

Abstract
Identification of new drug and cell therapy targets for disease treatment will be facilitated by a detailed molecular understanding of normal and disease development. Human pluripotent stem cells can provide a large in vitro source of human cell types and, in a growing number of instances, also three-dimensional multicellular tissues called organoids. The application of stem cell technology to discovery and development of new therapies will be aided by detailed molecular characterisation of cell identity, cell signalling pathways and target gene networks. Big data or ‘omics’ techniques—particularly transcriptomics and proteomics—facilitate cell and tissue characterisation using thousands to tens-of-thousands of genes or proteins. These gene and protein profiles are analysed using existing and/or emergent bioinformatics methods, including a growing number of methods that compare sample profiles against compendia of reference samples. This review assesses how compendium-based analyses can aid the application of stem cell technology for new therapy development. This includes via robust definition of differentiated stem cell identity, as well as elucidation of complex signalling pathways and target gene networks involved in normal and diseased states.
Keywords: Pluripotent stem cell, Bioinformatics, Compendium, Signalling, Growth factor, Pathway, Gene regulatory network
Introduction
All somatic cells in a multicellular organism such as humans contain the same DNA. However, each normal distinct cell type within the organism only expresses a subset of the available genome required for proper functioning of that particular cell type (Ralston and Shaw 2008). Expression of particular sets of target genes (TGs) is regulated by a range of transcriptional regulators (TRs) including transcription factors and histone modifiers (Hoopes 2008; Ralston and Shaw 2008). Disease states typically involve acquisition of abnormal cellular transcriptional profiles that, in turn, alter cell phenotypes and function, for instance, during tumorigenesis.
Maturation of cellular phenotype and function occurs through the interplay between environmental cues—sensed, for example, via growth factor receptors—and transcriptional changes that take place within the cell (Hoopes 2008; Ralston and Shaw 2008). For most cell type/external cue combinations, little molecular detail is known either of the molecular events that lead to transcriptional changes or the breadth of TGs changes that occur. Greater detail of these processes is recognised as a key frontier for the development of new therapies for a broad range of diseases (Berg 2016). Thus, there is a compelling need to identify TG sets that are regulated by particular signalling pathways and environmental factors, in order to better characterise the development and maintenance of cellular phenotypes, behaviours and biological processes. This information will also greatly facilitate improved understand of how these events become dysregulated in ageing and disease.
Stem cells enable molecular characterisation of human biology
Historically, the inability to access large amounts of normal and diseased human tissue—particularly during the early stages of disease initiation—significantly impeded efforts to define cell identity at a molecular level. The scarcity of human tissues has also hindered efforts to define how environmental cues alter cell biology and disease progression.
Significant genomic and functional similarities exist between human cells and tissues compared to those of other species. Consequently, many different animal models have been developed to try and progress investigation of normal and disease development. While valuable knowledge has been gained through decades of animal studies, the ability for animal models to specifically predict treatment responses in human patients is questionable (Shanks et al. 2009). This has led both academic researchers and the pharmaceutical industry to investigate human stem cells as an alternative source of information for both basic research and drug discovery (Cressey 2012; O'Connor 2013).
Human pluripotent stem (PS) cells offer a unique opportunity to rapidly progress our understanding of how environmental cues modulate signalling cascades and TG sets. This is due to key properties of human PS cells (O'Connor 2013; O'Connor et al. 2011a; Ungrin et al. 2007), including the ability to:
Self-renew (i.e., proliferate while retaining developmental potential), thereby enabling production of extremely large numbers of human cells in vitro
Differentiate into essentially any desired human cell type for research and clinical applications
Enable simple and highly targeted gene modification through technologies such as Crispr/Cas9
Obtain both normal and disease-specific human PS cells, either from donated IVF embryos (i.e., embryonic stem cells, or ES cells), by cell reprogramming (i.e., induced pluripotent stem cells) or by genome modification of these PS cell types
Directly model human biology without confounding species-specific differences that can arise through studies of animal models
As a result of these properties, use of human PS cell technology has become widespread. For example, in 2010 GE Healthcare announced the commercial availability of human ES cell-derived cardiomyocytes. These PS cell-derived cells provided a readily available and biologically relevant alternative to animal models and primary cells for cardiac drug discovery and toxicity testing.
Realising the full academic, industrial and clinical potential of human PS cells will require application of big data or ‘omics’ techniques to overcome major challenges that face the field. These challenges include (i) improving culture manipulations for optimal PS cell maintenance and directed differentiation, (ii) development of efficient cell purification strategies, and (iii) establishment of robust characterisation assays for differentiated cell types.
Overcoming these challenges will require defining the similarities between differentiated cell types and desired primary cell types. This will include assessment of the developmental maturity of differentiated cells as relates to their phenotypes and functions, as well as the molecular events required to achieve and maintain cell phenotypes and functions. Doing so will provide both minimal characterisation criteria for reproducible production of desired differentiated cell types, and also a molecular framework for disease investigation and drug target discovery.
