

Abstract
Objectives
We sought to investigate the tissue specificity of drug sensitivities in large-scale pharmacological studies and compare these associations to those found in drug clinical indications.
Materials and Methods
We leveraged the curated cell line response data from PharmacoGx and applied an enrichment algorithm on drug sensitivity values’ area under the drug dose-response curves (AUCs) with and without adjustment for general level of drug sensitivity.
Results
We observed tissue specificity in 63% of tested drugs, with 8% of total interactions deemed significant (false discovery rate <0.05). By restricting the drug-tissue interactions to those with AUC > 0.2, we found that in 52% of interactions, the tissue was predictive of drug sensitivity (concordance index > 0.65). When compared with clinical indications, the observed overlap was weak (Matthew correlation coefficient, MCC = 0.0003, P > .10).
Discussion
While drugs exhibit significant tissue specificity in vitro, there is little overlap with clinical indications. This can be attributed to factors such as underlying biological differences between in vitro models and patient tumors, or the inability of tissue-specific drugs to bring additional benefits beyond gold standard treatments during clinical trials.
Conclusion
Our meta-analysis of pan-cancer drug screening datasets indicates that most tested drugs exhibit tissue-specific sensitivities in a large panel of cancer cell lines. However, the observed preclinical results do not translate to the clinical setting. Our results suggest that additional research into showing parallels between preclinical and clinical data is required to increase the translational potential of in vitro drug screening.
BACKGROUND
Large projects such as the Cancer Genome Atlas1 and the International Cancer Genome Consortium2 have enabled the comprehensive characterization of molecular aberrations in multiple cancer types. The collection of mutations, copy number variations, gene expressions, and other features enables molecularly based patient stratification across diverse tumor types, potentially creating a shift from the traditional classification based on tissue type.3–5 However, tumors with similar genomic aberrations may respond differently to cytotoxic and targeted therapies, suggesting that tissue-of-origin is unlikely to be supplanted by molecular stratification.6
Testing drug potency in large populations of patients with different cancer types is an expensive and lengthy process.7 Cancer cell lines provide a safe and cost-efficient method by which to measure drug response in multiple cancer types.8 However, translation of these preclinical findings in animal studies9,10 and clinical settings11 is complex, as cancer cell lines may differ from the patient tumors they originate from.12,13 This discrepancy has several causes. Repeatedly culturing cell lines allows for the potential acquisition of genomic aberrations, causing the cell lines to diverge from their initial samples.14 In addition, mislabeling, simple clerical mistakes in cell line annotations, and cross-contamination can also cause skewing of drug screening results.15–17 Despite these drawbacks, cell lines are the only model systems currently enabling high-throughput drug screening and will therefore remain the model of choice for drug development and biomarker discovery.18–23
In a recent paper investigating a pharmacogenomic dataset of 59 cell lines (NCI60), Jaeger et al.24 observed that drugs designed for specific tissue types, such as lapatinib for breast cancer, had similar activity across all tested tissue types, rather than unique sensitivity patterns for targeted tissue types. Despite the small number of cell lines in NCI60, the authors concluded that cancer-specific drugs do not show higher efficacy in cell lines representing the tissue of interest, raising doubts about the relevance of in vitro screening for drug discovery and repurposing. If the results of this seminal study were generalized to a larger panel of cell lines, this would call for more curation of established cell lines to verify their tissues of origin, and for generation of new cell lines or organoids freshly derived from patients as better models for high-throughput drug screening.10,25–27
The recent release of multiple large-scale pharmacogenomic datasets enables analysis of sensitivities of thousands of cell lines to hundreds of drugs.18–21,23 Subsequent evaluation of these datasets, however, found only moderate inter-laboratory concordance in the drug response phenotypes,20,28–31 highlighting the need for meta-analysis of these complex yet valuable studies.32 Such meta-analysis is hindered by the lack of standardization in cell line and drug identifiers. We addressed this issue by developing the PharmacoGx platform, which provides a computational system to allow unified processing of pharmacogenomic datasets curated with standard cell line and drug identifiers.33
OBJECTIVES
We sought to investigate the tissue specificity of drug sensitivities in large-scale pharmacological studies and compare these associations to those found in drug clinical indications using our compendium of curated cell line response data. We also sought to identify novel tissue-drug associations, which may present new avenues for drug repurposing.
