

Abstract
Danforth’s short tail (Sd) mice provide an excellent model for investigating the underlying etiology of human caudal birth defects, which affect 1 in 10 000 live births. Sd animals exhibit aberrant axial skeleton, urogenital and gastrointestinal development similar to human caudal malformation syndromes including urorectal septum malformation, caudal regression, vertebral-anal-cardiac-tracheo-esophageal fistula-renal-limb (VACTERL) association and persistent cloaca. Previous studies have shown that the Sd mutation results from an endogenous retroviral (ERV) insertion upstream of the Ptf1a gene resulting in its ectopic expression at E9.5. Though the genetic lesion has been determined, the resulting epigenomic and transcriptomic changes driving the phenotype have not been investigated. Here, we performed ATAC-seq experiments on isolated E9.5 tailbud tissue, which revealed minimal changes in chromatin accessibility in Sd/Sd mutant embryos. Interestingly, chromatin changes were localized to a small interval adjacent to the Sd ERV insertion overlapping a known Ptf1a enhancer region, which is conserved in mice and humans. Furthermore, mRNA-seq experiments revealed increased transcription of Ptf1a target genes and, importantly, downregulation of hedgehog pathway genes. Reduced sonic hedgehog (SHH) signaling was confirmed by in situ hybridization and immunofluorescence suggesting that the Sd phenotype results, in part, from downregulated SHH signaling. Taken together, these data demonstrate substantial transcriptome changes in the Sd mouse, and indicate that the effect of the ERV insertion on Ptf1a expression may be mediated by increased chromatin accessibility at a conserved Ptf1a enhancer. We propose that human caudal dysgenesis disorders may result from dysregulation of hedgehog signaling pathways.
Introduction
The semidominant Danforth’s short tail (Sd) mutation results in severe developmental abnormalities of the urogenital and gastrointestinal systems and the lower spine. These phenotypes overlap with caudal malformation syndromes in humans, including Currarino syndrome (OMIM #176450), urorectal septum malformation sequence, vertebral-anal-cardiac-tracheo-esophageal fistula-renal-limb anomalies (VACTERL) association (OMIM #192350) and caudal regression syndrome (OMIM #600145). Caudal birth defects affect 1:10 000 live births but their genetic etiology, intermediate molecular consequences to the epigenome and transcriptome and resulting pathophysiology are largely unknown (1). The phenotypic overlap of the Sd mouse with multiple distinct human caudal dysgenesis disorders makes it an outstanding model to investigate the underlying etiology of human caudal dysgenesis.
Heterozygous (Sd/+) mice have a shortened tail, sacral vertebral anomalies and frequent kidney anomalies but are viable and survive into adulthood (2). Homozygous (Sd/Sd) mice exhibit cessation of the vertebral column at the lumbar level and complete absence of the tail (2). Urogenital defects include agenesis or hypoplasia of the kidneys with occasional formation of the bladder and urethra and persistence of the cloaca. Gastrointestinal anomalies include imperforate anus. Homozygous animals are born in Mendelian ratios but are not viable beyond 24 h (2). Sd/Sd and Sd/+ embryos are visually distinguishable from wild-type (WT) embryos at E11 by caudal truncation and hemorrhagic lesions in the tailbud (3). Defects are apparent slightly earlier in Sd/Sd embryos. Prior to identification of the genetic lesion, differentiation of Sd/Sd and Sd/+ embryos could only be made by the increased phenotypic severity in Sd/Sd mutants during gestation (4).
In 2013 our group and others identified the Sd mutation as an 8528 bp insertion of an endogenous retroviral element (ERV) at a point 12 463 bp upstream of the Pancreas specific transcription factor 1a (Ptf1a) gene (5–7). The ERV insertion results in ectopic expression of Ptf1a at E8.5 in the notochord, lateral plate mesoderm and tail mesenchyme that persists into the caudal notochord, tailbud mesenchyme, mesonephros and hindgut of E9.5 embryos (6). The critical role of PTF1A in causing the Sd phenotype was demonstrated by knockout (KO) of Ptf1a genomic sequences followed by replacement of Ptf1a coding sequences, which resulted in attenuation and recapitulation of the Sd phenotype, respectively (7).
Ptf1a is a basic helix-loop-helix (bHLH) transcription factor (TF) that is expressed in the pancreas and in neuronal progenitors in the cerebellum, hindbrain, neural tube (NT) and retina (8–11). Null mutations in Ptf1a in humans and mouse result in pancreatic and cerebellar agenesis (12,13). PTF1A interacts with an E-box protein and RBPJ to form the trimeric PTF1 complex. PTF1 binds a bipartite cognate site including a canonical E-box motif (CANNTG) and TC-box (TGGGAAA) (14). PTF1 drives initial pancreatic organogenesis as well as the specification of GABAergic neurons in the NT (13,15,16). In the pancreas, PTF1 subsequently drives differentiation of mature acinar cells (9). Masui et al. characterized a Ptf1a autoregulatory enhancer region, which contains two PTF1 motifs located 14.8 and 13.5 kb upstream of Ptf1a (17). Both enhancers drive LacZ expression in the dorsal and ventral pancreas and the NT (17). NT-specific enhancers have been identified 12.4 kb downstream of Ptf1a coding sequences (18,19). Two long non-coding RNAs (lncRNA’s), Gm13344 and Gm13336, have been annotated in the genomic region surrounding Ptf1a in the mouse. Interestingly, the transcription start site (TSS) for Gm13344 overlaps the region corresponding to the Ptf1a 13.5 kb upstream autoregulatory enhancer, while Gm13336 overlaps Ptf1a coding sequences (17).
Our previous studies of the Sd mutation implicated ectopic Ptf1a expression in the Sd phenotype. The genome-wide epigenome and transcriptome alterations induced by the Sd mutation are largely uncharacterized. To address this issue, we measured chromatin accessibility using ATAC-seq and transcriptional profiles using mRNA-seq on E9.5 WT and Sd/Sd tailbuds (20–22). We found minimal changes to the genome-wide landscape of chromatin accessibility—only one peak met our genome-wide 5% False Discovery Rate (FDR) threshold. Strikingly, the peak is adjacent to the Sd insertion site, which is located at the Ptf1a 13.5 kb upstream autoregulatory enhancer that overlaps with the Gm13344 TSS. This peak is more open in Sd mice compared to WT. The mRNA-seq results confirmed upregulation of Ptf1a and Gm13344 in Sd mutants and identified significantly (<5% FDR) reduced expression of genes within the Hedgehog signaling pathway. In situ hybridization confirmed downregulation of sonic hedgehog (Shh) as well as other notochordal markers. Further, dysregulation of caudal NT patterning was observed in Sd/Sd embryos, consistent with Shh downregulation. Finally, we observed increased apoptosis in the tailbud and caudal somites. Our findings suggest that ectopic Ptf1a expression, potentially driven by increased chromatin accessibility at an upstream enhancer, induces a cascade of transcriptional dysregulation of multiple genes in the Hedgehog signaling pathway. We propose that ectopic Ptf1a ultimately leads to downregulation of Shh signaling, degeneration of the notochord and increased apoptosis in the developing caudal region.
Results
ATAC-seq reveals localized changes in chromatin accessibility near the Sd insertion
TF binding and chromatin accessibility are strongly coupled. Closed chromatin and inaccessible DNA may prevent a TF from binding a target site; on the other hand, cooperative binding of TFs, or binding of certain ‘pioneer factors’, may increase or maintain chromatin accessibility. Therefore, the over- or under-expression of TFs may alter chromatin accessibility (23–26). Ptf1a is strongly overexpressed in Sd mice and is a direct regulator of other TFs. The Sd insertion is located adjacent to a Ptf1a autoregulatory enhancer region. In order to quantify changes in chromatin accessibility in Sd mutant embryos, we performed ATAC-seq on E9.5 WT and Sd/Sd tailbuds. We analyzed four WT and four Sd/Sd ATAC-seq libraries, pooling tailbuds from two embryos for each library to ensure sufficient input material. ATAC-seq peak calling identified 26620 reproducible peaks (defined as those peaks that appear in at least two of the eight libraries; see Materials and Methods). Correlation between all libraries was high with the mean Pearson’s r = 0.84 for all pairwise comparisons (Supplementary Material, Fig. S1), demonstrating the reproducibility and quality of the libraries. A differential peak analysis uncovered little change in ATAC-seq signal genome wide, revealing only one significantly differential peak (unadjusted P-value 2.2 × 10−7, adjusted P-value 5.8 × 10−3, 5% FDR threshold for the analysis) (27). This peak corresponds to the promoter of Gm13344, an lncRNA located 13.7 kb upstream of Ptf1a (Fig. 1A and B, Supplementary Material, Fig. S2). The peak called at the Ptf1a promoter region was not significantly different between WT and Sd/Sd E9.5 embryos at 5% FDR; however, the peak is skewed toward being more open in the WT (unadjusted P-value 4.5 × 10−4, adjusted P-value 0.61 Fig. 1B). This suggests that while there are few genome-wide changes in chromatin accessibility in the Danforth mouse, local chromatin accessibility near Ptf1a and the Sd insertion site is altered.
Figure 1.
