugent LF, Tan C, Wawrousek EF, Klinman DM et al Differential involvement of Th1 and Th17 in pathogenic autoimmune processes triggered by different TLR ligands. J Immunol 2013; 191:415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rudd BD, Brien JD, Davenport MP, Nikolich‐Žugich J. Cutting edge: TLR ligands increase TCR triggering by slowing peptide‐MHC class I decay rates. J Immunol 2008; 181:5199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mercier BC, Cottalorda A, Coupet C‐A, Marvel J, Bonnefoy‐Bérard N. TLR2 engagement on CD8 T‐cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol 2009; 182:1860–7. [DOI] [PubMed] [Google Scholar]
- 48. Bendigs S, Salzer U, Lipford GB, Wagner H, Heeg K. CpG‐oligodeoxynucleotides co‐stimulate primary T‐cells in the absence of antigen‐presenting cells. Eur J Immunol 1999; 29:1314–27. [DOI] [PubMed] [Google Scholar]
- 49. Miranda‐Hernandez S, Gerlach N, Fletcher JM, Biros E, Mack M, Körner H et al Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol 2011; 187:791–804. [DOI] [PubMed] [Google Scholar]
- 50. Reynolds JM, Pappu BP, Peng J, Martinez GJ, Chung Y, Ma L et al Toll‐like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 2010; 32:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Christensen SR, Shlomchika MJ. Regulation of lupus‐related autoantibody production and clinical disease by Toll‐like receptors. Semin Immunol 2007; 19:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thwaites R, Chamberlain G, Sacre S. Emerging role of endosomal Toll‐like receptors in rheumatoid arthritis. Front Immunol 2014; 5:5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barros PO, Dias ASO, Kasahara TM, Ornelas AMM, Aguiar RS, Leon SA et al Expansion of IL‐6+ Th17‐like cells expressing TLRs correlates with microbial translocation and neurological disabilities in NMOSD patients. J Neuroimmunol 2017; 307:82–90. [DOI] [PubMed] [Google Scholar]
- 54. Zastepa E, Fitz‐Gerald L, Hallett M, Antel J, Bar‐Or A, Baranzini S et al Naive CD4 T‐cell activation identifies MS patients having rapid transition to progressive MS. Neurology 2014; 82:681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ghoreschi K, Laurence A, Yang X, Tato CM, Mandy J, Konkel J et al Generation of pathogenic Th17 cells in the absence of TGF‐b signaling. Nature 2010; 467:967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce‐shaikh B, Blumenschein W et al Interleukin 23 receptor is essential for terminal differentiation of effector T helper type 17 cells in vivo . Nat Immunol 2009; 10:314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McGeachy MJ, Bak‐Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T et al TGF‐beta and IL‐6 drive the production of IL‐17 and IL‐10 by T‐cells and restrain T(H)‐17 cell‐mediated pathology. Nat Immunol 2007; 8:1390–7. [DOI] [PubMed] [Google Scholar]
- 58. Mufazalov IA, Schelmbauer C, Regen T, Kuschmann J, Wanke F, Gabriel LA et al IL‐1 signaling is critical for expansion but not generation of autoreactive GM‐CSF + Th17 cells. EMBO J 2017; 36:102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Samoilova EB, Horton JL, Hilliard B. Liu T‐ST, Chen Y. IL‐6‐deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL‐6 in the activation and differentiation of autoreactive. J Immunol 1998; 161:6480–6. [PubMed] [Google Scholar]
- 60. Ferreira TB, Hygino J, Barros PO, Teixeira B, Kasahara TM, Linhares UC et al Draft version. Endogenous interleukin‐6 amplifies interleukin‐17 production and corticoid‐resistance in peripheral T‐cells from patients with multiple sclerosis. Immunology 2014; 143:560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M et al Blockade of interleukin‐6 signaling augments regulatory T‐cell reconstitution and attenuates the severity of graft‐versus‐host disease. Blood 2009; 114:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y et al The influence of excessive IL‐6 production in vivo on the development and function of Foxp3+ regulatory T‐cells. J Immunol 2011; 186:32–40. [DOI] [PubMed] [Google Scholar]
- 63. Mortezagholi S, Babaloo Z, Rahimzadeh P, Namdari H, Ghaedi M, Gharibdoost F et al Evaluation of TLR9 expression on PBMCs and CpG ODN‐TLR9 ligation on IFN‐α production in SLE patients. Immunopharmacol Immunotoxicol 2017; 39:11–8. [DOI] [PubMed] [Google Scholar]
- 64. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll‐like receptor 4 signaling in T‐cells promotes autoimmune inflammation. Proc Natl Acad Sci USA 2012; 109:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B et al Dysregulation of the cCytokine GM‐CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity 2017; 46:245–60. [DOI] [PubMed] [Google Scholar]
- 66. Cree BAC, Spencer CM, Varrin‐Doyer M, Baranzini SE, Zamvil SS. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens . Ann Neurol 2016; 80:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Varrin‐Doyer M, Spencer CM, Schulze‐Topphoff U, Nelson PA, Stroud RM, Bruce BA et al Aquaporin 4‐specific T‐cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 2012; 72:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T et al Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLOS ONE 2015; 10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nakanishi Y, Sato T, Ohteki T. Commensal Gram‐positive bacteria initiates colitis by inducing monocyte/macrophage mobilization. Mucosal Immunol 2014; 8:1–9. [DOI] [PubMed] [Google Scholar]
- 70. Teixeira B, Bittencourt VC, Ferreira TB, Kasahara TM, Barros PO, Alvarenga R et al Low sensitivity to glucocorticoid inhibition of in‐vitro Th17‐related cytokine production in multiple sclerosis patients is related to elevated plasma lipopolysaccharide levels. Clin Immunol 2013; 148:209–18. [DOI] [PubMed] [Google Scholar]
- 71. Aquino DA, Capello E, Weisstein J, Sanders V, Lopez C, Tourtellotte WW et al Multiple sclerosis: altered expression of 70‐ and 27‐kDa heat shock proteins in lesions and myelin. J Neuropathol Exp Neurol 1997; 56:664–72. [PubMed] [Google Scholar]
- 72. Lund BT, Chakryan Y, Ashikian N, Mnatsakanyan L, Bevan CJ, Aguilera R et al Association of MBP peptides with Hsp70 in normal appearing human white matter. J Neurol Sci 2006; 249:122–34. [DOI] [PubMed] [Google Scholar]
- 73. Mansilla MJ, Costa C, Eixarch H, Tepavcevic V, Castillo M, Martin R et al Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLOS ONE 2014; 9:e105737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The levels of expression of Toll‐like receptor (TLR) on T‐cells from multiple sclerosis (MS) patients.
Figure S2. Representative flow cytometric histograms showing the percentage of peripheral cytokine‐producing CD4+ T‐cells able to express, or not, Toll‐like receptor (TLR)‐2, ‐4 and ‐9 in healthy and multiple sclerosis (MS) patients.
Figure S3. Representative flow cytometric histograms showing the percentage of peripheral cytokine‐producing CD8+ T‐cells able to express, or not, Toll‐like receptor (TLR)‐2, ‐4 and ‐9 in healthy and multiple sclerosis (MS) patients.