

Abstract
Background
Pathological complete response (pCR) after neoadjuvant chemotherapy (NAC) in triple-negative breast cancer (TNBC) varies between 30 and 40% approximately. To provide further insight into the prediction of pCR, we evaluated the role of an epigenetic methylation-based signature.
Methods
Epigenetic assessment of DNA extracted from biopsy archived samples previous to NAC from TNBC patients was performed. Patients included were categorized according to previous response to NAC in responder (pCR or residual cancer burden, RCB = 0) or non-responder (non-pCR or RCB > 0) patients. A methyloma study was performed in a discovery cohort by the Infinium HumanMethylation450 BeadChip (450K array) from Illumina. The epigenetic silencing of those methylated genes in the discovery cohort were validated by bisulfite pyrosequencing (PyroMark Q96 System version 2.0.6, Qiagen) and qRT-PCR in an independent cohort of TN patients and in TN cell lines.
Results
Twenty-four and 30 patients were included in the discovery and validation cohorts, respectively. In the discovery cohort, nine genes were differentially methylated: six presented higher methylation in non-responder patients (LOC641519, LEF1, HOXA5, EVC2, TLX3, CDKL2) and three greater methylation in responder patients (FERD3L, CHL1, and TRIP10). After validation, a two-gene (FER3L and TRIP10) epigenetic score predicted RCB = 0 with an area under the ROC curve (AUC) = 0.905 (95% CI = 0.805–1.000). Patients with a positive epigenetic two-gene score showed 78.6% RCB = 0 versus only 10.7% RCB = 0 if signature were negative.
Conclusions
These results suggest that pCR in TNBC could be accurately predicted with an epigenetic signature of FERD3L and TRIP10 genes. Further prospective validation of these findings is warranted.
Electronic supplementary material
The online version of this article (10.1186/s13148-019-0626-0) contains supplementary material, which is available to authorized users.
Keywords: Triple-negative breast cancer, Prediction, Epigenetic signature
Background
At the present time, chemotherapy (CT) is the only proven therapy for triple-negative breast cancer (TNBC) subtype. Anthracycline and taxane-based CT is still the standard of care for TNBC [1], with pathological complete response (pCR) rates ranging ~ 30–40% [2–6]. pCR rate in TNBC are associated with better outcomes while residual disease after neoadjuvant chemotherapy (NAC) have a higher relapse risk and poor prognosis [7]. With the advantage of high sequencing technology, several molecular signatures have been developed in the recent years to predict response to neoadjuvant chemotherapy (NAC). Oncotype-Dx [8], MammaPrint [9], Blue Print [10], Endopredict [11], or Prosigna [12] are some of them. Recently, an initial 199-gene signature, (E2F4 target gene signature), has shown accurate prediction of response to NAC even when reduced to 33-gene panel and has been validated in 1129 patients across five independent data sets [13]. However, all these predictive panels have shown to do better in ER positive breast cancer than in ER negative. Thus, accurate prediction of response in TNBC still remains a medical need. Other study addressed the issue of the prediction to NAC in 94 patients TNBC treated with paclitaxel and carboplatin according to the Lehman’s TNBC type 4 classification. Basal-like 1 (BL1) subtype showed a pCR rate of 65.2%, while basal-like 2 (BL2) was 47.4%, mesenchymal (M) 36.4%, and luminal androgen receptor (LAR) 21.4% [14]. The I-SPY 2 is an ongoing prospective trial of NAC in breast cancer including a cohort of TNBC patients. A 70-gene panel combined with DNA deficient biomarkers have shown a 75% pCR in a subset of TNBC patients treated with carboplatin and veliparib in a recent publication of this cohort [15]. All these panels have been designed based on arrays of gene-expression techniques. Nevertheless, epigenetic modifications of certain genes can lead to silence or activation of different genes [16, 17].
The epigenetic modifications of the DNA, such as methylation, can modulate gene expression with no DNA sequence modification and contribute to disease development [18]. In this context, epigenetic changes in tumor DNA before CT administration could potentially have a predictive role of response to this therapy [18]. The aim of this study was to identify a predictive epigenetic signature of pCR as defined by the residual cancer burden (RCB) index by Symmans et al. [19](RCB = 0) in patients with TNBC treated with NAC including anthracyclines and/or taxanes-based regimens.
Results
Clinical characteristics of TNBC patients
Fifty-four patients were included: 24 in the discovery cohort (DC) and 30 in the validation cohort (VC). The clinical characteristics of the patients are summarized in Table 1. After biopsy, all patients were treated with NAC based on a taxane and/or anthracycline regimen and all of them were considered TNBC according to immunohistochemistry for ER, PR, and HER2. Patients were classified in responders (R) if RCB = 0 or non-responders (NR) if RCB > 0.
Table 1.
Clinical characteristics of TNBC patients included in the study
| Variable | Whole cohort N = 54 |
Discovery cohort N = 24 |
Validation cohort N = 30 |
|---|---|---|---|
| Age; median (range) | 47.88 (27.19–78.92) | 46.88 (30.33–78.07) | 48.49 (27.19–78.92) |
| cT | |||
| cTx | 5 (9.3%) | 2 (8.3%) | 3 (10.0%) |
| cT1–2 | 40 (74.1%) | 16 (66.6%) | 24 (80.0%) |
| cT3–4 | 9 (16.6%) | 6 (25.1%) | 3 (10.0%) |
| cN | |||
| cNx | 4 (7.4%) | 2 (8.3%) | 2 (6.7%) |
| cN0 | 32 (59.3%) | 14 (58.4%) | 18 (60.0%) |
| cN+ | 18 (33.3%) | 8 (33.3%) | 10 (33.3%) |
| Ki67 in biopsy | |||
| Missing value | 10 (18.5%) | 7 (29.2%) | 3 (10.0%) |
| ki67 < =50% | 17 (31.6%) | 7 (29.2%) | 10 (33.3%) |
| ki67 > 50% | 26 (49.9%) | 10 (41.6%) | 17 (56.7%) |
| Type of NAC | |||
| Taxanes | 14 (25.9%) | 5 (20.8%) | 9 (30.0%) |
| Anthracyclines | 1 (1.9%) | 0 (0%) | 1 (3.3%) |
| Taxanes and anthracyclines | 39 (72.2%) | 19 (79.2%) | 20 (66.7%) |
| RCB | |||
| RCB = 0 | 19 (35.2%) | 10 (41.7%) | 9 (30.0%) |
| RCB > 0 | 35 (64.8%) | 14 (58.3%) | 21 (70.0%) |
| RCB: | |||
| RCB = 0 | 19 (35.2%) | 10 (41.7%) | 9 (30.0%) |
| RCB = 1 | 8 (14.8%) | 3 (12.5%) | 5 (16.7%) |
| RCB = 2 | 19 (35.3%) | 6 (25.0%) | 13 (43.3%) |
| RCB = 3 | 8 (14.8%) | 5 (20.8%) | 3 (10.0%) |
(NAC neoadjuvant treatment)
Analysis of DNA methylome in responder and non-responder patients: discovery cohort
A genome-wide DNA methylation study was performed in the DC (N = 24, 10 R (RCB = 0) and 14 NR (RCB > 0)). Figure 1a summarizes the whole process during this study. The analysis of methylation data showed 133 CpGs sites (71 genes) with differences in methylation levels ≥ 20% (p value < 0.05) that distinguished R patients (treatment sensitive) from NR patients (treatment resistant) (Fig. 1b). According to a Gene Ontology (GO) analysis, some of these genes were involved in biological functions and pathways such as DNA repair, cell adhesion, transcription regulation, or signaling mediated by GTPases (Fig. 1c, Additional file 1: Table S1) that have shown to be implicated in chemoresistance of cancer, including the response to anthracyclines and/or taxanes [20–23].
