

Abstract
Large-scale ‘omic’ studies investigating the pathophysiological processes that lead to Alzheimer's disease (AD) dementia have identified an increasing number of susceptibility genes, many of which are poorly characterized and have not previously been implicated in AD. Here, we evaluated the utility of human induced pluripotent stem cell-derived neurons and astrocytes as tools to systematically test AD-relevant cellular phenotypes following perturbation of candidate genes identified by genome-wide studies. Lentiviral-mediated delivery of shRNAs was used to modulate expression of 66 genes in astrocytes and 52 genes in induced neurons. Five genes (CNN2, GBA, GSTP1, MINT2 and FERMT2) in neurons and nine genes (CNN2, ITGB1, MINT2, SORL1, VLDLR, NPC1, NPC2, PSAP and SCARB2) in astrocytes significantly altered extracellular amyloid-β (Aβ) levels. Knockdown of AP3M2, CNN2, GSTP1, NPC1, NPC2, PSAP and SORL1 reduced interleukin-6 levels in astrocytes. Only knockdown of FERMT2 led to a reduction in the proportion of TAU that is phosphorylated. Further, CRISPR-Cas9 targeting of FERMT2 in both familial AD (fAD) and fAD-corrected human neurons validated the findings of reduced extracellular Aβ. Interestingly, FERMT2 reduction had no effect on the Aβ42:40 ratio in corrected neurons and a reduction of phospho-tau, but resulted in an elevation in Aβ42:40 ratio and no reduction in phospho-tau in fAD neurons. Taken together, this study has prioritized 15 genes as being involved in contributing to Aβ accumulation, phosphorylation of tau and/or cytokine secretion, and, as illustrated with FERMT2, it sets the stage for further cell-type-specific dissection of the role of these genes in AD.
Introduction
Recent emphasis on the use of large data sets to uncover mechanisms underlying complex pathologies, such as those of Alzheimer's disease (AD), has led to the identification of many genes that influence disease susceptibility and/or course. A critical step toward prioritizing and developing therapeutic targets from such a list of putative targets is characterizing the pathogenic roles of each gene. To accomplish this, genes that emerge from large ‘omic’ screens of human subjects need to be integrated and evaluated with practical and complementary in vitro strategies designed to link such genes to a molecular function. These strategies can identify pathways that may be preferred targets for cellular perturbations leading to AD and guide the initial evaluation of genes, which have little or no information linking them to a particular disease-related molecular phenotype.
AD is characterized by progressive cognitive decline due to the dysfunction and degeneration of neurons, which follows the accumulation of both extracellular amyloid-β (Aβ)-rich neuritic plaques and intracellular neurofibrillary tau-containing tangles over many years. In the familial or early-onset form of AD (fAD), causal mutations are found in APP, PSEN1 and PSEN2; the latter two genes encode the core component of γ-secretase that, with β-secretase, contributes to the generation of pathogenic Aβ peptides produced by the sequential proteolytic cleavage of the amyloid precursor protein (APP). Therefore, disease-causing mutations are found in both the substrate and the enzyme leading to Aβ generation, suggesting that Aβ plays an important role in fAD pathology. Further supporting a role of Aβ in disease pathogenesis, high concentrations of soluble Aβ lead to neurotoxic effects including inhibition of long-term potentiation, elevated neuron excitability, neuroinflammation and neuron loss (reviewed in 1). Genome-wide association studies (GWASs) have identified several additional genes and processes potentially contributing to late-onset AD. These pathways may act through Aβ or may act in an Aβ-independent fashion.
Induced pluripotent stem cells (iPSC) permit the use of AD-relevant human cell types and are a useful tool for probing the cellular and molecular mechanisms underlying disease (reviewed in 2). In this study, we employ a differentiation protocol that directly converts iPSCs to neurons via neurogenin-2 (NGN2) expression (3). These neurons, termed iNs, have a morphology and gene expression profile consistent with postmitotic neurons and spontaneously fire by days of differentiation in vitro (DIV) 14. While it is possible to obtain iPSC-derived human astrocytes as well, we chose here to utilize primary human astrocytes, available from ScienCell (Carlsbad, CA). Herein, we leverage these in vitro cell systems to identify genes with proposed connections to AD that can alter AD-associated phenotypes.
We selected genes for evaluation using several complementary strategies. First, we included susceptibility genes identified in GWAS of AD (4,5). Second, we selected genes implicated by DNA methylome-wide association studies (6). Third, we selected genes based on pathways previously implicated in AD. For example, genes that have been shown to bind to APP and affect its cleavage were included. In addition, endolysosomal trafficking has been implicated in Aβ and tau pathologies found in AD not only by GWAS but also by observations that (1) inhibition of endocytosis leads to a decrease in Aβ generation (7,8), (2) autophagic vacuoles and enlarged endosomes accumulate early in AD (reviewed in 9), (3) disruption of lysosomal function by Niemann–Pick type C (NPC) mutations leads to prominent neurodegeneration that includes the formation of tau tangles and the accumulation of Aβ (10–14) and (4) genetic variants of the lysosomal genes glucocerebrosidase (GBA) and Scavenger Receptor Class B Member 2 (SCARB2) are implicated in dementia with Lewy bodies (DLB), the second most common form of dementia whose pathological hallmarks include amyloid plaques and tau tangles in addition to Lewy bodies (15–17). Thus, in addition to genes emerging from large, unbiased screens, we selected key disease-related genes involved in the endolysosomal–autophagic pathway.
Here, we find differential expression of genes across the three model systems suggesting cell type-specific involvement for subsets of candidate genes, which can guide further study design. Indeed, shRNA-mediated knockdown of gene targets in astrocytes and iNs yields both overlapping and divergent pools of candidates capable of altering extracellular Aβ levels across the cell types. CNN2 and MINT2 knockdown altered extracellular Aβ levels in both astrocytes and iN cultures. GBA, GSTP1 and FERMT2 knockdown alter Aβ levels in iNs but not astrocytes, while reduced expression of ITGB1, SORL1, VLDLR, NPC1, NPC2, PSAP and SCARB2 modulate Aβ levels only in astrocyte cultures. Further, we have found that GSTP1 and FERMT2 knockdown reduce phosphorylated TAU levels in iNs. Finally, AP3M2, CNN2, GSTP1, NPC1, NPC2, PSAP and SORL1 reduce extracellular levels of the proinflammatory cytokine interleukin-6 (IL-6) in astrocyte cultures.
Interestingly, shRNA targeting of FERMT2 in iNs reduced both extracellular Aβ levels and the proportion of intracellular TAU that is phosphorylated. Validation experiments were performed using CRISPR-Cas9 targeting of fAD (18) and isogenic-corrected iPSC lines (fADcorr, 19). Reduction of FERMT2 protein levels in mutation-corrected, wild-type iNs replicated the phenotype observed with shRNA knockdown. Similarly, FERMT2 reduction in fAD iNs lowered total Aβ and tau levels. However, unlike their wild-type counterparts, reduction of FERMT2 in fAD iNs raised the ratio of Aβ42 to Aβ40 and did not reduce the proportion of TAU that is phosphorylated. These results suggest that lowering FERMT2 levels in non-fAD neurons may have a beneficial effect on amyloid and tau pathology. It was previously shown that single nucleotide polymorphisms (SNPs) at the FERMT2 locus are associated with late onset Alzheimer's disease (LOAD) (5), and here we also show an association of these SNPs with amyloid burden and VGF protein levels in the human postmortem brain. These results, in combination with previously published findings (20–24), support a role for FERMT2 in LOAD that is mediated by its effects on Aβ and TAU.
Taken together, these results highlight the different influences that cell types can exert in pathological phenotypes. The findings also point to the importance in AD pathogenesis of FERMT2 expression in neurons and to the endolysosomal pathway in both neurons and astrocytes. In addition to these biological findings that assemble Aβ susceptibility genes with shared functional consequences, this study is a resource for the use of human neurons and astrocytes for systematic screening of genes related to central nervous system disease; its moderate throughput protocols are a model of how to systematically approach the identification of optimal shRNA sequence (RNAseq) targets for successful gene knockdown, and we provide a publicly available reference RNAseq data for neurons and astroyctes to assess the level of expression of desired genes in these experimental systems (https://www.synapse.org/#!Synapse:syn2580853/wiki/409853).
Results
Target gene selection and assignment to an experimental system
Candidate genes were selected using a number of inclusion criteria (Fig. 1; Supplementary Material, Table S1). The majority of the candidate genes were included because of their identification in genome- and epigenome-wide AD studies (4–6). APP and MAPT also were included because they are the primary components of amyloid plaques and neurofibrillary tangles that characterize AD pathology. For the remaining genes, we focused on genes implicated in pathways of interest, prioritizing those genes associated with neurodegeneration and/or implicated in APP biology.
Figure 1.
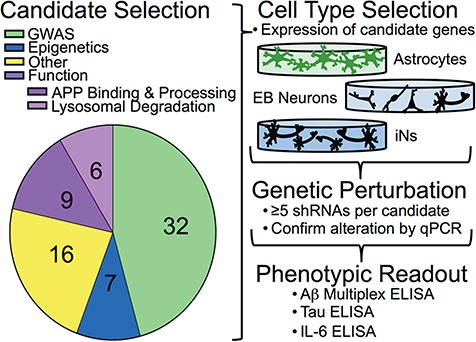
Screening strategy. Candidate genes were selected based either on their identification through large data studies (GWAS or epigenetics) or their functional role in known AD-related processes. Other genes were selected based on their relation to previously selected genes (Supplementary Material, Table S1). The appropriate cell type in which to screen candidates was determined based on the expression of candidates in each cell type. Genetic perturbation of candidates was performed by screening multiple shRNAs per gene, and knockdown was evaluated by qPCR (Supplementary Material, Tables S1 and S2). Those lentiviruses with which robust reduction of gene expression resulted were used to evaluate the effect of gene knockdown on AD-related phenotypic readouts.
Figure 2.