SIGNIFICANCE
While there is strong evidence that tissue context significantly impacts therapy response in the clinical setting,6 tissue specificity of drug sensitivity in immortalized cancer cell lines remains unclear, with contradictory reports.23,24 Our meta-analysis of 732 experimental and approved drugs screened in up to 1527 unique cancer cell lines originating from 20 different tissue types indicates that tissue of origin is strongly predictive of drug response in vitro. However, we found that, except for a few drugs, these preclinical associations did not concur with results from clinical trials, calling for further investigations of the relevance of cancer cell lines for drug sensitivities in specific tissue types.
MATERIALS AND METHODS
The overall analysis design is represented in Figure 1 .
Figure 1.

Schematic representation of data input and analysis pipeline.
Pharmacogenomic datasets
We curated the 4 largest pharmacogenomic datasets within our PharmacoGx platform33: the Cancer Cell Line Encyclopedia (CCLE),18 the Genomics of Drug Sensitivity in Cancer (GDSC1000),19,23,34 the Cancer Therapy Response Portal (CTRPv2),21,35 and the Genentech Cell Line Screening Initiative20 (Table 1). Cell lines were annotated using the Cellosaurus annotation database,36 while drugs were annotated using SMILES structures,37 PubChem IDs,38 and InChiKeys.39 All curated data were stored as PharmacoSet objects within our PharmacoGx platform (version 1.4.3).33
Table 1.
Characteristics of the pharmacogenomic datasets
| Pharmacogenomic datasets | CCLE | GDSC1000 | CTRPv2 | gCSI |
|---|---|---|---|---|
| No. of cell lines | 1061 | 1124 | 887 | 410 |
| No. of tissue types | 24 | 36 | 23 | 23 |
| No. of drugs | 24 | 251 | 545 | 16 |
| No. of drug dose-response curves | 11 670 | 225 480 | 395 263 | 6455 |
| Pharmacological Assay | CellTiter Glo | Syto60 | CellTiter Glo | CellTiter Glo |
| Data source | broadinstitute.org/ccle/ | cancerrxgene.org/ | broadinstitute.org/ctrp/ | research-pub.gene.com/gCSI-cellline-data |
| Reference | 18 | 23 | 21 | 20 |
Tissue of origin of cancer cell lines
We used the Catalog of Somatic Mutations in Cancer nomenclature to consistently annotate cancer cell lines with their tissues of origin.40 Tissues with <15 cancer cell lines were removed in each dataset to ensure sufficient sample numbers for subsequent analysis.
Drug sensitivity
To ensure consistent evaluation of drug sensitivity, we used our PharmacoGx platform to reprocess the drug dose-response curves in our compendium of pharmacogenomic datasets.33 All dose-response curves were fitted to the equation
where y = 0 denotes death of all cancer cells within a sample, y = y(0) = 1 denotes no effect of the drug dose on the cancer cell sample, EC50 is the concentration at which viability is reduced to half of the viability observed in the presence of an arbitrarily large concentration of drug, and HS (Hill slope) is a parameter describing the cooperativity of binding. HS < 1 denotes negative binding cooperativity, HS = 1 denotes noncooperative binding, and HS > 1 denotes positive binding cooperativity. The parameters of the curves were fitted using the least squares optimization framework. This fitting normalizes drug response data, reducing the effects of drug-dependent variables such as dosage and differences in administration. We used the area above the dose-response curve (AUC ∈ [0,1]) to quantify drug sensitivity across cell lines, as AUC is always defined (as opposed to IC50) and combines the potency and efficacy of a drug into a single parameter.41 In this work, high AUC is indicative of sensitivity to a given drug.
To adjust for the general level of drug sensitivity of each cell line, we corrected the AUC values using the approach proposed by Geeleher et al.42 Briefly, to correct the AUC values for drug d, the principal component of AUC values for the 25% least correlated drugs was computed for each cell line and subtracted from the original AUC values.