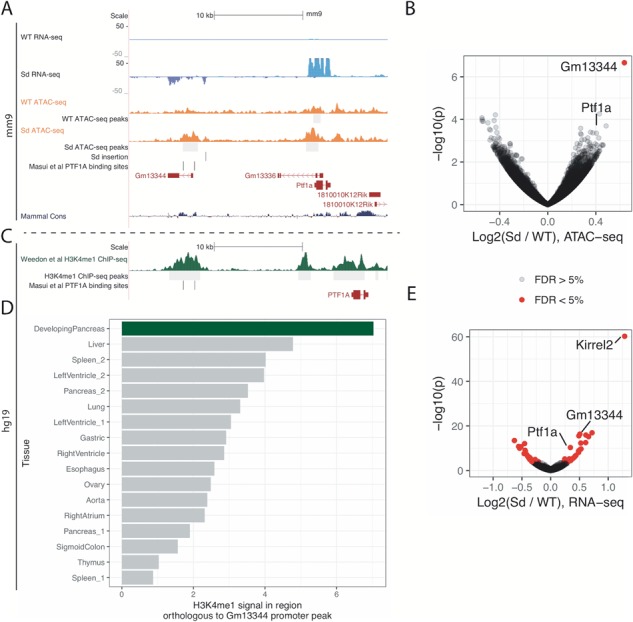
Chromatin accessibility and transcriptome changes occur at the Ptf1a locus of Sd/Sd mutant embryos. (A) UCSC Genome Browser (Kent et al.. 2002; http://genome.ucsc.edu) displaying ATAC-seq (orange) and RNA-seq signal (blue) near the Sd insertion (denoted by the vertical black line), Ptf1a and Gm13344, for one WT and one Sd/Sd library. Previously characterized PTF1A binding sites are denoted by vertical black lines near the Gm13344 promoter and intron. (B) Volcano plot for ATAC-seq data (n = 4 libraries per genotype). The gene represented by the nearest TSS for the significantly differential peak is indicated, as is the peak representing the Ptf1a promoter. Red dots denote significance at FDR <5%. (C) Weedon et al. ChIP-seq data (30), indicating a peak above the human region orthologous to the Gm13344 promoter. (D) The ChIP-seq signal for H3K4me1 in developing pancreas (from Weedon et al.) is greater than the signal in other tissues/at other time points. (E) Volcano plot for RNA-seq data (n = 3 libraries per genotype).
Gm13344 promoter is orthologous to a human pancreatic developmental enhancer
The mouse lncRNA Gm13344 is not present in standard human genome annotations such as Gencode and RefSeq. In a previous study, expression of a transgene containing the Sd ERV and Gm13344 in mice did not lead to the development of a short tail, indicating that Gm13344 overexpression alone is unlikely to cause the Danforth phenotype (7). LncRNAs are generally not conserved across species and may evolve from unstable transcription of regulatory elements (28,29). Masui et al. previously found that in the human genome, PTF1A binds an enhancer element ∼15 kb upstream of the PTF1A gene containing two PTF1A motif matches that are conserved in mouse (Fig. 1A, Supplementary Material, Fig. S2) (17). Interestingly, the orthologous mouse Ptf1a binding sites occur near the Gm13344 promoter, suggesting that the mouse Gm13344 lncRNA promoter may be functionally comparable to the human enhancer. In order to determine whether this human enhancer is active during pancreatic development, we uniformly processed and analyzed publicly available ChIP-seq data for H3K4me1, a histone mark that colocalizes with enhancers, from developing human pancreatic cells and other control tissues (control tissues downloaded from Roadmap Epigenomics) (30–32). Developmental pancreas H3K4me1 data show a clear H3K4me1 peak at the enhancer, consistent with the enhancer being active during human pancreatic development. (Fig. 1C). Notably, H3K4me1 ChIP-seq data from other tissues and time points display lower signal in the region orthologous to the mouse Gm13344 promoter (Fig. 1D). Collectively, these results suggest that the Sd-insertion-mediated increase in chromatin accessibility at the Gm13344 promoter acts as an enhancer for Ptf1a in E9.5 mice, and transcription of the lncRNA is at least partially a byproduct of this regulatory activity.
mRNA-seq reveals misexpression of Ptf1a target genes
To determine which genes show evidence of misregulation in Sd mutant embryos at E9.5, we performed mRNA-seq on three WT and three Sd/Sd E9.5 tailbud libraries (samples were pooled as necessary for sufficient input material to each library; see Materials and Methods and Supplementary Material, Table S3). The WT tailbud transcriptome can be found in the supplemental data (Supplementary Material, Table S1). We performed differential gene expression analysis and found 49 genes to be significantly differentially expressed (5% FDR; Figs. 1E and 2). Several of the differential genes discovered by mRNA-seq recapitulate previous findings. As expected, Ptf1a showed almost no expression in WT tailbuds but strong expression in Sd/Sd tailbuds (Figs. 1A and 2, Supplementary Material, Fig. S2). Similarly, Gm13344, the lncRNA upstream of Ptf1a, showed almost no expression in WT tailbuds but strong expression in the Sd mutant samples, consistent with previous reports (Figs. 1A and 2, Supplementary Material, Fig. S2) (7). Carboxypeptidase A1 (Cpa1), previously found to be underexpressed in E10.5 Ptf1a KO mice, is overexpressed in Sd mutant tailbuds (Fig. 2) (33). We found Foxa1 to be downregulated in Sd mutant samples (Fig. 2), consistent with the upregulation of Foxa1 in Ptf1a KO mice (33). Foxa2 also trended toward downregulation in Sd mice, although the adjusted P-value did not reach significance (unadjusted P-value 2.0 × 10−4, adjusted P-value 0.066) (Fig. 2). Two of the most strongly upregulated genes in Sd/Sd tailbuds were Kirrel2 and Nphs1 (Figs. 1E and 2), which share a bidirectional promoter to which Ptf1a is known to bind at later time points in NT and pancreas (34). Aplp1, positioned directly downstream of Kirrel2, is similarly upregulated in Sd mutant samples (Fig. 2). mRNA-seq also revealed upregulation of Sox8 in Sd mutant tailbuds (Fig. 2). SOX8 is a high mobility group TF that functions redundantly with SOX9 and SOX10 in neural crest development and its expression precedes that of SOX9 and SOX10 (35). No significant differences in the caudally expressed genes Wnt3a, Cyp26a1 and Cdx2 were identified in Sd mutant samples in our mRNA-seq data (Fig. 2).
Figure 2.
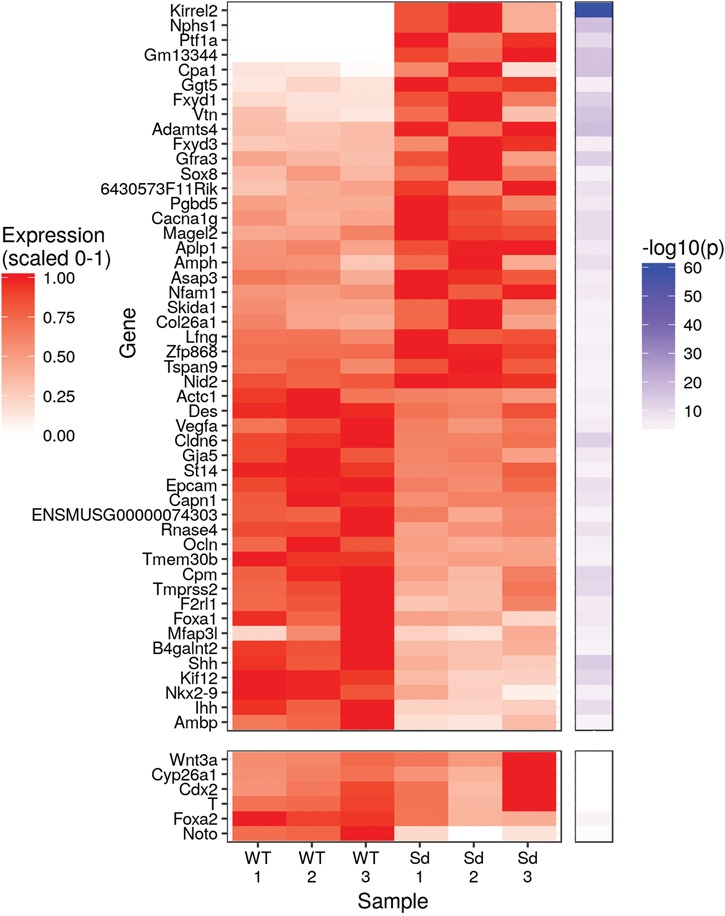
Ectopic Ptf1a expression drives global transcriptomic changes within Sd/Sd embryo tailbud. Heatmap displaying normalized expression and -log10(p) for differentially expressed genes. Several genes that did not meet the threshold of <5% FDR but are of interest due to their known expression in the tailbud or notochord, including Wnt3a, Cyp26a1, Cdx2, T, Foxa2 and Noto, are included at the bottom of the heatmap. P-values are from the differential gene expression analysis with DESeq2.
Whole mount in situ hybridization analysis validates mRNA-seq expression changes
To confirm expression changes in Sd mutant tailbuds identified by mRNA-seq analysis, we used whole mount in situ hybridization in E9.5 embryos. These studies confirmed ectopic expression of Ptf1a in the tailbud of E9.5 Sd/+ and Sd/Sd embryos (Supplementary Material, Fig S3). The ectopic Ptf1a expression was stronger in homozygous mutants compared to Sd/+embryos, and appeared highest in the presomitic mesoderm with lower levels in the notochord in the anterior thoracic region. In addition, we found increased expression of Gm13344 in the tailbud of Sd/+ and Sd/Sd embryos (Supplementary Material, Fig. S3). We also found upregulated expression of Kirrel2 (Fig. 3A–C) in Sd/+ and Sd/Sd mutants, as identified by mRNA-seq. Foxa1 expression was clearly reduced in Sd/Sd embryos (Fig. 3D–F), in accord with our mRNA-seq data. Foxa2 expression was also reduced in the tailbud by whole mount in situ hybridization (Fig. 3G–I). Although the unadjusted P-value for Foxa2 was significant in the mRNA-seq analysis, it did not survive genome-wide multiple testing correction using 5% FDR. This could be due to the relatively small proportion of Foxa2-expressing cells in the tailbud (36). We also examined expression of the T-box TF brachyury (T) in the tailbud of Sd/Sd embryos. T is expressed in notochord-derived cells and is required for notochord development, mesoderm differentiation and development of structures posterior to the forelimb level in mouse embryos (37–39). We found that T expression was unaffected in the tailbud of Sd mutant embryos, although reduced expression was observed in the more anterior part of the notochord of Sd/Sd embryos (Fig. 3J–L), which correlates with our mRNA-seq data (Fig. 2). Noto, a Not family homeobox gene, is expressed in the WT mouse tailbud and is essential for proper caudal notochord function (36). We did not detect Noto expression in the tailbud of Sd/Sd embryos, and expression was decreased in the Sd/+ embryos (Fig. 3M–O), which could indicate suppression of its expression, since T expression indicates the presence of the caudal notochord in Sd/Sd embryos at this timepoint.