Fig. 1.

Differentially methylated CpG sites from 450K array (Illumina) between the responder and non-responder group. a Workflow summary. b Hierarchical clustering heatmap of differentially methylated CpGs: 133 CpGs (71 genes) differentially methylated were found between responders (R) and non-responders (NR) group identified from 450K array analysis with mean differences in methylation levels ≥ 20% (p < 0.05). c Most representative biological processes of the 71 differentially methylated genes according to Gene Ontology analysis
Thirty-five CpGs located in promoters, islands, or shores from 23 genes were selected to further validations (Additional file 2: Table S2). Of these, taking into account an intra-group SD ≤0.2, 11 CpGs corresponding to 11 genes showed significant methylation differences (Additional file 3: Table S3), and 9 of these genes showed a consistent methylation profile on consecutive CpGs (Fig. 2). These candidate genes were LOC641519, LEF1, HOXA5, EVC2, TLX3, and CDKL2 with high methylation in NR group and genes FERD3L, CHL1, and TRIP10 with high methylation in R group.
Fig. 2.

Methylation profile of the selected CpGs in responder and non-responder group in the discovery cohort (DC). From the 450K array, ten CpGs corresponding to nine genes were found in CpG islands or in the promoter regions with a standard deviation intragroup ≤ 20% and with a consistent DNA methylation profile of consecutive CpGs. These candidate genes were LOC641519, LEF1, HOXA5, EVC2, TLX3, and CDKL2 with high methylation in non-responder group and genes FERD3L, CHL1, and TRIP10 with high methylation in responder group. Consecutive CpGs to the CpGs of interest were evaluated in each gene by the 450K array. Points represent mean methylation values from patients
Validation of methylation in candidate genes by pyrosequencing: validation cohort
A pyrosequencing study in the DC and in the VC (N = 30, 9 RCB = 0 and 21 RCB > 0) was performed to validate the candidate genes first technically and secondly in an independent cohort, respectively. Methylation was analyzed for each gene taking into account the differentially methylated CpGs identified in the 450K array and other CpGs located close to them in order to obtain a more consistent result (Additional file 4: Table S4).
In the DC, we replicated by pyrosequencing the methylation data obtained in 450K array for LOC641519/LEF1 gene (p value = 0.02) and HOXA5 gene (p value = 0.0001), where we also observed a methylation level significantly higher in NR patients than R patients, and in FERD3L gene (p value = 0.04), TRIP10 (p value = 0.003), and CHL1 (p value = 0.03), where methylation was also significantly higher in R patients than NR patients. However, in EVC2 gene (p value = 0.07), CDKL2 gene (p value = 0.05), and TLX3 gene (p value = 0.07), replication was not statistically significant but showed a trend towards a higher level of methylation in NR patients (Fig. 3a).
Fig. 3.

Validation of the differently methylated CpGs by pyrosequencing. a Validation results in the discovery cohort (DC, n = 24)). The array data were replicated in LEF1/ LOC641519 and HOXA5 genes, with a significant higher methylation in non-responders patients vs. responder patients (p < 0.05) and FERD3L, TRIP, and CHL1 genes, with a significantly higher methylation in responder patients vs. non-responder patients (p < 0.05). The CDKL2, EVC2, and TLX3 genes showed a trend for significance (p ≥ 0.05). b Validation results in the validation cohort (VC, n = 30). Methylation data from the 450K array were only replicated for FERD3L gene. Values were statistically different when compared non-responder vs. responder group showing low methylation in non-responder patients (p = 0.0087). The TRIP10 gene showed a non-significant trend towards a high methylation in responder group compared to non-responder group (p = 0.19)
Pyrosequencing in the VC validated the results for FERD3L gene (p value = 0.0087) with high methylation in R group versus NR group (Fig. 3b). Moreover, differences in TRIP10 methylation showed a trend towards significance between R and NR (p value = 0.19). Accordingly, by means of a biological pathway analysis of these genes using the publicly available resource Pathway Commons [24], we observed that FERD3L and TRIP10 are able to interact with several genes (Additional file 5: Figure S1) which have been previously associated with therapy resistance of breast cancer and other types of tumors [23, 25].
FERD3L methylation and gene expression in TNBC cell lines
In order to evaluate whether DNA methylation has a functional role in the transcriptional control of FERD3L, we evaluated FERD3L gene expression by qRT-PCR and methylation by pyrosequencing in a set of TNBC cell lines (Fig. 4a) with the aim of correlating FERD3L methylation levels with gene expression level and corroborate the results from patients. We observed that FERD3L gene was methylated in all the cell lines studied with levels always higher than 40%. The FERD3L expression inversely correlated with the methylation detected as was expected, showing a low gene expression when methylation was high. Thereby, MDA-MB-231 cell line showed the lowest methylation level and correlated with the highest level of gene expression. Conversely, HCC-1143 cell line that showed the higher methylation was the one with the lowest gene expression level.
Fig. 4.

FERD3L methylation and transcript levels in patients and cell lines. a FERD3L gene expression levels (qRT-PCR) and methylation levels (pyrosequencing) in a set of TNBC cell lines. FERD3L was methylated in all the cell lines studied (≥ 40%) correlating with the gene expression detected. b Variation in FERD3L methylation and gene expression in MDA-MB-436 cell line after AZA (5 uM) treatment. Cells showed a decrease in methylation level (p = 0.05) that correlated with a statistically significant increase in gene expression (p = 0.0022) when they were treated with AZA and compared with control cells not treated with AZA agent. c qRT-PCR results for FERD3L gene in TNBC patients (DC + VC). Non-responder group showed higher gene expression levels than responder group (p = 0.04) correlating with methylation levels obtained both in 450K array and pyrosequencing. d Spearman correlation between methylation and gene expression for FERD3L gene in a population of breast cancer patients (n = 713) from TCGA database. The result showed a correlation between a high gene methylation and a low gene expression when analyzed all the CpGs in the FERD3L gene promoter or only the cg10043037 identified in the 450K array
FERD3L demethylation assays and gene expression in TNBC cell lines
In order to check if changes in FERD3L methylation status also affect to gene expression level, we performed an assay with the MDA-MB-436 cell line treated with AZA demethylating agent. We observed that the treatment modified FERD3L methylation in MDA-MB-436 cell line inducing a decrease when compared with control cells (MDA-MB-436 cell line not treated with AZA) (p value = 0.05). As we expected, this change in FERD3L methylation was correlated with a significant increase (p value = 0.0022) in FERD3L expression (Fig. 4b).
