

Abstract
Primary cilia are hair-like organelles that play crucial roles in vertebrate development, organogenesis and when dysfunctional result in pleiotropic human genetic disorders called ciliopathies, characterized by overlapping phenotypes, such as renal and hepatic cysts, skeletal defects, retinal degeneration and central nervous system malformations. Primary cilia act as communication hubs to transfer extracellular signals into intracellular responses and are essential for Hedgehog (Hh) signal transduction in mammals. Despite the renewed interest in this ancient organelle of growing biomedical importance, the molecular mechanisms that trigger cilia formation, extension and ciliary signal transduction are still not fully understood. Here we provide, for the first time, evidence that the deubiquitinase ubiquitin-specific protease-14 (Usp14), a major regulator of the ubiquitin proteasome system (UPS), controls ciliogenesis, cilia elongation and Hh signal transduction. Moreover, we show that pharmacological inhibition of Usp14 positively affects Hh signal transduction in a model of autosomal dominant polycystic kidney disease. These findings provide new insight into the spectrum of action of UPS in cilia biology and may provide novel opportunities for therapeutic intervention in human conditions associated with ciliary dysfunction.
Introduction
Primary cilia are microtubule-based organelles anchored by the basal body and extending from the apical cell surface. The biogenesis and resorption of cilia are intimately associated with cell cycle, as primary cilia form during quiescence and resorb before mitosis (1,2). Proteins travel from the cytoplasm to their final destination via intra-flagellar transport (IFT), which consists of kinesin-mediated anterograde and dynein-powered retrograde microtubule-based transport of cargo within cilia (3).
Cilia play a prominent role in development, tissue maintenance and regeneration, and their abnormalities result in severe human developmental diseases, called ciliopathies, which include rare conditions such as the Oral-facial-digital type I [OFDI (MIM 311200)], Meckel–Gruber (MIM 611134), Bardet–Biedl [BBS (MIM 615992)] and Joubert syndromes [JBS (MIM 614464)] and more common disorders such as the autosomal dominant and recessive forms of polycystic kidney disease, ADPKD (MIM 173900) and ARPKD (MIM 263200), respectively (4). Ciliopathies show overlapping phenotypes, such as retinal degeneration, renal and hepatic cysts, skeletal defects, situs inversus, obesity, ciliary dyskinesia, mental retardation, central nervous system malformations and infertility (4,5). Mutations in ciliary proteins associated with ciliopathies can lead to shorter, longer, malformed or missing cilia. In some instances, ciliopathies are associated with morphologically normal cilia such in the case of ADPKD, caused by mutations in polycystins, PC1 and PC2, codified by PKD1 and PKD2, respectively, which localize to primary cilia but are not critical for ciliogenesis (6–8).
Primary cilia act as sensory organelles that integrate multiple signaling pathways, such as Wnt, Notch, PCP, Pdgfα and the Hedgehog (Hh) signaling (9). In particular, Hh signaling, which exerts an essential role in promoting development and tissue homeostasis, is fully dependent on primary cilia, unlike other signaling pathways (reviewed in 10). Key regulatory components of the Hh pathway show dynamic changes in their localization to cilia, depending on the presence or absence of specific inputs from the ligand Hh that are critical to execute Hh signal transduction (11–14). Thus, under Hh stimulus, the transcriptional effectors Gli2 and Gli3 become enriched at the tip of cilia (11) where they are activated by the transmembrane protein Smoothened (Smo) through an unknown mechanism and then migrate to the nucleus to activate target gene transcription (14,15). The negative regulators Sufu and Kif7 are also enriched at cilia tips under Hh stimulus (15–18). In particular Kif7, a ciliopathy protein (19,20) that belongs to the kinesin-4 family of proteins, displays an essential role in controlling both localization of Hh signaling molecules and cilia length, through its ability to regulate dynamics of microtubule plus ends (21).
The molecular mechanisms that trigger cilia formation, axonemal extension, ciliary signal transduction and trafficking of Hh mediators to and within cilia are not fully understood.
Interestingly, genomic RNAi screens (22,23), in addition to high-throughput affinity proteomics (24) and in silico network-based analysis (25), showed that components of the ubiquitin proteasome system (UPS) are involved in biogenesis and/or maintenance of the primary cilium. In addition, proteasomal degradation of specific targets such as trichoplein, a keratin intermediate filament scaffold protein, promotes ciliogenesis and, UPS-mediated degradation of NDE1, a centrosomal protein, affects ciliary length (26–28). In addition, we and others demonstrated that impaired degradation of ciliary signaling pathway mediators is associated with BBS and OFDI syndromes (29). It is thus likely that a close functional link between ciliary proteins and the proteasome does exist.
Proteomic studies, based on protein correlation profiles or proximity labeling on mammalian primary cilia (30,31) and on isolated cilia from different flagellated protozoa (32) show some common UPS components including ubiquitin-specific protease-14 (USP14), a well-characterized regulator of the UPS, which inhibits the activity of proteasomes by removing ubiquitin chains from specific proteasome-bound substrates, thus inhibiting their degradation (33–36).
Here we provide evidence that USP14 controls ciliogenesis, cilia elongation and Hh signal transduction. Moreover, we show that Hh signal transduction is impaired in mouse embryonic fibroblasts (MEFs) from an ADPKD murine model and that pharmacological Usp14 inhibition positively affects Hh signal transduction in this disease model.
These findings provide new insight into the spectrum of action of UPS in cilia biology and may provide novel opportunities for therapeutic intervention on human conditions associated with ciliary dysfunction.
Results
Usp14 controls ciliogenesis and cilia length
We hypothesized that the UPS component Usp14 could play a role in cilia biology. We propose that inhibition of Usp14 may stimulate ciliogenesis since proteasomal degradation of specific targets promotes ciliogenesis and affects ciliary length (26–28). To test this hypothesis Usp14 was silenced using siRNA in cycling MEFs. The efficiency of Usp14 silencing was confirmed (Fig. 1A), and the effects on ciliogenesis were determined after 96 h (Fig. 1B). Despite the cells being cultured in 10% FBS medium, and thus cycling, knockdown of Usp14 had a strong effect on cilium formation, as 14% of silenced cells had visible cilia, while only ∼4% of cycling serum-stimulated control MEFs were ciliated (Fig. 1B), as expected for non-quiescent cells.
Figure 1.
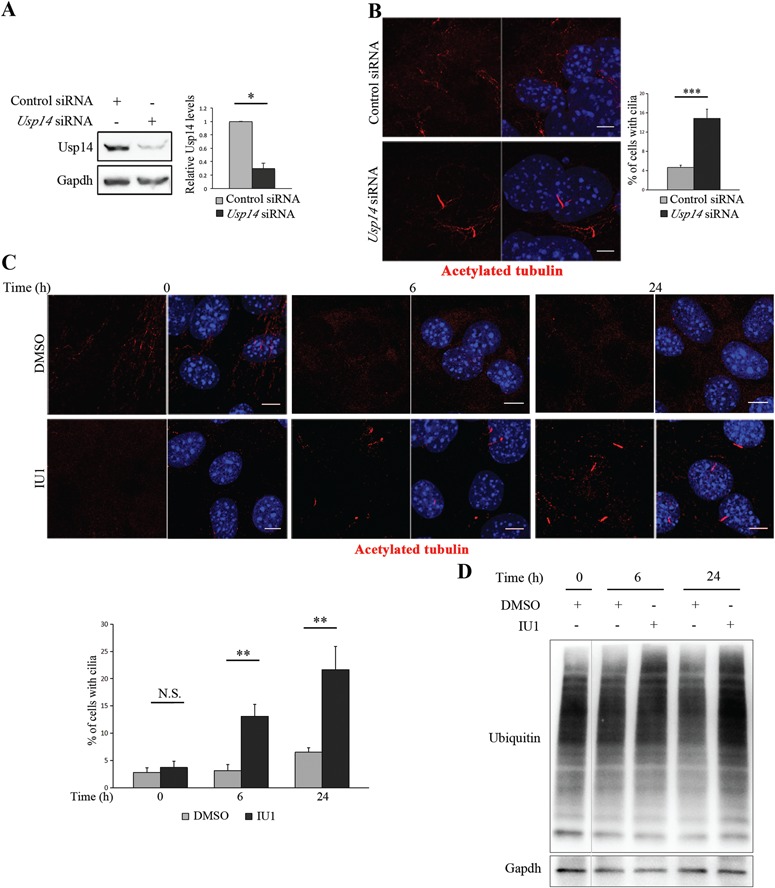
Usp14 controls ciliogenesis. (A) Validation by WB of Usp14 depletion in MEFs transfected with siRNAs targeting Usp14 (Usp14 siRNA) and non-targeting negative control siRNAs (Control siRNA). (B) Representative confocal images of primary cilia in cycling MEFs in complete medium (with FBS), transfected with Usp14 siRNA and control siRNA for 96 h. Acetylated tubulin (red) marks cilia. Hoechst (blue) labels nuclei. Histogram shows the quantification of percentage of ciliated cells in this culture condition. (C) Representative confocal images of primary cilia in cycling MEFs in complete medium, treated with IU1 or DMSO for 0, 6 and 24 h. Acetylated tubulin (red) marks cilia. Hoechst (blue) labels nuclei. Histogram, bottom left, shows the quantification of percentage of ciliated cells. (D) Analysis by WB to verify suppression of ubiquitin chain trimming by IU1 at 0, 6 and 24 h of treatment. The bands are at the same exposure, non-contiguous on the same gel. Data are expressed as mean ± SEM (n = 3 independent experiments); *P < 0.05; **P < 0.01; ***P < 0.001, one-tailed Student’s t-test (A) and the likelihood ratio test for negative binomial generalized linear models (B and C) were applied; ≥250 cells (B) and ≥140 cells (C) were analyzed per sample; Scale bar = 5 μm.