Molecular profiling using big data
Transcriptional changes that result from environmental cues occur via activation and/or repression of specific TG sets. Historically, investigations of signalling pathways and related TGs developed from the discovery of recombinant DNA technology (Cohen et al. 1973) and the ability to genetically modify mice and other organisms. Initial characterisation technologies for these studies included PCR, histology and electron microscopy. While these initial approaches yielded useful information, limited molecular detail of affected signalling pathways or TG sets was obtained.
The development of big data techniques for transcriptomics (from spotted arrays and microarrays to RNA-sequencing, also known as RNA-seq) (Bumgarner 2013) and proteomics (particularly mass spectrometry) (Han et al. 2008) enabled much higher resolution characterisation of the molecular changes that link environmental sensing, signal transduction and affected TG sets. Additionally, traditional immunoprecipitation techniques—that provide evidence of protein interactions through antibody-based protein capture—have been coupled with both microarray analysis and DNA sequencing. For example, chromatin immunoprecipitation (ChIP) techniques (termed ChIP-chip and ChIP-seq, respectively) enable interactions between proteins and DNA to be defined with high resolution of the chromosomal location (Furey 2012; Mardis 2007). Both ChIP-chip and ChiP-seq assays have been widely used with cell lines and animal tissue to determine the chromosomal location of post-translationally modified histones, histone variants, transcription factors and chromatin modifying enzymes (Bailey et al. 2013; Collas 2010).
Computational approaches have also been developed to investigate TG regulation by TRs. This has largely been driven by the capacity for genome-wide assessment of DNA-binding motifs within gene promoters, as a consequence of sequencing the human genome. Algorithms such as PASTAA (Roider et al. 2009), Homer (Heinz et al. 2010), GeoSTAN (Zacher et al. 2017), iRegulon (Janky et al. 2014) and compendium-based approaches (Banks et al. 2016) are examples of software that use different approaches to predict TG regulation by transcription factors. As these methods are evolved, the accuracy of TG predictions increases. Combinations of sequencing and computational-based approaches have also been developed for identification of TG regulation by TRs. For example, cap analysis of gene expression (CAGE) data generated through the Fantom5 consortium has provided sequencing data from the 5′ region of mRNA transcripts (as opposed to traditional 3′ sequencing approaches) for 975 human and 399 mouse cell samples (Andersson et al. 2014; Consortium et al. 2014). Computational analysis of this data has been used to predict TRs responsible for regulation of large sets of TGs across many cell types (Marbach et al. 2016).
Current big data analysis tools
The above technical and technological advances mean it is now possible to accurately and simultaneously measure the expression levels of essentially all genes for species that have had their genomes sequenced. It is also possible to begin interrogating the TRs involved in generating gene expression profiles, through ChIP-seq and or computational analyses. Alternatively, mass spectrometry enables simultaneous measurement of the levels of many thousands of proteins.
A variety of open source and proprietary software has been developed to analyse whole transcriptome expression data. For example, Gene Pattern (Broad Institute) (Reich et al. 2006) and GeneSpring (Agilent) for microarray data; limma for both microarray and RNA-seq gene expression data (Ritchie et al. 2015); and EdgeR (Robinson et al. 2010) for RNA-seq data. These different softwares enable identification of differentially expressed genes related to developmental and/or disease states. However, it should be noted that sequencing-based approaches, such as RNA-seq, tend to be better suited for identification of expressed vs. non-expressed genes, as opposed to identification of only differentially expressed genes. This is due to the digital nature of transcript detection by sequencing techniques, compared to the analogue nature of microarray based techniques (that typically rely on fluorescent-based methods for transcript detection, thereby making determination of absolute expression cut-off thresholds challenging).
Transcriptome analysis software can generate lists of expressed and/or differentially expressed genes from either new whole transcriptome data or reanalysis of published studies. These gene lists then provide insights into the signalling pathways and TGs involved in development or function of normal tissue, as well as pathways and TGs altered by disease states. A commonly used approach to investigate differentially expressed gene lists is identification of gene groupings via gene ontology (GO) analysis. Various GO analysis software are available including the DAVID Gene Ontology Functional Annotation Clustering tool (Huang et al. 2009a, b), Enricher (Kuleshov et al. 2016), GO-Bayes (Zhang et al. 2010), Babelomics (Medina et al. 2010), etc. Alternatively, assessment of expressed growth factor signalling pathway members can be performed by comparison of gene lists against the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway database (Kanehisa and Goto 2000).
Characterising pluripotency mechanisms using big data
Transcriptional, translational and ChIP profiling studies have been performed using cell lines and primary cells/tissue, and more recently using stem cells and their differentiated derivatives. For example, landmark studies have highlighted genes that are highly expressed across multiple human PS cell lines, thus identifying core transcriptional machinery consisting of the transcription factors OCT4/POU5F1, NANOG and SOX2 (Boyer et al. 2005; Cloonan et al. 2008; Hirst et al. 2007). These studies have also identified some TGs of these key pluripotency TRs. Additional studies have identified other human PS cell regulators including FOXD3, SALL4, Polycomb-group proteins, etc. (Lee et al. 2006; O'Connor et al. 2011b; Respuela et al. 2016).