High FERD3L expression levels correlates with low gene methylation in TNBC patients
The analysis of FERD3L gene expression in the 54 patients showed a significant difference (p value = 0.04) in FERD3L gene expression with high expression in NR patients versus R patients (Fig. 4c). Therefore, it suggests an inverse correlation between methylation and gene expression in NR patients and R patients. The Cancer Genome Atlas dataset (TCGA) analysis for 713 breast cancer patients showed negative correlation between methylation and expression, according with our data. It was detected both when all CpGs in the FERD3L gene promoter were included and when only analyzed the CpG cg10043037 validated for FERD3L gene in the study (Fig. 4d).
Statistical model to predict response to neoadjuvant treatment in TNBC patients
Due to FERD3L and TRIP10 showed the higher level of significance in the VC, we selected both genes for therapy response analysis. It is interesting to note that these two genes did not show statistical differences in methylation between R and NR in terms of age, tumor size, nodule affectation, or ki67 expression (Additional file 6: Table S5) indicating that these genes were not associated with any relevant clinicopathological prognostic factor. Importantly, we were able to create a statistical epigenomic predictive model of pathological response (RCB = 0) with the FERD3L and TRIP10 methylation and using the whole cohort. These two genes were selected as both showed the higher level of significance in the VC. The statistical model for the prediction of therapy response was based on the Akaike information criterion (AIC) and by constructing a receiver operating characteristic (ROC) curve. Based on this model, the following rule was constructed:
A × FERD3L methylation level (%) + B × TRIP10 methylation level (%) > 971 (A and B being constants)
Levels > 971 showed a high likelihood for RCB = 0 (8 out of 11 cases 78.6%), while levels ≤ 971 showed a poor probability for RCB = 0 (3 out of 28 patients 10.7%). Value based of the calculation of the ROC curve with AUC = 0.9056 (95% CI = 0.805–1.000) (Fig. 5a, b).
Fig. 5.
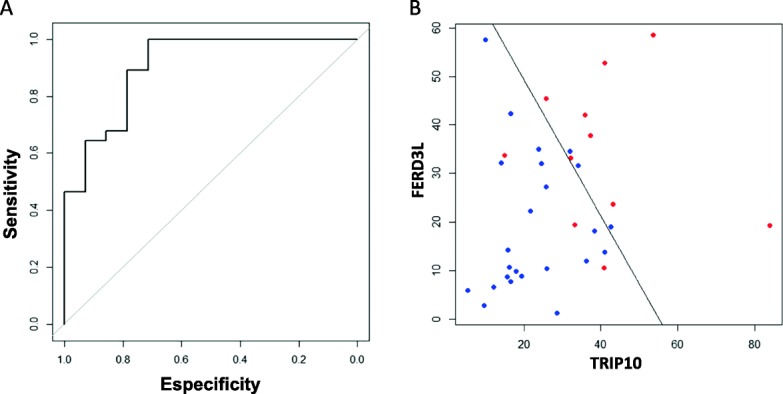
FERD3L and TRIP10 genes as predictive markers of pathological complete response (pCR) in TNBC patients. a The ROC curve for FERD3L and TRIP10 show graphically the connection/trade-off between clinical sensitivity and specificity. The area under the ROC curve (AUC) was 0.905 (95% CI = 0.805–1.000). b Based on the algorithm of the methylation status of FERD3L and TRIP10 in patients with TNBC, when the score was > 971, the probability to get a RCB = 0 was of 78.6%. For values lower than 971, the probability to get a RCB = 0 was of 10.7%. Red points represent patients with clinical RCB = 0 and in blue are indicated those patients with clinical RCB > 0. To the right of the line, patients has been classified as RCB = 0 and to the left as RCB > 0 by our algorithm
Discussion
This study evaluated the methylation profile of triple-negative breast cancer (TNBC) patients treated with neoadjuvant systemic chemotherapy (NAC) depending on the treatment response and identified a two-gene epigenetic signature for discriminating responders (R) from non-responders (NR). The discovery cohort included 24 TNBC patients and was analyzed by using Infinium Human Methylation 450 BeadChip array whereas the validation cohort included 30 TNBC patients and was analyzed by pyrosequencing for the significant and most relevant genes identified in the discovery cohort. For each case, DNA was obtained from core biopsies before the beginning of the treatment. The analysis of the discovery cohort identified nine genes differentially methylated. Six genes with higher methylation in NR patients (LOC641519, LEF1, HOXA5, EVC2, TLX3, CDKL2) and three genes with greater methylation in R patients (FERD3L, CHL1 and TRIP10). After technical and analytical validation by pyrosequencing in both discovery and validation cohorts, we identified a two-gene (FERD3L and TRIP10) signature able to predict response to NAC. The role of promoter methylation in the regulation of FERDL3 gene expression was evaluated in TNBC cell lines and in TNBC tissues, demonstrating an inverse correlation between methylation and expression levels. To our knowledge, the two genes epigenetic model shown in this study is the first epigenetic signature for prediction of response to NAC in TNBC patients. It should be noted that this model almost doubles the predictive potential described for the TNBC subtype by other approaches (~ 30–40% versus 78.6% with our model) [2–6].
Pathological complete response (pCR) is an effective surrogate marker for survival among patients with luminal B/HER2 negative, HER2 positive, and TNBC tumors. The results of a meta-analysis of 6377 patients with operable or advanced non-metastatic disease from six prospective neoadjuvant studies support this [26]. In the TNBC group of 911 patients who received anthracycline and taxane based NAC, 31% achieve pCR. Most recently, a retrospective analysis of 452 TNBC patients showed a pCR of 33% [3]. Overall and disease-free survival were significantly longer among patients achieving pCR versus residual disease [3]. In this context, the development of a more accurate predictive signature in TNBC could have an important clinical impact. To date, available gene panels for prediction of pCR to NAC are based in genomic signatures [8–10, 13, 27].