To test whether Usp14 antagonizes ciliogenesis through its catalytic activity we treated wild-type (wt) MEFs with the small molecule 1-[1-(4-fluorophenyl)-2,5-dimethylpyrrol-3-yl]-2-pyrrolidin-1-ylethanone (IU1), a specific inhibitor of the catalytic activity of Usp14, which enhances degradation of a subset of proteasomal substrates (35–47). The treatment with IU1 had no apparent effects on viability of MEFs. Cells were scored for the presence of cilia under serum stimulus at 6 and 24 h of IU1 treatment; the number of ciliated cells increased over time, while DMSO-treated controls remained unciliated without serum depletion (Fig. 1C). The suppression of ubiquitin chain removal by IU1 was verified by western blot (WB) analysis of whole cell lysates using antibodies to ubiquitin (Fig. 1D).
Proteasomal activity impacts different stages of ciliogenesis, promoting proper cilia biogenesis and elongation (26–28). We therefore examined whether depletion of Usp14 may influence axoneme elongation. Usp14 siRNA-treated MEFs were analyzed 30 h after serum starvation and displayed increased ciliary length compared with controls (Fig. 2A). To provide independent evidence, Usp14 wt and knockout MEFs (Usp14+/+and Usp14−/−, respectively) were plated in complete medium, grown to 100% density and then serum starved for 30 h; the average length of cilia was remarkably increased in Usp14−/− MEFs, compared with Usp14+/+ cells (Fig. 2B). An increased ciliary length was also observed in wt MEFs that were plated in complete medium, serum-starved for 30 h and treated with IU1 for 24 h, confirming that the effect of Usp14 on cilia elongation is mediated by its catalytic activity (Fig. 2C). Consistent with these observations, doxycycline-inducible Usp14 expression delivered through lentiviral infection in Usp14−/− MEFs, rescued cilia length (Fig. 2D). In addition, lentiviral delivered expression of an active site mutant of Usp14, Usp14-C114A (Usp14-CA) (35), did not show any effect on cilia length (Fig. 2E), further supporting that Usp14 catalytic activity is responsible for cilia length control. The significant induction of both Usp14 and Usp14-CA expression in response to doxycycline treatment was verified by WB analysis (Supplementary Material, Fig. S1).
Figure 2.
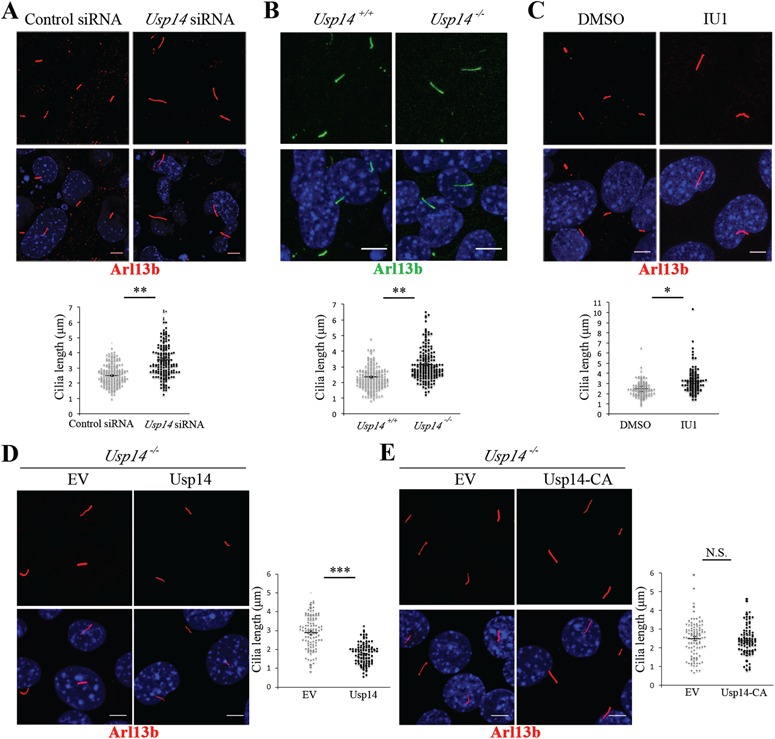
Usp14 controls cilia length. (A) Representative confocal images of primary cilia in Usp14 depleted (Usp14 siRNA) and non-targeting negative control siRNAs (Control siRNA) transfected MEFs. Arl13b marks primary cilia (red) and Hoechst (blue) labels nuclei. (B) Staining of primary cilia with Arl13b (green) and of nuclei with Hoechst dye (blue) in Usp14+/+and Usp14−/− MEFs. (C) Staining of primary cilia with Arl13b (red) and of nuclei with Hoechst (blue) in wt MEFs treated with IU1 or DMSO for 24 h. (D) Staining of primary cilia with Arl13b (red) and of nuclei with Hoechst (blue) in Usp14−/− MEFs expressing lentiviral delivered with empty vector (EV) and wt Usp14. (E) Staining of primary cilia with Arl13b (red) and of nuclei with Hoechst (blue) in Usp14−/− MEFs expressing lentiviral delivered EV and the active site mutant Usp14-CA. For each condition cells were serum-starved for 30 h. Charts show quantification of primary cilia length. Data are expressed as mean ± SEM (n = 3 independent experiments); one-tailed Student’s t-test *P < 0.05; **P < 0.01; ≥150 cells (A), ≥190 cells (B) and ≥ 100 cells (C–E) were analyzed per each sample; Scale bar = 5 μm. N.S. = not significative.
The length of cilia and the proportion of a cell population with cilia are increased in quiescent (G0) rather than cycling cells (1,2). To investigate whether there was a correlation between increased number of cilia, longer cilia and cellular growth rate, we assayed cell proliferation by counting viable cells at different time points (1, 2, 3 and 4 days) (Supplementary Material, Fig. S2). Our results demonstrated that Usp14−/− MEFs show an increased proliferation rate compared with controls supporting the idea that the ciliary phenotype observed in Usp14 depleted cells is not associated to a slower mitotic index. In addition, the clear requirement of Usp14 in restraining cilia elongation hinders the dissection of the contribution of this protein in cilia disassembly. We showed that, similarly to control cells, Usp14−/− MEFs undergo cilium disassembly following serum stimulation (2 h), confirming that Usp14 controls cilia assembly and is dispensable for cilia disassembly (Supplementary Material, Fig. S3).
Taken together, the above results indicate a role for Usp14 in the control of ciliogenesis in vitro and highlight the capability of pharmacological proteasomal degradation induction to positively affect ciliogenesis and cilia elongation.
Usp14 controls localization of Hh mediators at cilia
To analyze the influence of Usp14 on cilia function, we focused on Hh signal transduction pathway, which is entirely dependent on primary cilia, although its activation is not affected by cilia length and does not influence cilia morphology (10,48,49). A major parameter associated with active Hh signaling is the increased levels of pathway mediators along the primary cilium under Hh stimulus (15). We first determined whether localization of Hh signaling components was affected in cilia of Usp14−/− MEFs. We assayed the localization of Gli2 and Gli3 by fluorescence staining with endogenous antibodies, and the fluorescence was quantified in unstimulated conditions or after 24 h of stimulation with either the Smoothened agonist (SAG) or the ligand Sonic Hedgehog (Shh) recombinant protein. Gli2 staining was significantly enriched in cilia of mutant Usp14−/− MEFs upon induction with SAG or Shh compared with Usp14+/+ cells (Fig. 3A). Similarly, the fluorescence intensity of ciliary Gli3 was stronger in Usp14−/− mutant cells compared with wt cells under both SAG and Shh stimuli (Supplementary Material, Fig. S4). We also assayed localization of endogenous Gli2 and Gli3 with pericentrin, a basal body marker, after 24 h of stimulation with SAG. This experiment revealed that Gli2/3 staining was enriched at the tip of cilia, where they usually localize under stimulus (Supplementary Material, Fig. S5).
Figure 3.