Through comparison with mouse ES cell transcriptional data, these human studies provided a molecular framework for understanding the different culture requirements for PS cells obtained from different species. For instance, while mouse and human ES cells are both obtained from fertilised embryos, maintenance of mouse ES cells is LIF-dependent and FGF-independent. Conversely, human ES cells are LIF-independent and FGF-dependent. Transcriptional profiling studies have helped provide an explanation for these observations. The initially isolated mouse ES cell state is now recognised as a developmentally earlier state termed the ‘naïve’ pluripotency state. In contrast, the initially isolated human ES cell state is now termed the ‘primed’ pluripotency state that is analogous to pluripotent cells that can be isolated from the mouse epiblast. Naïve human ES cells can be transitioned between the naïve and primed pluripotency states (Chen et al. 2015; Duggal et al. 2015; Warrier et al. 2017), raising the possibility of obtaining naïve human ES cells directly from blastocysts (Van der Jeught et al. 2015). As naïve PS cells may enable better control of differentiated cell production, the transcriptomics studies described here provide evidence that big data might facilitate improvement and application of stem cell technology.
Big data repositories for defining cell identity
A major challenge for the stem cell field is the reliable production and characterisation of desired differentiated cell types. Cell-type identification via a whole transcriptome gene expression profile can provide a relatively rapid, broad and reasonably cost-effective approach. Accurate cell-type identification is needed to enable better manipulation of differentiated cells in culture (e.g., by identifying growth factor requirements), and also to provide a framework for understanding the molecular events that occur in a disease state.
Transcriptional and/or translational analyses typically involve characterisation of a control sample with or without comparison to treated sample(s) generated through chemical or genomic perturbations. Time-course components are also often included. The vast number of transcriptional and translational studies performed over the past 15 years has led to the establishment of large data repositories to facilitate public access to gene and protein expression data. Examples of public repositories for gene expression data include the Gene Expression Omnibus (GEO) that accepts data from any species (Barrett et al. 2013); human and mouse data available via the ENCODE consortium (Consortium TEP 2012; Consortium TME 2012); and human data available via GTEx (Consortium GT 2013). Protein data repositories include UniProt (Consortium TU 2007) and STRING (von Mering et al. 2003). These public gene and protein expression data repositories can provide compendia for more comprehensive/more robust cell-type identification for differentiated PS cell progeny.
Compendium-based methods for defining cell identity
Discovery of new biology by comparison of a test gene expression profile against a larger collection (i.e., compendium) of expression profiles has been used for almost two decades (Fig. 1a). However, compendium-based analyses have not yet been widely used by the stem cell field, despite the opportunity for robust cell type identification through compendia (Fig. 1a–c).
Fig. 1.
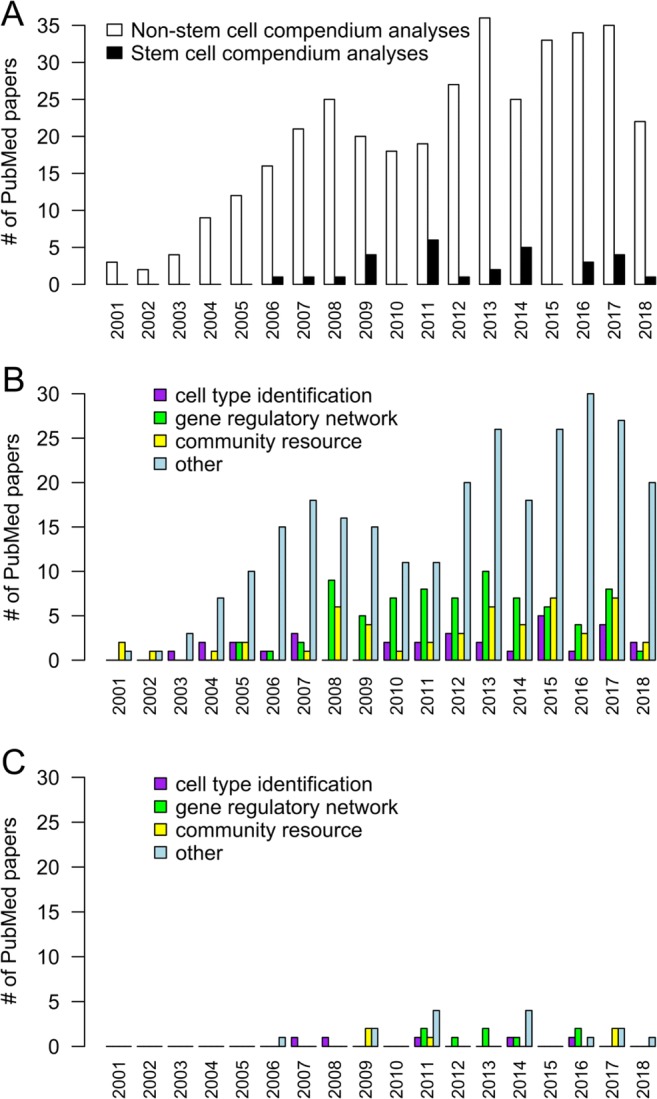
Increases in the number of published articles making use of compendium-based analyses (as identified via PubMed searches). a The number of articles using compendium-based analyses for both non-stem cell types (white bars) and stem cell types (black bars). b An indication of the number of publications using particular compendium-based applications for analysis of non-stem cell types. c Publications using particular compendium-based applications for analysis of stem cell types
Two general approaches have been used for compendium-based cell-type identification: those that use a somewhat limited gene set as the query and those that use a larger expression profile as the query (DeFreitas et al. 2016). Compendium-based approaches can also be further divided into those that enable within-species comparisons and (less frequent) those that enable cross-species comparisons. For example, SPELL enables within-species identification (only for yeast) from a limited gene set against large gene expression microarray compendia (Hibbs et al. 2007). Alternatively, GEMINI uses a large transcriptome profile to query for similar profiles but only within “The Cancer Genome Atlas” database (DeFreitas et al. 2016). GEMINI uses a principal component analysis to reduce the dimensionality of the query transcriptome, and then uses a distance function to search for the closest match within the compendium. It does not support cross-species comparisons.