Epigenetics changes affect cellular processes such as gene expression and have clearly been related to the development of diseases such as breast cancer. Few methylation studies have been specifically done on TNBC subtype. Stizarken et al. identified differentially methylated regions that could separate TNBC and non-TNBC patients and classified those according to prognosis. This provided the first evidence that changes in the methylation profile of DNA could be useful to identify and stratify TNBC patients [28]. Recently, Mathe et al. performed a study of gene expression and DNA methylation in the same population, demonstrating that DNA methylation contributes to the deregulation of gene expression [29]. In this context, our results suggest that response to NAC can be predicted accurately with an algorithm of the methylation status of FERD3L and TRIP10 genes in patients with TNBC.
The FERD3L (Fer3-like bHLH transcription factor) gene, also named NATO3 or N-TWIST, is a gene located on chromosome 7 and is a basic helix-loop-helix (bHLH) transcription factor. These factors play an essential role in multiple developmental processes, mainly in neurogenesis, where its regulation is essential for the right development [30]. FERD3L is a member of the TWIST genes family that is implicated in epithelial-mesenchymal transition (EMT) in cancer cells, a process also related to metastasis and may lead to chemo-resistance in TNBC [31].
The TWIST genes induce cell dedifferentiation and cell migration [32] and are also related to the inhibition of apoptosis [33], the cancer stem cell phenotype [34], and chemotherapy resistance [35].
Several studies have shown that in metastatic carcinomas including aggressive and metastatic breast cancer, there are an overexpression of TWIST [36]. The inactivation of TWIST by siRNA technology or chemotherapeutic approaches has proved successful [36–38], so it is presented as a potential therapeutic target for metastatic breast cancer. There is only one work in the literature that relates the FERD3L gene to cancer, specifically neuroblastoma [39]. Promoter CpG islands in FERD3L gene was found to be highly methylated in neuroblastoma cell lines causing gene silencing and poor prognosis. However, to our knowledge, there is no previous evidence of relation between FERD3L expression and response to CT in the clinical setting or any relation with breast cancer.
The TRIP10 (thyroid hormone receptor interactor 10 gene) is located in chromosome 19 and belongs to the minor histocompatibility antigens family and codifies the Cdc42-interacting protein 4 (CIP4). This protein interacts with the GTPase Cdc42 that is related with actin formation and has been implicated in cytoskeleton organization [40]. Paclitaxel, a type of taxane, promotes microtubule stabilization and polymerization leading to a cell cycle arrest and apoptosis [41]. Actin-microtubule crosstalk is particularly important for cell shape and polarity during cell migration and division [42]. In this context, TRIP10 hypermethylation could increase the efficacy of paclitaxel effects on cytoskeleton. Furthermore, TRIP10 has an important role in the cellular motility and cohesion control since it is implicated in E-cadherin regulation [43]. In fact, previous studies have related TRIP10 with cell invasion in TNBC cells in vitro [44]. Interestingly, another study demonstrated also that TRIP10 gene controls EMT [43]. They described a pro-metastatic role of TRIP10, in concordance with the in vitro and in vivo data from Cerqueira et al. [45]. In addition, TRIP10 expression has shown to be regulated by DNA methylation in mesenchymal stem cell differentiation [46] and in several types of cancer cell lines and tumors [47]. In particular, in breast cancer, it has been also confirmed that TRIP10 expression can be regulated by epigenetic mechanisms such as DNA methylation [47, 48].
Basing a signature on the methylation status of only two genes could be a limitation of this study. However, the identification of this signature was provided by a very stringent analysis to select the most suitable candidate genes and they were further validated in an independent cohort of patients. In fact, there are several examples of other two-gene signatures, including methylation based signatures, that have previously shown clinical utility for cancer in different types of tumors [49–56]. Additionally, the sample size could be considered small; however, it is important to highlight that the group of patients is very homogeneous in order to eliminate potential confusion factors. In our analysis, we compared methylation levels among TNBC patients who responded and did not respond to neoadjuvant chemotherapy. Both groups were homogeneous also regarding to staging and severity, basing the difference only in the response to chemotherapy. In fact, no statistically significant differences were observed in methylation levels according to staging and severity. Therefore, the strict selection of patients could be a strength that give support and power to these results. The low number of cases analyzed could limit the immediate translational relevance but represents a very good start point for future studies in the field.
Conclusions
Triple-negative breast cancer has a high relapse rate after conventional chemotherapy treatment. To date, no predictors of treatment effectiveness have been identified. In this study, we propose an epigenetic signature based on the methylation levels of the FERD3L and TRIP10 genes. Our algorithm has a complete pathological response prediction potential of 78.6% and increases the predictive potential described by other approaches. This is especially relevant if we consider that it could be a predictive tool in clinical practice that will allow selecting the appropriate treatment as well as better stratification of patients for clinical trials.
Methods
Patients treatment and tumor samples
Patients treated with anthracyclines and/or taxanes NAC in the Hospital Clínico of Valencia and diagnosed with an early TNBC between 2005 and 2015 (Table 1) were retrospectively selected for the study according to clinical inclusion/exclusion criteria (Additional file 7: Table S6). Tumor samples were obtained before exposure to any systemic anticancer treatment using ultrasound-guided core needle biopsy. The cores were placed on OCT and stored at − 80 °C, or included in FFPE. Tumor percentage, histology and ER, PR, HER2, and Ki67 expression were determined. ER and PR status were considered negative when nuclear staining is < 10%. For the assessment of HER2, ASCO/CAP recommendations were used [57]. Diagnosis of TNBC was done according to IHC results. The pathological response after NAC was evaluated by the Symmans method (residual cancer burden; RCB) [19]. A value of RCB = 0 implies pCR, whereas values of RCB > 0 indicates that there is still residual tumor. From an initial analysis of the database, 70 patients were identified. Only those that had a tumor percentage > 25% and reached 500 ng after DNA extraction were used. Fifty-four patients were included in the study; of these, a group of 24 patients (10 RCB = 0 and 14 RCB > 0) were selected for the discovery cohort (DC) and 30 patients (9 RCB = 0 and 21 RCB > 0) were included in the validation cohort (VC).
Sample size calculation
Sample size calculation was based on data from a recent series reporting that TNBC patients treated with NAC obtaining a RCB = 0 was around 30% [4]. If our methylation data would be able to predict two groups of responders (R) versus non-responders (NR) and we estimate that the proportion of patients with RCB = 0 could be 45% in R while the proportion of RCB = 0 in the NR group would be 10%, with an alpha error of 5% and a power of 80%, the sample size needed to identify this difference between both proportions is 44 patients (22 patients/group). According to this, the theoretical sample size, including a 15% drop-out, should be 51 patients, (https://select-statistics.co.uk/calculators/sample-size-calculator-two-proportions/).