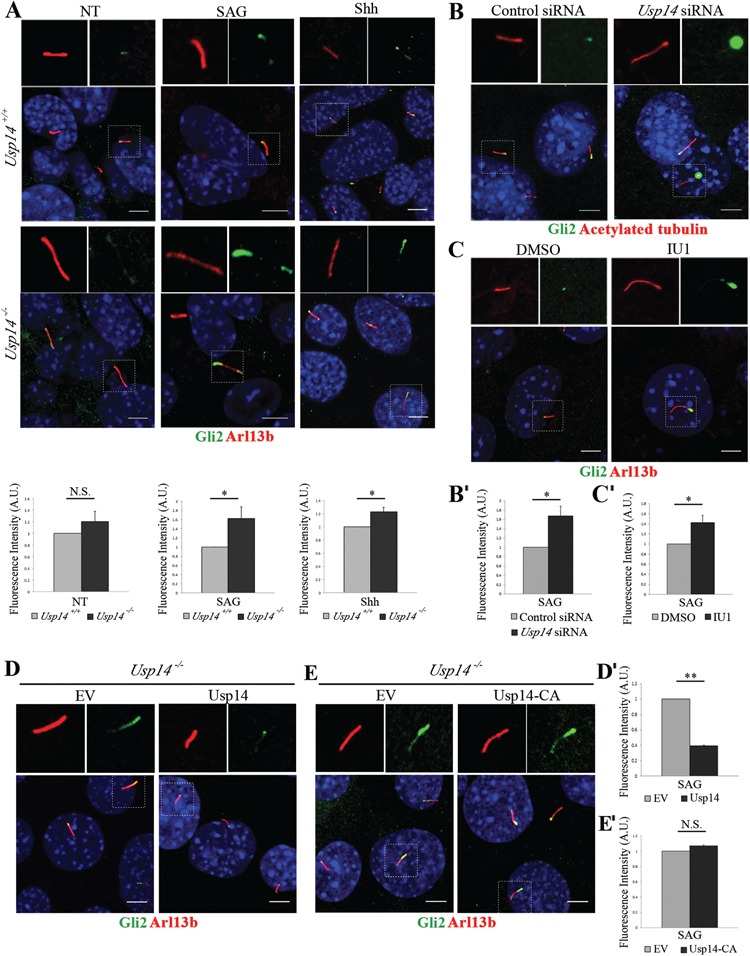
Usp14 controls localization of Gli2 at cilia. (A) Representative confocal images of endogenous Gli2 (green) localization to cilia in Usp14+/+ and Usp14−/− MEFs in basal condition (NT) and under SAG or Shh stimuli. Arl13b marks primary cilia (red), nuclei were stained with Hoechst dye (blue). Histograms below each panel show the quantification of the relative fluorescence intensity of ciliary Gli2 for NT, SAG and Shh stimulated MEFs, respectively. (B) Immunofluorescence staining of endogenous Gli2 (green) and acetylated tubulin (red) on Usp14 siRNA depleted MEFs and controls under SAG stimulus for 24 h. Hoechst labels nuclei (blue). Histogram (B') shows the quantification of Gli2 relative fluorescence intensity. (C) Immunofluorescence staining of endogenous Gli2 (green) on wt MEFs under SAG stimulus and treated with IU1 inhibitor or DMSO for 24 h. Arl13b marks primary cilia (red). Histogram (C') shows the quantification of Gli2 relative fluorescence intensity. (D) Staining of endogenous Gli2 (green) on Usp14−/− MEFs expressing lentiviral delivered EV and wt Usp14. (E) Staining of endogenous Gli2 (green) on Usp14−/− MEFs expressing lentiviral EV and the active site mutant Usp14-CA. Arl13b marks primary cilia (red). Cells are under SAG stimulus. (D' and E') Histograms show quantification of Gli2 relative fluorescence intensity. Data are expressed as mean ± SEM (n = 3 independent experiments); one-tailed Student’s t-test *P < 0.05; **P < 0.01. ≥80 cells (A), ≥300 cells (B) and ≥130 cells (C) and ≥100 (D and E) were analyzed for each sample; Scale bar = 5 μm. SAG = Smoothened agonist, Shh = recombinant N-Shh, A.U. = arbitrary units, N.S. = not significative, NT = not treated (not stimulated).
To provide independent evidence of Usp14 controlling the localization of Hh mediators at cilia, Usp14 was silenced by siRNA in wt MEFs, and cells were stimulated with SAG for 24 h. Immunofluorescence experiments showed that also transient silencing of Usp14 had a strong effect on Gli2 ciliary localization compared with controls (Fig. 3B and B'). We then asked whether Usp14 inhibition promotes recruitment of Hh signaling effectors to cilia through its catalytic activity. Wt MEFs were plated in complete medium, serum-starved for 30 h and treated with IU1 for 24 h. Confocal analysis revealed increased ciliary fluorescence intensity for Gli2 confirming that the catalytic activity of Usp14 acts also on the ciliary localization of Hh signaling components (Fig. 3C and C'). Consistent with these observations, doxycycline-inducible lentiviral delivered expression of Usp14 in Usp14−/− MEFs rescued Gli2 ciliary localization (Fig. 3D and D'). Similarly, lentiviral delivered expression of active site mutant Usp14-CA did not show any effects on Gli2 ciliary localization (Fig. 3E and E').
Moreover, we demonstrated that enhanced recruitment of Gli2/3 proteins to cilia consequent either to genetic loss of Usp14 or to its pharmacological inhibition is not associated with increased levels of Gli2/3 proteins, which are even lower compared with controls, as revealed by WB analysis (Supplementary Material, Fig. S6).
These data suggest that the efficiency of ciliary recruitment and transport of Hh regulatory proteins is controlled by Usp14.
Usp14 controls Gli2 nuclear translocation
The enhanced ciliary localization of Gli proteins observed in Usp14 depleted models could also be associated with defective retrograde transport. We thus monitored Gli2, the main Hh transcriptional activator, which is normally translocated to the nucleus to activate gene targets transcription under Hh stimulus (15). IU1 treatment of SAG-stimulated wt MEFs revealed that the nuclear amounts of Gli2 are increased compared with amounts detected in untreated cells, and conversely, levels of cytoplasmic Gli2 protein decreased compared with untreated cells (Fig. 4A). The same findings were obtained for SAG-stimulated Usp14 siRNA-treated MEF cells and for Usp14−/− MEF cells (Fig. 4A). Accordingly, transfection of 3xFLAG-Usp14 in Usp14−/− SAG-stimulated MEFs resulted in increased cytoplasmic levels and reduced nuclear amounts of Gli2 compared with cells transfected with an empty vector (EV), as the protein is retained in the cytoplasm (Fig. 4B).
Figure 4.
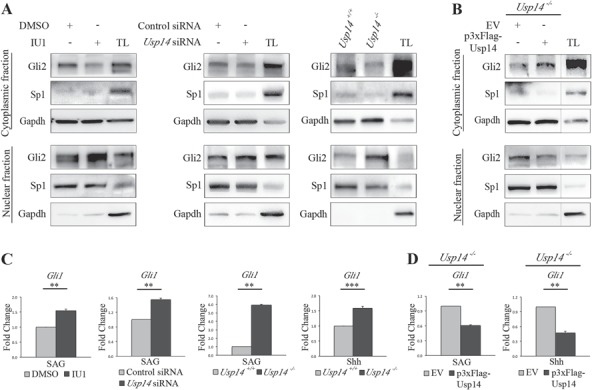
Usp14 controls migration of Gli2 to the nucleus. (A) WB analysis of Gli2 protein levels in cytoplasmic and nuclear fractions from (left) IU1 or DMSO-treated MEFs, (central) Usp14 siRNA depleted MEFs and control and (right) Usp14+/+and Usp14−/− MEFs lysates. The quality of the fractions was analyzed by using Gapdh as cytoplasm marker and Sp1 as nuclear marker. For all three conditions cells were stimulated with SAG for 6 h. (B) WB analysis on Gli2 protein levels in cytoplasmic and nuclear fractions from Usp14−/− cells transiently transfected with EV or with p3xFlag-Usp14 for 48 h. Cells were stimulated with SAG for 6 h. The bands were non-contiguous but on the same gel, at the same exposure. (C) qRT-PCR analysis of mRNA expression levels of Gli1 in IU1 or DMSO-treated wt MEFs under SAG treatment for 24 h (first panel), in Usp14 siRNA depleted MEFs and controls (second panel) and in Usp14+/+and Usp14−/− MEFs (third panel). qRT-PCR expression analysis of Gli1 in Usp14+/+and Usp14−/− MEFs under Shh treatment for 24 h is shown in the fourth panel. (D)Usp14−/− cells transiently transfected for 48 h with EV or p3xFlag-Usp14 and stimulated with SAG (left) and Shh (right) for 24 h. The qRT-PCR expression analysis is expressed as fold change after normalization (Hprt mRNA). Data are expressed as mean ± SEM (n = 3 independent experiments); one-tailed Student’s t-test **P < 0.01, ***P < 0.001. SAG = Smoothened agonist, Shh = recombinant N-Shh, TL = total lysate.
To confirm the functional activity of enhanced levels of nuclear Gli2 retrieved in the absence of Usp14, we evaluated the levels of activation of Gli1, the main downstream direct target. MEFs were exposed to both SAG and Shh stimuli for 24 h, and Gli1 transcriptional levels were monitored by quantitative real time quantitative reverse transcription polymerase chain reaction (qRT-PCR). Our results indicated that levels of Gli1 were significantly higher in IU1-treated cells compared with DMSO-treated controls (Fig. 4C). These findings were confirmed both in Usp14−/− MEFs and Usp14 siRNA depleted cells (Fig. 4C). Consistent with these observations, transfection of 3xFLAG-Usp14 into Usp14−/− MEFs rescued Gli1 mRNA expression levels compared with cells transfected with an EV (Fig. 4D), suggesting that Usp14 acts as an inhibitor of the Hh signal transduction pathway.
The above-described results reveal a critical role for Usp14 and for activation of proteasomal degradation, not only in the control of ciliogenesis and cilia elongation, but also in Hh signaling.
Kif7 is a target of Usp14
To our knowledge Kif7 is the only Hh inhibitor that controls both localization of Hh mediators and axonemal length (17,18,21). We thus hypothesized that Usp14 may act on Hh signaling through degradation of the Kif7 protein.
WB analysis of whole cell lysates from wt MEFs treated with IU1 showed that protein levels of endogenous Kif7 were significantly decreased in IU1-treated wt cells (Fig. 5A), Usp14 siRNA-transfected MEFs (Fig. 5B) and Usp14−/− MEFs (Fig. 5C) compared with respective controls. Conversely, 3xFLAG-Usp14 overexpression increased Kif7 protein levels both in wt and Usp14−/− MEFs compared with cells transfected with an EV (Fig. 5D). Taken together these findings strongly suggest that Kif7 is a target of Usp14.
Figure 5.