Figure 3.
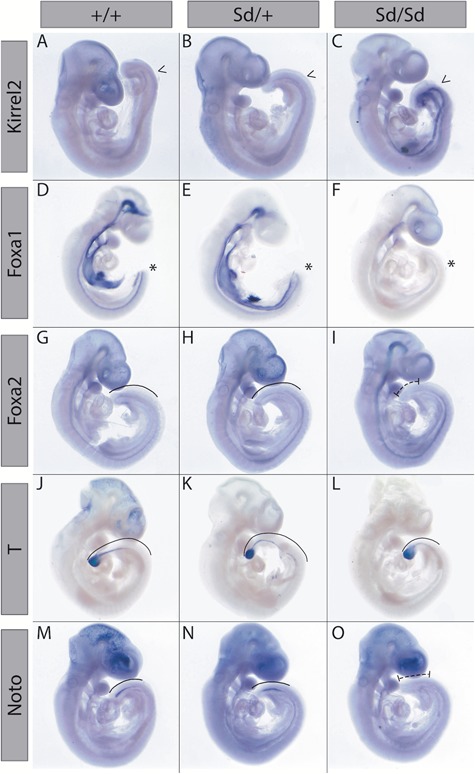
Whole mount in situ hybridization confirms transcriptomic changes in Sd/Sd embryos. Whole mount in situ hybridization with antisense probes for Kirrel2(A–C), Foxa1(D–F), Foxa2(G–I), T(J–L) and Noto(M–O) in E9.5 WT, Sd/+ and Sd/Sd embryos. Arrow heads (A–C) and asterisks (D–F) indicate increased and decreased, respectively, expression patterns in the tailbud of Sd/Sd embryos. Solid lines indicate staining within the tailbud (G, H, J, K, L, M, N) and dashed lines (I, O) indicate extent of reduced staining in Sd/Sd tailbuds. N > 3 for each genotype per probe.
Pathway analysis shows dysregulated hedgehog signaling
To determine which developmental signaling pathways are disrupted in Sd mutant tailbuds, we performed a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using the P-values of all genes included in the differential gene expression analysis. Several pathways related to cellular adhesion and migration show significant enrichment for disrupted gene expression (Fig. 4A). Interestingly, the only developmental pathway to show significant enrichment for altered gene expression was the Hedgehog signaling pathway (Fig. 4A). Shh and Indian Hedgehog (Ihh) are two of the most strongly downregulated genes in Sd mutant samples (Fig. 2A), and in total seven genes annotated to hedgehog signaling show an unadjusted P-value for differential expression of <0.05 [Shh (P = 4.43 × 10−14), Ihh (P = 6.62 × 10−10), Gli1 (P = 0.018), Gli3 (P = 0.019), Ptch1 (P = 0.021), Ptch2 (P = 0.002), Bmp4 (P = 0.006)] with changes in the direction expected for downregulation of this pathway (Supplementary Material, Table S2). Whole mount in situ hybridization confirmed that Shh expression was absent in the tailbud of Sd/Sd embryos at E9.5, and its expression decreased in a reciprocal manner to the ectopic expression of Ptf1a in the tailbud (Fig. 4B–D). NKX2.9 is a homeobox TF expressed in close proximity to SHH in NT structures along the entire neuraxis and is regulated by SHH (40,41). Nkx2.9 expression was significantly reduced in Sd/Sd embryos by mRNA-seq and confirmed by whole mount in situ hybridization (Fig. 2A and Fig. 4E–G). Gli1, a major downstream effector of SHH signaling, was reduced by LacZ staining in mice from a cross between Sd/+ and Gli1-LacZ reporter mice (Fig. 4H, I) in accordance with SHH downregulation.
Figure 4.
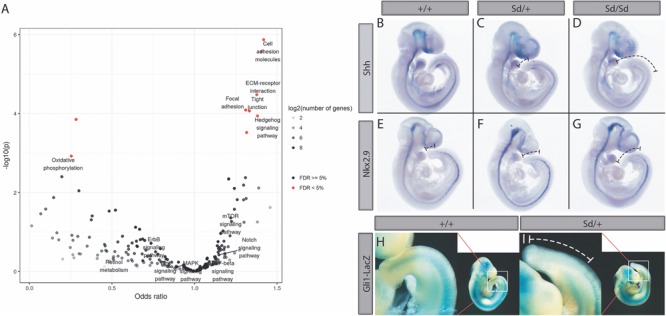
KEGG pathway analysis of Sd/Sd embryos identifies dysregulated SHH signaling. (A) Volcano plot for enrichment of disrupted gene expression in KEGG pathways. Notable developmental signaling pathways are labeled. Hedgehog signaling is the only developmental signaling pathway showing enrichment for differential gene expression. (B–D) Whole mount in situ hybridization with Shh and Nkx2.9(E–G) antisense probes in E9.5 WT, Sd/+ and Sd/Sd embryos. Dashed lines in C, F, D and G indicate extent of reduced staining in Sd/+ and Sd/Sd tailbuds, respectively. (H, I) X-gal staining of E10.5 WT and Sd/+ embryos from a cross between Sd/+ mice and Gli1-lacZ reporter mice. White dashed line in tailbud enlargement (I) shows extent of reduced lacZ reporter gene expression in the tailbud of Sd/+ embryo. N > 3 for each genotype of stained embryos.
The absence of Shh results in aberrant caudal NT patterning in Sd/Sd embryos
We hypothesized that dysregulation of SHH signaling in developing Sd mutant embryos would result in aberrant NT patterning. To test this hypothesis, we conducted immunofluorescence experiments on E10.5 WT, Sd/+ and Sd/Sd mice. Embryos were sectioned in the transverse plane through the NT and notochord at the level of both the forelimb and hindlimb bud. Staining with an antibody against SHH revealed aberrant notochordal morphology in Sd/+ embryos and absence of the notochord in Sd/Sd embryos (Fig. 5A–F). Floorplate expression of SHH was maintained throughout Sd/+ embryos; however, it was lost at the hindlimb level of Sd/Sd embryos. Floorplate-derived SHH is induced following FOXA2 initiation in the floorplate by notochordal-derived SHH (42–46). Thus, SHH loss in the floorplate is likely due to the downregulation of FOXA2 in the floorplate (Fig. 5G–L, E’, F’). Ventralization of the NT relies on SHH signals; thus, loss of the V3 domain marker NKX2.2 in the Sd/Sd hindlimb NT is consistent with SHH loss (Fig. 5M–R, G’, H’) (47). Interestingly, the more widely expressed marker NKX6.1 is expressed but ventrally constricted in Sd/Sd mutant embryos (Fig. 5S–X). Consistent with the loss of SHH signaling from the floorplate and notochord, the dorsally restricted TF PAX6 exhibits ventral expansion at the hindlimb level of Sd/Sd embryos (Fig. 5Y–D’). Reduced expression of ventral markers as well as expansion of dorsal NT markers at the hindlimb level in the NT of Sd/Sd embryos indicates the notochord of Sd/Sd mutant embryos degenerates before correct patterning of the presumptive floorplate can be established.
Figure 5.
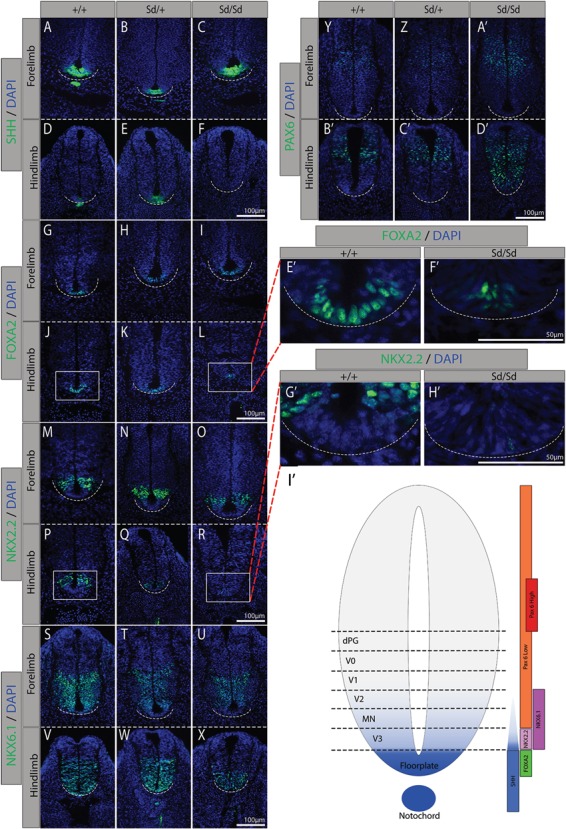
Immunofluorescence staining confirms dysregulated SHH expression in Sd/Sd embryos and identifies aberrant NT patterning. Immunofluorescence studies of SHH expression (green) in WT, (A, D)Sd/+, (B, E) and Sd/Sd(C, F) embryos as well as downstream targets FOXA2, (G–L) NKX2.2, (M–R) NKX6.1 (S–X) in transverse forelimb (top pane) and hindlimb (bottom pane) sections. Dorsal marker PAX6 (Y–D’) indicates the competing WNT/BMP gradient. Enlarged FOXA2 (E’, F’) and NKX2.2 (G’, H’) floorplate images of WT (J, P) and Sd/Sd (L, R) hindlimb images, respectively. (I’) WT expression domains of these TFs. All sections co-stained with DAPI for histological reference (blue). Dashed line indicates ventral border of NT. N > 3 for each genotype per antibody.