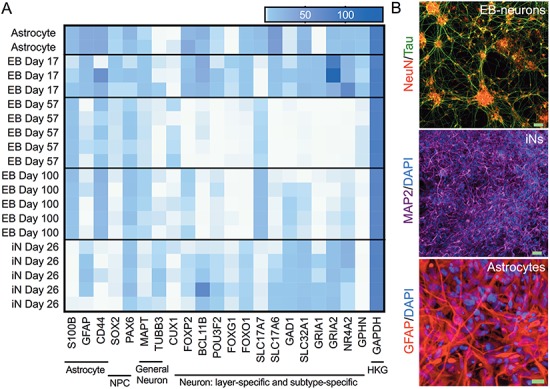
Cell type characterization. (A) Purified RNA of cell lysates collected from astrocytes (day 6), EB-neurons at 3 differentiation time points (days 17, 57 and 100) and iNs (day 26) were analyzed by RNAseq. Genes were selected that mark subsets of neuronal and glial cells and a heat map was created using Prism. NPC, neural progenitor cell; HKG, housekeeping gene. (B) Representative images of immunocytochemistry of each cell culture type with markers for neuron (NeuN and MAP2) and astrocytes (GFAP). Scale bars: EB-neurons and iNs, 50 μm; astrocytes, 20 μm.
To begin, the appropriate cell model for gene targeting was identified for each gene. Three cell culture models were used in our evaluations: (1) iPSC-derived neurons using an NGN2 neuronal induction procedure (iN) (3), (2) iPSC-derived neurons using the embryoid aggregate procedure (EB) (18,25,26) and (3) primary astrocytes isolated from the human cerebral cortex, purchased and cultured according to manufacturer's specifications (ScienCell). The two neuronal models produce distinct cell populations: with the EB protocol, iPSC colonies form aggregates in suspension, which transition through a neuroectodermal state and then neural ‘rosette’, and aggregate structures prior to plating resultant neurons in a monolayer. After 40 days of differentiation, these neurons adopt forebrain cortical neuronal fates, primarily glutamatergic projection neurons with a lower percentage of GABAergic interneurons. By contrast, iN neurons were produced by transduction of iPSCs with lentivirus encoding NGN2 (3), which yields neuronal morphologies and transcriptional profiles consistent with layer 2/3 excitatory projection neurons within 2 weeks (3,27,28). Puromycin selection of the iNs minimizes cellular heterogeneity, while the EB-neurons are inherently mixtures of multiple neural cell fates. RNAseq profiles were created highlighting cell type and subtype markers to characterize the cells used in this study (Fig. 2A). Expression profiles differ between the neurons generated by each differentiation protocol and between EB-neurons at different phases of differentiation. Cell type identity also was confirmed by immunocytochemistry using markers for neurons (NEUN and MAP2) or astrocytes (GFAP) (Fig. 2B). RNAseq was used to determine the expression level of each candidate gene in our model cell types. A heat map is shown with normalization within a gene (Fig. 3A) to show differential expression between cell models. While EB-derived neurons and iNs are both neuronal in fate, certain candidate genes are differentially expressed between these cell culture models. These data highlight the importance of consideration of cellular fate in choosing a cell culture model.
Figure 3.
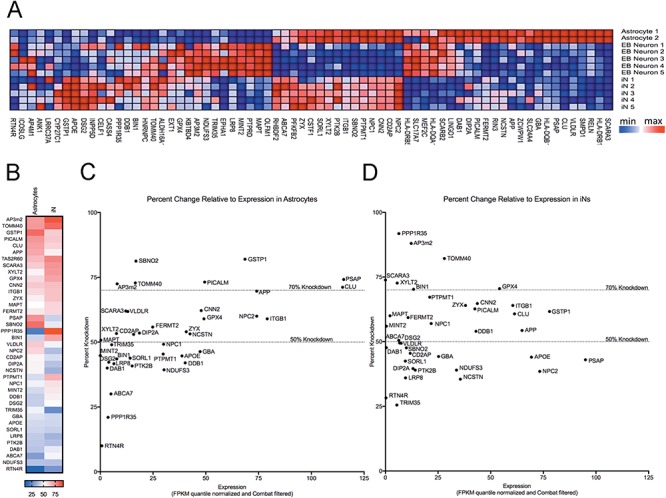
Relative expression of target genes in the three cell culture experimental systems. Cell lysates were collected and pooled from 3 wells of a 96-well plate for each sample. Astrocytes were collected 6 days after plating, while EB-derived neurons and iNs were collected at DIV 57 and DIV 26, respectively. RNA was purified and analyzed by RNAseq. Quantile normalization method was used on FPKM values followed by Combat to remove experimental batch effects. Heat maps were created using Gene Pattern (Broad Institute). Samples were row normalized (A). (B) In this heat map, for each tested gene (rows) in astrocytes (left column) and iNs (right column), we report the highest average knockdown achieved by an shRNA construct. A color key is provided at the bottom of the panel. In (C–D), each dot represents one gene and we plot the results for the shRNA construct with the highest average knockdown (y-axis) for that gene relative to the gene's level of expression (x-axis) in astrocytes (C) and iNs (D).
Figure 4.
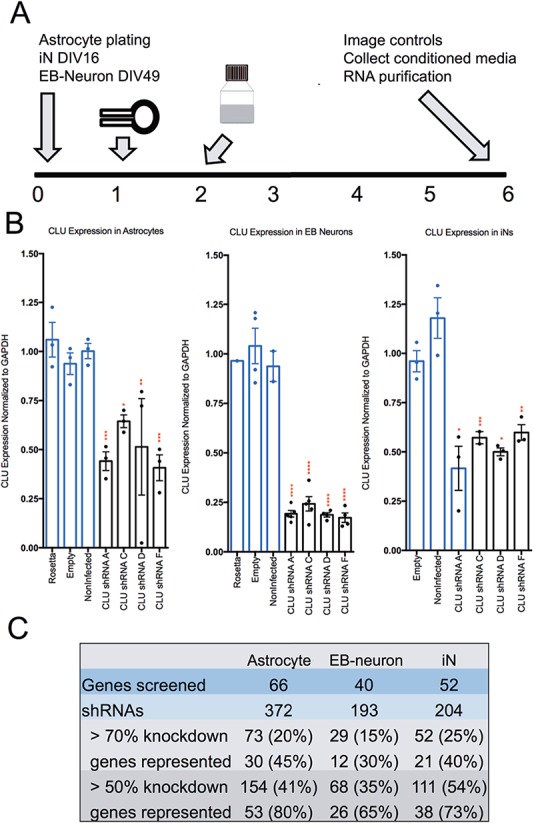
Overview of shRNA screen of AD-related candidate genes. (A) Schematic representation of lentiviral shRNA transduction protocol. On day 1, lentivirus packaged shRNA is applied in fresh media. The following day (day 2) media is removed and replaced. On day 6, GFP-transduced cells are imaged to examine morphology and transduction efficiency, conditioned media collected and RNA purified from all wells. (B) Representative graphs of data collected from each cell type from a single experiment. Relative expression of CLU, the gene encoding clusterin, in human primary astrocytes (left), EB-derived neurons (middle) and iNs (right) following transduction of lentivirus encoding shRNAs against CLU as measured by qPCR. Results are normalized to GAPDH and presented as percent change from averaged controls normalized to 1. Each dot shows data from one well. Statistical analysis performed by ANOVA. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. (C) Summary of numbers of genes and shRNAs screened in astrocytes, EB-derived neurons and iNs.
Initially, it was not clear what an appropriate expression cut-off would be that would enable meaningful testing in our experimental systems. If baseline expression of a particular gene is ‘too low’ and not biologically relevant, one could imagine that lowering of expression with shRNA targeting would not be possible. Figure 3B–D shows the average maximum percent knockdown per gene relative to expression of that gene in astrocytes (C) and iNs (D). Based on this data, the top ∼10 000 expressed genes can potentially be targeted in these cellular systems [based on fragments per kilobase of transcript per million mapped reads (FPKM) values: astrocytes FPKM, >5.1; EB-derived neurons FPKM, >5.3; iN FPKM, ≥5.23].
Following optimization of cell density, virus concentration and viral infection timeline, a standard operating procedure for gene knockdown was determined for each cell type (Fig. 4A). Human primary astrocytes (ScienCell) were plated 24 h prior to infection with a 1:5 dilution of lentivirus (titer ∼7 × 107 ± 3 × 107 SD viral particles/ml). EB-derived neurons and iNs were cultured as previously described (3,18,25,26) and infected with a 1:1 dilution of lentivirus at DIV 50 and 17, respectively. For each viral transduction, ∼1.75 million particles were applied to 25 000 cells for an 18 h period prior to washout. Attempts to reduce this concentration, such as by polybrene treatment, proved ineffective and/or deleterious in our cell model systems. For each target gene, we tested an average of five shRNAs targeting different regions (Supplementary Material, Table S2);constructs were selected from the Broad Institute Genetic Perturbation Platform. As controls in each experiment, subsets of cells were left untransduced or transduced with viruses expressing an empty vector (empty). Astrocytes and EB-derived neurons also were transduced with a green fluorescent protein expressing lentivirus (‘rosetta’) to permit visualization of cells as a means to confirm effective transduction (iNs already expressed green fluorescent protein, GFP). Media were changed ∼18 h after viral transduction, and, following ∼114 h incubation period, conditioned media were collected and cells lysed for RNA purification. RT-qPCR (reverse transcription-quantitative polymerase chain reaction) then was performed on each sample to determine the extent of target gene knockdown by each tested shRNA construct (reported as percent change from the average of controls; see example in Fig. 4B). No significant change in lactate dehydrogenase activity between control and shRNA treated as iNs and astrocytes was detected, indicating that lentiviral infection did not affect overall cell health (Supplementary Material, Fig. S1). These results were confirmed by a parallel calcein assay that revealed no qualitative difference between infected and uninfected cells, with uptake of the live-cell marker calcein observed in >90% of iNs and astrocytes (data not shown).
Figure 5.