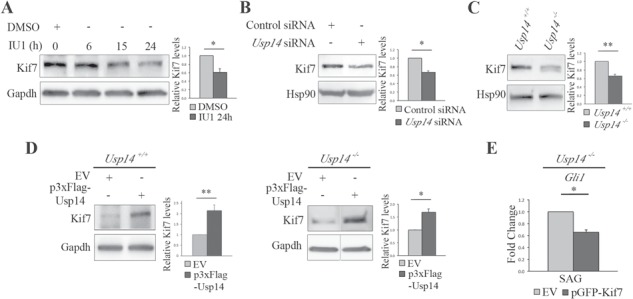
Usp14 inhibition promotes Kif7 degradation. (A) WB analysis of Kif7 protein levels in wt MEFs treated with IU1 at different time points. Histogram on the right shows quantification of the relative band intensities for Kif7 normalized versus Gapdh after 24 h of treatment. (B–D) WB analysis of Kif7 protein levels in (B) Usp14 siRNA depleted MEFs and control, (C) Usp14+/+and Usp14−/− MEFs and (D) Usp14+/+ (left) and Usp14−/− (right) MEFs transiently transfected with EV or p3xFlag-Usp14 for 48 h. The bands are at the same exposure, non-contiguous on the same gel (D, panel on the right). For all conditions, MEFs were stimulated by SAG for 24 h. Histograms show quantification of relative band intensities for Kif7 normalized versus Hsp90 (B and C) or versus Gapdh (D). (E) qRT-PCR analysis on mRNA expression levels of Gli1 under SAG treatment in Usp14−/− cells transiently transfected for 48 h with the EV vector or the pGFP-Kif7 construct, expressed as fold change after normalization (Hprt mRNA). Data are expressed as mean ± SEM (n = 3 independent experiments); one-tailed Student’s t-test *P < 0.05; **P < 0.01. SAG = Smoothened agonist, TL = total lysate, IP = immunoprecipitation.
Furthermore, we asked whether the reduced amount of Kif7 could be connected to the increase of Hh signaling activation observed in Usp14 depleted cells. Usp14−/− MEFs were transfected with EV alone or the GFP-Kif7 plasmid for 48 h, followed by qPCR to monitor Gli1 transcriptional levels. Our results indicated that levels of Gli1 were rescued in Kif7 overexpressing cells compared with controls (Fig. 5E), suggesting the existence of a novel mechanism by which the Hh pathway is regulated by the UPS.
Usp14 inhibition rescues Gli2 ciliary localization in Pkd1 mutant MEFs
Our findings show that the catalytic activity of Usp14 modulates ciliogenesis and Hh signaling transduction, suggesting that Usp14 is intimately related to cilia biology. We thus evaluated whether Usp14 inhibition might modulate the consequences of the Hh impairment observed in ciliopathies. Given the requirement of normal cilia for Hh signaling (10–14), we turned to a ciliated model system to study Hh signaling. We thus selected Pkd1−/− MEFs, an in vitro model that, in contrast to the majority of ciliopathy mutants, displays morphologically normal cilia, in agreement with the role in ciliary function rather than in ciliogenesis proposed for PC1 (7,8,50). Pkd1 null mice die at E13.5 and exhibit kidney, liver and pancreatic cystic disease associated to osteochondrodysplasia and delayed bone formation (51).
We first confirmed the presence of apparently normal cilia in Pkd1−/− MEFs and found that ∼95% of mutant and control confluent MEFs were ciliated, and that the length of cilia was comparable between Pkd1−/− and control samples (data not shown). The response of Pkd1−/− cells to exogenous Hh stimulation has never been tested and we observed that, despite the presence of cilia, under Hh stimulus Pkd1−/− cells do not display induction of Gli1 gene transcription that was only seen in wt MEFs (Fig. 6A). We thus evaluated the ciliary localization of Gli2 in Pkd1−/− cells by fluorescence staining with the antibody to endogenous Gli2 and quantified the fluorescence under SAG stimulus. The results indicated that Gli2 levels are dramatically decreased in Pkd1−/− mutant cilia (Fig. 6B). We also sought to determine whether the levels of Usp14 and Kif7 were affected in Pkd1−/− cells; WB analysis of whole cell lysates from wt and Pkd1−/− MEFs showed that both Usp14 and Kif7 were significantly increased in Pkd1−/− mutant MEFs compared with controls (Fig. 6C), probably contributing to loss of activation of Hh signaling. Consistent with these observations, treatment of Pkd1−/− MEFs with IU1 rescued concomitantly Kif7 protein levels (Fig. 6D) and Gli2 ciliary localization (Fig. 6E) and partially rescued Gli1 mRNA expression levels (Fig. 6F). The capability of Usp14 inhibition to rescue Gli2 ciliary localization and, partially, Gli1 expression levels in Pkd1−/− MEFs, was also confirmed in Usp14 siRNA-treated cells (Supplementary Material, Fig. S7).
Figure 6.
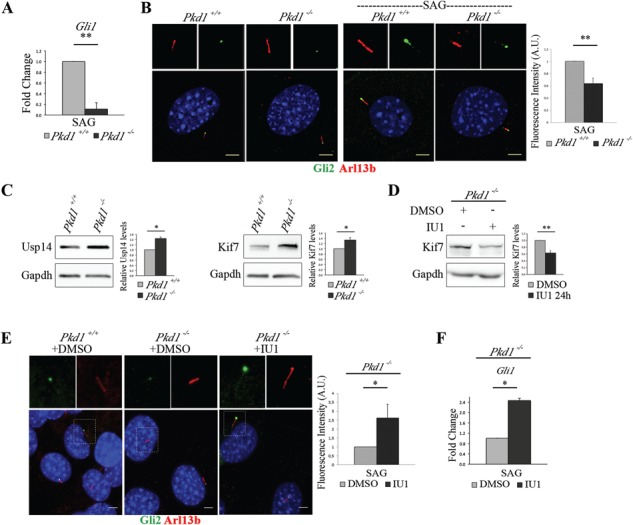
Usp14 inhibition rescues Gli2 ciliary localization in Pkd1 mutant MEFs. (A) mRNA expression levels of Gli1 in Pkd1+/+ and Pkd1−/− MEFs under 24 h of SAG stimulus expressed as fold change after normalization with the Hprt mRNA. (B) Immunofluorescence staining of endogenous Gli2 (green) and Arl13 (red) on unstimulated Pkd1+/+and Pkd1−/– MEFs (left panel), and on Pkd1+/+ and Pkd1−/− MEFs stimulated with SAG for 24 h (right panel). Hoechst labels nuclei (blue). Histogram shows the quantification of Gli2 relative fluorescence intensity on cilia in Pkd1+/+ and Pkd1−/− MEFs under SAG treatment. (C) Usp14, Kif7 and Gapdh were detected by WB analysis. Band intensities for Usp14 and Kif7 versus Gapdh were normalized and the relative quantification was represented in histograms as fold change. (D) WB analysis of Kif7 protein levels in Pkd1−/− MEFs treated with IU1 for 24 h. Histogram on the right shows quantification of the relative band intensities for Kif7 normalized versus Gapdh. (E) Immunofluorescence staining of endogenous Gli2 (green) and Arl13 (red) on Pkd1+/+ (DMSO, left panel), Pkd1−/− (DMSO, central panel) and Pkd1−/− treated with IU1 for 24 h (right panel). Hoechst labels nuclei (blue). Histogram shows the quantification of Gli2 relative fluorescence intensity on cilia of Pkd1−/− MEFs treated with IU1 and controls. Each sample was under SAG stimulus for 24 h. (F) qRT-PCR analysis revealed mRNA expression levels of Gli1 under SAG treatment for 24 h in IU1 or DMSO-treated Pkd1−/− MEFs. Data are expressed as mean ± SEM (n = 3 independent experiments); one-tailed Student’s t-test *P < 0.05; **P < 0.01. ≥100 cells were analyzed per sample. Scale bar = 5 μm. SAG = Smoothened agonist.
Altogether these results indicate that Pkd1 is required for Hh signal transduction and that Usp14 inhibition positively affects Hh signal transduction in MEFs from an ADPKD model.
Discussion
We report that deubiquitinase Usp14, a major regulator of the UPS (35,36), is involved in a novel signaling mechanism controlling ciliogenesis, ciliary length and Hh pathway activation. Ciliary defects are associated with severe developmental diseases, collectively called ciliopathies. Overlapping phenotypes in ciliopathies are associated with mutations in genes coding for proteins that are localized to primary cilia and/or control cilia elongation and functionality.
The majority of ciliopathies are associated with ciliary deficiency (abnormally short, absent or bulbous malformed cilia). Conversely some ciliopathies, such as certain forms of JBS, display abnormally long cilia (21,52–54). Longer cilia with an intact microtubular structure have also been detected in renal tubules from different ciliopathy murine models (55–59). Alternatively, mutations in ciliary proteins might not influence cilia morphology and instead affect cilia capability to transduce intracellular signaling pathways; thus, in the case of BBS2, BBS3 and BBS4, mutant proteins do not overtly affect cilia structure but interfere with ciliary entry or exit of signaling molecules (60,61). Another example is given by PC1, which was shown to be dispensable for correct ciliogenesis (8) but necessary for transduction of extracellular mechanical stimuli into a Ca2+ signaling response (7,50).
Recent data suggest that UPS is one of the core mechanisms in regulating formation and maintenance of primary cilia (22,23). Interestingly, UPS removes the trichoplein protein, leading to ciliogenesis (26), while affecting ciliary length by promoting degradation of NDE1, a suppressor of later stages of cilium formation (27,28). A role for proteasome-mediated degradation in cilia biology is also supported by the presence of UPS components in networks generated through proteomics (24) and in silico analyses (25). Finally, we and others demonstrated that ciliopathy proteins interact with regulatory subunits of the proteasome holoenzyme (29,62) and that perturbed UPS-mediated degradation of Shh, Wnt and Notch signaling mediators is associated with BBS and OFDI syndromes (29). Despite this body of evidence, the role of the proteasome complex in ciliogenesis and in signal transduction remains poorly understood.