TNBC cellular lines and treatments
Five TNBC cell lines from American Type Culture Collection (ATCC) were cultured (HCC-1937, HCC-1143, HCC-38, MDA-MB-231, and MDA-MB-436) following standard culture conditions. Treatment with the demethylating agent 5-aza-2′-deoxycytidine (AZA) (Sigma, St. Louis, MO, USA) was performed at 5 uM during 72 h. In this assay, results were performed in triplicates and data were compared with the corresponding non-treated cell line.
DNA extraction and bisulfite conversion
Both OCT or FFPE TNBC tissue were used depending on availability and DNA extraction was performed using DNA purification protocol with NaCl or the kit “DNA PPPE QIAamp Tissue” (Qiagen) respectively. DNA from cells lines was extracted using Trizol reagent. DNA samples were quantified using PicoGreen method (Invitrogen) and quality was evaluated using Nanodrop (Thermo Scientific) and electrophoresis gels. For DNA bisulfite modification, 500 ng of DNA were used and modification was performed with EZ-96 DNA Methylation (Zymo Research Corp.)
DNA methylation analysis by Infinium Human Methylation 450 BeadChip array
Microarray-based DNA methylation analysis was conducted with the Infinium Human Methylation 450 BeadChip (450K array; Illumina, San Diego, CA), that covers > 450,000 CpG sites along the human genome [58]. After bisulfite conversion, hybridization was performed following the Illumina Infinium HD methylation protocol.
Methylation score of each CpG was represented as β value that ranged between 0 (unmethylated) and 1 (completely methylated). Color balance adjustment and normalization were performed using GenomeStudio Illumina software (V2010.3). After filtering, differentially methylated CpG sites (DMCpGs) between R and NR groups were identified following this flowchart (Fig. 1a): for each probe/CpG, the sets of methylation β values belonging to both groups were compared to obtain (1) DMCpGs with a significant p value < 0.05 (1030 CpGs); (2) DMCpGs with average β values between R and NR groups ≥ 0.20 (133 out of 1030 CpGs); (3) DMCpGs localized in island or shore regions of promotors (35 out of 133 CpGs); (4) DMCpGs with intragroup standard deviation (SD) ≤ 0.20 in order to select the most relevant positions for validation (11 out of 35 CpGs); (5) DMCpGs with a consistent methylation profile in consecutive CpGs (10 out of 11). This final filter with ten CpGs (9 genes) yielded the best candidates for validation.
DNA methylation analysis by bisulfite pyrosequencing
Quantitative DNA methylation analysis was performed by bisulfite pyrosequencing of consecutive cytosines located in islands or shores of promoter regions of candidate genes using a Pyro Gold SQA™ Reagent Kit (Qiagen) in a PyroMark Q96 System version 2.0.6 (Qiagen) according to the manufacturer’s instructions. CpG site methylation quantification was obtained using Pyro Q-CpG 1.0.9 (Qiagen). Primer sequences (Additional file 8: Table S7) were designed with PyroMark Assay Design 2.0 (Qiagen).
Gene expression studies and correlation with methylation levels
Total RNA was isolated from OCT/ FFPE samples by mirVana Isolation Kit (Ambion) and from cell lines using Trizol (Invitrogen) according to the manufacturer’s protocol. The RNA (500 ng) were retrotranscribed using the High-Capacity cDNA Reverse Transcription kit (Applied biosystems) according to the manufacturer. Quantitative RT-PCR (qRT-PCR) reactions were performed in triplicate on an Applied Biosystems 7900HT Fast Real-Time PCR system using TaqMan expression assays (FERD3L Hs00541737_s1; TRIP10 Hs00182848_m1; GAPDH Hs03929097_g1 (Applied Biosystems). Gene expression was assayed with GAPDH as endogenous control and using the delta delta Ct method.
Gene ontology and The Cancer Genome Atlas (TCGA) database analysis
A gene ontology (GO) analysis was performed to estimate the enrichment of the DMCpGs identified in particular biological processes [59]. This analysis detects the significant over-representation of GO terms in one of the sets with respect to the other for the entire genome. GO terms with p value < 0.05 were considered significant. DNA methylation and expression data from patients with invasive breast carcinoma were obtained from The Cancer Genome Atlas (TCGA) using the MethHc database (http://methhc.mbc.nctu.edu.tw/php/index.php). Paired DNA methylation and expression data from 713 patients obtained from Infinium 450K array and RNA-Seq, respectively, were used. Based in the methylation results of our study, we considered the methylation data of FERD3L promoter region and the methylation of an individual CpG (cg10043037) located at the promoter region.
Statistical analysis
In the DC, data were summarized by mean, SD, or median. To identify consistent patterns of differentially methylated CpG sites between responders (R) versus non-responders (NR), a non-parametric Wilcoxon rank sum test was performed in the DC. This test demonstrates quite robust results even for a small number of subjects. Globally, a two-tailed p value of less than 0.05 was considered to indicate statistical significance. All statistical analyses were performed using GraphPad Prism 7 and R software (version 3.2.0).
In the VC, differences in DNA methylation and transcript levels of the identified genes between R and NR and gene expression changes in the cell lines after the demethylating treatment were assessed by the non-parametric Mann-Whitney U test. The correlation between methylation and transcript levels was assessed by Spearman’s rank correlation coefficient.
Receiver operating characteristic (ROC) curves were used to assess the diagnostic predictive capacity of the candidate biomarkers. The area under the curve (AUC) was computed for each ROC curve, and 95% confidence intervals (CI) were also estimated by bootstrapping with 1000 iterations. Sensitivity and specificity were estimated at the optimal cut-off point according to Youden criteria.
Data were summarized by mean, SD, or median. Globally, a two-tailed p value of less than 0.05 was considered to indicate statistical significance. All statistical analyses were performed using GraphPad Prism 7 and R software (version 3.2.0).
Additional files
List of all the biological processes enriched for the 71 differentially methylated genes between responder and non-responder patients according to the Gene Ontology analysis (DOCX 32 kb)
Thirty-five differentially methylated CpGs between responders and non-responders group selected from 450k array (delta value ≥ 0.2) corresponding to 23 genes located in promoter and island/shore (PPT 172 kb)
Eleven differentially methylated CpGs, corresponding to 11 genes, showed significant methylation differences between non-responder and responder patients: 6 genes (LOC641518; LEF1; HOXA5; EVC2; CDKL2; TLX3) presented a methylation increase in non-responders group vs responders, and 5 genes (ZFHX4; LOC100192378; FERD3L; CHL1; TRIP10) decreased methylation level in non-responder patients compared to those who responded to NAC treatment (PPT 225 kb)
CpGs studied by pyrosequencing in the DC and in the VC to validate methylation in the candidate genes identified in the 450k array (Illumina). In bold, CpGs from 450k array. Normal type, consecutive CpGs (PPT 140 kb)
Representation of the pathway interaction network of FERD3L and TRIP10 with other genes using Pathway Commons. FERD3L and TRIP10 are able to interact with different genes that have shown to be implicated in cancer drug resistance (PPT 452 kb)
Mean differences in methylation levels according to clinicopathological prognostic factors in both cohorts (DC+VC). cT, clinical tumor size; cN, clinical nodule affectation (PPTX 48 kb)
Clinical inclusion and exclusion criteria followed to select TNBC patients for the methylation study (PPT 89 kb)
Sequence of primers used by pyrosequencing in the validation assay of candidate genes obtained from 450k array (PPT 143 kb)
Acknowledgements
The authors thank sample donors and workers who supported the data collection.