Tissue specificity of drug sensitivity
Identification of drug-tissue associations using enrichment analysis
For each drug, cell lines were first ranked based on their drug sensitivity (original and adjusted AUC values, separately) in each dataset. We then adapted the gene set enrichment analysis43 implemented in the piano package44 to test whether this ranked list was enriched in sensitive cell lines belonging to specific tissue types (Supplementary Figure S1). Our tissue enrichment analysis (TEA) therefore allowed us to compute the significance of the association between each tissue and drug sensitivity using 10 000 cell line permutations in the tissue set for each drug separately. It is worth noting that TEA compares the tissue-specific distributions of drug sensitivity data (AUC) and, as such, is not restricted to detection of the largest mode and lowest variance of AUC values. However, TEA is a conservative approach, as drugs exhibiting high sensitivity for all tissue types will not yield any significant drug-tissue associations, although they may show therapeutic effects in multiple cancer types.
Meta-analysis of drug-tissue associations
Applying TEA to each dataset generates a set of P values for each drug-tissue association. These P values were combined using the weighted Z method45 implemented in the combine.test function of our survcomp package (version 1.24.0).46 Weights were defined as the number of cell lines in a given tissue type in each dataset from which the P value has been computed. These combined P values were subsequently corrected for multiple testing using the false discovery rate procedure47 for all drugs. To focus on the drug-tissue associations that are reproducible across 2 or more datasets, the associations found in only 1 dataset were discarded from further analysis.
Predictability of significant drug-tissue associations
To estimate the predictive value of tissue t for sensitivity to drug d, we created a binary variable b set to 1 for cell lines belonging to tissue t and 0 otherwise. We then assessed the predictive value of the variable b by computing the concordance index48 between b and the adjusted AUC values, as implemented in the Hmisc package (version 4.0.2). Drug-tissue associations with concordance index ≥0.65 are considered predictable.
Clinical drug-tissue associations
Known clinical drug applications were mined from DrugBank (version 5.0)49 using the XML R package (version 3.98‐1.5). We downloaded all the drug entries in DrugBank (www.drugbank.ca/releases/5‐0‐6/downloads/target-all-uniprot-links) to uniquely map each drug to a unique DrugBank identifier. These identifiers can be appended to the URL www.drugbank.ca/drugs/ to get the web page showing the drugs of interest, which we then scraped using the XML R package to extract the corresponding clinical indications, an example indication being “Metastatic Non-Small Cell Lung Cancer.” Anticancer drugs were selected by matching their clinical indications to the list of cancer terms provided in Supplementary File S1. The list of anticancer drugs was then restricted to clinical indications matching the tissue types present in PharmacoGx (Supplementary File S2).
Comparison of drug-tissue associations between preclinical and clinical settings
To test whether drug-tissue associations extracted from clinical indications were recapitulated in vitro, we compared the sets of preclinical and clinical associations by restricting our analysis to the associations tested in our meta-analysis of the pharmacogenomic data. We visualized the associations observed in preclinical or clinical settings or both as a network with colored edges in a Circos plot.50 The MCC51 was used to quantify the level of concordance between preclinical and clinical drug-tissue associations, and the significance was computed using a permutation test as implemented in the PharmacoGx R package.33
Research reproducibility
This study complies with the standards of research reproducibility published by Sandve et al.52 The datasets are freely available through our PharmacoGx platform.33 The code to replicate the analysis results, figures, and tables is open access and available on GitHub (github.com/bhklab/DrugTissue). In addition, we have set up a Docker virtual environment53 online with all required R packages and tools preinstalled to facilitate reproduction of the study results. Detailed descriptions of the software environment and the main steps to replicate the figures and tables are provided in Supplementary Information.
RESULTS
Given the increasingly prominent use of high-throughput in vitro testing in biomedical research, we sought to test whether cancer cell lines originating from specific tissues responded differently to a large set of cytotoxic and targeted therapies. Such associations between drugs and tissues based on in vitro sensitivity data can be derived based on 2 pharmacological aspects: (1) the associated tissues are enriched in cell lines specifically sensitive to the drug of interest, while other tissues are not, or (2) the cell line in an associated tissue could be highly sensitive to most of the drugs, and the association is therefore not limited to the drug of interest. While discriminating between these 2 categories of drug-tissue associations is difficult in the clinical setting, such discrimination can be made using preclinical model systems screened with a large number of drugs, as is the case for the pharmacogenomic datasets used in this study. We designed an analysis pipeline to identify drug-tissue associations in each category and assessed the overlap of these associations with the clinical indications provided in the DrugBank database.