Sd mutant embryos exhibit increased apoptosis in the tailbud
Previous studies have revealed hemorrhaging within the regressing tailbud of E12.5 Sd mutant embryos; however, degeneration of the tailbud in Sd mutant embryos has not been closely investigated (4). No visible phenotypic differences were apparent at E9.5. However, we found that hematomas are clearly evident in Sd/Sd embryos at E10.5, which likely give way to severe hemorrhaging as development progresses (Fig. 6A–C). Further investigation of the tailbud degeneration using immunofluorescence with an antibody to cleaved caspase-3 revealed increased apoptosis in Sd/Sd tailbud tissues relative to WT and Sd/+ littermates (Fig. 6D–F). Increased apoptosis could be a result of loss of hedgehog patterning from the notochord in Sd/Sd embryos (48–52).
Figure 6.
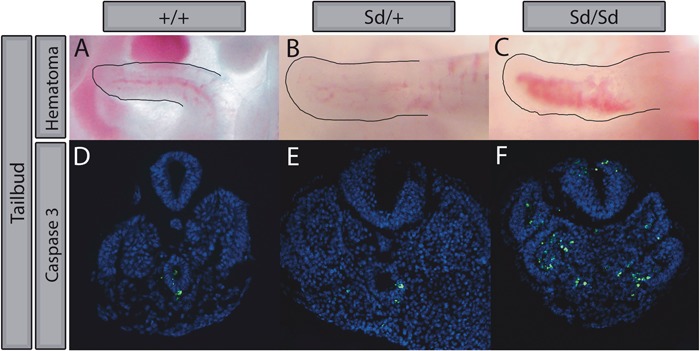
Sd/Sd tailbuds exhibit gross hematomas and increased apoptosis. WT (A), Sd/+(B) and Sd/Sd(C) tailbuds imaged immediately following dissection in PBS. Hematoma is evident in Sd/Sd embryo tailbud. Increased apoptosis was observed by immunofluorescence staining for Caspase 3 (green) in Sd/Sd tailbud (F) compared to WT (D) and Sd/+(E) tissues. All sections co-stained with DAPI (blue). N > 3 for each genotype.
Discussion
Our group and others previously identified the Sd mutation as an ERV insertion upstream of Ptf1a that leads to its ectopic expression during caudal development, resulting in the Sd mutant phenotype (5–7). The underlying mechanisms that lead to upregulation of Ptf1a and the downstream developmental pathways dysregulated by ectopic Ptf1a expression were not previously determined. In the current study, we used ATAC-seq to analyze genome-wide chromatin accessibility and mRNA-seq to gain additional insight into these underlying mechanisms.
Our ATAC-seq data showed a significant change in chromatin accessibility near the Ptf1a genomic locus, mapping to the promoter region of Gm13344, an lncRNA that has been annotated in the mouse genome. This lncRNA overlaps with a previously characterized autoregulatory enhancer of Ptf1a (17). According to our mRNA-seq data, Gm13344 is transcribed and overexpressed in Sd/Sd tailbuds relative to WT tailbuds. Previous studies also detected overexpression of Gm13344 in Sd mutant embryos, although a transgene containing the Sd ERV and Gm13344 without Ptf1a did not recapitulate the Sd phenotype, while a transgene containing the Sd ERV and Ptf1a did recapitulate some, but not all, of the Sd phenotype (7). We previously generated a transgene containing 31.9 kb of genomic sequence surrounding Ptf1a with and without the Sd ERV, which did not recapitulate the Sd phenotype (5). This transgene contained part of the Gm13344 sequence, including the PTF1A binding sites. The discrepancy between these transgene experiments suggests that the genomic regulatory context is critical for appropriate localization of ectopic Ptf1a expression and may have been strongly influenced by positional integration effects of the transgenes. Thus, the exact effects of Gm13344 on the Sd phenotype are not known.
We found no evidence of an annotated human lncRNA that is orthologous to the mouse Gm13344 lncRNA. However, the promoter region is conserved in humans and maps to a region that contains high H3K4me1 signal in developing pancreas relative to other tissues, including adult pancreas (30). This finding suggests that the enhancer properties of the Gm13344 orthologous promoter sequence in humans are conserved. Our results suggest that the genomic sequence corresponding to the Gm13344 promoter in mouse functions as an enhancer, consistent with previous studies, and the resulting RNA produced from Gm13344 is a byproduct of this enhancer activity, as has been shown for other developmentally regulated genes (17,29,53). In addition, we speculate that it is the combination of TFs binding to the Sd ERV and the Gm13344 enhancer region that is required for the tissue and temporal specificity of ectopic Ptf1a expression in Sd mutant embryos. A corollary of this hypothesis is that ectopic Ptf1a feeds into this autoregulatory loop to further increase ectopic Ptf1a expression. One intriguing question that remains unanswered is the mechanism underlying the exquisite specificity of ectopic Ptf1a expression in the tailbud. We speculate that this could either be due to sequences within the ERV that direct expression of Ptf1a in combination with Gm13344 enhancer sequences, or due to enhancer sequences within the surrounding genomic region, possibly within the TAD containing Ptf1a, that form a regulatory loop to direct this tailbud specificity. 4C-seq or Capture C experiments could shed light on this question in the future (54,55).
Our transcriptome analysis revealed 49 genes with significant differential expression at 5% FDR. As expected, Ptf1a and Gm13344 were significantly upregulated, as were known PTF1A downstream targets, including Kirrel2, Nphs1 and Cpa1. Kirrel2 and Nphs1 share a bidirectional promoter, which contains known PTF1A binding sites. They were originally identified as components of the interdigitating podocyte foot processes of the glomerular filter but are also expressed in the developing nervous system (56). Expression of both Kirrel2 and Nphs1 are lost in Ptf1a null mice, while forced expression of Ptf1a in the cerebral cortex induced ectopic expression of both genes (57). Therefore, the transcriptomic changes we identified were widespread but characterized by expected changes due to upregulation of Ptf1a expression.
It is notable that a considerable number of genes were differentially expressed while only a single region of differential chromatin accessibility was identified. Chromatin accessibility can be an important factor in regulating gene expression, but there are many other factors (such as miRNA binding, mRNA stability/degradation and TF availability) and the correlation between chromatin accessibility and gene expression is known to be poor (58). Changes in TF binding at a promoter can often occur without changes in the ATAC-seq signal; recent work shows that the majority of TFs do not have a noticeable impact on chromatin openness (59). As Ptf1a overexpression does not lead to extensive changes in chromatin accessibility, we speculate that PTF1A is one such TF. Given the above findings, it is unsurprising that many changes in gene expression occur in the absence of changes in chromatin accessibility.
One of the most striking findings revealed by our mRNA-seq analysis was that the hedgehog signaling pathway is altered in Sd mutant embryos. Both Shh and Ihh were significantly downregulated, and KEGG pathway enrichment analysis revealed the Hedgehog pathway as significantly dysregulated compared to other known developmental signaling pathways. These findings were confirmed by in situ hybridization analysis with Shh, as well as a cross of Sd/+ mice to the Shh reporter strain Gli1-LacZ, which revealed decreased β-galactosidase expression in Sd/+ tailbuds. We also confirmed reduced expression of Nkx2.9 and Foxa2, which are regulated by SHH along the neural axis (60).
Our mRNA-seq data did not demonstrate significant differences in expression of Cdx2, T, Wnt3a and Cyp26a1, and the KEGG pathway enrichment analysis did not identify the Wnt or Retinoic acid signaling pathways as being significantly dysregulated. Because T is expressed in notochord-derived cells and is required for notochord development, we performed in situ hybridization analysis for T expression (39,61,62). While we did observe reduced T expression in the anterior notochord in Sd/Sd embryos, expression of T in the tailbud was unchanged. Although our data conflict with a previous study, our analysis of tailbud tissue and use of more specific methodology (RNA-seq and in situ hybridization compared to quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR) in RNA from whole embryos) could explain this apparent discrepancy (7). While our use of tailbud tissue for our analyses is limited by the heterogeneous nature of the tissue, our rationale was to enrich for the entire region of ectopic Ptf1a expression. Although this reduces our statistical power to detect differential expression of genes expressed in only a small subset of cells within the tailbud, such as Foxa2, our use of additional qualitative methods to evaluate changes in gene expression, such as in situ hybridization and immunofluorescence, minimizes these concerns.
We observed increased expression of Lfng in Sd/Sd tailbuds by mRNA-seq. Lfng is a downstream effector of Notch1 signaling. Although RBPJ is an integral component of the PTF1 complex, its expression was unchanged in Sd mutant tailbuds (unadjusted P-value = 0.56), and the Notch signaling pathway was not found to be dysregulated by KEGG pathway analysis. Since different E-box proteins are used as the third component of PTF1 complex depending on the context, it is difficult to predict which E-box proteins might be dysregulated in Sd mutant embryos. In addition to T, we also examined expression of Noto as a marker of notochord expression (36,63,64). Not surprisingly, we found that Noto expression was absent in the caudal notochord of Sd/Sd embryos. It is possible that the absence of Noto expression is due to degeneration of notochord cells in this region. However, the persistence of T expression in the caudal notochord suggests that Noto expression is suppressed either directly or indirectly by ectopic Ptf1a.