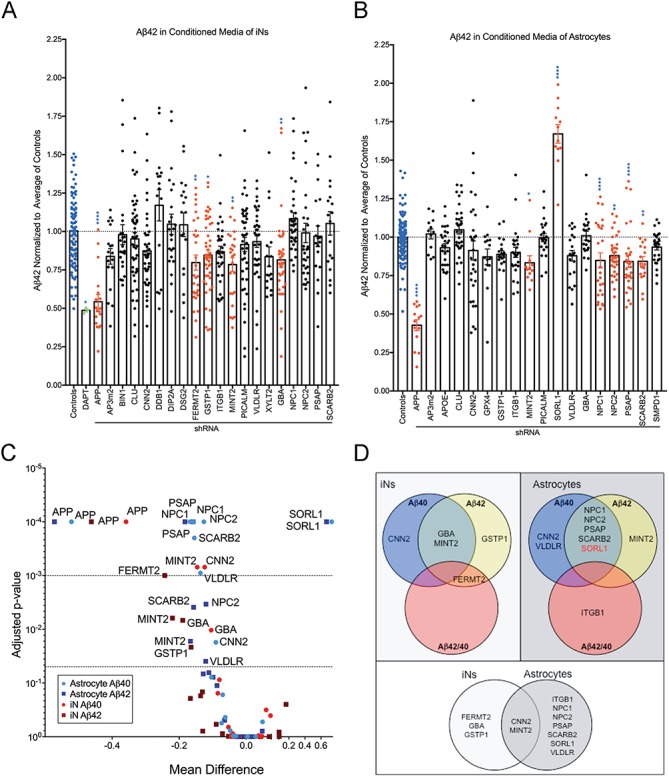
Knockdown of selected gene targets significantly reduces extracellular Aβ42 concentration targeted astrocytes and iNs. Conditioned media from shRNA targeted iNs (A) or astrocytes (B) were analyzed by Aβ triplex ELISA. Results were normalized to the average of control conditions for each experiment before multiple experiments were combined. Each dot represents data from an independent well. Blue bars highlight controls; green bars highlight treatment with a gamma-secretase inhibitor (DAPT); red bars highlight conditions reaching statistical significance. Shown is the mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001 by ordinary one-way ANOVA. (C) The adjusted P-value plotted as a function of the mean difference for Aβ40 and Aβ42 in astrocytes and iNs. (D) Summary of Aβ phenotypes observed in astrocytes and iNs. Only SORL1 knockdown (red) showed an increase, all others a decrease.
Figure 6.
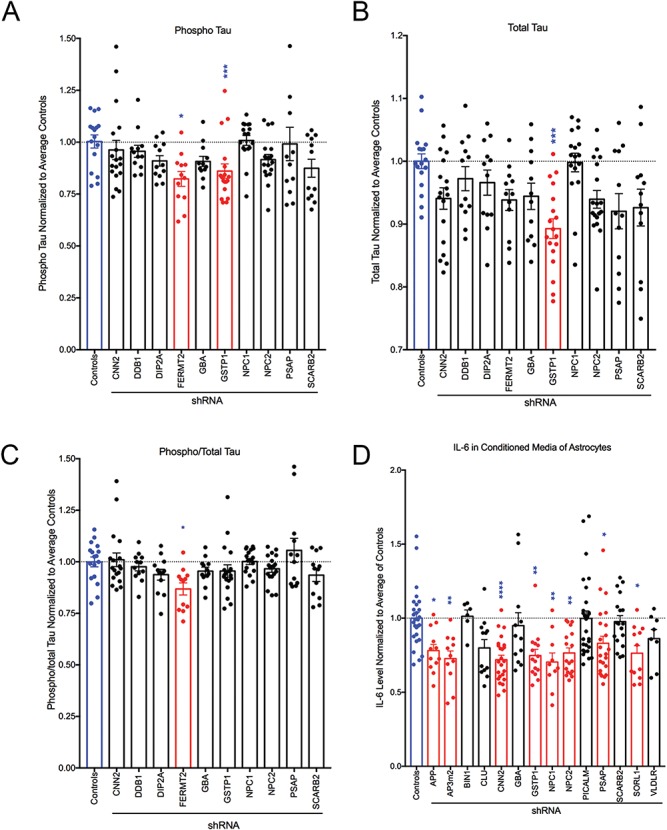
Knockdown of selected gene targets alters phosphorylated tau and IL-6 levels. (A–C) Lysates collected from iNs targeted by shRNA were analyzed by phospho Thr231/total tau multiplex ELISA (MSD). Results were normalized to the average of controls for each experiment and combined across experiments. (D). IL-6 levels were measured in conditioned media from astrocyte cultures via ELISA (MSD). Results were normalized to average of controls for each experiment and combined across experiments. For all graphs, blue bars indicate controls and red bars indicate conditions in which significant changes were detected. Each dot represents data from an independent well. Shown is the mean ± SEM. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001 by Kruskal–Wallis test with Dunn's multiple comparisons test.
Screening of shRNA target sequences
Our screen consisted of two sequential stages. In Stage 1, we identified those genes that could be appropriately investigated: we required at least 2 different shRNAs targeting a given gene with knockdown efficiency of >50% for that gene to be considered in Stage 2 where we measure our primary outcome measure, extracellular Aβ levels. Transduction efficiency was determined by calculating the number of cells expressing GFP following transduction with GFP-encoding lentivirus (rosetta) relative to the total number of cells positive for DAPI staining. EB-derived neurons had a transfection efficiency of 87 ± 6% (mean ± SD), while astrocytes have an efficiency of 85 ± 9% (mean ± SD). For each experiment, three wells of a given cell type were infected with the same aliquot of an shRNA-bearing virus, and the qPCR expression results were averaged across the three wells. Multiple shRNAs were used to knock down each gene, each targeting a different sequence within the gene (Supplementary Material, Table S2), and knockdown efficiency was typically variable between constructs targeting the same gene. For effective shRNAs, multiple independent experiments were performed for the same viral production lot and across multiple lots, and the greatest possible knockdown rate for that construct was recorded (Supplementary Material, Table S1). On average, variability between constructs (as measured by SD) was 2-fold lower in astrocytes than in iPSC-derived neurons. This was true across wells within an experiment for a given condition, across lots of virus and across independent experiments (Supplementary Material, Table S1).
Of the 372 shRNAs tested in astrocytes, 20% were capable of knocking down their gene target by at least 70%, while 41% were able to reduce gene expression by at least 50% (Fig. 4C). In EB-derived neurons, 15% of the shRNAs were able to reduce gene expression by 70%, and 35% of shRNAs reduced expression by 50%. Candidate gene-targeted shRNAs in iNs showed that 25% were capable of 70% knockdown and 54% were capable of 50% knockdown. Of note, lentiviral-mediated gene knockdown was more efficient in iNs than in EB-derived neurons, perhaps owing to the more homogenous nature of the iN cultures. Results for perturbation of selected candidate genes are summarized in Figure 3C, and full results are available in Supplementary Material, Table S2.
Identification of shRNAs that alter AD phenotypes
To begin Stage 2, we measured Aβ in culture supernatants using an Aβ triplex ELISA (MesoScale Discoveries). EB-derived neurons were excluded from this stage of the study due to reduced knockdown efficiency and higher levels of cell-type heterogeneity between differentiation rounds and to facilitate rapid screening. Results were normalized to the average level of secreted Aβ in control non-infected and empty vector conditions in each experiment. The results from all shRNAs targeting the same gene were pooled to give a summary, gene-level view of the consequence of gene perturbation. As a positive control, we included a condition where we inhibited γ-secretase, the protease that cleaves the Aβ peptide from APP, using DAPT. As expected, DAPT treatment led to a robust decrease in all amyloid peptides identified in conditioned media (Aβ38, Aβ40 and Aβ42) (Fig. 5A; Supplementary Material, Fig. S2). APP knockdown also led to a predictable, robust reduction in extracellular Aβ (Fig. 5A; Supplementary Material, Fig. 2), validating our approach using lentiviral delivery of shRNA constructs.
Figure 7.
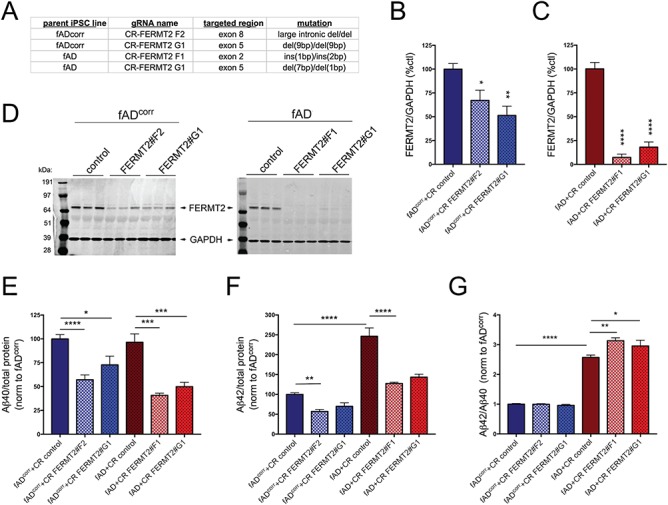
CRISPR/Cas9 targeting of FERMT2 in fAD and fADcorr iPSC lines confirms that lowering FERMT2 levels in human neurons results in lower extracellular Aβ levels. The FERMT2 locus was targeted in iPSC lines using gRNAs recognizing the exons listed, and the resultant indel mutations introduced are shown (A). Following targeting, iPSC lines were differentiated to neuronal fates and protein lysates collected at day 21 of differentiation. Example western blots and quantifications are shown for fADcorr iNs (B, D) and fAD iNs (C, D). ‘CR control’ refers to iPSC subclones that underwent a mock targeting, whereby Cas9 and empty gRNA vector were transfected, and monoclonal subclones isolated and analyzed in parallel to subclones whereby FERMT2 was targeted. At day 21 of differentiation, 48 h conditioned media were collected and cells lysed. Aβ40 and 42 levels were measured via multiplexed ELISA (MSD), normalized to total protein levels in the cell lysate and then normalized to fADcorr CR control for each differentiation (E, F). Data in (G) shows Aβ42:40 ratios for each condition. Quantifications for B and C are from 2–3 independent differentiations, for fAD n = 9, 6, 9; fADcorrn = 6, 5, 6. One-way ANOVA with Holm–Sidak multiple comparisons tests performed; *P < 0.05; **P < 0.01; ****P < 0.0001. For E–G, in order from left to right, n = 9, 9, 9, 6, 6, 6. One-way ANOVA with Holm–Sidak multiple comparisons tests performed; *P < 0.05, **P < 0.01, ***P < 0.0005, ****P < 0.0001.