Here we provide experimental evidence that Usp14 controls ciliogenesis and cilia length elongation, while also modulating the Hh pathway through its catalytic activity. The relationship between primary cilia and mammalian Hh signaling is fascinating, complex and directly relevant to ciliopathies, since important aspects of the disease phenotypes, such as skeletal and neural tube defects, can be explained by deregulation of Hh signaling (9). A combination of genetics, cell biology and developmental biology provided overwhelming evidence that mammalian Hh signal transduction depends on primary cilia and defined how ciliary regulators and components control the movement of Hh players (10). IFT is integral to Hh transduction by trafficking signaling components within cilia. Loss of most complex B proteins, which mediates anterograde IFT, causes absence of cilia and inability to transduce Hh signal (63–65), while loss of complex B IFT25 or IFT27 results in perturbed trafficking of Gli2 and Smo in the presence of normally structured cilia (66–68). In contrast, loss of IFT complex A proteins, IFT139 and IFT122, which largely mediate retrograde IFT, sequesters proteins at distal tips of shortened cilia and causes inappropriate activation of the Hh pathway (69,70). To our knowledge, KIF7, a member of the kinesin-4 family and an evolutionarily conserved component of the core Hh signal transduction pathway, is, to date, the only molecule known to control both cilia length and Hh signaling (21). Mutations in KIF7 have been reported in different ciliopathies associated with perinatal lethality, including fetal hydrolethalus, Acrocallosal syndrome and JBS (19,20,71–76). Intriguingly, fibroblasts from Acrocallosal syndrome patients show upregulation of direct targets of Hh signaling and longer cilia (20). Based on these observations, we propose a model where Usp14-mediated degradation of Kif7 regulates Hh signaling. It is tempting to hypothesize that Kif7 Usp14-mediated degradation contributes also to its role in ciliogenesis although additional experiments will be needed to validate our model. Our findings are also supported by recent data exploring the functional relationship between USP14 and other signaling pathways, such as JNK and WNT pathways (77,78).
Moreover, we show, for the first time, that drug-induced proteasomal degradation leads to cilia elongation and Hh signaling activation. Small molecules capable of activating or enhancing proteasome activity are rare and not well studied. Lee et al. discovered IU1, a small molecule acting as UPS inhibitor by specifically inhibiting the proteasome bound form of USP14, that accelerate proteasomal degradation of specific substrates in vitro (35–47). In addition, IU1 has recently been shown to enhance proteasome functionality also in vivo after treatment in mammalian models, thus extending the potential therapeutic applications of proteasome activators such as IU1 (79).
In accordance with our model, we show that inhibition of the catalytic activity of Usp14 rescues localization of Hh mediators to cilia in Pkd1−/−. Hh signaling has a key role in embryonic development, including vertebrate skeletal patterning and neural tube formation, which are commonly found altered in ciliopathies either individually or in combination with other conditions affecting kidneys, liver, eyes and other organs and tissues. Pkd1 loss of function in mice results in embryonic lethality associated with renal cystogenesis, cardiac abnormalities (80), vascular lesions (81) and skeletal malformations, consistent with the ubiquitous expression of the gene (82). In particular, in these mice, histology of long bones showed that the hypertrophic chondrocyte zone is reduced in length, similar to what seen in mice lacking Indian Hh, a member of the Hh family expressed in chondrocytes of early cartilaginous skeletal elements (83), indicating that loss of polycystin-1 can lead to defective chondrocyte differentiation and maturation, and is required for normal mouse skeletogenesis (80,84). Endochondral bone formation defects associated with Hh impairment were described in ciliopathy mutants such as OFDI (85), short-rib polydactyly syndrome and Jeune asphyxiating thoracic dystrophy (86), Ellis-van Creveld syndrome (87) and Sensenbrenner syndrome (88). Contrary to the majority of ciliary mutants, Pkd1−/− MEFs display normal cilia, and this model was thus chosen because of our need of a ciliopathy model in which cilia are fully formed, in order to investigate Hh signaling activation. Hh signaling has never been analyzed in Pkd1−/− MEFs; surprisingly we showed that Pkd1 is required for proper Hh signal transduction and for Gli2 localization to cilia tips under Hh stimulus. In agreement with our model, protein levels of both Usp14 and Kif7 are increased in Pkd1−/− MEFs, and Usp14 inhibition rescues the following: (a) Kif7 protein levels, (b) Gli2 ciliary localization and (c) at least partially, Gli1 target gene expression. The molecular mechanisms underlying these observations need to be determined as well as whether the increased Usp14 levels influence or simply accompany the progression of Hh downregulation in Pkd1−/− MEFs. We speculate that downregulation of Hh response in Pkd1−/− MEFs could be associated with the embryonic lethality and the defective chondrocyte differentiation and maturation observed in Pkd1 null mice.
One of the biggest challenges in ADPKD is to discover the specific local polycystin-regulated signaling processes that occur in renal cilia, and one paradigm may be found in the mammalian Hh signaling pathway. Experimental evidence indicate that Gli1 is upregulated in cystic kidneys of ciliary mutants (89–91). However, a recent paper showed that Hh signaling is intact in ADPKD primary renal epithelial cells, which correctly responds to Hh modulation (92) suggesting that further analysis of Hh signaling in appropriate in vivo models will be needed to determine the functional role for the Hh pathway in renal cystogenesis.
In conclusion, our findings not only support that UPS degradation is involved in ciliogenesis but also contribute to a comprehensive understanding of the role of Usp14 in governing a connection between ciliogenesis and Hh signal transduction. In addition, our findings that IU1 treatment has the ability to promote primary ciliogenesis and Hh activation may provide new insights in the spectrum of actions of proteasome activating compounds and may warrant further investigation into application of these molecules in future therapeutic approaches for ciliopathies.
Materials and Methods
Cell culture, treatments and proliferation assay
Usp14+/+ and Usp14−/− MEFs were provided by Professor Scott Wilson and were previously described (93). Pkd1−/− and Pkd1+/+ MEFs were kindly provided by Dr Alessandra Boletta and previously characterized (94). MEFs were cultured in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% FBS, 1% l-glutamine, 1% sodium pyruvate and 1% antibiotics (penicillin/streptomycin) at 37°C and 5% CO2. To induce ciliogenesis, MEFs were grown to 100% density and brought to quiescence by serum starvation with medium containing 0.5% FBS for 30 h. To activate Hh pathway cells were treated with either Shh-N recombinant protein 100 ng/ml (R&D Systems, Minneapolis, Minnesota) or SAG 200 nm (Millipore, Burlington, Massachusetts) for 24 h. Drug treatment with IU1 (Cayman Chemical, Ann Arbor, Michigan) was carried out at a final concentration of 40 μm.
For proliferation assay, cells were washed and counted (Trypan Blue staining of viable cells) using the LUNA™ automated cell counter (Logos Biosystems, Gyeonggi-do, South Korea) and then seeded in 6 well plates (130.000/well). Cells were allowed to grow at 37°C and 5% CO2, and each well was counted every day for 4 days after trypsinization. Three independent experiments were performed.
Constructs and cell transfections
Full-length murine Usp14 was tagged at the C-terminus with a 3xFLAG tag by cloning Usp14 into the 3xFLAG-pCMV vector. The generated plasmid was sequence-verified. The pEGFP-N3-Kif7 construct was previously described (21) and provided by Professor Kathryn V. Anderson. Cells (∼550.000/well) were transfected into 6 well plates, in suspension with TransIT®-LT1 Transfection Reagent (Mirus Bio, Madison, Wisconsin) following manufacturer’s instruction; after cells reached 100% confluency, they were brought to quiescence by serum starvation (0.5% FBS) for 30 h, treated with Shh/SAG for 24 h and collected 48 h after transfection. Full-length cDNA of wt Usp14 and of the catalytically inactive mutant Usp14-CA (bearing the mutation Cys114Ala) were cloned into a doxycycline-inducible lentiviral backbone pCW57-1 (Addgene plasmid #41393, Watertown, Massachusetts).
Lentivirus production and infection
For the production of lentivirus, Usp14-wt/CA-pCW57-1 plasmid was co-transfected with a mixture of lentiviral packaging plasmids (pHDM-Hgpm2, pMD-Tat, pRC/CMV-rev, pHDM-G) into human HEK293T cells using Polyethylenimine. Forty-eight hours post-transfection, media containing virus was collected and filtered with 0.45 μm filters. To generate stable cell lines, 75 000 Usp14−/− MEFs were infected with either wt Usp14 or Usp14-CA virus in the presence of 4 μg/ml of polybrene. Stably infected cell lines were produced under 2 μg/ml of puromycin selection, and isogenic clones were generated using limited dilutions. Several clones of each genotype were selected and expanded for further testing. Usp14−/− MEFs stably carrying empty pCW57-1 vector, which underwent the same process, were also created as controls.
RNAi
MEFs (∼180.000) were transfected with 100 nm of the ON-TARGETplus SMARTpool against Usp14 (L-060789-01-0005; Thermo Scientific Dharmacon) or ON-TARGETplus Non-Targeting Pool (D-001810-10-20; Thermo Scientific Dharmacon), using Interferin (Polyplus), per well into 6 well plates, following manufacturer’s recommendations. Cells were incubated in complete medium, and they were grown to 100% density, when they were starved with 0.5% FBS containing medium for 30 h, treated with Shh/SAG for 24 h. Cells were then collected 96 h after transfection for WB and qRT-PCR analyses, or they were fixed for immunofluorescence staining.