FOXA2 induces Shh expression in both notochordal and floorplate tissues, and importantly, floorplate-derived SHH is sufficient to pattern the NT (42–46,65). Immunofluorescence staining through the transverse plane of forelimb and hindlimb level NT revealed normal expression of SHH in the floorplate of Sd/+ embryos, which is sufficient to correctly maintain the expression domains of FOXA2, NKX2.2, NKX6.1 and PAX6 in the E10.5 NT, despite aberrant notochord morphology and reduced SHH signaling from this organizing tissue. Similarly, in Sd/Sd mutant embryos, normal expression of SHH from the floorplate is sufficient to pattern the NT, however, only at the forelimb level. In Sd/Sd embryos, SHH is completely absent from the presumptive floorplate at the hindlimb level and as a result, dorsal NT TF domains of FOXA2, NKX2.2 and NKX6.1 are reduced, and expression of the ventrally constricted TF PAX6 is expanded dorsally. Taken together, these experiments demonstrate that dysregulated SHH signaling adversely affects patterning of the NT in Sd/Sd embryos. If dysregulated SHH signaling drives the Sd phenotype, then the increased phenotypic severity of Sd/Sd embryos could result from the degeneration of the notochord before redundancy between the floorplate and notochord has been established in the caudal Sd/Sd embryo.
Numerous published studies in mouse models have demonstrated that caudal and urogenital malformations resembling the malformations observed in Sd mutants can be secondary to disrupted SHH signaling. The Shh KO mouse (66) exhibits abnormal axial structures including the floorplate, ventral NT and sclerotome. Additional mouse models with disrupted SHH signaling have been demonstrated to have phenotypes of anorectal, renal and vertebral malformations that are observed in Sd mutant mice, resembling VACTERL association (67–69). Runck et al. (70) studied cloacal development in Shh KO mice and in human patients with cloacal malformations and found striking similarities between the two. KO of Shh in the notochord or floorplate using either ShhCreERT2 or Foxa2CreERT2 mice exhibits a phenotype that is strikingly similar to Sd mutant mice only in the notochord KO (71). KO of both Foxa1 and Foxa2 in the notochord results in severe defects in formation of the axial skeleton and dorsal–ventral patterning of the NT secondary to reduced Shh expression (72), also phenocopying Sd mutant mice. Finally, in vivo knockdown of T in the notochord results in reduced Shh expression in the notochord and also phenocopies the Sd mutant phenotype (62). Thus, the caudal phenotype observed in Sd mutant mice is consistent with previous reports of disrupted SHH signaling in the notochord.
Collectively, our results show that the Sd insertion leads to widespread transcriptional dysregulation of the hedgehog developmental signaling pathway. The effect of the Sd insertion may be mediated by a nearby conserved Ptf1a autoregulatory enhancer, which shows increased chromatin accessibility in Sd mutant mice. More generally, our results suggest a cascade of molecular events where a single non-coding regulatory mutation propagates to perturb a critical signaling pathway. Further, our findings suggest that regulatory mutations in hedgehog signaling pathway genes could explain some human caudal malformations without known genetic etiologies. Future studies of viable mouse models, such as the Sd mouse, with alterations in SHH signaling, will be critical to dissecting the timing and impact of key molecular events and will help to further our understanding of human caudal malformations.
Materials and Methods
Animals
All animals were housed in environmentally controlled conditions with 14 h light and 10 h dark cycles with food and water provided ad libitum. All protocols were approved by the Institutional Animal Care & Use Committee at the University of Michigan and comply with policies, standards and guidelines set by the State of Michigan and the United States Government. Sd/+ animals were maintained on an outbred CD-1 background (Charles River Laboratories, Wilmington, MA).
Timed pregnancies
Matings for timed embryo isolation were set up using standard animal husbandry techniques. Noon on the day of vaginal plug observation was considered E0.5. Embryos were isolated at E9.5–E10.5. Yolk sac DNA was isolated via the HotSHOT extraction method for genotyping. Genotyping was performed as previously described (5).
For RNA-seq and ATAC-seq, embryo tailbuds were dissected just prior to the terminal somite, snap frozen in liquid nitrogen and stored at −80°C.
mRNA-seq experiments
RNA was collected from the tailbuds using a dounce homogenizer and RNAeasy mini extraction kit (Qiagen, Hilden, Germany). First, samples were placed into a dounce homogenizer with 350 uL of RLT buffer and homogenizing with five strokes. The extraction procedure was followed as specified by the kit, including the optional DNAse treatment. Samples were eluted in 35 uL of RNAse free water. Libraries were prepared at the University of Michigan DNA Sequencing Core using the Illumina TruSeq stranded mRNA-seq kit.
ATAC-seq experiments
All procedures were done at 4°C to minimize degradation of nuclei, and following the previously published protocol except as noted here (20). Each tailbud was disrupted in 200 uL of NIB with 0.5% Triton-X buffer by pipetting up and down. Then the samples were spun, supernatant removed and was followed by a second wash with RSB buffer. Each pellet of nuclei was then transposed using a home-made Tn5 enzyme (21,73). After 30 min of incubation at 37°C, each sample was cleaned up with MinElute PCR purification kit (Qiagen, Hilden, Germany).
Sequencing data
Libraries were multiplexed and sequenced on an Illumina HiSeq 2500 instrument. All raw and processed data have been deposited to Gene Expression Omnibus under the accession GSE108804.
ATAC-seq data processing
Adapters were trimmed using cta (v. 0.1.2). Reads were aligned to mm9 using bwa mem (v. 0.7.15-r1140; flags: -M) (74). Picard MarkDuplicates (v. 2.8.1; http://broadinstitute.github.io/picard) was used for duplicate removal (options: VALIDATION_STRINGENCY = LENIENT) and samtools (v. 1.3.1) was used to filter for autosomal, properly paired and mapped read pairs with mapping quality ≥30 (samtools view -b -h -f 3 -F 4 -F 8 -F 256 -F 1024 -F 2048 -q 30) (75). Peak calling was performed using MAC2 callpeak (v. 2.1.1.20160309; options: --nomodel --broad --shift -100 --extsize 200 --keep-dup all) (76). Peaks were filtered against a blacklist (downloaded from http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm9-mouse/mm9-blacklist.bed.gz). For figures displaying ATAC-seq coverage, we normalized the signal to account for differences in library size. This was done by dividing each line in the ‘treat_pileup’ bedgraph files generated during peak calling with MACS2 by the number of tags in the treatment (‘total tags in treatment’ as output by MACS2) and multiplying by 10 million. The bedgraph files were then converted to bigwig format using bedGraphToBigWig (v. 4) (77). ATAC-seq quality control was performed using ataqv (v. 1.0.0; https://github.com/ParkerLab/ataqv). The resulting interactive HTML quality control report is available at https://theparkerlab.med.umich.edu/data/porchard/qc/danforth_ataqv_master_peaks/. To generate this report, we used the list of peaks used for differential peak calling, the TSS file (mm9.tss.refseq.housekeeping.ortho.bed.gz in the ataqv GitHub repository and the mm9 blacklist that was used for peak filtering).
Differential peak calling
The list of reproducible peaks for downstream analysis was generated by taking the union of all 5% FDR broad peaks across all eight samples, and keeping the union peaks that overlapped with 5% FDR peak calls from at least two of the eight ATAC-seq libraries. We additionally tested two alternate thresholds, keeping only the union peaks that overlapped with 5% FDR peak calls from at least three or four of the eight ATAC-seq libraries, rather than from at least two; these alternate thresholds similarly resulted in only a single differential peak, the same peak identified using our selected threshold. Differential peak calling was performed across all the replicates using DESeq2 (v. 1.14.1) (27). Read counts for each sample and each peak were collected using bedtools’ coverageBed (v. 2.26.0; coverageBed -counts) (78). The number of somites in each sample (Supplementary Material, Table S3) was used as a covariate (in the case that the sample consisted of pooled embryos, the number of somites for the sample was set to the mean number of somites in the embryos). Differential peaks were called at 5% FDR.
RNA-seq processing and differential gene expression analysis
Reads were aligned to mm9 (Gencode vM1 comprehensive gene annotation) using the STAR splice-aware aligner (v. 2.5.2b; --outSAMUnmapped Within KeepPairs) (79,80). Aligned reads were filtered to autosomal reads with mapping quality 255 (samtools view -b -h -f 3 -F 4 -F 8 -F 256 -F 2048 -q 255). We used QoRTs (v. 1.0.7) to gather read counts per sample per gene for differential gene expression analysis (81). Differential gene expression analysis was performed using DESeq2 (5% FDR); only genes with at least one read in at least one library were included (i.e. we excluded those genes that were completely unexpressed in all libraries). The number of somites in each sample (Supplementary Material, Table S3) was used as a covariate. For figures displaying RNA-seq coverage, we normalized the signal to account for differences in library size. To do this, the filtered RNA-seq bam files were converted to wiggle format using QoRTs’ bamToWiggle (with options --negativeReverseStrand --stranded --sizefactor XXX), where XXX represents the corresponding sample’s size factor (from QoRTs function get.size.factors).