Funding
This work was supported by the project GV037/2016, from Consellería D′Educació, Investigació, Cultura i Esport, and to CIBERONC (CB16/12/00481), PI15/01617 and PI18/01219 from ISCIII and FEDER founds. ADL. is funded by a contract “Juan Rodés” (JR17/00016) from ISCIII. ABC. is funded by a contract “Miguel Servet” (CP17/00088) from ISCIII.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AIC
Akaike information criterion
- ASCO/CAP
American Society of Clinical Oncology/College of American Pathologists
- ATCC
American Type Culture Collection
- AUC
Area under the curve
- AZA
5-aza-2′-deoxycytidine
- bHLH
Basic helix-loop-helix
- BL-1
Basal-like 1
- BL-2
Basal-like 2
- CDKL2
Cyclin-dependent kinase like 2
- CHL1
Cell adhesion molecule L1 like
- CI
Confidence intervals
- CpG
Cytosine and guanine separated by a phosphate
- CT
Chemotherapy
- Ct
Cycle threshold
- DC
Discovery cohort
- DMCpGs
Differentially methylated CpG sites
- DNA
Deoxyribonucleic acid
- E2F4
E2F transcription factor 4
- EMT
Epithelial-mesenchymal transition
- ER
Estrogen receptors
- EVC2
EvC ciliary complex subunit 2
- FERD3L
Fer3-like bHLH transcription factor
- FFPE
Formalin-fixed paraffin-embedded
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GO
Gene ontology
- HER2
Erb-b2 receptor tyrosine kinase 2
- HOXA5
Homeobox A5
- IHC
Immunohistochemistry
- LAR
Luminal androgen receptor
- LEF1
Lymphoid enhancer binding factor 1
- M
Mesenchymal
- MethHc
A database of DNA Methylation and gene expression in Human Cancer
- NAC
Neoadjuvant chemotherapy
- Non-pCR
Non-pathological complete response
- NR
Non-responders
- pCR
Pathological complete response
- PR
Progesterone receptor
- qRT-PCR
Quantitative reverse transcriptase polymerase chain reaction
- R
Responders
- RCB
Residual cancer burden
- RNA-seq
RNA sequencing (RNA: ribonucleic acid)
- ROC
Receiver operating characteristic
- RT-PCR
Reverse transcriptase polymerase chain reaction
- SD
Standard deviation
- siRNA
Small interfering RNA
- TCGA
The cancer genome atlas
- TLX3
T cell leukemia homeobox 3
- TN
Triple negative
- TNBC
Triple-negative breast cancer
- TRIP10/CIP4
Thyroid hormone receptor interactor 10
- TWIST
Twist family bHLH transcription factor 1
- VC
Validation cohort
Authors’ contributions
ALL and PE contributed to the conception and design. BP, ADL, and ABC contributed to the development of the methodology. JAPF, OB, IGB, and JS contributed to the acquisition of data. PE, JAPF, ADL, JS, and BP contributed to the analysis and interpretation of data. ADL, PE, BP, JAPF, ABC, ME, ALL, and JS contributed to the writing, review, and/or revision of the manuscript. OB, IGB, and ABC contributed to the administrative, technical, or material support. ADL, ME, ALL, and PE contributed to the study supervision. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the local clinical Research Ethics Committee of the Hospital Clínico Universitario de Valencia /INCLIVA with the reference number 2014/178. Patients were requested to sign informed consent for tumor tissue molecular analysis and biobanking.
Consent for publication
Not applicable
Competing interests
Authors BP, ADL, JA PF, ME, ALL, and PE are currently applying for a patent (application number EP18382390.5) relating the content of the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Begoña Pineda, Email: bepime@hotmail.com.
Angel Diaz-Lagares, Email: adiazla@gmail.com.
José Alejandro Pérez-Fidalgo, Email: japfidalgo@msn.com.
Octavio Burgués, Email: octavioburgues@gmail.com.
Inés González-Barrallo, Email: igbarrallo@gmail.com.
Ana B. Crujeiras, Email: anabelencrujeiras@hotmail.com
Juan Sandoval, Email: epigenomica@iislafe.es.
Manel Esteller, Email: mesteller@idibell.cat.
Ana Lluch, Email: lluch_ana@gva.es.
Pilar Eroles, Email: pilar.eroles@uv.es.