Data sets for Aβ38, Aβ40 and Aβ42 measured for iNs and astrocytes are summarized in Supplementary Material, Figs S2 and S3. As Aβ42 possesses a propensity for aggregation and is hypothesized to be the most pathogenic and readily detected of the Aβ species, these results are highlighted in Figure 5. Of the 19 candidate genes screened in iNs, only 4 (FERMT2, GSTP1, MINT2 and GBA) significantly reduced Aβ42 in the conditioned media of cultured cells (Fig. 5A and C). In astrocytes, 6 (MINT2, NPC1, NPC2, PSAP, SCARB2 and SORL1) of 17 genes screened significantly altered Aβ42 levels (Fig. 5B and C). Interestingly, only SORL1 knockdown increased Aβ42, consistent with previous findings that APP processing is increased in hippocampal extracts from Sorl1 knockout mice carrying a human APP transgene (29).
In order to determine the specificity of each gene for altering extracellular Aβ42 levels, we compared the list of Aβ42 altering genes to those capable of affecting Aβ40 (Fig. 5C, summarized in Fig. 5D). Knockdown of GSTP1 and FERMT2 alter Aβ42 but not Aβ40 in iNs, while only MINT2 exclusively affects Aβ42 generation in astrocytes. An additional gene, CNN2, significantly reduces Aβ40 without affecting Aβ42 levels in both iNs and astrocytes. CNN2 and MINT2 are the only genes whose knockdown reduces an Aβ species in both iNs and astrocytes, providing a window into the potentially unique mechanisms regulating extracellular Aβ levels in neuron versus astrocyte cultures. The finding that MINT2 knockdown reduces extracellular Aβ levels is consistent with a reduction in amyloid plaques deposition in Mint2 knockout mice (30), while this role for CNN2 has not previously been demonstrated.
Familial AD mutations favor production of Aβ42 over Aβ40, suggesting that an imbalance in the ratio of these two species is important for AD pathogenesis (31,32). We therefore determined whether altered expression of any of our candidates leads to alterations in the proportion of Aβ42 relative to Aβ40. Knockdown of FERMT2 in iNs resulted in a decrease in the ratio of Aβ42 to 40, suggesting a propensity toward Aβ40 release driven by a decrease in Aβ42 levels. The ratio of Aβ42/40 also is reduced by the knockdown of ITGB1 in astrocytes, suggesting that knockdown of this gene may benefit pathology either by reducing the production or increasing the degradation of pathogenic Aβ42 in this cell type.
We further evaluated the effect of gene knockdown on AD-relevant phenotypes by examining the protein encoding another pathological hallmark of AD, TAU. The hyperphosphorylation of this microtubule protein facilitates intracellular tangle formation. We measured both total and phospho-tau levels in the lysates of cultured iNs by ELISA (MesoScale Discoveries). These experiments were restricted to iNs as astrocytes do not express significant levels of MAPT, the gene encoding TAU protein. Of note, the only phospho-epitope measured by this assay is Thr231. TAU phosphorylation at this site by GSK3β is critical in regulating TAU binding (33) and is thought to play a role in tangle formation. Of 10 genes screened, only GSTP1 knockdown significantly reduced both total and phospho-tau (Fig. 6A–B). Knockdown of FERMT2 reduced only phospho-tau, leading to an overall reduction in the ratio of phospho-tau to total tau (Fig. 6C). Integrating these results with previously identified linkages of these genes to AD highlights FERMT2 and GSTP1 as potential mediators of tau tangle formation.
Figure 8.
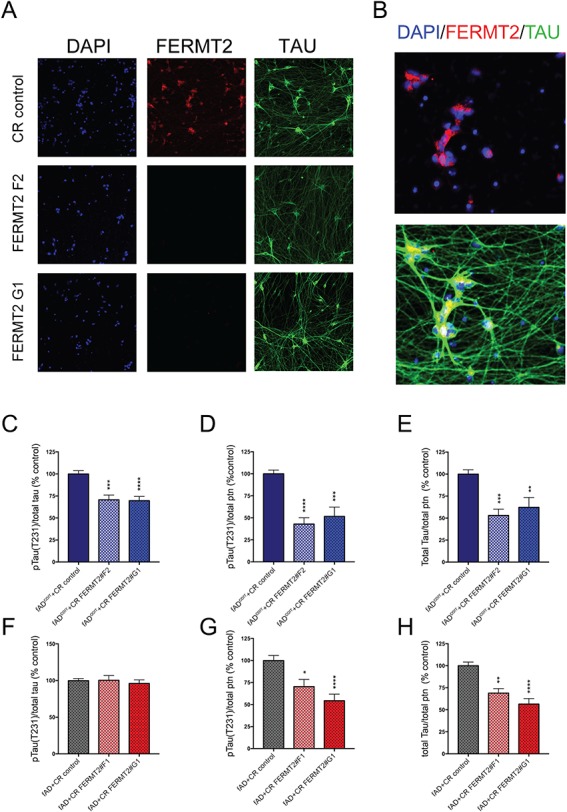
FERMT2 targeting results in lower phospho- to total-TAU levels in fADcorr but not fAD iNs. FERMT2 targeted iPSC lines were differentiated to iN fates. At day 21 of differentiation, cells were either fixed and immunostained (A–B) or else lysed and TAU analyzed by ELISA (C–H). (A, B) Example immunocytochemistry for FERMT2 and TAU in fADcorr iNs. (A) FERMT2 targeting lowers FERMT2 immunostaining. (B) Magnified view of immunostaining of CR control to show subcellular localization of FERMT2 in untargeted human neurons. (C–H) Data from a multiplexed ELISA (MSD) showing pTAU(Thr231) and total TAU are shown for fADcorr (C–E) and fAD (F–H) iNs. Quantification is from 3 independent differentiations, for (C–E) n = 15, 15, 15 and for (F–H) n = 15,8,15. One-way ANOVA with Dunnett's multiple comparisons tests performed; *P < 0.05, **P < 0.01, ***P < 0.0005, ****P < 0.0001.
The Aβ plaques and tau tangles that are the hallmarks of AD are accompanied by chronic neuroinflammation. Activated astrocytes and microglia are often situated near plaques and produce proinflammatory signaling molecules including chemokines, growth factors, complement molecules and cytokines (reviewed in (34)). We therefore measured the concentration of one such cytokine, IL-6, as a consequence of candidate gene knockdown in the conditioned media of cultured astrocytes. Eight genes (APP, AP3M2, CNN2, GSTP1, NPC1, NPC2, PSAP and SORL1) all produced significant reductions in IL-6 (Fig. 6D). Viewed together, these results show phenotypic and cell-type-specific consequence to targeted gene knockdown (Table 1).
Table 1.
Summary of phenotypic results: the level of significance (P-value) in the perturbation of a given phenotype by targeting a given gene is listed in the table
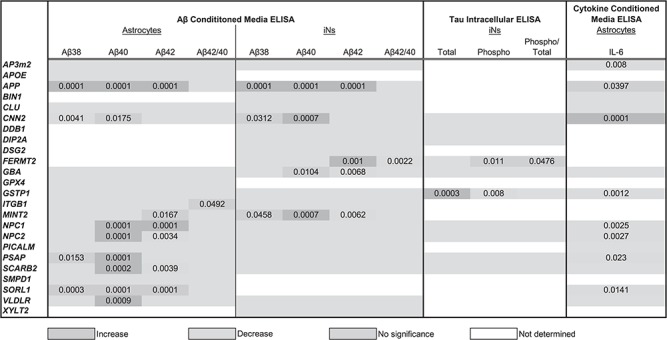
|
Validation of a role for FERMT2 in LOAD-relevant phenotypes
FERMT2 arose as the only gene targeted that affected both the proportion of tau phosphorylated and Aβ levels in iN cultures. Interestingly, FERMT2 also was identified in a screen for modulators of TAU toxicity in Drosophila (20) and in a screen of mediators of Aβ generation in HEK cells (22). Here, we observe that FERMT2 knockdown with shRNA reduces both extracellular Aβ levels and the proportion of TAU that is phosphorylated. In order to validate these findings, we next used CRISPR-Cas9 to target FERMT2 in two additional iPSC lines: one derived from a patient with early-onset AD carrying the APPV717I mutation (‘fAD’) (18) and, the other, its isogenic mutation-corrected control line (‘fAD’) (19). Targeting of each line resulted in different indels within FERMT2 (Fig. 7A) and in varying levels of reduction of FERMT2 protein when these lines were differentiated to iNs (Fig. 7B–D). Both targeting events in the fAD line showed a strong, >50% reduction in FERMT2 protein levels (Figs 7C and D), while targeting of fADcorr lines reduced FERMT2 levels significantly, but to a lesser degree (Figs B and D). Consistent with these results, FERMT2 reduction in fAD iNs resulted in a strong and consistent reduction in extracellular Aβ levels (Fig. 7E and F), while Aβ levels were reduced more modestly with targeting of fADcorr (Fig. 7E and F). As previously reported (19), fAD iNs showed an elevated ratio of Aβ42 to Aβ40 (Fig. 7G). Interestingly, reduction of FERMT2 in fAD iNs showed a further increase in this ratio, while the ratio was unaffected with FERMT2 reduction in fADcorr iNs (Fig. 7G).
Figure 9.