Quantitative RT-PCR
Total RNA extracted from MEFs was isolated by using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer’s instructions. During the extraction protocol, RNAs were digested with DNase I to remove DNA contaminating. RNA (1 μg) was then reverse-transcribed into cDNA by QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) with random hexamers oligo. For qRT-PCR, cDNAs were analyzed with Roche Light Cycler 480 system using a LightCycler 480 SYBR Green I Master mix (Roche, Basel, Switzerland). The final concentration of primers was of 0.5 μm.Quantification data were expressed as cycle threshold (Ct). The Ct values were averaged for each in-plate technical triplicate. The averaged Ct was normalized as difference in Ct values (ΔCt) between each sample and the Hprt gene, which was used as endogenous reference. The ΔCt values were normalized with respect to the ΔCt values of the control (ΔΔCt). The variation was reported as fold change (2-ΔΔCt). All the results are shown as means ± Standard Error of the Mean (SEM) of at least three independent biological replicates. Primer sequences for Gli1 and Hprt were previously reported (95,96).
WB analysis
MEFs were lysed in RIPA buffer containing protease inhibitors (Sigma-Aldrich, Saint Louis, Missouri), phosphatase inhibitors (PhosSTOP, Roche, Basel, Switzerland) and MG132 (Millipore, Burlington, Massachusetts). Protein extract concentrations were determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, Massachusetts) and a spectrophotometer reader (Bio-Rad, Hercules, California) at a wavelength of 595 nm. Protein samples were separated by 5% SDS-PAGE or 4–15% Precast Protein gel (Bio-Rad, Hercules, California) and transferred to polyvinylidene difluoride membranes (Immobilon-P, Merck Millipore, Burlington, Massachusetts). Membranes were blocked in Tris-buffered saline (TBS) containing 5% non-fat dry milk (Cell Signaling Technology) and incubated with the following commercial primary antibodies: anti-GAPDH 1:10 000 (sc-32233, Santa Cruz Biotechnology); anti-HSP90 1:1000 (4874S, Cell Signaling Technology, Danvers, Massachusetts); anti-GLI2 1:200 (AF3635, R&D Systems); anti-GLI3 1:400 (AF3690, R&D Systems, Minneapolis, Minnesota); anti-KIF7 1:1000 (ab95884, Abcam, Cambridge, England); anti-Ubiquitin 1:500 (sc-8017, Santa Cruz Biotechnology, Dallas, Texas); anti-Flag 1:1000 (F3165, Sigma-Aldrich, Saint Louis, Missouri); anti-USP14 1:3000 (A300-920A, Bethyl Laboratories, Montgomery, Texas); anti-Sp1 1:300 (sc-14027, Santa Cruz Biotechnology, Dallas, Texas). Proteins of interest were detected with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG antibody (1:3000, GE Healthcare, Chicago, Illinois) and rabbit anti-goat IgG antibody (1:3000, Millipore, Burlington, Massachusetts), and they were visualized with the Luminata Crescendo substrate (Merck Millipore, Burlington, Massachusetts) or the Super Signal West Femto substrate (Thermo Fisher Scientific, Waltham, Massachusetts), according to manufacturer’s protocol. WB images were acquired using the ChemiDoc imaging system (UVP) and densitometric analysis was performed with ImageJ, Madison, Wisconsin 1.51v software. Signals for each protein staining were quantified and then normalized for Gapdh or Hsp90 in the same sample. These normalized values were then compared with the values in the control sample. The average of the normalized values from three different biological replicates is reported as the relative fold change.
Nuclear-cytoplasmic extraction
All subcellular fractionation experiments were performed on ice with freshly harvested cells. Nuclear and cytoplasmic fractions from MEFs were isolated using NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Waltham, Massachusetts) according to manufacturer’s instructions. Fractions for each sample were quantified before taking samples for gel electrophoresis to ensure that equal amounts of nuclear fractions (20 μg) and cytoplasmic fractions (30 μg) from each sample were loaded on the gel.
Primary cilia assembly and disassembly assays
Cells were plated on glass coverslips and grown until they reached 100% confluency, then they were brought to quiescence by serum starvation with medium containing 0.5% FBS for 30 h, treated with Shh/SAG, and with IU1 or DMSO for 24 h, and fixed for immunofluorescence staining. For cilia disassembly assay, confluent cells were starved with medium containing 0.5% FBS for 30 h to induce cilia formation, followed by addition of medium containing 10% FBS for 2 h and then fixed for immunofluorescence staining.
To evaluate Usp14 silencing effects on cilia, MEFs (∼180.000) transfected with siRNAs against murine Usp14 and non-targeting RNA were seeded into 6 well dishes containing slides and incubated in fresh complete medium. Cells were grown to 100% density, starved with 0.5% FBS containing medium for 30 h and treated with Shh/SAG for 24 h, for a total of 96 h after transfection. The slides were then transferred into 24 well dishes, and cells were fixed for immunofluorescence staining. The remaining cells from each well were used for WB analysis to evaluate silencing efficiency.
Immunofluorescence
MEFs were grown on glass coverslips and treated as described above. Cells were fixed for 10 min in 4% paraformaldehyde in PBS, next they were washed three times with PBS and permeabilized with 2.5% BSA + 0.2% Triton X-100 in PBS (blocking buffer) for 45 min. Cells were incubated with the following primary antibodies either at 4°C overnight or at room temperature for 2 h: anti-GLI2 1:50 (AF3635, R&D Systems, Minneapolis, Minnesota); anti-GLI3 1:100 (AF3690, R&D Systems, Minneapolis, Minnesota); anti-Acetylated tubulin 1:10 000 (T6793, Sigma-Aldrich, Saint Louis, Missouri); anti-ARL3b 1:400 (17711-1-AP, Proteintech, Rosemont, Illinois). Cells were then washed three times with PBS and incubated with the appropriate secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen) for 1 h at room temperature. DNA was stained with Hoechst (33342, Sigma-Aldrich, Saint Louis, Missouri). After three washings with PBS, slides were mounted on coverslips with Mowiol (Millipore, Burlington, Massachusetts).
Confocal fluorescence microscopy and image processing
For confocal imaging, samples were examined under LSM700, LSM710 or LSM800 High-resolution confocal laser-scanning microscopes (Zeiss, Oberkochen, Germany). Optical sections were obtained under an X63 oil-immersion objective at a definition of 1024 × 1024 pixels, adjusting the pinhole diameter to 1 Airy unit for each emission channel. To perform quantitative image analysis, randomly chosen fields were scanned, using the same setting parameters (i.e. laser power and detector amplification) below pixel saturation between the samples of interests and controls. Images were taken as Z-stacks and then flattened by maximum intensity projections.
To quantify fluorescence intensity of Gli2 on cilia, the perimeter of each cilium was defined using Arl13b/acetylated tubulin cilia marker, and the mean intensity of fluorescence per cilia was determined using ImageJ, Madison, Wisconsin 1.51v. The background was subtracted from each fluorescence value, and all of the fluorescence pixel values above background levels were quantified. The average intensities along the axoneme were measured for each experiment, and the mean from different experiments was calculated for each condition and plotted as fold change in the graphs. All the experiments were performed at least three times and representative images were shown. For analysis of IF data ≥100 cells were counted for each experiment. The significance of the results was calculated by one-tailed Student’s t-test and reported as P-value.
Statistical analysis
In experiments requiring counts of cilia and in the cell proliferation assay the likelihood ratio test for negative binomial was applied. In all remaining experiments, statistical significance between two groups was evaluated by one-tailed Student’s t-test, P < 0.05 was considered significant. Quantitative data are presented as the mean ± SEM.
Supplementary Material
Acknowledgements
We thank Professor Lucia Di Marcotullio (Department of Molecular Medicine, University of Rome La Sapienza, Italy) for helpful discussion and Drs Iaconis, Indrieri and Napolitano (TIGEM) for critical reading of the manuscript. We thank Professor Scott Wilson (Department of Neurobiology and Civitan International Research Center, University of Alabama at Birmingham, Birmingham, AL, USA) for the gift of the Usp14–/– MEFs, Professor Kathryn V. Anderson (Developmental Biology Program, Sloan Kettering Institute, New York) for providing the pEGFP-N3-Kif7 construct and Dr Alessandra Boletta (Division of Genetics and Cell Biology Dibit-San Raffaele, Milan, Italy) for the gift of the Pkd1–/– and Pkd1+/+ MEFs.
Conflict of Interest statement. The authors (B.H.L. and D.F.) declare competing financial interests; a patent on IU1 has been filed by Harvard University on behalf of the authors.
Funding
Italian Telethon Foundation (TGM11CB3 to B.F.); Programme STAR, supported by UniNA and Compagnia di San Paolo (L1 2014 to M.M.); National Institutes of Health (R01GM043601 to D.F.).