A small number of compendium-based approaches that enable cross-species cell-type identification have recently been described. For example, the web server ProfileChaser mines only the curated GEO datasets for gene expression profiles that differentially regulate the same transcriptional programs as the query profiles (Engreitz et al. 2011). Another web server that matches query gene sets (to a maximum of 100 differentially expressed genes) by searching the GEO database is ExpressionBlast. Required inputs are the limited query gene list together with their expression comparison values, a species type, a desired output species type and a distance metric (Euclidean/correlation/anti-correlatin/anti-Euclidean). The algorithm then uses text analysis methods to perform similarity matching of the query gene set against GEO datasets. ExpressionBlast then outputs the relevant GEO datasets that similarly express the same genes as the query gene list (Zinman et al. 2013). The web server Cell Montage permits searching for similar gene expression profiles compared to a query gene profile (Fujibuchi et al. 2007). The method is platform specific (i.e., specific to similar microarray platforms) and also only allows users to query against GEO datasets that contain raw expression values.
Compendium-based analyses for stem cell research
A small number of groups have started utilising the compendium-based approach for stem cell research (Fig. 1c). For example, Germanguz et al. used a compendium-based approach consisting of 17 cell state-specific gene expression data (including PS cells) to identify genes that uniquely define cell states and developmental stages. They also identified core genes (including transcription factors) that can drive and maintain the cell states (Germanguz et al. 2016). StemCellNet is a web server for interactive network analysis and visualisation in the context of stem cell biology (Pinto et al. 2014). HAEMCODE is a repository of transcription factor binding maps for mouse blood cells generated by ChIP-seq (Ruau et al. 2013). Asp et al. generated a dataset of genome-wide locations for ten key histone marks and transcription factors. By using mouse myoblasts and terminally differentiated myotubes, they were able to discover key epigenetic changes underlying myogenesis (Asp et al. 2011). Hannah et al. described a ChIP-Seq compendium to discover transcriptional mechanisms operating in the haematopoietic system (Hannah et al. 2011). Sharov et al. identified a reliable set of direct TGs for Pou5f1, Sox2 and Nanog by utilising a compendium of published and new microarray data (Sharov et al. 2008). Hackney and Moore built a compendium of information and data derived from biological and molecular studies relating to haematopoietic stem cell regulation (Hackney and Moore 2005).
The above compendium-based stem cell studies tended to either compare multiple cell types or identify a specific cell type. These approaches are not optimised for identification of an unknown cell type. In comparison, a new open source R package developed by our group, termed C3, allows cross-species identification of any cell type. C3 uses a large transcriptomic profile rather than a limited gene list, and is compatible with a wide variety of input compendia (Kabir et al. 2018a). The cross-species comparison enabled by C3 makes use of a recently developed cross-species gene set analysis method called XGSA (Djordjevic et al. 2016). C3 can identify unknown cell types for a wide variety of species by comparing gene expression profiles with a large compendium of public human and mouse gene expression datasets. This approach is suitable for identification of poorly characterised cell types obtained from stem cell differentiation strategies (Murphy et al. 2018). In this way, C3 fits well into the pipeline of cell analyses needed by the stem cell field (Fig. 2).
Fig. 2.

Schematic diagram showing how compendium-based analyses can be used to accelerate application of stem cell technology to identification and testing of new drug and cell-based therapies
In addition to identification and characterisation of differentiated stem cell progeny, transcriptional profiles are also being used to guide stem cell differentiation strategies. For example, a recently published algorithm called MOGRIFY uses gene expression data to predict TRs responsible for generating cell type-specific transcriptional profiles (and thus cell-specific phenotypes and functions) (Rackham et al. 2016). These cell type-specific combinations of TRs can then be used to guide overexpression studies aimed at directly converting (i.e., trans-differentiating) one cell type into another.