GO enrichment
Gene Ontology (GO) enrichment analysis using the P-values for all genes included in the differential gene expression analysis was performed using RNA-Enrich (with the KEGG database; code downloaded from supplemental materials of 82). Gene sets with less than five genes or more than 500 genes were excluded from the analysis; otherwise, default parameters were used. The ‘baseMean’ column in the DESeq2 differential gene expression output, representing the gene-wise average read count across samples after normalizing for sequencing depth, was used as the ‘avg_readcount’ values for RNA-Enrich.
The results of the GO enrichment are dependent on the output of the differential gene expression analysis. As outlined in the ‘RNA-seq processing and differential gene expression analysis’ section of the Materials and Methods, we included in the differential gene expression analysis all genes that showed at least one read in at least one library. To verify that the GO enrichment results were robust to this threshold, we tested several alternative thresholds for the differential gene expression analysis, in each case feeding the new results into the GO enrichment analysis. The GO enrichment analysis results under these alternative thresholds are displayed in Supplementary Material, Fig. S4. GO enrichment results were highly robust to the thresholds used; in each case, the only developmental pathway to show significant enrichment for altered gene expression was the Hedgehog signaling pathway.
ChIP-seq processing
The accession numbers of the data we used from Weedon et al. and Roadmap Epigenomics are listed in Supplementary Material, Table S4 (30,32). We aligned this data using bwa aln (default parameters). All reads were trimmed to 36 bps (using fastx_trimmer from FASTX toolkit v 0.0.14; http://hannonlab.cshl.edu/fastx_toolkit) to prevent read length from confounding comparisons. Picard MarkDuplicates was used for duplicate removal (options: VALIDATION_STRINGENCY = LENIENT) and samtools was used to filter to autosomal, properly paired and mapped read pairs with mapping quality ≥30 (samtools view -b -h -F 4 -F 256 -F 1024 -F 2048 -q 30). Peak calling was performed using MAC2 callpeak (options: --broad --keep-dup all). Peaks were filtered against hg19 blacklists (downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz and http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDukeMapabilityRegionsExcludable.bed.gz). We normalized the signal for each experiment to 10 M reads to prevent sequencing depth from confounding comparisons. Quality control was performed using phantompeakqualtools (v.2.0); all samples included in the analysis had relative strand cross-correlation coefficient (RSC) > 0.8 and normalized strand cross-correlation coefficient (NSC) > 1.05. (83,84). One of the two Weedon et al. H3K4me1 samples (ERR361008) appeared as a QC outlier (NSC = 1.31, RSC = 2.67) relative to other samples (mean NSC = 1.10, mean RSC = 1.47) and was therefore excluded (30).
To determine the human sequence orthologous to the Gm13344 promoter peak, we used bnMapper with an mm9 to hg19 chain file from University of California, Santa Cruz (UCSC) (downloaded from http://hgdownload-test.cse.ucsc.edu/goldenPath/mm9/liftOver/mm9ToHg19.over.chain.gz) to map the Gm13344 promoter peak onto the human genome (85). The resulting closely spaced stretches of human orthologous sequence were then merged together (bedtools merge –d 30) in order to eliminate small gaps between them, and the signal for this region in each tissue’s H3K4me1 ChIP-seq experiment was used for the comparison in (Fig. 1C).
Reproducibility of computational analyses
We have created a GitHub repo that has all the code and steps necessary to reproduce the analyses in this work. The repo is located at https://github.com/ParkerLab/danforth-2018.
X-gal staining
X-gal experiments were conducted on Gli1tm2Alj/J (Jackson Laboratories, Bar Harbor, Maine, #008211) mice crossed to Sd/+ mice. Embryos were dissected in phosphate buffered saline (PBS) and fixed with glutaraldehyde solution (1.25%, 1 M EGTA; .2%: 1 M MgCl2; 2%: 25% glutaraldehyde in PBS) and rinsed in wash buffer (.2%, 1 MgCl2; .4%, 5% NP-40 in sodium phosphate buffer). Embryos were incubated in X-gal stain solution containing ferric and ferrocyanide ions for 2 h. Embryos were then fixed with 4% paraformaldehyde (PFA) then rinsed and stored in PBS. (n ≥ 3 for all conditions). Images were taken with a Leica MZ10F dissecting microscope with a Fostec EKE ACE external light source.
Whole mount in situ hybridization
Embryos were dissected in cold diethyl pyrocarbonate (DEPC) treated PBS, fixed in 4% PFA in DEPC treated PBS, washed in PBST, dehydrated through a graded methanol series (25, 50, 75 and 100%) and processed for in situ hybridization as described previously (86,87). Embryos at each developmental stage for each probe were processed together, and images were captured under identical settings for comparison of staining intensity among WT, Sd/+ and Sd/Sd genotypes, n ≥ 3 embryos of each genotype were studied with all probe conditions.
Ptf1a probes were generated from IMAGE clone: 8861527, (MGC:170132, GenBank BC138507.1) in PCR4-TOPO. Sense probe was generated using digoxygenin labeling kit (Roche, Indianapolis, Indiana), T3 polymerase and Not I digested plasmid. Antisense probe was generated using T7 polymerase and Spe I digested plasmid. For the Gm13344 probe, a 511 bp fragment (Chr2: 19,351,134–19,351,644) of Gm13344 lncRNA was amplified using primers GGGTGTATCACCCAGCAATC (Forward) and AAGAGGAGGAACCCAGGTGT (Reverse) from BAC DNA RP24-347 M17, and cloned into pGEMT-easy vector. The antisense digoxygenin-AP probe was generated using SP6 polymerase from the Nco I digested pGEMT clone while the sense probe using T7 polymerase from Nde I digested clone. Probes for Shh (88–90), Noto (64), Kirrel2/Neph3 (91), Nkx 2–9 (40), Foxa1, Foxa2 (92,93) and T (94) were previously described.
Immunofluorescence
Embryos were dissected into fresh, ice cold 4% PFA (Fluka: 76240) in PBS for 30 min then cryoprotected overnight in solution of 10% sucrose and 2 mM MgCl2 and cut immediately rostral to the forelimb and hindlimb buds. The tissues were embedded in optimum cutting temperature (OCT) compound and stored at −80°. Frozen sections were taken through the transverse plane and stored at −80°. Tissue sections were blocked with PBS with 3% Bovine Serum Albumin (BSA), 1% Goat Serum, 0.1% Triton X-100, brought to a final pH of 7.4 for 1 h at room temperature in a humidified container. Blocking solution was replaced with primary antibody diluted in blocking buffer. Primary antibodies were purchased from the Developmental Studies Hydronima Bank [SHH (5E1, AB_2188307), FOXA2 (4C7, AB_2278498), NKX2.2 (74.5A5, AB_2314952), NKX6.1 (F55A10, AB_532378), PAX6 (PAX6, AB_528427)]. Tissue sections from n ≥ 3 embryos per genotype were incubated with each primary antibody overnight at 4°C at the following concentrations: 1:20 for SHH, FOXA2, NKX2.2, NKX6.1 and 1:2500 for PAX6. Sections were stained with DAPI (Kirkegaard & Perry Laboratories, Gaithersburg, Maryland: 71-03-00), incubated for 1 h with Alexa fluor 488 goat anti-mouse secondary antibody (A11001), then coverslipped with aqueous mounting medium (Thermo Scientific, Waltham, Massachusetts: Immu-Mount: 990402). Fluorescent imaging was conducted on a Leitz DMRB microscope, with a Leica EL6000 fluorescent lamp.
Caspase 3 immunofluorescence
Embryos were processed for Paraffin embedding with a Tissue Tek VIP (Miles Scientific, Newark, Delaware) and embedded in paraffin blocks. Blocks were sectioned with a Spencer Microtome (American Optical, Bethlehem, Pennsylvania) at a thickness of 7 μm and stored at room temperature. Paraffin was removed with Xylene followed by EtOH and PBS for rehydration. Antigen retrieval was achieved by citrate boiling and slides were then blocked with suppressor solution (5% goat serum, 3% BSA, 0.5% Tween 20) for 20 min. Sections (n > 3 per genotype) were incubated with Caspase 3 primary antibody (Cleaved Caspase-3 anti-rabbit, D175: 9661S Cell Signaling Technology) overnight at 4°C at a concentration of 1:400. Sections were stained with DAPI (Kirkegaard & Perry Laboratories: 71-03-00), incubated for 1 h with Alexa fluor 488 goat anti-rabbit secondary antibody (A11008), rinsed with PBS, then coverslips were applied with aqueous mounting medium (Thermo Scientific, Waltham, Massachusetts: Immu-Mount: 990402). Fluorescent imaging was conducted on a Leitz DMRB microscope, with a Leica EL6000 fluorescent lamp.
Supplementary Material
Acknowledgements
The authors thank Drs Sally Camper and Miriam Meisler for critical review of this manuscript. We acknowledge Dr Andreas Kispert for the Kirrel2/Neph3 probe, Dr Hans-Henning Arnold for the Nkx2-9 probe, Dr Andreas Schedl for the Nephrin/NPHS1 probe, Dr Klaus Kaestner for Foxa1 and Foxa2 probes and Dr Cheryle Seguin for the Noto probe. We thank the University of Michigan DNA Sequencing Core for assistance with sequencing.
Conflict of Interest statement. None declared.
Funding
March of Dimes (6-FY14-403 to C.E.K.); the American Diabetes Association Pathway to Stop Diabetes (1-14-INI-07 to S.C.J.P.); and the National Institutes of Health (T32 HG00040 to P.O.).