References
- 1.Sousa B, Cardoso F. Neoadjuvant treatment for HER-2-positive and triple-negative breast cancers. Ann Oncol. 2012;23(Suppl 10):x237–x242. doi: 10.1093/annonc/mds348. [DOI] [PubMed] [Google Scholar]
- 2.Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- 3.Biswas T, Efird JT, Prasad S, Jindal C, Walker PR. The survival benefit of neoadjuvant chemotherapy and pCR among patients with advanced stage triple negative breast cancer. Oncotarget. 2017;8(68):112712–112719. doi: 10.18632/oncotarget.22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee HJ, Park IA, Song IH, Kim SB, Jung KH, Ahn JH, et al. Comparison of pathologic response evaluation systems after anthracycline with/without taxane-based neoadjuvant chemotherapy among different subtypes of breast cancers. PLoS One. 2015;10(9):e0137885. doi: 10.1371/journal.pone.0137885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance) J Clin Oncol. 2015;33(1):13–21. doi: 10.1200/JCO.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gamucci T, Pizzuti L, Sperduti I, Mentuccia L, Vaccaro A, Moscetti L, et al. Neoadjuvant chemotherapy in triple-negative breast cancer: a multicentric retrospective observational study in real-life setting. J Cell Physiol. 2018;233(3):2313–2323. doi: 10.1002/jcp.26103. [DOI] [PubMed] [Google Scholar]
- 7.Penault-Llorca F, Viale G. Pathological and molecular diagnosis of triple-negative breast cancer: a clinical perspective. Ann Oncol. 2012;23(Suppl 6):vi19–vi22. doi: 10.1093/annonc/mds190. [DOI] [PubMed] [Google Scholar]
- 8.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 9.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 10.Whitworth P, Stork-Sloots L, de Snoo FA, Richards P, Rotkis M, Beatty J, et al. Chemosensitivity predicted by BluePrint 80-gene functional subtype and MammaPrint in the Prospective Neoadjuvant Breast Registry Symphony Trial (NBRST) Ann Surg Oncol. 2014;21(10):3261–3267. doi: 10.1245/s10434-014-3908-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertucci F, Finetti P, Viens P, Birnbaum D. EndoPredict predicts for the response to neoadjuvant chemotherapy in ER-positive, HER2-negative breast cancer. Cancer Lett. 2014;355(1):70–75. doi: 10.1016/j.canlet.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Prat A, Galvan P, Jimenez B, Buckingham W, Jeiranian HA, Schaper C, et al. Prediction of response to neoadjuvant chemotherapy using core needle biopsy samples with the Prosigna assay. Clin Cancer Res. 2016;22(3):560–566. doi: 10.1158/1078-0432.CCR-15-0630. [DOI] [PubMed] [Google Scholar]
- 13.Mark KMK, Varn FS, Ung MH, Qian F, Cheng C. The E2F4 prognostic signature predicts pathological response to neoadjuvant chemotherapy in breast cancer patients. BMC Cancer. 2017;17(1):306. doi: 10.1186/s12885-017-3297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Echavarria I, Lopez-Tarruella S, Picornell A, Garcia-Saenz JA, Jerez Y, Hoadley K, et al. Pathological response in a triple-negative breast cancer cohort treated with neoadjuvant carboplatin and docetaxel according to Lehmann's refined classification. Clin Cancer Res. 2018;24(8):1845–1852. doi: 10.1158/1078-0432.CCR-17-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolf DM, Yau C, Sanil A, Glas A, Petricoin E, Wulfkuhle J, et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2017;3:31. doi: 10.1038/s41523-017-0025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aznar MA, Labiano S, Diaz-Lagares A, Molina C, Garasa S, Azpilikueta A, et al. CD137 (4-1BB) Costimulation modifies DNA methylation in CD8(+) T cell-relevant genes. Cancer Immunol Res. 2018;6(1):69–78. doi: 10.1158/2326-6066.CIR-17-0159. [DOI] [PubMed] [Google Scholar]
- 17.Diaz-Lagares A, Crujeiras AB, Lopez-Serra P, Soler M, Setien F, Goyal A, et al. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc Natl Acad Sci U S A. 2016;113(47):E7535–E7E44. doi: 10.1073/pnas.1608585113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stefansson OA, Esteller M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am J Pathol. 2013;183(4):1052–1063. doi: 10.1016/j.ajpath.2013.04.033. [DOI] [PubMed] [Google Scholar]
- 19.Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol. 2007;25(28):4414–4422. doi: 10.1200/JCO.2007.10.6823. [DOI] [PubMed] [Google Scholar]
- 20.Chekhun VF, Kulik GI, Yurchenko OV, Tryndyak VP, Todor IN, Luniv LS, et al. Role of DNA hypomethylation in the development of the resistance to doxorubicin in human MCF-7 breast adenocarcinoma cells. Cancer Lett. 2006;231(1):87–93. doi: 10.1016/j.canlet.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Liu X, Wu Y, Fang Z, Wu Q, Wu C, et al. MicroRNA-34a attenuates metastasis and chemoresistance of bladder cancer cells by targeting the TCF1/LEF1 axis. Cell Physiol Biochem. 2018;48(1):87–98. doi: 10.1159/000491665. [DOI] [PubMed] [Google Scholar]
- 22.Liu X, Luo X, Wu Y, Xia D, Chen W, Fang Z, et al. MicroRNA-34a attenuates paclitaxel resistance in prostate cancer cells via direct suppression of JAG1/Notch1 axis. Cell Physiol Biochem. 2018;50(1):261–276. doi: 10.1159/000494004. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Zhang Y, Dagher MC, Shacter E. Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res. 2005;65(14):6054–6062. doi: 10.1158/0008-5472.CAN-05-0175. [DOI] [PubMed] [Google Scholar]
- 24.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, et al. Pathway commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39(Database issue):D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang M, Li Y, Shen X, Ruan Y, Lu Y, Jin X, et al. CLDN6 promotes chemoresistance through GSTP1 in human breast cancer. J Exp Clin Cancer Res. 2017;36(1):157. doi: 10.1186/s13046-017-0627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012;30(15):1796–1804. doi: 10.1200/JCO.2011.38.8595. [DOI] [PubMed] [Google Scholar]
- 27.Ueno T, Masuda N, Yamanaka T, Saji S, Kuroi K, Sato N, et al. Evaluating the 21-gene assay recurrence score (R) as a predictor of clinical response to 24 weeks of neoadjuvant exemestane in estrogen receptor-positive breast cancer. Int J Clin Oncol. 2014;19(4):607–613. doi: 10.1007/s10147-013-0614-x. [DOI] [PubMed] [Google Scholar]
- 28.Stirzaker C, Zotenko E, Song JZ, Qu W, Nair SS, Locke WJ, et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun. 2015;6:5899. doi: 10.1038/ncomms6899. [DOI] [PubMed] [Google Scholar]
- 29.Mathe A, Wong-Brown M, Locke WJ, Stirzaker C, Braye SG, Forbes JF, et al. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci Rep. 2016;6:33435. doi: 10.1038/srep33435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verzi MP, Anderson JP, Dodou E, Kelly KK, Greene SB, North BJ, et al. N-twist, an evolutionarily conserved bHLH protein expressed in the developing CNS, functions as a transcriptional inhibitor. Dev Biol. 2002;249(1):174–190. doi: 10.1006/dbio.2002.0753. [DOI] [PubMed] [Google Scholar]