Investigating extracellular regulation of cell behaviour
A second major challenge for the stem cell field, and disease research in general, is to define how extracellular signalling pathways regulate transcriptional events required for cell development, environmental sensing and disease progression (Berg 2016; Zhang and Mallick 2013). At the genome level, gene transcription is often activated or repressed by the action of transcription factors (also referred to as trans-regulatory factors) that bind to promoter regions generally upstream (i.e., 5′) of a gene’s transcription start site (termed cis-regulatory elements). The specific DNA sequences within the genome to which transcription factors bind are called DNA-binding motifs and are often described via position weight matrices (Babu et al. 2004; Boeva 2016; Spitz and Furlong 2012).
Transcriptional and translational profiles represent molecular snapshots that result from the combined action of an array of transcriptional, post-transcriptional and translational regulators, often under extracellular control via signalling pathways. Individual gene transcript abundance is largely determined by the net activity of the transcription factors bound to a gene’s promoter (Beer and Tavazoie 2004; Chen and Rajewsky 2007; Kim and O'Shea 2008)—though other regulators of transcript abundance can also be involved such as transcriptional regulators acting at more distance (e.g., enhancer) sites and post-transcriptional regulators (such as micro-RNA). Overall, the ability of any particular transcription factor to activate or repress gene expression is dependent upon the interplay between the intracellular context and regulatory cues received from the extracellular environment, for instance via growth factor signalling pathways.
Signal transduction pathways and target genes
As discussed above, a range of computational tools have been developed to elucidate gene regulatory networks by defining transcription factor/TG interactions (e.g., PASTAA, Homer, GeoSTAN, iRegulon, etc.). Sequencing approaches that target the 5′ end of mRNA transcripts, such CAGE, have also been developed. Significant recent progress has been made by applying these approaches within large, international collaborative efforts. For example, the Fantom5 consortium generated CAGE data across 975 human and 399 mouse samples, including primary cells, tissues and cancer cell lines (Andersson et al. 2014; Consortium et al. 2014). From these data, TG sets for transcription factors expressed by 394 human cell samples have been defined via analysis of DNA-binding motifs within gene promoters and enhancers (Marbach et al. 2016).
While the above approaches have provided a wealth of information on transcription factor/TG interactions, there are relatively few open source or proprietary algorithms that exist for comprehensively linking signal pathways to TG sets. A typical signal transduction pathway for transmitting extracellular cues involves growth factors binding to specific cell surface receptors, subsequent modulation of intracellular kinase activities, and ultimately altered transcription factor activity and consequent changes in TG expression (Wang et al. 2011). The coordinated activity of different signalling pathways within and between multiple cell types is the basis of many important biological processes, such as development, tissue repair and immunity (Zhao and Li 2017; Zhao et al. 2008). Activation of different signalling pathways can lead to numerous physiological or cellular responses, such as cell proliferation, differentiation, metabolism and death—key processes relevant to stem cells and their progeny both in vitro and in vivo.
Bioinformatic determination of signal pathways
Various resources have been created to assist in defining signalling pathways. The collection of manually drawn pathway diagrams available via KEGG provides a starting point for understanding particular receptor-mediated signalling pathways. However, their use can be limiting when attempting to define cell type-specific signalling pathways. Conversely, the STRING database contains millions of known protein-protein interactions (PPIs); however, accessing cell type-specific subsets of these interactions can be challenging.
Several bioinformatics methods have been described that reconstruct known signalling pathways from PPI data, with or without inclusion of gene expression data (Bebek and Yang 2007; Gil et al. 2017; Ritz et al. 2016; Wang et al. 2011). CASCADE_SCAN uses a steepest descent method to build a specific pathway from a list of protein molecules (Wang et al. 2011). Pathlinker creates signal pathways by using input receptors and transcriptional regulators to interrogate PPI databases (Gil et al. 2017; Ritz et al. 2016). PathFinder uses characteristics of known signal pathways together with related association rules to find pathways from a receptor to a transcription factor in PPI networks (Bebek and Yang 2007). Gitter et al. proposed a method to handle the orientation problem (i.e., orienting protein interaction edges using directionless PPI data) in weighted protein interaction graphs (Gitter et al. 2011). Mei et al. proposed a multi-label, multi-instance transfer learning method to simultaneously reconstruct 27 human signalling pathways (Mei and Zhu 2015). Scott et al. proposed a method to reconstruct known signalling pathways by applying a colour coding algorithm (Scott et al. 2006). Tuncbag et al. formulated a forest approach (defined as a disjointed union of trees) to simultaneously reconstruct multiple pathways from biological networks that are altered in a particular condition (Tuncbag et al. 2013). Other methods identify known signalling pathways using gene expression datasets to calculate edge weights for PPI data (Liu and Zhao 2004; Steffen et al. 2002; Zhao and Li 2017; Zhao et al. 2008).