References
- 1. Pauli R.M. (1994) Lower mesodermal defects: a common cause of fetal and early neonatal death. Am. J. Med. Genet., 50, 154–172. [DOI] [PubMed] [Google Scholar]
- 2. Dunn L.C., Gluecksohn-Schoenheimer S. and Bryson V. (1940) A new mutation in the mouse affecting spinal column and urogenital system. J. Hered., 31, 343–348. [Google Scholar]
- 3. Grüneberg H. (1953) Genetioal studies on the skeleton of the mouse. J. Genet., 51, 317–326. [PubMed] [Google Scholar]
- 4. Gluecksohn-Schoenheimer S. (1945) The embryonic development of mutants of the Sd-strain in mice. Genetics, 30, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vlangos C.N., Siuniak A.N., Robinson D., Chinnaiyan A.M., Lyons R.H. Jr., Cavalcoli J.D. and Keegan C.E. (2013) Next-generation sequencing identifies the Danforth’s short tail mouse mutation as a retrotransposon insertion affecting Ptf1a expression. PLoS Genet., 9, e1003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lugani F., Arora R., Papeta N., Patel A., Zheng Z., Sterken R., Singer R.A., Caridi G., Mendelsohn C., Sussel L. et al. (2013) A retrotransposon insertion in the 5′ regulatory domain of Ptf1a results in ectopic gene expression and multiple congenital defects in Danforth's short tail mouse. PLoS Genet., 9, e1003206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Semba K., Araki K., Matsumoto K., Suda H., Ando T., Sei A., Mizuta H., Takagi K., Nakahara M., Muta M. et al. (2013) Ectopic expression of Ptf1a induces spinal defects, urogenital defects, and anorectal malformations in Danforth’s short tail mice. PLoS Genet., 9, e1003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krapp A., Knofler M., Frutiger S., Hughes G.J., Hagenbuchle O. and Wellauer P.K. (1996) The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. EMBO J., 15, 4317–4329. [PMC free article] [PubMed] [Google Scholar]
- 9. Willet S.G., Hale M.A., Grapin-Botton A., Magnuson M.A., MacDonald R.J. and Wright C.V. (2014) Dominant and context-specific control of endodermal organ allocation by Ptf1a. Development, 141, 4385–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Obata J., Yano M., Mimura H., Goto T., Nakayama R., Mibu Y., Oka C. and Kawaichi M. (2001) p48 subunit of mouse PTF1 binds to RBP-Jkappa/CBF-1, the intracellular mediator of Notch signalling, and is expressed in the neural tube of early stage embryos. Genes Cells, 6, 345–360. [DOI] [PubMed] [Google Scholar]
- 11. Glasgow S.M., Henke R.M., Macdonald R.J., Wright C.V. and Johnson J.E. (2005) Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development, 132, 5461–5469. [DOI] [PubMed] [Google Scholar]
- 12. Sellick G.S., Barker K.T., Stolte-Dijkstra I., Fleischmann C., Coleman R.J., Garrett C., Gloyn A.L., Edghill E.L., Hattersley A.T., Wellauer P.K. et al. (2004) Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat. Genet., 36, 1301–1305. [DOI] [PubMed] [Google Scholar]
- 13. Krapp A., Knofler M., Ledermann B., Burki K., Berney C., Zoerkler N., Hagenbuchle O. and Wellauer P.K. (1998) The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev., 12, 3752–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cockell M., Stevenson B.J., Strubin M., Hagenbuchle O. and Wellauer P.K. (1989) Identification of a cell-specific DNA-binding activity that interacts with a transcriptional activator of genes expressed in the acinar pancreas. Mol. Cell. Biol., 9, 2464–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R.J. and Wright C.V. (2002) The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet., 32, 128–134. [DOI] [PubMed] [Google Scholar]
- 16. Beres T.M., Masui T., Swift G.H., Shi L., Henke R.M. and MacDonald R.J. (2006) PTF1 is an organ-specific and Notch-independent basic helix-loop-helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol. Cell. Biol., 26, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Masui T., Swift G.H., Hale M.A., Meredith D.M., Johnson J.E. and Macdonald R.J. (2008) Transcriptional autoregulation controls pancreatic Ptf1a expression during development and adulthood. Mol. Cell. Biol., 28, 5458–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meredith D.M., Masui T., Swift G.H., MacDonald R.J. and Johnson J.E. (2009) Multiple transcriptional mechanisms control Ptf1a levels during neural development including autoregulation by the PTF1-J complex. J. Neurosci., 29, 11139–11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mona B., Avila J.M., Meredith D.M., Kollipara R.K. and Johnson J.E. (2016) Regulating the dorsal neural tube expression of Ptf1a through a distal 3′ enhancer. Dev. Biol., 418, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y. and Greenleaf W.J. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scott L.J., Erdos M.R., Huyghe J.R., Welch R.P., Beck A.T., Wolford B.N., Chines P.S., Didion J.P., Narisu N., Stringham H.M. et al. (2016) The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat. Commun., 7, 11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varshney A., Scott L.J., Welch R.P., Erdos M.R., Chines P.S., Narisu N., Albanus R.D., Orchard P., Wolford B.N., Kursawe R. et al. (2017) Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc. Natl. Acad. Sci. U. S. A., 114, 2301–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lamparter D., Marbach D., Rueedi R., Bergmann S. and Kutalik Z. (2017) Genome-wide association between transcription factor expression and chromatin accessibility reveals regulators of chromatin accessibility. PLoS Comput. Biol., 13, e1005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cirillo L.A., Lin F.R., Cuesta I., Friedman D., Jarnik M. and Zaret K.S. (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell, 9, 279–289. [DOI] [PubMed] [Google Scholar]
- 25. Chen R. and Gifford D.K. (2017) Differential chromatin profiles partially determine transcription factor binding. PLoS One, 12, e0179411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherwood R.I., Hashimoto T., O'Donnell C.W., Lewis S., Barkal A.A., Hoff J.P., Karun V., Jaakkola T. and Gifford D.K. (2014) Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol., 32, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love M.I., Huber W. and Anders S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Washietl S., Kellis M. and Garber M. (2014) Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res., 24, 616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paralkar V.R., Taborda C.C., Huang P., Yao Y., Kossenkov A.V., Prasad R., Luan J., Davies J.O., Hughes J.R., Hardison R.C. et al. (2016) Unlinking an lncRNA from its associated cis element. Mol. Cell, 62, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weedon M.N., Cebola I., Patch A.M., Flanagan S.E., De Franco E., Caswell R., Rodriguez-Segui S.A., Shaw-Smith C., Cho C.H., Allen H.L. et al. (2014) Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat. Genet., 46, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heintzman N.D., Stuart R.K., Hon G., Fu, Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet., 39, 311–318. [DOI] [PubMed] [Google Scholar]
- 32. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson N., Gesina E., Scheinert P., Bucher P. and Grapin-Botton A. (2012) RNA profiling and chromatin immunoprecipitation-sequencing reveal that PTF1a stabilizes pancreas progenitor identity via the control of MNX1/HLXB9 and a network of other transcription factors. Mol. Cell. Biol., 32, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meredith D.M., Borromeo M.D., Deering T.G., Casey B.H., Savage T.K., Mayer P.R., Hoang C., Tung K.C., Kumar M., Shen C. et al. (2013) Program specificity for Ptf1a in pancreas versus neural tube development correlates with distinct collaborating cofactors and chromatin accessibility. Mol. Cell. Biol., 33, 3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Donnell M., Hong C.S., Huang X., Delnicki R.J. and Saint-Jeannet J.P. (2006) Functional analysis of Sox 8 during neural crest development in Xenopus. Development, 133, 3817–3826. [DOI] [PubMed] [Google Scholar]
- 36. Abdelkhalek H.B., Beckers A., Schuster-Gossler K., Pavlova M.N., Burkhardt H., Lickert H., Rossant J., Reinhardt R., Schalkwyk L.C., Muller I. et al. (2004) The mouse homeobox gene Not is required for caudal notochord development and affected by the truncate mutation. Genes Dev., 18, 1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Di Gregorio A., Harland R.M., Levine M. and Casey E.S. (2002) Tail morphogenesis in the ascidian, Ciona intestinalis, requires cooperation between notochord and muscle. Dev. Biol., 244, 385–395. [DOI] [PubMed] [Google Scholar]
- 38. Schulte-Merker S., Eeden F.J., Halpern M.E., Kimmel C.B. and Nusslein-Volhard C. (1994) No tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development, 120, 1009–1015. [DOI] [PubMed] [Google Scholar]
- 39. Wilson V., Manson L., Skarnes W.C. and Beddington R.S. (1995) The T gene is necessary for normal mesodermal morphogenetic cell movements during gastrulation. Development, 121, 877–886. [DOI] [PubMed] [Google Scholar]
- 40. Pabst O., Herbrand H. and Arnold H.H. (1998) Nkx2-9 is a novel homeobox transcription factor which demarcates ventral domains in the developing mouse CNS. Mech. Dev., 73, 85–93. [DOI] [PubMed] [Google Scholar]
- 41. Pabst O., Herbrand H., Takuma N. and Arnold H.H. (2000) NKX2 gene expression in neuroectoderm but not in mesendodermally derived structures depends on sonic hedgehog in mouse embryos. Dev. Genes Evol., 210, 47–50. [DOI] [PubMed] [Google Scholar]
- 42. Weinstein D.C., Ruiz, Altaba i., A., Chen W.S., Hoodless P., Prezioso V.R., Jessell T.M. and Darnell J.E. Jr. (1994) The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell, 78, 575–588. [DOI] [PubMed] [Google Scholar]
- 43. Epstein D.J., McMahon A.P. and Joyner A.L. (1999) Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development, 126, 281–292. [DOI] [PubMed] [Google Scholar]
- 44. Jeong Y. and Epstein D.J. (2003) Distinct regulators of Shh transcription in the floor plate and notochord indicate separate origins for these tissues in the mouse node. Development, 130, 3891–3902. [DOI] [PubMed] [Google Scholar]