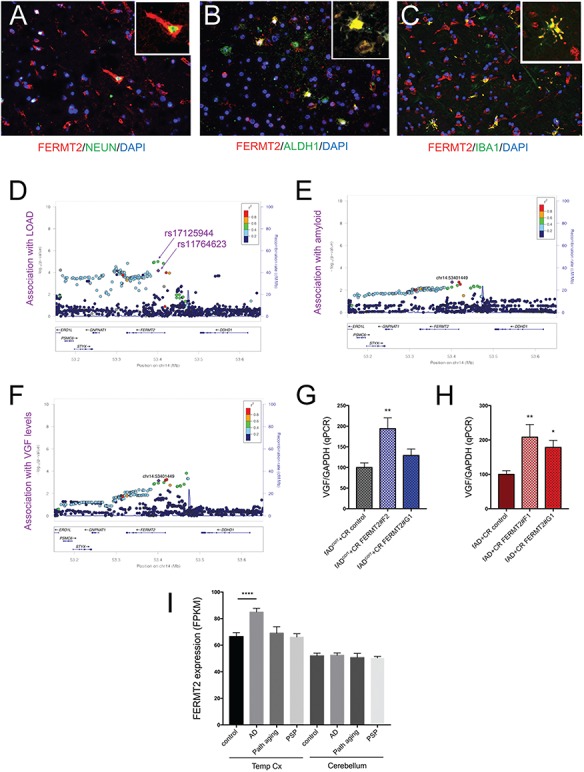
FERMT2 expression and association with LOAD (A–C) Postmortem human cerebral cortex from a non-AD subject was embedded, paraffin sectioned and immunostained for FERMT2, NEUN, ALDH1 and IBA1, with DAPI counterstaining nuclei. (D–F) Regional genetic association plots for FERMT2 and LOAD (D), amyloid burden (E) and VGF protein levels (F) in the postmortem brain. In these regional plots, association results are presented for all SNPs (dots) within the FERMT2 region of the genome. The lead SNP (rs117646236; chr14:53401449) in this region is shown in purple, and other SNPs are colored based upon the extent of linkage disequilibrium with the lead SNP, following the color key in the upper right panel. The x-axis denotes the physical position of the SNP, and the y-axis reports −log10(P-value) for each SNP. The blue line denotes the recombination rate in this region in European participants from the 1000 Genomes Project. The location of the gene is present at the bottom of each panel. (G, H) VGF expression was measured via qRT-PCR in human iNs with and without FERMT2 targeting. Quantification is from 3 independent differentiations: (G), n = 8, 9, 8; (H), n = 9, 6, 9. One-way ANOVA with Dunnett's multiple comparisons tests performed; *P < 0.05, **P < 0.01, ***P < 0.0005, ****P < 0.0001. (I) Publicly available RNA-seq analyses (Mayo Clinic Brain Bank) from post-mortem human temporal cortex (Temp Cx) and cerebellum (Cb) is shown for FERMT2. n = 275 and 276 for Temp Cx and Cb, respectively. One-way ANOVA with Dunnett's multiple comparisons tests performed; ****P < 0.0001.
All lines with FERMT2 targeting showed a reduction in total tau and phospho-tau levels when normalized to total protein (Fig. 8). However, while FERMT2 targeting in fADcorr showed a reduction in the proportion of TAU that is phosphorylated (Fig. 8C), fAD iNs with FERMT2 reduction did not show such a reduction in phospho-tau relative to total tau (Fig. 8F). Thus, fAD iNs differ from fADcorr in two phenotypes: fAD iNs display an elevation in the extracellular Aβ 42:40 ratio with FERMT2 reduction, and no rescue of the phospho-tau to total tau ratio is observed with FERMT2 reduction.
FERMT2 in the human brain
Our data suggest that FERMT2 expression in human neurons has effects on both Aβ and TAU. We first confirmed that FERMT2 protein is indeed expressed in the human brain in neurons by coimmunostaining with a neuronal marker, NeuN (Fig. 9A). It is important to note, however, that FERMT2 protein also colocalizes in cells that express astrocyte (ALDH1) and microglial (IBA1) markers in the human brain (Fig. 9B and C). SNPs in the FERMT2 locus were previously identified to be associated with LOAD (5), and we had previously shown an association of FERMT2 SNPs with a pathological diagnosis of AD (21). Here, using published postmortem pathological data derived from the Religious Order Study (ROS) and Memory and Aging Project (MAP) ROS and MAP cohorts (35), we find that an SNP in the intron between exons 2 and 3 of FERMT2 (rs117646236) is associated with amyloid levels in the postmortem brain (P = 0.001). Further, we interrogated available proteomic data from postmortem dorsolateral prefrontal cortex (DLPFC) of ROS and MAP participants (36) to examine if the level of any of the 67 proteins measured are associated with rs117646236. Levels of one protein previously implicated in AD in several network analyses (http://agora.ampadportal.org/), VGF, were significantly associated with the presence of rs117646236 (P = 0.000696) (Fig. 9F). VGF also has been shown by several groups to be a potential biomarker for AD (reviewed in 37). In order to determine whether modulating FERMT2 levels might affect VGF expression, we performed qPCR on our human iN experimental system, and we found that VGF expression levels were significantly elevated in iNs with FERMT2 reduction (Fig. 9G and H). Lastly, interrogation of publicly available RNAseq data of temporal cortex and cerebellum of humans (38) showed that FERMT2 RNA levels are elevated in LOAD in the temporal cortex but not the cerebellum, suggesting a possible regionalization of effect. Further, the absence of this differential expression in temporal cortex of individuals with either ‘pathological aging’ or progressive supranuclear palsy (Fig. 9I) suggests that FERMT2 may have a role that is specific to the AD context.
Discussion
Through evaluation of GWAS, epigenetic and functional studies, we prioritized 72 genes with potential roles in AD pathogenesis. The identification of AD-contributing genes necessitates the evaluation and validation of these targets and, importantly, models in which to perform additional studies. The use of human iPSC-derived neurons and astrocytes facilitates evaluation in a species-relevant, cell type-specific manner to probe the mechanisms through which these factors act to impact disease susceptibility. Functional and expression studies of astrocytes and neurons allow comparison of the relative contributions of gene function across cell types.
This screening was accomplished through knockdown of candidate genes by shRNAs packaged into lentiviral vectors. Efficient transduction of iPSC-derived neurons required multiple optimization steps. The day of neuron infection (EB-derived neurons at DIV 50 and iNs at DIV 17) was selected based on the ability of neurons to demonstrate spontaneous synaptic activity and to present morphological and expression markers consistent with differentiated neurons while minimizing the amount of time required for maintenance of cell culture to maximize scalability. When neurons are maintained longer and transduced later, this may contribute to phenotypic drift and less consistency between wells and differentiation rounds. For each viral transduction, ∼1.75 million viral particles were applied to 25 000 cells for an 18 h period prior to washout. Attempts to reduce this concentration, such as by polybrene treatment, proved ineffective and/or deleterious in our cell model systems.
It is important to note the limitations of this experimental system. First, knockdown was the sole method of genetic perturbation in this study despite the directionality of risk alleles potentially differing amongst candidate genes. For many SNPs, it is unknown whether an increase or decrease in expression, splicing or some other mechanism is responsible for risk. Knockdown allowed us to compare phenotypic responses across cell types. However, overexpression or targeting of particular splice variants also would be of value, and the system described herein would be amenable to these types of studies. Secondly, the differentiated cells that are generated using currently available protocols have expression profiles that are more similar to fetal brain cells than to adult brain cells, and the in vitro connectivity of different brain regions is not recapitulated in our cultures. In spite of these drawbacks, our results suggest that these experimental systems can be useful for examining molecular signaling pathways that occur at the level of the cell, and that different cell biological outcomes in response to gene perturbation can be observed when different cell types are examined in parallel.
Many of the candidate genes that we examined have proposed associations with amyloid levels, either through gene-trait associations or through studies in other model systems. Aβ can be found in two pools: intracellular and extracellular. The majority of Aβ is secreted: in our iPSC-derived neuronal system and in primary astrocytes, greater than 10-fold higher levels of Aβ are detected in the conditioned media relative to the cellular lysate (18). Aβ deposits are found extracellularly in the AD brain, and Aβ secretion into the extracellular space is thought to be a major contributing factor to the toxicity at the synapse observed in AD (39,40). We therefore focused our measurements on secreted, extracellular Aβ present in the conditioned media of cultured cells. In a limited number of experiments, intracellular Aβ was measured. However, intracellular Aβ42 levels were near or below the limit of detection of our assay for many samples. Therefore, these data could not be reliably interpreted across gene perturbations.
Through our screening, we identify 12 genes that, when targeted, affect extracellular Aβ levels in either cell type tested (Table 1). A total of 3 of the 12 genes (MINT2, VLDLR and SORL1) have previously been linked to APP trafficking. In this study, both MINT2 and VLDLR reduce Aβ secretion in astrocytes and iNs, consistent with their known role in APP trafficking. It was previously shown that conditional knockdown of the MINT proteins leads to a reduction in APP internalization in mouse primary hippocampal cultures, and knockdown of Mint2 in transgenic mice reduces Aβ plaque deposition (30,41). Expression of Vldlr significantly increases cell surface levels of APP and α-secretase cleavage products in primary murine hippocampal neurons (42). SORL1 knockdown increased extracellular Aβ levels in astrocytes in this study, consistent with the reduction in Sorl1 leading to an increase in Aβ in cortical extracts from Sorl1−/− mice (43). SORL1 is downregulated in sporadic AD (44), again demonstrating the link between SORL1 gene expression and disease. These results validate our models by reinforcing published results and underscore the importance of APP trafficking in Aβ pathology.
A striking number of the genes that altered extracellular Aβ levels in astrocyte cultures function in lysosomal degradation (PSAP, SCARB2, NPC1 and NPC2), and in iNs, GBA knockdown altered extracellular Aβ levels (Table 1). GBA encodes glucocerebrosidase, the lysosomal enzyme responsible for cleavage of glucosylceramide (45). SCARB2 encodes the protein responsible for shuttling GBA to the lysosome (46) and PSAP encodes prosaposin, a precursor protein that, when cleaved, yields saposin C, an activator of glucocerebrosidase (47). Mutations in GBA cause Gaucher's disease, which is marked by a sustained inflammatory reaction, plasma protein abnormalities, fatigue, increased metabolic rate, enlargement of the spleen and Parkinsonism (reviewed in (48)). Mutations in GBA also are now understood to be a common genetic factor in Parkinson's disease (PD), found in 3–20% of patients with PD (49). Multiple recent studies have shown that mutations in GBA can lead to DLB, which is hallmarked by both Lewy bodies and the characteristic plaques and tangles of AD (15–17). Further, PSEN1, a critical component of γ-secretase, is required for lysosomal proteolysis (50). Additionally, TRPML1, a lysosomal ion channel, is defective in AD pathogenesis (51) and with the AD-like cognitive deficits exhibited in HIV (52). Finally, glucocerebrosidase protein and enzyme activity is decreased in sporadic AD (53), demonstrating a potential link between lysosomal dysfunction and AD.