Linking signal pathways and TG sets
All the above methods for signal pathway analysis generate topological structures for known signalling pathways. One potential limitation is that most of the methods were assessed and applied only to yeast data, with few methods designed for complex mammalian data. Recent work from our group has demonstrated a novel approach—termed SPAGI (Signal Pathway Analysis for Gene regulatory network Identification)—that systematically identifies biologically relevant signalling pathways for mammalian cells (Kabir et al. 2018c). The SPAGI approach starts with a whole transcriptome expression profile and uses it to construct a comprehensive catalogue of signalling pathways from PPI data. Application of the SPAGI approach to mouse and human cell RNA-seq data, including from differentiated progeny of human PS cells, identified known critical signalling pathways relevant to the cell types used. Subsequent research using human lens epithelial cell gene expression data has coupled each of the SPAGI-generated receptor-defined paths to TG sets obtained from the Fantom5 consortium data (Kabir et al. 2018b). The resulting lens epithelial cell gene expression framework (or lens transcriptional blueprint) describes growth factor-mediated control of transcriptional programs important to lens epithelial cell biology. Initial validation studies have shown that known gene regulatory interactions were identified, and predicted new transcriptional regulators were validated via Western blotting. This approach directly addresses a major challenging in the stem cell and disease research fields, namely, the need for large-scale generation of discrete and testable molecular hypotheses that describe the influence of environmental factors during tissue development and disease progression (Fig. 2).
Defining disease mechanisms by integrating signal pathways and disease genes
A key motivation driving the establishment of integrated signalling pathways and TG networks is the need to better define disease processes to enable identification of novel drug targets (Butcher et al. 2004; Davidson et al. 2002). Information relating to genes and gene variants involved in disease phenotypes can be found within the Online Mendelian Inheritance in Man (OMIM) database (Hamosh et al. 2005). Tissue-specific disease gene databases also exist for numerous tissues including the kidney, heart, muscle, brain, lens, etc. By correlating the abovementioned lens transcriptional blueprint with the Cat-Map database of lens-related disease genes (Shiels et al. 2010), our group has been able to identify both known and novel gene regulation events and map them to growth factor signalling pathways (Kabir et al. 2018b). This approach can also be applied to other cell types, including differentiated stem cell derivatives, to define candidate drug targets—and therefore candidate novel therapeutics—for human diseases (as outlined in Fig. 2).
Conclusion
Stem cells provide an opportunity to examine normal and disease human biology on a scale not possible with primary cells and tissues. Realising these opportunities requires overcoming specific challenges relating to determination of cell type identity, and definition of how environmental cues including growth factor signalling pathways regulate gene transcription involved in tissue development, repair/regeneration and disease. Compendium-based analyses hold promise for rapid and robust identification of first-reported differentiated stem cell types, as well as batch-produced cells for industry or cell therapy applications. Bioinformatic methods that generate comprehensive and integrated combinations of signalling pathways and gene regulatory networks are starting to provide specific molecular disease hypotheses that can be investigated using human PS cell-derived cell types. Thus compendium-based big data approaches to stem cell research present significant opportunities for the development of novel cell and drug therapies.
Author contributions
M.H.K drafted the manuscript. M.H.K and M.D.O’C revised and approved the manuscript.
Funding
M.H.K was supported by WSU Postgraduate Research Awards. M.D.O’C was supported by The Medical Advances Without Animals Trust.
Conflict of interest
Md Humayun Kabir declares that he has no conflict of interest. Michael D. O’Connor declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects performed by any of the authors.
Footnotes
This article is part of a Special Issue on ‘Big Data’ edited by Joshua WK Ho and Eleni Giannoulatou.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp P, et al. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A. 2011;108:E149–E158. doi: 10.1073/pnas.1102223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol. 2004;14:283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Bailey T, et al. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol. 2013;9:e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks CJ, Joshi A, Michoel T. Functional transcription factor target discovery via compendia of binding and expression profiles. Sci Rep. 2016;6:20649. doi: 10.1038/srep20649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebek G, Yang J. PathFinder: mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinformatics. 2007;8:335. doi: 10.1186/1471-2105-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- Berg J. Gene-environment interplay. Science. 2016;354:15. doi: 10.1126/science.aal0219. [DOI] [PubMed] [Google Scholar]