We collected and curated the 4 largest pharmacogenomic datasets published to date, namely CCLE, GDSC1000, CTRPv2, and gCSI (Table 1), and integrated them into our PharmacoGx platform.33 These datasets contain 732 drugs, 1527 cancer cell lines, and 20 tissue types represented by at least 15 cell lines across all datasets (Figure 2 A and Table 1). Importantly, our curation28,31,33 revealed that these studies investigated many identical cell lines and drugs, including 303 cell lines and 3 drugs – erlotinib, paclitaxel, and crizotinib – screened in all 4 datasets (Figure 2B and Supplementary File S3).
Figure 2.

Composition and overlap of our compendium of pharmacogenomic datasets. (A) Number of cell lines representing each tissue type with respect to their source dataset. Tissue types represented by <5 cell lines in a given dataset were removed for the dataset. (B) Overlap for drugs, cell lines, and tissue types across datasets.
We leveraged our compendium of pharmacogenomic datasets to identify statistically significant drug-tissue associations in vitro using our TEA (Supplementary Figure S1). To control for the general level of drug sensitivity of each cancer cell line, we used the approach recently proposed in42 and adjusted the drug sensitivity data (AUC) accordingly. As previous studies reported that cell lines originating from the hematopoietic and lymphoid tissue are highly sensitive to chemical perturbations,54,55 we discarded this tissue from subsequent analyses to avoid bias in our enrichment analysis. TEA was performed with original and adjusted AUC values for each drug-tissue association (Supplementary File S4). Given the high level of noise in the drug sensitivity data,20,28,29,31 we restricted our analysis to the set of drugs and tissues that were assessed in at least 2 datasets to focus on the associations that are reproducible across datasets. This filtering resulted in a set of 85 drugs and 18 tissue types Supplementary File S4). Out of these 85 drugs, we found that 63% (54) yielded significantly higher sensitivities in at least 1 tissue type, with 8% of all the drug-tissue associations assessed in our study (170/2226) being significant (false discovery rate <5%; Supplementary File S4). Among the drug-tissue associations identified in vitro, we found targeted therapies associated with the tumor types enriched for the corresponding drug target. For example, erlotinib, which targets the epidermal growth factor receptor (EGFR),56,57 is associated with non–small-cell lung cancer, where mutations in EGFR are frequent.58 Moreover, we observed an association between breast cancer and lapatinib, a dual tyrosine kinase inhibitor that interrupts the HER2/neu and EGFR pathways commonly used in HER2-positive breast cancer.59 The association of imatinib with the large intestine is another example of concordance between in vitro associations and clinical indications, as imatinib is widely used to treat gastrointestinal stromal tumors.60 These results support the relevance of our TEA for discovery of drug-tissue associations in a preclinical setting that are potentially relevant for clinical use.
We investigated whether these significant associations in vitro were uniformly distributed across tissue types. Skin and small-cell lung cancer had the largest numbers of associated drugs, totaling >21% of the significant interactions (Figure 3 ). Interestingly, when controlling for the general level of drug sensitivity of each cell line, the majority of tissue types lost their association with drugs (stomach, esophagus, central nervous system, non–small-cell lung cancer, autonomic ganglia, bone, and soft tissues), suggesting that these cell lines undergo a nonspecific response to chemical perturbations. On the other hand, kidney, breast, upper aerodigestive tract, large intestine, small-cell lung cancer, and skin tissues were associated with drugs only when drug sensitivity was adjusted, indicating that their response to (class of) therapeutic compounds is more specific. There was no significant correlation between the number of significant drug-tissue associations and the number of cell lines in each tissue type (Spearman correlation coefficient ρ = 0.006, P = .78, although TEA controls for the size of tissue sets during the permutation testing procedure).
Figure 3.