- 45. Litingtung Y. and Chiang C. (2000) Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci., 3, 979–985. [DOI] [PubMed] [Google Scholar]
- 46. Ribes V., Balaskas N., Sasai N., Cruz C., Dessaud E., Cayuso J., Tozer S., Yang L.L., Novitch B., Marti E. et al. (2010) Distinct Sonic Hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev., 24, 1186–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Briscoe J., Sussel L., Serup P., Hartigan-O’Connor D., Jessell T.M., Rubenstein J.L. and Ericson J. (1999) Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature, 398, 622–627. [DOI] [PubMed] [Google Scholar]
- 48. Noguchi K.K., Cabrera O.H., Swiney B.S., Salinas-Contreras P., Smith J.K. and Farber N.B. (2015) Hedgehog regulates cerebellar progenitor cell and medulloblastoma apoptosis. Neurobiol. Dis., 83, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharma N., Nanta R., Sharma J., Gunewardena S., Singh K.P., Shankar S. and Srivastava R.K. (2015) PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget, 6, 32039–32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang X., Wei S., Zhao Y., Shi C., Liu P., Zhang C., Lei Y., Zhang B., Bai B., Huang Y. et al. (2017) Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett., 385, 128–136. [DOI] [PubMed] [Google Scholar]
- 51. Zhao W., Pan X., Li T., Zhang C. and Shi N. (2016) Lycium barbarum polysaccharides protect against trimethyltin chloride-induced apoptosis via Sonic Hedgehog and PI3K/Akt signaling pathways in mouse Neuro-2a cells. Oxid. Med. Cell. Longev., 2016, 9826726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen K.Y., Chiu C.H. and Wang L.C. (2017) Anti-apoptotic effects of Sonic hedgehog signalling through oxidative stress reduction in astrocytes co-cultured with excretory-secretory products of larval Angiostrongylus cantonensis. Sci. Rep., 7, 41574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Latos P.A., Pauler F.M., Koerner M.V., Senergin H.B., Hudson Q.J., Stocsits R.R., Allhoff W., Stricker S.H., Klement R.M., Warczok K.E. et al. (2012) Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science, 338, 1469–1472. [DOI] [PubMed] [Google Scholar]
- 54. Davies J.O., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R. and Hughes J.R. (2016) Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods, 13, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Denker A. and Laat W. (2016) The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev., 30, 1357–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ristola M. and Lehtonen S. (2014) Functions of the podocyte proteins nephrin and Neph3 and the transcriptional regulation of their genes. Clin. Sci., 126, 315–328. [DOI] [PubMed] [Google Scholar]
- 57. Nishida K., Hoshino M., Kawaguchi Y. and Murakami F. (2010) Ptf1a directly controls expression of immunoglobulin superfamily molecules Nephrin and Neph3 in the developing central nervous system. J. Biol. Chem., 285, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S. and Crawford G.E. (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell, 132, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baek S., Goldstein I. and Hager G.L. (2017) Bivariate genomic footprinting detects changes in transcription factor activity. Cell Rep., 19, 1710–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jessell T.M. (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet., 1, 20–29. [DOI] [PubMed] [Google Scholar]
- 61. Herrmann B.G., Labeit S., Poustka A., King T.R. and Lehrach H. (1990) Cloning of the T gene required in mesoderm formation in the mouse. Nature, 343, 617–622. [DOI] [PubMed] [Google Scholar]
- 62. Pennimpede T., Proske J., Konig A., Vidigal J.A., Morkel M., Bramsen J.B., Herrmann B.G. and Wittler L. (2012) In vivo knockdown of Brachyury results in skeletal defects and urorectal malformations resembling caudal regression syndrome. Dev. Biol., 372, 55–67. [DOI] [PubMed] [Google Scholar]
- 63. Plouhinec J.L., Granier C., Le Mentec C., Lawson K.A., Saberan-Djoneidi D., Aghion J., Shi D.L., Collignon J. and Mazan S. (2004) Identification of the mammalian Not gene via a phylogenomic approach. Gene Expr. Patterns, 5, 11–22. [DOI] [PubMed] [Google Scholar]
- 64. McCann M.R., Tamplin O.J., Rossant J. and Seguin C.A. (2012) Tracing notochord-derived cells using a Noto-cre mouse: implications for intervertebral disc development. Dis. Model. Mech., 5, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ang S.L. and Rossant J. (1994) HNF-3 beta is essential for node and notochord formation in mouse development. Cell, 78, 561–574. [DOI] [PubMed] [Google Scholar]
- 66. Chiang C., Litingtung Y., Lee E., Young K.E., Corden J.L., Westphal H. and Beachy P.A. (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature, 383, 407–413. [DOI] [PubMed] [Google Scholar]
- 67. Mo R., Kim J.H., Zhang J., Chiang C., Hui C.C. and Kim P.C. (2001) Anorectal malformations caused by defects in sonic hedgehog signaling. Am. J. Pathol., 159, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kimmel S.G., Mo R., Hui C.C. and Kim P.C. (2000) New mouse models of congenital anorectal malformations. J. Pediatr. Surg., 35, 227–230discussion 230-221. [DOI] [PubMed] [Google Scholar]
- 69. Kim P.C.W., Mo R. and Hui C.-c. (2001) Murine models of VACTERL syndrome: role of sonic hedgehog signaling pathway. J. Pediatr. Surg., 36, 381–384. [DOI] [PubMed] [Google Scholar]
- 70. Runck L.A., Method A., Bischoff A., Levitt M., Pena A., Collins M.H., Gupta A., Shanmukhappa S., Wells J.M. and Guasch G. (2014) Defining the molecular pathologies in cloaca malformation: similarities between mouse and human. Dis. Model. Mech., 7, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Choi K.-S., Lee C. and Harfe B.D. (2012) Sonic hedgehog in the notochord is sufficient for patterning of the intervertebral discs. Mech. Dev., 129, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maier J.A., Lo Y. and Harfe B.D. (2013) Foxa1 and Foxa2 are required for formation of the intervertebral discs. PLoS One, 8, e55528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Picelli S., Bjorklund A.K., Reinius B., Sagasser S., Winberg G. and Sandberg R. (2014) Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res., 24, 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kent W.J., Zweig A.S., Barber G., Hinrichs A.S. and Karolchik D. (2010) BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics, 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quinlan A.R. (2014) BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics, 47, 11.12.1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aken B.L., Ayling S., Barrell D., Clarke L., Curwen V., Fairley S., Fernandez Banet J., Billis K., García Girón C., Hourlier T. et al. (2016) The Ensembl gene annotation system. Database, 2016, baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. (2012) Landscape of transcription in human cells. Nature, 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hartley S.W. and Mullikin J.C. (2015) QoRTs: a comprehensive toolset for quality control and data processing of RNA-seq experiments. BMC Bioinformatics, 16, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee C., Patil S. and Sartor M.A. (2016) RNA-enrich: a cut-off free functional enrichment testing method for RNA-seq with improved detection power. Bioinformatics, 32, 1100–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res., 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kharchenko P.V., Tolstorukov M.Y. and Park P.J. (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol., 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Denas O., Sandstrom R., Cheng Y., Beal K., Herrero J., Hardison R.C. and Taylor J. (2015) Genome-wide comparative analysis reveals human-mouse regulatory landscape and evolution. BMC Genomics, 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Keegan C.E., Hutz J.E., Else T., Adamska M., Shah S.P., Kent A.E., Howes J.M., Beamer W.G. and Hammer G.D. (2005) Urogenital and caudal dysgenesis in adrenocortical dysplasia (ACD) mice is caused by a splicing mutation in a novel telomeric regulator. Hum. Mol. Genet., 14, 113–123. [DOI] [PubMed] [Google Scholar]
- 87. Vlangos C.N., O’Connor B.C., Morley M.J., Krause A.S., Osawa G.A. and Keegan C.E. (2009) Caudal regression in adrenocortical dysplasia (ACD) mice is caused by telomere dysfunction with subsequent p53-dependent apoptosis. Dev. Biol., 334, 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Harfe B.D., Scherz P.J., Nissim S., Tian H., McMahon A.P. and Tabin C.J. (2004) Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell, 118, 517–528. [DOI] [PubMed] [Google Scholar]
- 89. Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A. and McMahon A.P. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell, 75, 1417–1430. [DOI] [PubMed] [Google Scholar]
- 90. Everson J.L., Fink D.M., Yoon J.W., Leslie E.J., Kietzman H.W., Ansen-Wilson L.J., Chung H.M., Walterhouse D.O., Marazita M.L. and Lipinski R.J. (2017) Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development, 144, 2082–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Volker L.A., Petry M., Abdelsabour-Khalaf M., Schweizer H., Yusuf F., Busch T., Schermer B., Benzing T., Brand-Saberi B., Kretz O. et al. (2012) Comparative analysis of Neph gene expression in mouse and chicken development. Histochem. Cell Biol., 137, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Monaghan A.P., Kaestner K.H., Grau E. and Schutz G. (1993) Postimplantation expression patterns indicate a role for the mouse forkhead/HNF-3 alpha, beta and gamma genes in determination of the definitive endoderm, chordamesoderm and neuroectoderm. Development, 119, 567–578. [DOI] [PubMed] [Google Scholar]
- 93. Kaestner K.H., Hiemisch H., Luckow B. and Schütz G. (1994) The HNF-3 gene family of transcription factors in mice: gene structure, cDNA sequence, and mRNA distribution. Genomics, 20, 377–385. [DOI] [PubMed] [Google Scholar]
- 94. Wilson V., Rashbass P. and Beddington R.S. (1993) Chimeric analysis of T (Brachyury) gene function. Development, 117, 1321–1331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.