References
- 1. Kim S. and Tsiokas L. (2011) Cilia and cell cycle re-entry: more than a coincidence. Cell Cycle, 10, 2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sung C.H. and Li A. (2011) Ciliary resorption modulates G1 length and cell cycle progression. Cell Cycle, 10, 2825–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim S. and Dynlacht B.D. (2013) Assembling a primary cilium. Curr. Opin. Cell Biol., 25, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reiter J.F. and Leroux M.R. (2017) Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol., 18, 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kagan K.O., Dufke A. and Gembruch U. (2017) Renal cystic disease and associated ciliopathies. Curr. Opin. Obstet. Gynecol., 29, 85–94. [DOI] [PubMed] [Google Scholar]
- 6. Yoder B.K., Hou X. and Guay-Woodford L.M. (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol., 13, 2508–2516. [DOI] [PubMed] [Google Scholar]
- 7. Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J. et al. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet., 33, 129–137. [DOI] [PubMed] [Google Scholar]
- 8. Wodarczyk C., Rowe I., Chiaravalli M., Pema M., Qian F. and Boletta A. (2009) A novel mouse model reveals that polycystin-1 deficiency in ependyma and choroid plexus results in dysfunctional cilia and hydrocephalus. PLoS One, 4, e7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goetz S.C. and Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bangs F. and Anderson K.V. (2017) Primary cilia and mammalian Hedgehog signaling. Cold Spring Harb. Perspect. Biol., 9, a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rohatgi R., Milenkovic L. and Scott M.P. (2007) Patched 1 regulates hedgehog signaling at the primary cilium. Science, 317, 372–376. [DOI] [PubMed] [Google Scholar]
- 12. Zeng H., Jia J. and Liu A. (2010) Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One, 5, e15900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Corbit K.C., Aanstad P., Singla V., Norman A.R., Stainier D.Y. and Reiter J.F. (2005) Vertebrate smoothened functions at the primary cilium. Nature, 437, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 14. Santos N. and Reiter J.F. (2014) A central region of Gli2 regulates its localization to the primary cilium and transcriptional activity. J. Cell Sci., 127, 1500–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haycraft C.J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E.J. and Yoder B.K. (2005) Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet., 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liem K.F. Jr., He M., Ocbina P.J. and Anderson K.V. (2009) Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. U. S. A., 106, 13377–13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ingham P.W., Nakano Y. and Seger C. (2011) Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet., 12, 393–406. [DOI] [PubMed] [Google Scholar]
- 18. Ingham P.W. and McMahon A.P. (2009) Hedgehog signalling: Kif7 is not that fishy after all. Curr. Biol., 19, R729–R731. [DOI] [PubMed] [Google Scholar]
- 19. Dafinger C., Liebau M.C., Elsayed S.M., Hellenbroich Y., Boltshauser E., Korenke G.C., Fabretti F., Janecke A.R., Ebermann I., Nurnberg G. et al. (2011) Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest., 121, 2662–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Putoux A., Thomas S., Coene K.L., Davis E.E., Alanay Y., Ogur G., Uz E., Buzas D., Gomes C., Patrier S. et al. (2011) KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet., 43, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He M., Subramanian R., Bangs F., Omelchenko T., Liem K.F. Jr., Kapoor T.M. and Anderson K.V. (2014) The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat. Cell Biol., 16, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wheway G., Schmidts M., Mans D.A., Szymanska K., Nguyen T.T., Racher H., Phelps I.G., Toedt G., Kennedy J., Wunderlich K.A. et al. (2015) An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol., 17, 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim J.H., Ki S.M., Joung J.G., Scott E., Heynen-Genel S., Aza-Blanc P., Kwon C.H., Kim J., Gleeson J.G. and Lee J.E. (2016) Genome-wide screen identifies novel machineries required for both ciliogenesis and cell cycle arrest upon serum starvation. Biochim. Biophys. Acta, 1863, 1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boldt K., Reeuwijk J., Lu Q., Koutroumpas K., Nguyen T.M., Texier Y., Beersum S.E., Horn N., Willer J.R., Mans D.A. et al. (2016) An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun., 7, 11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amato R., Morleo M., Giaquinto L., Bernardo D. and Franco B. (2014) A system biology approach to dissect the cilia/centrosome complex interactome. BMC Genomics, 15, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kasahara K., Kawakami Y., Kiyono T., Yonemura S., Kawamura Y., Era S., Matsuzaki F., Goshima N. and Inagaki M. (2014) Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun., 5, 5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim S., Zaghloul N.A., Bubenshchikova E., Oh E.C., Rankin S., Katsanis N., Obara T. and Tsiokas L. (2011) Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat. Cell Biol., 13, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maskey D., Marlin M.C., Kim S., Kim S., Ong E.C., Li G. and Tsiokas L. (2015) Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length. EMBO J., 34, 2424–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu Y.P., Tsai I.C., Morleo M., Oh E.C., Leitch C.C., Massa F., Lee B.H., Parker D.S., Finley D., Zaghloul N.A. et al. (2014) Ciliopathy proteins regulate paracrine signaling by modulating proteasomal degradation of mediators. J. Clin. Invest., 124, 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ishikawa H., Thompson J., Yates J.R. and Marshall W.F. (2012) Proteomic analysis of mammalian primary cilia. Curr. Biol., 22, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mick D.U., Rodrigues R.B., Leib R.D., Adams C.M., Chien A.S., Gygi S.P. and Nachury M.V. (2015) Proteomics of primary cilia by proximity labeling. Dev. Cell, 35, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sigg M.A., Menchen T., Lee C., Johnson J., Jungnickel M.K., Choksi S.P., Garcia G. III, Busengdal H., Dougherty G.W., Pennekamp P. et al. (2017) Evolutionary proteomics uncovers ancient associations of cilia with signaling pathways. Dev. Cell, 43, 744–762e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Borodovsky A., Kessler B.M., Casagrande R., Overkleeft H.S., Wilkinson K.D. and Ploegh H.L. (2001) A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J., 20, 5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koulich E., Li X. and DeMartino G.N. (2008) Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol. Biol. Cell, 19, 1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee B.H., Lee M.J., Park S., Oh D.C., Elsasser S., Chen P.C., Gartner C., Dimova N., Hanna J., Gygi S.P. et al. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature, 467, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee B.H., Lu Y., Prado M.A., Shi Y., Tian G., Sun S., Elsasser S., Gygi S.P., King R.W. and Finley D. (2016) USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature, 532, 398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boselli M., Lee B.H., Robert J., Prado M.A., Min S.W., Cheng C., Silva M.C., Seong C., Elsasser S., Hatle K.M. et al. (2017) An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem., 292, 19209–19225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sareen-Khanna K., Papillon J., Wing S.S. and Cybulsky A.V. (2016) Role of the deubiquitinating enzyme ubiquitin-specific protease-14 in proteostasis in renal cells. Am. J. Physiol. Renal Physiol., 311, F1035–F1046. [DOI] [PubMed] [Google Scholar]
- 39. Nakashima A., Ohnuma S., Kodani Y., Kaneko Y.S., Nagasaki H., Nagatsu T. and Ota A. (2016) Inhibition of deubiquitinating activity of USP14 decreases tyrosine hydroxylase phosphorylated at Ser19 in PC12D cells. Biochem. Biophys. Res. Commun., 472, 598–602. [DOI] [PubMed] [Google Scholar]
- 40. Liao Y., Liu N., Hua X., Cai J., Xia X., Wang X., Huang H. and Liu J. (2017) Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis., 8, e2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liao Y., Xia X., Liu N., Cai J., Guo Z., Li Y., Jiang L., Dou Q.P., Tang D., Huang H. et al. (2018) Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene, 37, 1896–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Y., Zhang Y., Sui Z., Zhang Y., Liu M. and Tang H. (2017) USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget, 8, 48725–48736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song C., Ma R., Yang X. and Pang S. (2017) The deubiquitinating enzyme USP14 regulates leukemic chemotherapy drugs-induced cell apoptosis by suppressing ubiquitination of Aurora kinase B. Cell. Physiol. Biochem., 42, 965–973. [DOI] [PubMed] [Google Scholar]
- 44. Chen M., Meng Q., Qin Y., Liang P., Tan P., He L., Zhou Y., Chen Y., Huang J., Wang R.F. et al. (2016) TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell, 64, 105–119. [DOI] [PubMed] [Google Scholar]
- 45. Xu D., Shan B., Sun H., Xiao J., Zhu K., Xie X., Li X., Liang W., Lu X., Qian L. et al. (2016) USP14 regulates autophagy by suppressing K63 ubiquitination of beclin 1. Genes Dev., 30, 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wei J., Dong S., Bowser R.K., Khoo A., Zhang L., Jacko A.M., Zhao Y. and Zhao J. (2017) Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci. Signal., 10eaak9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Min Y., Lee S., Kim M.J., Chun E. and Lee K.Y. (2017) Ubiquitin-specific protease 14 negatively regulates toll-like receptor 4-mediated signaling and autophagy induction by inhibiting ubiquitination of TAK1-binding protein 2 and beclin 1. Front. Immunol., 8, 1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bonnafe E., Touka M., AitLounis A., Baas D., Barras E., Ucla C., Moreau A., Flamant F., Dubruille R., Couble P. et al. (2004) The transcription factor RFX3 directs nodal cilium development and left-right asymmetry specification. Mol. Cell. Biol., 24, 4417–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bangs F.K., Schrode N., Hadjantonakis A.K. and Anderson K.V. (2015) Lineage specificity of primary cilia in the mouse embryo. Nat. Cell Biol., 17, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nauli S.M., Rossetti S., Kolb R.J., Alenghat F.J., Consugar M.B., Harris P.C., Ingber D.E., Loghman-Adham M. and Zhou J. (2006) Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol., 17, 1015–1025. [DOI] [PubMed] [Google Scholar]