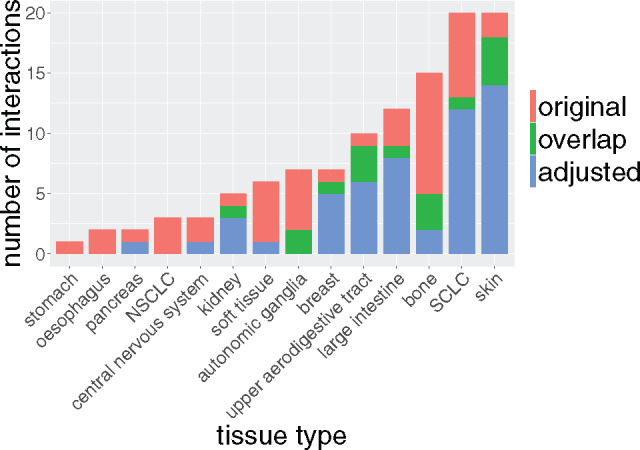
Distribution of in vitro drug-tissue associations. Number of significantly associated drugs for each tissue type in our compendium.
Although our meta-analysis leverages the 4 largest pharmacogenomic studies published to date, these datasets vary in terms of the number of drug dose-response curves actually measured (Table 1). We therefore assessed which dataset contributed the most to the discovery of statistically significant in vitro drug-tissue associations. As expected, the 2 largest datasets, GDSC1000 and CTRPv2, contributed several times more associations than gCSI and CCLE (Figure 4 ). Importantly, a substantial proportion of associations were not significant in each individual dataset but were selected during the meta-analysis phase based on their consistent trend to significance (Figure 4). These results support the benefit of combining multiple pharmacogenomic datasets in a meta-analysis framework.
Figure 4.

Number of in vitro drug-tissue associations in each pharmacogenomic dataset and meta-analysis. The associations that are significant in a dataset and in the meta-analysis are in blue. The associations found significant in a dataset but not selected after meta-analysis are in red. The associations found nonsignificant in a dataset but ending up selected after meta-analysis are in green.
Given the significant tissue specificity of most drugs in vitro, we sought to assess whether these associations were consistent with clinical observations regarding the efficacy of drugs in specific tissue types. For drugs to be considered for further clinical testing, they must yield sufficient growth inhibition in a subset of preclinical models. We therefore selected drug-tissue associations where at least 25% of the cell lines exhibited a minimum level of sensitivity (AUC > 0.2) and where the drug had at least 1 clinical indication for treatment of cancer, as extracted from DrugBank (see Materials and Methods section). This selection resulted in a set of 10 drugs significantly associated with 9 tissues in vitro (Supplementary File S5). We further assessed how well drug sensitivity could be predicted from the associated tissue. We computed, for each drug-tissue association, the concordance index between the binary value representing the tissue of interest and drug sensitivity (Supplementary File S5). In 70 drug-tissue associations (52%), the tissue was predictive of drug sensitivity (concordance index ≥ 0.65). We then extracted clinical indications for the set of anticancer drugs from DrugBank49 and identified 5 drug-tissue associations that were consistent across the preclinical and clinical settings (Table 2 and Figure 5 ). However, the observed overlap was weak at best, as this represents a very small set of associations (MCC = 0.0003, P > .10; Supplementary Figure S2A and Table 2). We also compared our results with the drug-tissue associations reported by Jaeger et al.24 but only found a small overlap (Supplementary Figure S2B)
Table 2.
List of drug-tissue associations conserved across in vitro and clinical settings
| Tissue | Drug | In vitro FDR | In vitro efficacy | In vitro predictability | DrugBank |
|---|---|---|---|---|---|
| Non–small-cell lung cancer | Erlotinib | 0.011 | 0.18 | 0.60 | DB00530 |
| Breast | Lapatinib | 0.009 | 0.62 | 0.58 | DB01259 |
| Soft tissue | Etoposide | 0.006 | 0.43 | 0.64 | DB00773 |
| Skin | Etoposide | 1.3e-8 | 0.91 | 0.71 | DB00773 |
| Large intestine | Imatinib | 0.0002 | 0.56 | 0.67 | DB00619 |
In vitro FDR: false discovery rate computed from the x for all drug-tissue associations investigated in our study; In vitro efficacy: upper quartile of the AUC distribution for the drug of interest in the associated tissue type (≤0.2 indicative of lack of in vitro efficacy); In vitro predictability: concordance index of the binary value representing the tissue in the association, testing how well the associated tissue can predict drug sensitivity; DrugBank: link to the DrugBank database for the drug of interest.