- Boeva V. Analysis of genomic sequence motifs for deciphering transcription factor binding and transcriptional regulation in eukaryotic. Cells Front Genet. 2016;7:24. doi: 10.3389/fgene.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumgarner R. Overview of DNA microarrays: types, applications, and their future. Curr Protoc Mol Biol. 2013;Chapter 22:Unit 22 21. doi: 10.1002/0471142727.mb2201s101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher EC, Berg EL, Kunkel EJ. Systems biology in drug discovery. Nat Biotechnol. 2004;22:1253–1259. doi: 10.1038/nbt1017. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- Chen H, et al. Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat Commun. 2015;6:7095. doi: 10.1038/ncomms8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Cohen SN, Chang AC, Boyer HW, Helling RB. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci U S A. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P. The current state of chromatin immunoprecipitation. Mol Biotechnol. 2010;45:87–100. doi: 10.1007/s12033-009-9239-8. [DOI] [PubMed] [Google Scholar]
- Consortium F et al A promoter-level mammalian expression atlas. Nature. 2014;507:462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TEP An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TME An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13:418. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium TU The universal protein resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressey D (2012) Stem cells take root in drug development. Nat News
- Davidson EH, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- DeFreitas T, Saddiki H, Flaherty P. GEMINI: a computationally-efficient search engine for large gene expression datasets. BMC Bioinf. 2016;17:102. doi: 10.1186/s12859-016-0934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic D, Kusumi K, Ho JW. XGSA: a statistical method for cross-species gene set analysis. Bioinformatics. 2016;32:i620–i628. doi: 10.1093/bioinformatics/btw428. [DOI] [PubMed] [Google Scholar]
- Duggal G, et al. Alternative routes to induce naive pluripotency in human embryonic stem cells. Stem Cells. 2015;33:2686–2698. doi: 10.1002/stem.2071. [DOI] [PubMed] [Google Scholar]
- Engreitz JM, Chen R, Morgan AA, Dudley JT, Mallelwar R, Butte AJ. ProfileChaser: searching microarray repositories based on genome-wide patterns of differential expression. Bioinformatics. 2011;27:3317–3318. doi: 10.1093/bioinformatics/btr548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujibuchi W, Kiseleva L, Taniguchi T, Harada H, Horton P. CellMontage: similar expression profile search server. Bioinformatics. 2007;23:3103–3104. doi: 10.1093/bioinformatics/btm462. [DOI] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germanguz I, Listgarten J, Cinkornpumin J, Solomon A, Gaeta X, Lowry WE. Identifying gene expression modules that define human cell fates. Stem Cell Res. 2016;16:712–724. doi: 10.1016/j.scr.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil DP, Law JN, Murali TM. The PathLinker app: connect the dots in protein interaction networks. F1000Res. 2017;6:58. doi: 10.12688/f1000research.9909.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitter A, Klein-Seetharaman J, Gupta A, Bar-Joseph Z. Discovering pathways by orienting edges in protein interaction networks. Nucleic Acids Res. 2011;39:e22. doi: 10.1093/nar/gkq1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney JA, Moore KA. A functional genomics approach to hematopoietic stem cell regulation. Methods Mol Med. 2005;105:439–452. doi: 10.1385/1-59259-826-9:439. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Aslanian A, Yates JR., 3rd Mass spectrometry for proteomics. Curr Opin Chem Biol. 2008;12:483–490. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah R, Joshi A, Wilson NK, Kinston S, Gottgens B. A compendium of genome-wide hematopoietic transcription factor maps supports the identification of gene regulatory control mechanisms. Exp Hematol. 2011;39:531–541. doi: 10.1016/j.exphem.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs MA, Hess DC, Myers CL, Huttenhower C, Li K, Troyanskaya OG. Exploring the functional landscape of gene expression: directed search of large microarray compendia. Bioinformatics. 2007;23:2692–2699. doi: 10.1093/bioinformatics/btm403. [DOI] [PubMed] [Google Scholar]
- Hirst M, et al. LongSAGE profiling of nine human embryonic stem cell lines. Genome Biol. 2007;8:R113. doi: 10.1186/gb-2007-8-6-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes L (2008) Introduction to the gene expression and regulation topic room. Nat Educ 1(1)
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Janky R, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014;10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir MH, Djordjevic D, O’Connor MD, Ho JWK. C3: an R package for cross-species compendium-based cell-type identification. Comput Biol Chem. 2018;77:187–192. doi: 10.1016/j.compbiolchem.2018.10.003. [DOI] [PubMed] [Google Scholar]
- Kabir MH, Murphy P, Lim S, Ho JWK, O’Connor MD (2018b) Large scale profiling of lens epithelial cell signalling pathways and target genes reveals regulatory networks for cataract-associated genes. Exp Eye Res (under review)
- Kabir MH, Patrick R, Ho JWK, O’Connor MD (2018c) Identification of active signaling pathways by integrating gene expression and protein interaction data. BMC Syst Biol in press [DOI] [PMC free article] [PubMed]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, O'Shea EK. A quantitative model of transcription factor-activated gene expression. Nat Struct Mol Biol. 2008;15:1192–1198. doi: 10.1038/nsmb.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhao H. A computational approach for ordering signal transduction pathway components from genomics and proteomics. Data BMC Bioinf. 2004;5:158. doi: 10.1186/1471-2105-5-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, Bergmann S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat Methods. 2016;13:366–370. doi: 10.1038/nmeth.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. ChIP-seq: welcome to the new frontier. Nat Methods. 2007;4:613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- Medina I, et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38:W210–W213. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S, Zhu H. Multi-label multi-instance transfer learning for simultaneous reconstruction and cross-talk modeling of multiple human signaling pathways. BMC Bioinf. 2015;16:417. doi: 10.1186/s12859-015-0841-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy P et al (2018) Light-focusing human micro-lenses generated from pluripotent stem cells model lens development and drug-induced cataract in vitro. Development 145. 10.1242/dev.155838 [DOI] [PMC free article] [PubMed]