- 31.Glackin CA. Targeting the twist and Wnt signaling pathways in metastatic breast cancer. Maturitas. 2014;79(1):48–51. doi: 10.1016/j.maturitas.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Li S, Kendall SE, Raices R, Finlay J, Covarrubias M, Liu Z, et al. TWIST1 associates with NF-kappaB subunit RELA via carboxyl-terminal WR domain to promote cell autonomous invasion through IL8 production. BMC Biol. 2012;10:73. doi: 10.1186/1741-7007-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, et al. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999;13(17):2207–17. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009;11(12):1318–1328. doi: 10.1593/neo.91084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P, et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012;31(27):3223–3234. doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Wang WS, Yang XS, Xia M, Jiang HY, Hou JQ. Silencing of twist expression by RNA interference suppresses epithelial-mesenchymal transition, invasion, and metastasis of ovarian cancer. Asian Pac J Cancer Prev. 2012;13(9):4435–4439. doi: 10.7314/apjcp.2012.13.9.4435. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res. 2006;66(9):4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. [DOI] [PubMed] [Google Scholar]
- 39.Abe M, Watanabe N, McDonell N, Takato T, Ohira M, Nakagawara A, et al. Identification of genes targeted by CpG island methylator phenotype in neuroblastomas, and their possible integrative involvement in poor prognosis. Oncology. 2008;74(1–2):50–60. doi: 10.1159/000139124. [DOI] [PubMed] [Google Scholar]
- 40.Gururaj AE, Rayala SK. Kumar R. p21-activated kinase signaling in breast cancer. Breast Cancer Res. 2005;7(1):5–12. doi: 10.1186/bcr961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dogterom M, Koenderink GH. Actin-microtubule crosstalk in cell biology. Nat Rev Mol Cell Biol. 2019;20(1):38–54. doi: 10.1038/s41580-018-0067-1. [DOI] [PubMed] [Google Scholar]
- 43.Rolland Y, Marighetti P, Malinverno C, Confalonieri S, Luise C, Ducano N, et al. The CDC42-interacting protein 4 controls epithelial cell cohesion and tumor dissemination. Dev Cell. 2014;30(5):553–568. doi: 10.1016/j.devcel.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Pichot CS, Arvanitis C, Hartig SM, Jensen SA, Bechill J, Marzouk S, et al. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASp. Cancer Res. 2010;70(21):8347–8356. doi: 10.1158/0008-5472.CAN-09-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cerqueira OL, Truesdell P, Baldassarre T, Vilella-Arias SA, Watt K, Meens J, et al. CIP4 promotes metastasis in triple-negative breast cancer and is associated with poor patient prognosis. Oncotarget. 2015;6(11):9397–9408. doi: 10.18632/oncotarget.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsiao SH, Lee KD, Hsu CC, Tseng MJ, Jin VX, Sun WS, et al. DNA methylation of the Trip10 promoter accelerates mesenchymal stem cell lineage determination. Biochem Biophys Res Commun. 2010;400(3):305–312. doi: 10.1016/j.bbrc.2010.08.048. [DOI] [PubMed] [Google Scholar]
- 47.Hsu CC, Leu YW, Tseng MJ, Lee KD, Kuo TY, Yen JY, et al. Functional characterization of Trip10 in cancer cell growth and survival. J Biomed Sci. 2011;18:12. doi: 10.1186/1423-0127-18-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leu YW, Yan PS, Fan M, Jin VX, Liu JC, Curran EM, et al. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004;64(22):8184–8192. doi: 10.1158/0008-5472.CAN-04-2045. [DOI] [PubMed] [Google Scholar]
- 49.Jatkoe TA, Karnes RJ, Freedland SJ, Wang Y, Le A, Baden J. A urine-based methylation signature for risk stratification within low-risk prostate cancer. Br J Cancer. 2015;112(5):802–808. doi: 10.1038/bjc.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meseure D, Vacher S, Alsibai KD, Nicolas A, Chemlali W, Caly M, et al. Expression of ANRIL-polycomb complexes-CDKN2A/B/ARF genes in breast tumors: identification of a two-gene (EZH2/CBX7) signature with independent prognostic value. Mol Cancer Res. 2016;14(7):623–633. doi: 10.1158/1541-7786.MCR-15-0418. [DOI] [PubMed] [Google Scholar]
- 51.Sun C, Cheng X, Wang C, Wang X, Xia B, Zhang Y. Gene expression profiles analysis identifies a novel two-gene signature to predict overall survival in diffuse large B-cell lymphoma. Biosci Rep. 2019;39(1). 10.1042/BSR20181293. [DOI] [PMC free article] [PubMed]
- 52.Sun Y, Hou L, Yang Y, Xie H, Yang Y, Li Z, et al. Two-gene signature improves the discriminatory power of IASLC/ATS/ERS classification to predict the survival of patients with early-stage lung adenocarcinoma. Onco Targets Ther. 2016;9:4583–4591. doi: 10.2147/OTT.S107272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Lu Y, Feng W, Chen Q, Guo H, Sun X, et al. A two kinase-gene signature model using CDK2 and PAK4 expression predicts poor outcome in non-small cell lung cancers. Neoplasma. 2016;63(2):322–329. doi: 10.4149/220_150817N448. [DOI] [PubMed] [Google Scholar]
- 54.Xu Q, Tan C, Ni S, Wang Q, Wu F, Liu F, et al. Identification and validation of a two-gene expression index for subtype classification and prognosis in diffuse large B-cell lymphoma. Sci Rep. 2015;5:10006. doi: 10.1038/srep10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu X, Li M, Hu J, Chen Z, Yu J, Dong Y, et al. Expression profile analysis identifies a two-gene signature for prediction of head and neck squamous cell carcinoma patient survival. J Cancer Res Ther. 2018;14(7):1525–1534. doi: 10.4103/jcrt.JCRT_557_18. [DOI] [PubMed] [Google Scholar]
- 56.Zhao F, Olkhov-Mitsel E, Kamdar S, Jeyapala R, Garcia J, Hurst R, et al. A urine-based DNA methylation assay, ProCUrE, to identify clinically significant prostate cancer. Clin Epigenetics. 2018;10(1):147. doi: 10.1186/s13148-018-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. Arch Pathol Lab Med. 2014;138(2):241–256. doi: 10.5858/arpa.2013-0953-SA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6(6):692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 59.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012;40(Web Server issue):W478–W483. doi: 10.1093/nar/gks402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of all the biological processes enriched for the 71 differentially methylated genes between responder and non-responder patients according to the Gene Ontology analysis (DOCX 32 kb)
Thirty-five differentially methylated CpGs between responders and non-responders group selected from 450k array (delta value ≥ 0.2) corresponding to 23 genes located in promoter and island/shore (PPT 172 kb)
Eleven differentially methylated CpGs, corresponding to 11 genes, showed significant methylation differences between non-responder and responder patients: 6 genes (LOC641518; LEF1; HOXA5; EVC2; CDKL2; TLX3) presented a methylation increase in non-responders group vs responders, and 5 genes (ZFHX4; LOC100192378; FERD3L; CHL1; TRIP10) decreased methylation level in non-responder patients compared to those who responded to NAC treatment (PPT 225 kb)
CpGs studied by pyrosequencing in the DC and in the VC to validate methylation in the candidate genes identified in the 450k array (Illumina). In bold, CpGs from 450k array. Normal type, consecutive CpGs (PPT 140 kb)
Representation of the pathway interaction network of FERD3L and TRIP10 with other genes using Pathway Commons. FERD3L and TRIP10 are able to interact with different genes that have shown to be implicated in cancer drug resistance (PPT 452 kb)
Mean differences in methylation levels according to clinicopathological prognostic factors in both cohorts (DC+VC). cT, clinical tumor size; cN, clinical nodule affectation (PPTX 48 kb)
Clinical inclusion and exclusion criteria followed to select TNBC patients for the methylation study (PPT 89 kb)
Sequence of primers used by pyrosequencing in the validation assay of candidate genes obtained from 450k array (PPT 143 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.