Figure 5.
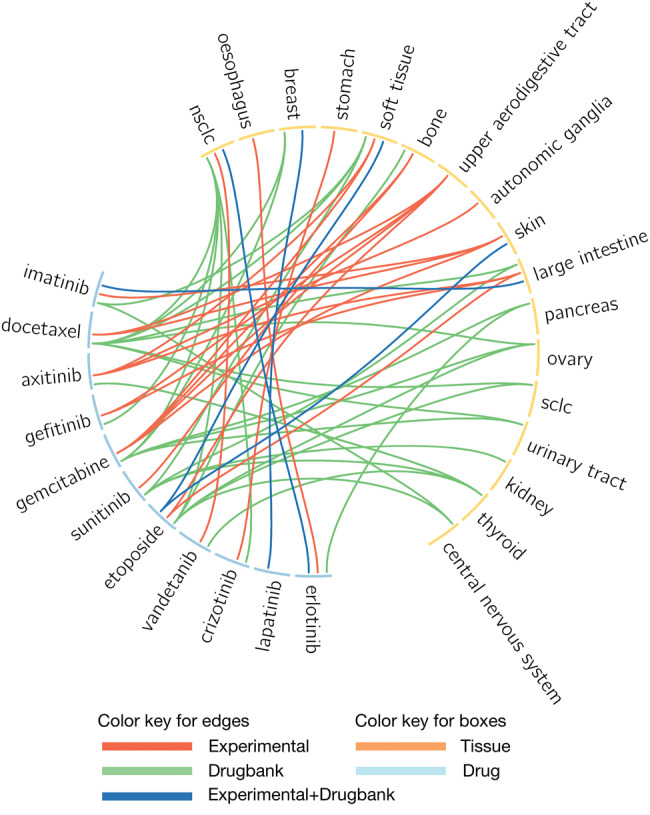
Circos plot representing the significant associations for drugs with clinical trial evidence. Light blue and orange boxes represent drugs and tissues, respectively. Red lines represent drug-tissue associations observed only in vitro (referred to as experimental). Pink lines indicate experimental relationships with no clinical relevance. Green lines indicate a clinical application not recognized in preclinical analysis. Blue lines indicate in vitro drug-tissue associations supported by clinical indications.
DISCUSSION
One of the main challenges in precision cancer medicine is to select drugs likely to yield responses for individual patients. Most of the current treatment regimens for cancer are based on the tissue of origin, as therapies are being designed for specific tissues.6,61 Recent high-throughput in vitro drug screening studies investigating large panels of cancer cell lines from multiple tissues18–21,23 provide a unique opportunity to assess the association between drug sensitivity and tissue types.61 However, it remains unclear to what extent cancer cell lines originating from different tissue types respond to a variety of cytotoxic and targeted drugs.24,29,54,55,62,63 If these drug-tissue associations recapitulated the differential drug response across tissues observed in the clinic, this would open a new avenue of research for tissue-based drug repurposing. In this study, we addressed this issue in the largest meta-analysis of pan-cancer in vitro drug screening data to date.
Our large compendium of drugs and cancer cell lines, combined with our TEA, allowed us to identify a large number of in vitro drug-tissue associations that are reproducible across independent datasets (113 associations; Figure 4 and Supplementary File S5). Our results indicate that the majority of our diverse set of drugs (71%) yielded higher sensitivity in at least 1 tissue type. Our meta-analysis shed new light on the recent controversy regarding the tissue specificity of drug sensitivity screens, where studies have reported substantial tissue-specific drug response,54,55 and the contrary.24 This apparent contradiction is partly due to the lack of a consensus definition of tissue specificity. In our study and the previous work from Klijn et al.54 and Gupta et al.,55 tissue specificity is defined as an association between drug sensitivity and any tissue type, while Jaeger et al.24 only considered associations with the tissues the drugs were developed for. The latter set of associations is therefore a subset of all drug-tissue associations that can be identified in vitro. Using the broader definition of tissue specificity combined with a meta-analysis of multiple pharmacogenomic datasets allowed us not only to identify more drug-tissue associations, but also to discard the drug-tissue associations that were not consistent across multiple datasets, increasing the robustness of our results.