- 51. Bhunia A.K., Piontek K., Boletta A., Liu L., Qian F., Xu P.N., Germino F.J. and Germino G.G. (2002) PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell, 109, 157–168. [DOI] [PubMed] [Google Scholar]
- 52. Sanders A.A., Vrieze E., Alazami A.M., Alzahrani F., Malarkey E.B., Sorusch N., Tebbe L., Kuhns S., Dam T.J., Alhashem A. et al. (2015) KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol., 16, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Srivastava S., Ramsbottom S.A., Molinari E., Alkanderi S., Filby A., White K., Henry C., Saunier S., Miles C.G. and Sayer J.A. (2017) A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet., 26, 4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stayner C., Poole C.A., McGlashan S.R., Pilanthananond M., Brauning R., Markie D., Lett B., Slobbe L., Chae A., Johnstone A.C. et al. (2017) An ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations. Sci. Rep., 7, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mokrzan E.M., Lewis J.S. and Mykytyn K. (2007) Differences in renal tubule primary cilia length in a mouse model of Bardet–Biedl syndrome. Nephron Exp. Nephrol., 106, e88–e96. [DOI] [PubMed] [Google Scholar]
- 56. Sohara E., Luo Y., Zhang J., Manning D.K., Beier D.R. and Zhou J. (2008) Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol., 19, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Smith L.A., Bukanov N.O., Husson H., Russo R.J., Barry T.C., Taylor A.L., Beier D.R. and Ibraghimov-Beskrovnaya O. (2006) Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol., 17, 2821–2831. [DOI] [PubMed] [Google Scholar]
- 58. Hopp K., Ward C.J., Hommerding C.J., Nasr S.H., Tuan H.F., Gainullin V.G., Rossetti S., Torres V.E. and Harris P.C. (2012) Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Invest., 122, 4257–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jin X., Muntean B.S., Aal-Aaboda M.S., Duan Q., Zhou J. and Nauli S.M. (2014) L-type calcium channel modulates cystic kidney phenotype. Biochim. Biophys. Acta, 1842, 1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berbari N.F., Lewis J.S., Bishop G.A., Askwith C.C. and Mykytyn K. (2008) Bardet–Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl. Acad. Sci. U. S. A., 105, 4242–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Q., Nishimura D., Seo S., Vogel T., Morgan D.A., Searby C., Bugge K., Stone E.M., Rahmouni K. and Sheffield V.C. (2011) Bardet–Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc. Natl. Acad. Sci. U. S. A., 108, 20678–20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gerhardt C., Lier J.M., Burmuhl S., Struchtrup A., Deutschmann K., Vetter M., Leu T., Reeg S., Grune T. and Ruther U. (2015) The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol., 210, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L. and Anderson K.V. (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature, 426, 83–87. [DOI] [PubMed] [Google Scholar]
- 64. Huangfu D. and Anderson K.V. (2005) Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. U. S. A., 102, 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu A., Wang B. and Niswander L.A. (2005) Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development, 132, 3103–3111. [DOI] [PubMed] [Google Scholar]
- 66. Eguether T., San Agustin J.T., Keady B.T., Jonassen J.A., Liang Y., Francis R., Tobita K., Johnson C.A., Abdelhamed Z.A., Lo C.W. et al. (2014) IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell, 31, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Keady B.T., Samtani R., Tobita K., Tsuchya M., San Agustin J.T., Follit J.A., Jonassen J.A., Subramanian R., Lo C.W. and Pazour G.J. (2012) IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell, 22, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liew G.M., Ye F., Nager A.R., Murphy J.P., Lee J.S., Aguiar M., Breslow D.K., Gygi S.P. and Nachury M.V. (2014) The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell, 31, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V. et al. (2008) THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet., 40, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qin J., Lin Y., Norman R.X., Ko H.W. and Eggenschwiler J.T. (2011) Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc. Natl. Acad. Sci. U. S. A., 108, 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ali B.R., Silhavy J.L., Akawi N.A., Gleeson J.G. and Al-Gazali L. (2012) A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J. Rare Dis., 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Walsh D.M., Shalev S.A., Simpson M.A., Morgan N.V., Gelman-Kohan Z., Chemke J., Trembath R.C. and Maher E.R. (2013) Acrocallosal syndrome: identification of a novel KIF7 mutation and evidence for oligogenic inheritance. Eur. J. Med. Genet., 56, 39–42. [DOI] [PubMed] [Google Scholar]
- 73. Karaer K., Yuksel Z., Ichkou A., Calisir C. and Attie-Bitach T. (2015) A novel KIF7 mutation in two affected siblings with acrocallosal syndrome. Clin. Dysmorphol., 24, 61–64. [DOI] [PubMed] [Google Scholar]
- 74. Tunovic S., Baranano K.W., Barkovich J.A., Strober J.B., Jamal L. and Slavotinek A.M. (2015) Novel KIF7 missense substitutions in two patients presenting with multiple malformations and features of acrocallosal syndrome. Am. J. Med. Genet. A, 167A, 2767–2776. [DOI] [PubMed] [Google Scholar]
- 75. Ibisler A., Hehr U., Barth A., Koch M., Epplen J.T. and Hoffjan S. (2015) Novel KIF7 mutation in a Tunisian boy with Acrocallosal syndrome: case report and review of the literature. Mol. Syndromol., 6, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barakeh D., Faqeih E., Anazi S. and M, S.A.-D., Softah, A., Albadr, F., Hassan, H., Alazami, A.M. and Alkuraya, F.S. (2015) The many faces of KIF7. Hum. Genome Var., 2, 15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vaden J.H., Bhattacharyya B.J., Chen P.C., Watson J.A., Marshall A.G., Phillips S.E., Wilson J.A., King G.D., Miller R.J. and Wilson S.M. (2015) Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener., 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jung H., Kim B.G., Han W.H., Lee J.H., Cho J.Y., Park W.S., Maurice M.M., Han J.K., Lee M.J., Finley D. et al. (2013) Deubiquitination of dishevelled by Usp14 is required for Wnt signaling. Oncogene, 2, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Min J.W., Lu L., Freeling J.L., Martin D.S. and Wang H. (2017) USP14 inhibitor attenuates cerebral ischemia/reperfusion-induced neuronal injury in mice. J. Neurochem., 140, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lu W., Shen X., Pavlova A., Lakkis M., Ward C.J., Pritchard L., Harris P.C., Genest D.R., Perez-Atayde A.R. and Zhou J. (2001) Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet., 10, 2385–2396. [DOI] [PubMed] [Google Scholar]
- 81. Kim K., Drummond I., Ibraghimov-Beskrovnaya O., Klinger K. and Arnaout M.A. (2000) Polycystin 1 is required for the structural integrity of blood vessels. Proc. Natl. Acad. Sci. U. S. A., 97, 1731–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Geng L., Segal Y., Pavlova A., Barros E.J., Lohning C., Lu W., Nigam S.K., Frischauf A.M., Reeders S.T. and Zhou J. (1997) Distribution and developmentally regulated expression of murine polycystin. Am. J. Physiol., 272, F451–F459. [DOI] [PubMed] [Google Scholar]
- 83. Bitgood M.J. and McMahon A.P. (1995) Hedgehog and Bmp genes are coexpressed at many diverse sites of cell–cell interaction in the mouse embryo. Dev. Biol., 172, 126–138. [DOI] [PubMed] [Google Scholar]
- 84. Boulter C., Mulroy S., Webb S., Fleming S., Brindle K. and Sandford R. (2001) Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl. Acad. Sci. U. S. A., 98, 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bimonte S., De Angelis A., Quagliata L., Giusti F., Tammaro R., Dallai R., Ascenzi M.G., Diez-Roux G. and Franco B. (2011) Ofd1 is required in limb bud patterning and endochondral bone development. Dev. Biol., 349, 179–191. [DOI] [PubMed] [Google Scholar]
- 86. Wang C., Yuan X. and Yang S. (2013) IFT80 is essential for chondrocyte differentiation by regulating Hedgehog and Wnt signaling pathways. Exp. Cell Res., 319, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ruiz-Perez V.L., Blair H.J., Rodriguez-Andres M.E., Blanco M.J., Wilson A., Liu Y.N., Miles C., Peters H. and Goodship J.A. (2007) Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development, 134, 2903–2912. [DOI] [PubMed] [Google Scholar]
- 88. Cortellino S., Wang C., Wang B., Bassi M.R., Caretti E., Champeval D., Calmont A., Jarnik M., Burch J., Zaret K.S. et al. (2009) Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol., 325, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Attanasio M., Uhlenhaut N.H., Sousa V.H., O'Toole J.F., Otto E., Anlag K., Klugmann C., Treier A.C., Helou J., Sayer J.A. et al. (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet., 39, 1018–1024. [DOI] [PubMed] [Google Scholar]
- 90. Jonassen J.A., San Agustin J., Baker S.P. and Pazour G.J. (2012) Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol., 23, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tran P.V., Talbott G.C., Turbe-Doan A., Jacobs D.T., Schonfeld M.P., Silva L.M., Chatterjee A., Prysak M., Allard B.A. and Beier D.R. (2014) Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J. Am. Soc. Nephrol., 25, 2201–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Silva L.M., Jacobs D.T., Allard B.A., Fields T.A., Sharma M., Wallace D.P. and Tran P.V. (2018) Inhibition of Hedgehog signaling suppresses proliferation and microcyst formation of human autosomal dominant polycystic kidney disease cells. Sci. Rep., 8, 4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wilson S.M., Bhattacharyya B., Rachel R.A., Coppola V., Tessarollo L., Householder D.B., Fletcher C.F., Miller R.J., Copeland N.G. and Jenkins N.A. (2002) Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet., 32, 420–425. [DOI] [PubMed] [Google Scholar]
- 94. Distefano G., Boca M., Rowe I., Wodarczyk C., Ma L., Piontek K.B., Germino G.G., Pandolfi P.P. and Boletta A. (2009) Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol. Cell. Biol., 29, 2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Niewiadomski P. and Rohatgi R. (2015) Measuring expression levels of endogenous Gli genes by immunoblotting and real-time PCR. Methods Mol. Biol., 1322, 81–92. [DOI] [PubMed] [Google Scholar]
- 96. Barbato S., Marrocco E., Intartaglia D., Pizzo M., Asteriti S., Naso F., Falanga D., Bhat R.S., Meola N., Carissimo A. et al. (2017) MiR-211 is essential for adult cone photoreceptor maintenance and visual function. Sci. Rep., 7, 17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.