Targeted knockdown of NPC1 and NPC2 in astrocytes lowered both extracellular Aβ levels and IL-6 levels (Table 1). NPC is a lysosomal storage disease caused by mutations in NPC1 or NPC2 and is marked by cholesterol accumulation in the lysosome resulting in deficits in free cholesterol esterification. As in AD, NPC presents with progressive neurodegeneration, involves cholesterol dysfunction, hyperphosphorylation of tau, and Aβ accumulation (reviewed in 54). NPC1 and NPC2 encode proteins involved in the transport of cholesterol and are impaired in NPC. In mouse Npc1 knockout models overexpressing human APP, peptide clearance is reduced leading to accumulation of Aβ and APP C-terminal fragments in endosomes and in the lysosomal/autophagic pathway (55). This would suggest that the reduction in extracellular Aβ levels in astrocyte cultures shown here could result from elevated intracellular accumulation of Aβ, which results in lowered extracellular levels of Aβ.
The remaining hits span a variety of functions but are less well studied in relationship to AD. GSTP1 encodes glutathione S-transferase pi, a protein that catalyzes detoxification. Increased oxidative stress is a known AD-related phenotype and is caused by a decrease in brain glutathione (56). GSTP1 was one of the two genes identified to be downregulated in the brains of MAPT transgenic mice (57), connecting GSTP1 and AD pathology. In addition, previous studies in cancer cell models have shown that IL-6 can affect GSTP1 mRNA and protein levels in a concentration dependent manner (58,59). Our results suggest that an inverse relationship also may exist with GSTP1 affecting IL-6 levels. Another hit, CNN2, encodes calponin, an actin filament-associated regulatory protein predominately found and functioning in smooth muscle (60). Macrophages from CNN2/APOE knockout mice have lower levels of pro-inflammatory cytokines than wild-type, attenuating development of atherosclerosis, a cardiovascular disease resulting in the build-up of plaques in arteries (61). Given its role in actin filament stabilization, it is possible that CNN2 affects other tau phenotypes, but we observed no significant effect of CNN2 knockdown on the tau phospho-epitope that we examined here.
ITGB1 encodes β1 integrin, a protein that physically interacts with APP and is involved in APP-dependent neurite outgrowth (62). Expression of β1 integrin increased β-secretase cleavage products of APP in HEK 293 cells (63), consistent with our observed decrease of Aβ levels with ITGB1 knockdown.
Targeted reduction of FERMT2 in human neurons either by viral-mediated shRNA or by genome editing both resulted in a reduction in extracellular Aβ40 and Aβ42 levels and a reduction in total and phospho-tau. Meta-analysis of GWAS first linked FERMT2 to LOAD (5). In a Drosophila screen of AD susceptibility genes, Fit1 and Fit2, the FERMT2 homologs, modified tau toxicity (20), demonstrating a link between FERMT2 and AD pathology. In addition, studies from our group showed that a SNP at the FERMT2 locus was significantly associated with the pathological hallmarks of AD (plaques and tangles) (21). More recently, it was shown that SNPs in the FERMT2 locus are associated with brain amyloidosis (as measured by florbetapir uptake) in a stage-dependent manner, with the strongest association at the mild cognitive impairment stage (24). Further, in a genome-wide RNAi screen for modulators of Aβ levels in HEK cells, FERMT2 was identified as a strong regulator of APP metabolism (22). In contrast to the results presented here, however, in that study RNAi-mediated reduction of FERMT2 levels resulted in an elevation in Aβ levels through modulation of cell surface levels of APP. The observed differences may be due to the differences in the experimental systems utilized (HEK cells versus human neurons) and/or differences in the methods and genomic location of FERMT2 targeting.
FERMT2 is a member of the kindlin family, which contain a FERM domain (named for its presence in Band4.1 protein, ezrin, radixin and merlin) and a pleckstrin homology domain. The most well-studied role of FERMT2 is its central role in regulating integrin activation (reviewed in 64), although additional functions have been identified in linking cortical actin structures, plasma membrane tension and focal adhesion function (65). FERMT2 binds to integrins, actin and phospholipids, acting as a molecular link between the extracellular environment, plasma membrane and cytoskeletal network. The work described here suggests that reduction of FERMT2 protein levels in a controlled human neuronal model results in effects on both Aβ and phospho-tau levels, directly linking FERMT2 to the two pathological hallmarks of AD in human neurons. Additional studies will be necessary to determine whether these effects are mediated through the known functions of FERMT2 or through an as of yet undescribed mechanisms of action.
Herein, we demonstrated the ability to screen gene targets for AD-related phenotypes in vitro in iPSC-derived neurons and human astrocytes on a moderate scale. We have generated publicly available reference RNAseq data for each cell type in this study to assess the level of expression of desired genes in these experimental systems (available for reference at https://www.synapse.org/#!Synapse:syn2580853/wiki/409853). The shRNA constructs we identified as effectively targeting candidate genes are available as bacterial glycerol stocks, plasmid DNA or lentiviral particles by Sigma-Aldrich (St. Louis, MO) or as bacterial glycerol stocks by Open Biosystems (Lafayette, CO). A total of 9 of the 12 genes that modulate Aβ levels do so in astrocytes, and this system provides a cellular context for addressing the mechanisms by which astrocytes affect extracellular Aβ levels. Future studies involving the coculture of iPSC-derived neurons and astrocytes would allow evaluation of the interplay between these cell types, and such a system should improve our reductionist models of the complex cellular systems of the brain. The ultimate hope is that genetic manipulation of these cell types both separately and in concert could identify pathways that, when targeted with small molecules, reduce pathological phenotypes. As more gene targets are identified from large data sets, the need for a system to readily screen these targets will become more critical. This study represents an important step in the development of high-throughput, human cell-type based screening, demonstrating not only the ability to identify genes that act in pathological pathways but also the importance of screening across multiple cell types.
Materials and Methods
Cell culture
The human iPSC line YZ1 was obtained from the University of Connecticut stem cell core facility and were maintained in media containing 400 ml DMEM/F12, 100 ml knockout serum replacement, 5 ml penicillin/streptomycin/glutamine, 5 ml MEM-NEAA and 500μl 2-mercaptoethanol (all from Invitrogen (Waltham, MA)) with fresh addition of 10 μg/ml bFGF (MilliporeSigma). Differentiations were performed following one of the two protocols. EB-derived neuron differentiation was carried out as described (27,28). EB-derived neurons were plated at DIV 24 on Matrigel coated 96-well plates and maintained in media containing 480 ml Neurobasal media (Gibco), 5 ml NEAA, 5 ml N2 supplement and 10 ml B27 supplement with fresh IGF, cAMP, BDNF and GDNF. The iN differentiation was accomplished as previously reported with minor modifications (3). iNs were plated at DIV 4 on Matrigel coated 96-well plates at a density of 25 000 cells/well and maintained in media consisting of Neurobasal medium with dextrose, Glutamax, MEM-NEAA, B27, 2 μg of doxycycline, 5 μg/ml of puromycin, BDNF (10 ng/ml), CNTF (10 ng/ml) and GDNF (10 ng/ml).
Human fetal astrocytes from the cerebral cortex were purchased from ScienCell and cultured in media containing 2% FBS, 1% penicillin/streptomycin and 1% astrocyte growth supplement (ScienCell). Astrocytes were trypsinized and plated in 96-well plates (40 000 cells/well). Astrocytes were used at less than 10 passages.
Lentiviral transduction
Astrocytes were plated and allowed to recover for 12–24 h. Cells were transduced by addition of a 1:4 ratio of lentivirus-packaged shRNA (titer range 1.4 × 107–3.43 × 108 virus particles/ml) to conditioned media. For each experiment, a subset of cells was infected with a lentivirus expressing the pLKO vector without an shRNA (‘empty’). For astrocyte experiments, a subset of cells was infected with a GFP expressing construct (‘rosetta’). EB-derived neurons and iNs were transduced at DIV 50 and DIV 17, respectively, with 1:1 ratio of media to lentivirus. Following ~18 h incubation, virus-containing media was removed and replaced with fresh media to incubate for an additional 96 h. Conditioned media were collected and stored at −20°C, and cells were lysed for RNA purification.
qPCR
Samples were prepared for qPCR using the Power SYBR Green Cells-to-Ct kit (Ambion (Waltham, MA)) according to manufacturer's instructions. Fast SYBR Green Master Mix (Applied Biosystems (Waltham, MA)) was used to assess three technical repeats with a ViiA 7 System (Applied Biosystems). ΔΔCT was calculated as a measure for relative expression (66) and results normalized to GAPDH.
RNAseq
RNA was purified using a PureLink RNA mini-kit (Life Technologies, Waltham, MA). RNA-Seq Libraries were constructed by the Broad Institute's Genomics Platform using their Large Insert Illumina Strand Specific TruSeq protocol, which includes insert sizes on average between 450 and 550 bp. Target coverage was 75 M reads in pairs. RNA-Seq data were processed by our parallelized and automatic pipeline. This pipeline includes trimming the beginning and ending bases from each read, identifying and trimming adapter sequences from reads, detecting and removing rRNA reads and aligning reads to a reference genome. We used the non-gapped aligner Bowtie to align reads to transcriptome reference (hg19 build with gencode v14 annotation) and then applied RSEM to estimate expression levels for all transcripts. The FPKM values were the outcome of our data RNA-Seq pipeline.
Aβ ELISA
Concentration of Aβ present in conditioned media was detected by 6E10 Aβ peptide panel multiplex ELISA (MesoScale Discovery, Rockville, MD) following manufacturer's instructions. Conditioned media from transduced cells were incubated in preblocked wells along with detection antibody solution. Plates were read using MSD SECTOR Imager 2400 and resulting peptide concentrations normalized to the average of control conditions for each experiment.
Tau ELISA
Protein was extracted from iNs by lysing in NP-40 Lysis buffer (1% NP40, 0.5 M EDTA, 5 M NaCl and 1 M Tris) containing complete protease inhibitors and phosSTOP (Roche, Indianapolis, IN). Lysates were subjected to Multi-spot Phospho (Thr 231)/Total Tau ELISA (MesoScale Discovery) following manufacturer's instructions. Lysates were incubated in preblocked wells for 1 h prior to detection antibody solution application for 1 hr. Plates were read using MSD SECTOR Imager 2400 and resulting concentrations normalized to the average of control concentrations for each experiment.