While the tissue specificity of drug sensitivity in vitro is relevant for drug development in the preclinical setting, its translational potential in the clinical setting remains unclear. In this regard, the study from Jaeger et al.24 was seminal, as the authors compared in vitro drug response patterns to clinical observations in breast, colorectal, and prostate cancer and found no concordance. Given that our results indicate strong tissue specificity of in vitro drug sensitivity, we tested the concordance of preclinical and clinical observations in 9 tissue types. Although we found 5 drug-tissue associations that were both significant in vitro and approved for clinical use (Table 2), we found no significant global overlap in drug-tissue associations between the preclinical and clinical settings (Supplementary Figure S2). One possible explanation for the lack of overlap is the failure of some drugs to progress beyond clinical trials due to a lack of additional benefits beyond the gold standard treatments in clinical settings or unforeseen side effects in patients. In addition, discrepancies between cell lines and patient tumors because of underlying biological and experimental factors could cause further disconnects, leading to the observed lack of global overlap. Ultimately, concurring with Jaeger et al.,24 our results call into question the translational potential of the in vitro results.
This study has several potential limitations. First, we and others have shown that pharmacological profiles are not consistent across studies for all drugs.20,28,29,31 To mitigate this important issue, we implemented a meta-analysis framework integrating 4 large pharmacogenomic datasets and limited our study to the drug-tissue associations assessed in at least 2 independent studies to improve reproducibility across datasets. Second, the annotation of cell lines is problematic, as these lines can be misidentified15 and there exists no standard ontology for their metadata.16 We leveraged our PharmacoGx platform33 to check the DNA identity of the cancer cell lines in each dataset31 and annotate these cell lines using the Catalog of Somatic Mutations in Cancer40 and Cellosaurus36 resources. Although these resources provide valuable metadata regarding all the cell lines investigated in this study, there is no consensus regarding tissue annotations. We recognize that alternative tissue nomenclature may affect the results of our study. Our analysis provides a foundation for further exploration of this important question. Lastly, we relied solely on DrugBank49 to extract known clinical indications for the anticancer drugs in our study. Our results could be further improved by mining other databases and manually curating the scientific literature and clinical trial databases, such as ClinicalTrials.gov, although such an analysis is beyond the scope our study.
We have come to recognize that cancer cell lines do not fully recapitulate the molecular features of patient tumors they originate from,11,64 which may hinder the translation of in vitro drug development to clinical settings.63,65–68 It is hoped that large panels of cancer cell lines will enable faithful representation of the molecular diversity observed in patient tumors.18,19,23 However, recent studies have identified cell lines exhibiting molecular phenotypes that are not observed in patients,12,13 casting doubt on the relevance of these model systems for biological investigation and drug screening. Moreover, there is no consensus regarding the experimental protocols used in large-scale in vitro drug screening studies,32 with drugs being tested using different pharmacological assays and concentration ranges, increasing heterogeneity across datasets. Another fundamental problem in cancer cell line studies is the lack of a standard nomenclature to uniquely annotate cell lines to their tissue of origin,16,69,70 even though ontologies are under active development.36,71 Lastly, cancer cell lines lack the tumor microenvironment, which has recently been shown to have a substantial effect on drug response and resistance.72,73 For example, a drug that might show sensitivity in a brain cancer cell line might be completely ineffective in vivo due to the blood-brain barrier, or a drug effective against liver cancer cell lines might be irrelevant to patients due to fundamental differences in metabolism. Patient-derived organoids and xenografts are new models of choice for drug screening, and their usage might alleviate the current limitations of cancer cell lines.9,10,25,27 These are key factors that are likely to contribute to the discrepancy between preclinical and clinical observations highlighted in this study. Although our meta-analysis provides the largest repository of in vitro drug-tissue associations to date, our results call for further investigations to improve the translational potential of cancer cell lines.