- O'Connor MD. The 3R principle: advancing clinical application of human pluripotent stem cells. Stem Cell Res Ther. 2013;4:21. doi: 10.1186/scrt169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MD, Kardel MD, Eaves CJ. Functional assays for human embryonic stem cell pluripotency. Methods Mol Biol. 2011;690:67–80. doi: 10.1007/978-1-60761-962-8_4. [DOI] [PubMed] [Google Scholar]
- O'Connor MD, et al. Retinoblastoma-binding proteins 4 and 9 are important for human pluripotent stem cell maintenance. Exp Hematol. 2011;39:866–879. doi: 10.1016/j.exphem.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Pinto JP, Reddy Kalathur RK, Machado RS, Xavier JM, Braganca J, Futschik ME. StemCellNet: an interactive platform for network-oriented investigations in stem cell biology. Nucleic Acids Res. 2014;42:W154–W160. doi: 10.1093/nar/gku455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham OJ, et al. A predictive computational framework for direct reprogramming between human cell types. Nat Genet. 2016;48:331–335. doi: 10.1038/ng.3487. [DOI] [PubMed] [Google Scholar]
- Ralston A, Shaw K (2008) Gene expression regulates cell differentiation. Nat Educ 1(1)
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Respuela P, Nikolic M, Tan M, Frommolt P, Zhao Y, Wysocka J, Rada-Iglesias A. Foxd3 promotes exit from naive pluripotency through enhancer decommissioning and inhibits germline specification cell. Stem Cell. 2016;18:118–133. doi: 10.1016/j.stem.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz A, et al. Pathways on demand: automated reconstruction of human signaling networks. NPJ Syst Biol Appl. 2016;2:16002. doi: 10.1038/npjsba.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roider HG, Manke T, O'Keeffe S, Vingron M, Haas SA. PASTAA: identifying transcription factors associated with sets of co-regulated genes. Bioinformatics. 2009;25:435–442. doi: 10.1093/bioinformatics/btn627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruau D, et al. Building an ENCODE-style data compendium on a shoestring. Nat Methods. 2013;10:926. doi: 10.1038/nmeth.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J, Ideker T, Karp RM, Sharan R. Efficient algorithms for detecting signaling pathways in protein interaction networks. J Comput Biol. 2006;13:133–144. doi: 10.1089/cmb.2006.13.133. [DOI] [PubMed] [Google Scholar]
- Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos Ethics Humanit Med. 2009;4:2. doi: 10.1186/1747-5341-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, et al. Identification of Pou5f1, Sox2, and Nanog downstream target genes with statistical confidence by applying a novel algorithm to time course microarray and genome-wide chromatin immunoprecipitation data. BMC Genomics. 2008;9:269. doi: 10.1186/1471-2164-9-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–2015. [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Steffen M, Petti A, Aach J, D'Haeseleer P, Church G. Automated modelling of signal transduction networks. BMC Bioinf. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbag N, et al. Simultaneous reconstruction of multiple signaling pathways via the prize-collecting steiner forest problem. J Comput Biol. 2013;20:124–136. doi: 10.1089/cmb.2012.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungrin M, O'Connor M, Eaves C, Zandstra PW. Phenotypic analysis of human embryonic stem cells. Curr Protoc Stem Cell Biol. 2007;Chapter 1:Unit 1B 3. doi: 10.1002/9780470151808.sc01b03s2. [DOI] [PubMed] [Google Scholar]
- Van der Jeught M, et al. Application of small molecules favoring naive pluripotency during human embryonic stem cell derivation. Cell Reprogram. 2015;17:170–180. doi: 10.1089/cell.2014.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, et al. CASCADE_SCAN: mining signal transduction network from high-throughput data based on steepest descent method. BMC Bioinf. 2011;12:164. doi: 10.1186/1471-2105-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier S, et al. Direct comparison of distinct naive pluripotent states in human embryonic stem cells. Nat Commun. 2017;8:15055. doi: 10.1038/ncomms15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacher B, Michel M, Schwalb B, Cramer P, Tresch A, Gagneur J. Accurate promoter and enhancer identification in 127 ENCODE and roadmap epigenomics cell types and tissues by GenoSTAN. PLoS One. 2017;12:e0169249. doi: 10.1371/journal.pone.0169249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Mallick BK. Inferring gene networks from discrete expression data. Biostatistics. 2013;14:708–722. doi: 10.1093/biostatistics/kxt021. [DOI] [PubMed] [Google Scholar]
- Zhang S, Cao J, Kong YM, Scheuermann RH. GO-Bayes: Gene Ontology-based overrepresentation analysis using a Bayesian approach. Bioinformatics. 2010;26:905–911. doi: 10.1093/bioinformatics/btq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XM, Li S. HISP: a hybrid intelligent approach for identifying directed signaling pathways. J Mol Cell Biol. 2017;9:453–462. doi: 10.1093/jmcb/mjx054. [DOI] [PubMed] [Google Scholar]
- Zhao XM, Wang RS, Chen L, Aihara K. Uncovering signal transduction networks from high-throughput data by integer linear programming. Nucleic Acids Res. 2008;36:e48. doi: 10.1093/nar/gkn145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinman GE, Naiman S, Kanfi Y, Cohen H, Bar-Joseph Z. ExpressionBlast: mining large, unstructured expression databases. Nat Methods. 2013;10:925–926. doi: 10.1038/nmeth.2630. [DOI] [PubMed] [Google Scholar]