IL-6 ELISA
IL-6 concentration was measured using V-PLEX Human IL-6 Kit (MesoScale Discovery). Conditioned media from transfected astrocytes were incubated for 2 h prior to detection antibody solution application. Plates were read using MSD SECTOR Imager 2400 and resulting concentrations normalized to the average of control concentrations.
iPSC CRISPR/Cas9 CRISPR editing and analysis
SgRNAseqs were designed using the CRISPR design tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) or else using the design tool of the Zhang laboratory (67). The sgRNAseqs were cloned into plasmid pXPR-003 (Addgene #52963).
fAD and fADcorr iPSCs (1 × 105) were plated on growth factor reduced Matrigel (Corning #354230) with StemFlex medium (Invitrogen). The next day, SpCas9 plasmid (pXPR_BRD111-Cas9v2; Addgene #78166) with FERMT2 sgRNA plasmids or empty vector was cotransfected into iPSCs with Lipofectamine 2000. Two days post transfection, the transfected iPSCs were selected with puromycin (5 μg/ml) and blasticidin (4 μg/ml). Genomic DNA were extracted from a portion of cells for a mismatch assay (GeneArt Genomic Cleavage Detection Kit, Invitrogen) to evaluate the editing efficiency. Limited dilution cloning was used to monoclonally isolate edited iPSCs. Monoclonal lines were examined by PCR-sequencing around the edited region.
FERMT2 CRISPR-edited iPSC cells were differentiated using NGN2 to iN fate (described above). INs were plated at day 4 on poly-ornithine/laminin coated 24-well plates at a density of 1.5 × 105 cells per well and maintained in Neurobasal medium with dextrose, Glutamax, MEM-NEAA, B27, 2 μg of doxycycline, 5 μg/ml of puromycin, BDNF (10 ng/ml), CNTF (10 ng/ml) and GDNF (10 ng/ml). Half media changes every other day until day 18. Then full media change was performed at day 18 and 4 days after conditioned media and lysates were collected.
Induced neurons were washed with cold phosphate-buffered saline (PBS) and lysis buffer (1% NP40 lysis, 50 mm Tris pH 7.6; 150 mm NaCl; 10 mM EDTA; 1× complete protease inhibitor mixture) added to cells and kept on ice for 15 min. A total of 15 μg protein per well were loaded on a NuPAGE Novex 4–12% Bis-Tris gel with MOPS running buffer and transferred onto nitrocellulose. The membranes were blocked in Odyssey PBS blocking buffer for 1 h at RT, followed by incubation overnight at 4°C with primary antibody for FERMT2 (GTX84507, GeneTex, Irvine, CA) and GAPDH (Proteintech, Rosemont, IL). Membranes were rinsed and incubated for 1 h with fluorescence-conjugated goat anti-mouse IgG. Blots were scanned and analyzed using Licor Odyssey Imaging system.
Immunocytochemistry of human postmortem brain samples
A total of 6 μm sections of formalin-fixed paraffin-embedded tissue from the human DLPFC were used to stain FERMT2 (Sigma), ALDH1L1 (eBioscience, Waltham, MA), Iba1 (Wako Richmond, VA) and NeuN (MilliporeSigma). Immunohistochemistry was performed using citrate as antigen retrieval. The sections were blocked with blocking medium (3% of BSA) and incubated overnight at 4°C with primary antibodies.
Sections were washed with PBS and incubated with fluorochrome conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA) and coverslipped with anti-fading reagent containing DAPI (P36931, Life technologies). Photomicrographs are captured at 20× magnification using Zeiss Axio Observer Z1 fluorescence microscope and exported to ImageJ imaging software (National Institutes of Health (NIH), Maryland, USA).
Statistical analysis of amyloid, SNP and proteomics associations in ROSMAP
Ascertainment of ROSMAP genotyping information was previously described (68). Methods for determining amyloid scores from postmortem brain of ROS and MAP participants were previously described (37). Proteomic profiles from postmortem brain samples of ROS and MAP were previously described (38). A GWAS was performed on amyloid (n = 984), modeling amyloid as the dependent variable; genotype as the independent variable; and cohort, age at death, sex and the first three genetic covariance matrix (EV1–3) as covariates. Using PLINK version 1.08p, a linear model assuming additive genetic effects was used, and separate analyses were performed according to genotyping platform. These results were meta-analyzed using PLINK to mitigate potential confounding effects due to the combination of platforms. SNPs with an INFO score less than 0.3 and a MAF of <0.01 were removed from analysis. The SNP with the most significant association with residual cognition within each independent locus was selected as the lead SNP for that locus.
GWAS was performed on proteomic data, due to a smaller sample size (n = 553), the ROS and MAP cohorts were combined and analyzed genome wide in R, adjusting for cohort, genotype chip, genotype call location, age at death and the first three genetic principal components, in a linear regression model. SNPs with an INFO score of <0.3 or with a MAF of <0.03 were removed from the results.
Supplementary Material
Acknowledgements
We would like to thank Amy He for technical assistance and Dennis Selkoe, David Root, Richard Pearse, Matt LaVoie and the U01 AMP-AD working group for advice and guidance. RNAseq data from postmortem brain obtained from Sage Synapse, Accelerating Medicines Partnership AD Knowledge Portal (https://www.synapse.org/#!Synapse:syn2580853/wiki/66722), under synapse ID syn3163039 (Mayo Clinic RNAseq).
Conflict of Interest statement. None declared.
Funding
National Institute on Aging (NIA) (U01AG046152 to P.L.D. and D.B., R01AG055909 and R33AG049864 to T.Y.P.); BrightFocus Foundation (T.Y.P.).
Author contributions
S.E.S., M.L., M.T., V.N.L. and R.V.S. conducted experiments and acquired data. S.E.S., M.L., C.W. and T.L.Y.P. analyzed data. V.N.L. and M.T. performed immunostaining and cell imaging. J.X. analyzed RNAseq data. D.B. provided advice and guidance, and edited the manuscript. S.E.S., P.L.D. and T.L.Y.P. wrote the manuscript.
References
- 1. Selkoe D.J. and Hardy J. (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med., 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sullivan S.E. and Young-Pearse T.L. (2017) Induced pluripotent stem cells as a discovery tool for Alzheimer's disease. Brain Res., 1656, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J. et al. (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertram L., McQueen M.B., Mullin K., Blacker D. and Tanzi R.E. (2007) Systematic meta-analyses of Alzheimer's disease genetic association studies: the AlzGene database. Nat. Genet., 39, 17–23. [DOI] [PubMed] [Google Scholar]
- 5. Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B. et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet., 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Jager P.L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L.C., Yu L., Eaton M.L., Keenan B.T., Ernst J., McCabe C. et al. (2014) Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci., 17, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cataldo A.M., Barnett J.L., Pieroni C. and Nixon R.A. (1997) Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J. Neurosci., 17, 6142–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koo E.H. and Squazzo S.L. (1994) Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem., 269, 17386–17389. [PubMed] [Google Scholar]
- 9. Nixon R.A. and Yang D.S. (2011) Autophagy failure in Alzheimer's disease—locating the primary defect. Neurobiol. Dis., 43, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Auer I.A., Schmidt M.L., Lee V.M., Curry B., Suzuki K., Shin R.W., Pentchev P.G., Carstea E.D. and Trojanowski J.Q. (1995) Paired helical filament tau (PHFtau) in Niemann–Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol., 90, 547–551. [DOI] [PubMed] [Google Scholar]
- 11. Love S., Bridges L.R. and Case C.P. (1995) Neurofibrillary tangles in Niemann–Pick disease type C. Brain, 118 (Pt 1), 119–129. [DOI] [PubMed] [Google Scholar]
- 12. Ohmi K., Kudo L.C., Ryazantsev S., Zhao H.Z., Karsten S.L. and Neufeld E.F. (2009) Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. U. S. A., 106, 8332–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saito Y., Suzuki K., Nanba E., Yamamoto T., Ohno K. and Murayama S. (2002) Niemann–Pick type C disease: accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann. Neurol., 52, 351–355. [DOI] [PubMed] [Google Scholar]
- 14. Suzuki K., Parker C.C., Pentchev P.G., Katz D., Ghetti B., D'Agostino A.N. and Carstea E.D. (1995) Neurofibrillary tangles in Niemann–Pick disease type C. Acta Neuropathol., 89, 227–238. [DOI] [PubMed] [Google Scholar]
- 15. Bras J., Guerreiro R., Darwent L., Parkkinen L., Ansorge O., Escott-Price V., Hernandez D.G., Nalls M.A., Clark L.N., Honig L.S. et al. (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet., 23, 6139–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geiger J.T., Ding J., Crain B., Pletnikova O., Letson C., Dawson T.M., Rosenthal L.S., Pantelyat A., Gibbs J.R., Albert M.S. et al. (2016) Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol. Dis., 94, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsuang D., Leverenz J.B., Lopez O.L., Hamilton R.L., Bennett D.A., Schneider J.A., Buchman A.S., Larson E.B., Crane P.K., Kaye J.A. et al. (2012) GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology, 79, 1944–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muratore C.R., Rice H.C., Srikanth P., Callahan D.G., Shin T., Benjamin L., Walsh D.M., Selkoe D.J. and Young-Pearse T.L. (2014) The familial Alzheimer's disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet., 23, 3523–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muratore C.R., Zhou C., Liao M., Fernandez M.A., Taylor W.M., Lagomarsino V.N., Pearse R.V. 2nd, Rice H.C., Negri J.M., He A. et al. (2017) Cell-type dependent Alzheimer's disease phenotypes: probing the biology of selective neuronal vulnerability. Stem Cell Rep., 9, 1868–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shulman J.M., Imboywa S., Giagtzoglou N., Powers M.P., Hu Y., Devenport D., Chipendo P., Chibnik L.B., Diamond A., Perrimon N. et al. (2014) Functional screening in Drosophila identifies Alzheimer's disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet., 23, 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Farfel J.M., Yu L., Buchman A.S., Schneider J.A., De Jager P.L. and Bennett D.A. (2016) Relation of genomic variants for Alzheimer disease dementia to common neuropathologies. Neurology, 87, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chapuis J., Flaig A., Grenier-Boley B., Eysert F., Pottiez V., Deloison G., Vandeputte A., Ayral A.M., Mendes T., Desai S. et al. (2017) Genome-wide, high-content siRNA screening identifies the Alzheimer's genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol., 133, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deming Y., Li Z., Kapoor M., Harari O., Del-Aguila J.L., Black K., Carrell D., Cai Y., Fernandez M.V., Budde J. et al. (2017) Genome-wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers. Acta Neuropathol., 133, 839–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Apostolova L.G., Risacher S.L., Duran T., Stage E.C., Goukasian N., West J.D., Do T.M., Grotts J., Wilhalme H., Nho K. et al. (2018) Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol., 75, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muratore C.R., Srikanth P., Callahan D.G. and Young-Pearse T.L. (2014) Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS One, 9, e105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zeng H., Guo M., Martins-Taylor K., Wang X., Zhang Z., Park J.W., Zhan S., Kronenberg M.S., Lichtler A., Liu H.X. et al. (2010) Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One, 5, e11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nehme R., Zuccaro E., Ghosh S.D., Li C., Sherwood J.L., Pietilainen O., Barrett L.E., Limone F., Worringer K.A., Kommineni S. et al. (2018) Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep., 23, 2509–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Srikanth P., Lagomarsino, V.N., Pearse R.V., Liao M., Ghosh S., Nehme R., Seyfried N., Eggan K. and Young-Pearse T.L. (2018) Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl. Psychiatry, 9 article #245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rohe M., Carlo A.S., Breyhan H., Sporbert A., Militz D., Schmidt V., Wozny C., Harmeier A., Erdmann B., Bales K.R. et al. (2008) Sortilin-related receptor with A-type repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J. Biol. Chem., 283, 14826–14834. [DOI] [PubMed] [Google Scholar]
- 30. Ho A., Liu X. and Sudhof T.C. (2008) Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease. J. Neurosci., 28, 14392–14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bentahir M., Nyabi O., Verhamme J., Tolia A., Horre K., Wiltfang J., Esselmann H. and De Strooper B. (2006) Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem., 96, 732–742. [DOI] [PubMed] [Google Scholar]
- 32. Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W. et al. (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med., 2, 864–870. [DOI] [PubMed] [Google Scholar]
- 33. Cho J.H. and Johnson G.V. (2004) Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J. Biol. Chem., 279, 54716–54723. [DOI] [PubMed] [Google Scholar]
- 34. Rubio-Perez J.M. and Morillas-Ruiz J.M. (2012) A review: inflammatory process in Alzheimer's disease, role of cytokines. ScientificWorldJournal, 2012, 756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bennett D.A., Schneider J.A., Wilson R.S., Bienias J.L. and Arnold S.E. (2005) Education modifies the association of amyloid but not tangles with cognitive function. Neurology, 65, 953–955. [DOI] [PubMed] [Google Scholar]
- 36. Yu L., Petyuk V.A., Gaiteri C., Mostafavi S., Young-Pearse T., Shah R.C., Buchman A.S., Schneider J.A., Piehowski P.D., Sontag R.L. et al. (2018) Targeted brain proteomics uncover multiple pathways to Alzheimer's dementia. Ann. Neurol., 84, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bartolomucci A., Pasinetti G.M. and Salton S.R. (2010) Granins as disease-biomarkers: translational potential for psychiatric and neurological disorders. Neuroscience, 170, 289–297. [DOI] [PubMed] [Google Scholar]
- 38. Carrasquillo M.M., Allen M., Burgess J.D., Wang X., Strickland S.L., Aryal S., Siuda J., Kachadoorian M.L., Medway C., Younkin C.S. et al. (2017) A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer's disease risk and increased TREML1 and TREM2 brain gene expression. Alzheimers Dement., 13, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cleary J.P., Walsh D.M., Hofmeister J.J., Shankar G.M., Kuskowski M.A., Selkoe D.J. and Ashe K.H. (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci., 8, 79–84. [DOI] [PubMed] [Google Scholar]
- 40. Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A. et al. (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med., 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chaufty J., Sullivan S.E. and Ho A. (2012) Intracellular amyloid precursor protein sorting and amyloid-beta secretion are regulated by Src-mediated phosphorylation of Mint2. J. Neurosci., 32, 9613–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dumanis S.B., Chamberlain K.A., Jin Sohn Y., Jin Lee Y., Guenette S.Y., Suzuki T., Mathews P.M., Pak D., Rebeck G.W., Suh Y.H. et al. (2012) FE65 as a link between VLDLR and APP to regulate their trafficking and processing. Mol. Neurodegener., 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersen O.M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., Arnim C.A., Breiderhoff T., Jansen P., Wu X. et al. (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A., 102, 13461–13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dodson S.E., Gearing M., Lippa C.F., Montine T.J., Levey A.I. and Lah J.J. (2006) LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J. Neuropathol. Exp. Neurol., 65, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baris H.N., Cohen I.J. and Mistry P.K. (2014) Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr. Endocrinol. Rev., 12 (Suppl 1), 72–81. [PMC free article] [PubMed] [Google Scholar]
- 46. Reczek D., Schwake M., Schroder J., Hughes H., Blanz J., Jin X., Brondyk W., Van Patten S., Edmunds T. and Saftig P. (2007) LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell, 131, 770–783. [DOI] [PubMed] [Google Scholar]
- 47. Tamargo R.J., Velayati A., Goldin E. and Sidransky E. (2012) The role of saposin C in Gaucher disease. Mol. Genet. Metab., 106, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cox T.M. and Cachon-Gonzalez M.B. (2012) The cellular pathology of lysosomal diseases. J. Pathol., 226, 241–254. [DOI] [PubMed] [Google Scholar]
- 49. Gan-Or Z., Leblond C.S., Mallett V., Orr-Urtreger A., Dion P.A. and Rouleau G.A. (2015) LRRK2 mutations in Parkinson's disease; a sex effect or lack thereof? A meta-analysis. Parkinsonism Relat. Disord., 21, 778–782. [DOI] [PubMed] [Google Scholar]
- 50. Lee J.H., Yu W.H., Kumar A., Lee S., Mohan P.S., Peterhoff C.M., Wolfe D.M., Martinez-Vicente M., Massey A.C., Sovak G. et al. (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell, 141, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee J.H., McBrayer M.K., Wolfe D.M., Haslett L.J., Kumar A., Sato Y., Lie P.P., Mohan P., Coffey E.E., Kompella U. et al. (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep., 12, 1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bae M., Patel N., Xu H., Lee M., Tominaga-Yamanaka K., Nath A., Geiger J., Gorospe M., Mattson M.P. and Haughey N.J. (2014) Activation of TRPML1 clears intraneuronal Abeta in preclinical models of HIV infection. J. Neurosci., 34, 11485–11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi S., Kim D., Kam T.I., Yun S., Kim S., Park H., Hwang H., Pletnikova O., Troncoso J.C., Dawson V.L. et al. (2015) Lysosomal enzyme glucocerebrosidase protects against Abeta1-42 oligomer-induced neurotoxicity. PLoS One, 10, e0143854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malnar M., Hecimovic S., Mattsson N. and Zetterberg H. (2014) Bidirectional links between Alzheimer's disease and Niemann–Pick type C disease. Neurobiol. Dis., 72 (Pt A), 37–47. [DOI] [PubMed] [Google Scholar]
- 55. Maulik M., Peake K., Chung J., Wang Y., Vance J.E. and Kar S. (2015) APP overexpression in the absence of NPC1 exacerbates metabolism of amyloidogenic proteins of Alzheimer's disease. Hum. Mol. Genet., 24, 7132–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saharan S. and Mandal P.K. (2014) The emerging role of glutathione in Alzheimer's disease. J. Alzheimers Dis., 40, 519–529. [DOI] [PubMed] [Google Scholar]
- 57. Chang S.H., Jung I.S., Han G.Y., Kim N.H., Kim H.J. and Kim C.W. (2013) Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer's disease. Biochem. Biophys. Res. Commun., 430, 670–675. [DOI] [PubMed] [Google Scholar]
- 58. Mizutani Y., Bonavida B., Koishihara Y., Akamatsu K., Ohsugi Y. and Yoshida O. (1995) Sensitization of human renal cell carcinoma cells to cis-diamminedichloroplatinum(II) by anti-interleukin 6 monoclonal antibody or anti-interleukin 6 receptor monoclonal antibody. Cancer Res., 55, 590–596. [PubMed] [Google Scholar]
- 59. Wang Y., Niu X.L., Qu Y., Wu J., Zhu Y.Q., Sun W.J. and Li L.Z. (2010) Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett., 295, 110–123. [DOI] [PubMed] [Google Scholar]
- 60. Liu R. and Jin J.P. (2016) Calponin isoforms CNN1, CNN2 and CNN3: regulators for actin cytoskeleton functions in smooth muscle and non-muscle cells. Gene, 585, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu R. and Jin J.P. (2016) Deletion of calponin 2 in macrophages alters cytoskeleton-based functions and attenuates the development of atherosclerosis. J. Mol. Cell. Cardiol., 99, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Young-Pearse T.L., Chen A.C., Chang R., Marquez C. and Selkoe D.J. (2008) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev., 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rice H.C., Young-Pearse T.L. and Selkoe D.J. (2013) Systematic evaluation of candidate ligands regulating ectodomain shedding of amyloid precursor protein. Biochemistry, 52, 3264–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lai-Cheong J.E., Parsons M. and McGrath J.A. (2010) The role of kindlins in cell biology and relevance to human disease. Int. J. Biochem. Cell Biol., 42, 595–603. [DOI] [PubMed] [Google Scholar]
- 65. Yasuda-Yamahara M., Rogg M., Frimmel J., Trachte P., Helmstaedter M., Schroder P., Schiffer M., Schell C. andHuber T.B. (2018) FERMT2 links cortical actin structures, plasma membrane tension and focal adhesion function to stabilize podocyte morphology. Matrix Biol., 68–69,263–279. [DOI] [PubMed] [Google Scholar]
- 66. Livak K.J. and Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 67. Sanjana N.E., Shalem O. and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods, 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. White C.C., Yang H.S., Yu L., Chibnik L.B., Dawe R.J., Yang J., Klein H.U., Felsky D., Ramos-Miguel A., Arfanakis K. et al. (2017) Identification of genes associated with dissociation of cognitive performance and neuropathological burden: multistep analysis of genetic, epigenetic and transcriptional data. PLoS Med, 14, e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.