CONCLUSION
Our meta-analysis of pan-cancer in vitro drug screening datasets indicates that most approved and experimental drugs exhibit tissue-specific sensitivities in a large panel of cancer cell lines. However, it is equally clear that the preclinical results do not translate to the clinical setting, as the vast majority of in vitro drug-tissue associations are not recapitulated in clinical trials. Our results suggest that additional research in showing parallels between preclinical and clinical data is required to increase the translational potential of in vitro drug screening.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Drs Samira Jaeger and Patrick Aloy for their constructive and insightful feedback on our study. We would also like to thank the editor and the reviewers, whose suggestions significantly improved our analysis pipeline and subsequent results.
FUNDING
This study was conducted with the support of the Cancer Research Society and the Ontario Institute for Cancer Research, through funding provided by the government of Ontario. SAAT was supported by a Connaught International Scholarship. ZS was supported by the Cancer Research Society (Canada). NE-H was supported by the Ministry of Economic Development, Employment and Infrastructure, and the Ministry of Innovation of the government of Ontario. BHK was supported by the Gattuso-Slaight Personalized Cancer Medicine Fund at Princess Margaret Cancer Centre and the Canadian Institutes of Health Research.
COMPETING INTERESTS
The authors declare no competing financial interests.
CONTRIBUTIONS
FY, ZS, and SAAT contributed equally to this work. FY, ZS, and SAAT wrote the code, performed the analysis, and interpreted the results. ZS, PS, and MF collected and fitted the drug dose-response curves. NE-H curated the cell lines and drug annotations. FY and VSKM performed the TEA. BHK supervised the study. FY, SAAT, and VSKM wrote the first version of the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material is available at Journal of the American Medical Informatics Association online.
References
- 1. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;4557216:1061–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. International Cancer Genome Consortium, Hudson TJ, Anderson W, et al. . International network of cancer genome projects. Nature. 2010;4647291:993–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Z, Zhang S. Tumor characterization and stratification by integrated molecular profiles reveals essential pan-cancer features. BMC Genomics. 2015;16:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang B, Mezlini AM, Demir F, et al. . Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;113:333–37. [DOI] [PubMed] [Google Scholar]
- 5. Hoadley KA, Yau C, Wolf DM, et al. . Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;1584:929–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen RL, Settleman J. From cancer genomics to precision oncology: tissue’s still an issue. Cell. 2014;1577:1509–14. [DOI] [PubMed] [Google Scholar]
- 7. Barlas S. The clinical trial model is up for review: time, expense, and quality of results are at issue, as is the relationship to drug pricing. Pharm Therapeutics. 2014;3910:691–94. [PMC free article] [PubMed] [Google Scholar]
- 8. Macarron R, Banks MN, Bojanic D, et al. . Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 2011;103:188–95. [DOI] [PubMed] [Google Scholar]
- 9. Gao H, Korn JM, Ferretti S, et al. . High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;2111:1318–25. [DOI] [PubMed] [Google Scholar]
- 10. Bruna A, Rueda OM, Greenwood W, et al. . A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;1671:260–74.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gillet J-P, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 2013;1057:452–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sinha R, Schultz N, Sander C. Comparing cancer cell lines and tumor samples by genomic profiles. bioRxiv. 2015:0281591–31. [Google Scholar]
- 14. Hughes P, Marshall D, Reid Y, Parkes H, Gelber C. The costs of using unauthenticated, over-passaged cell lines: how much more data do we need? Biotechniques. 2007;435:575, 577–78, 581–82 passim. [DOI] [PubMed] [Google Scholar]
- 15. Identity crisis. Nature. 2009;4577232:935–36. [DOI] [PubMed] [Google Scholar]
- 16. Yu M, Selvaraj SK, Liang-Chu MMY, et al. . A resource for cell line authentication, annotation and quality control. Nature. 2015;5207547:307–11. [DOI] [PubMed] [Google Scholar]
- 17. Masters JR. False cell lines: the problem and a solution. Cytotechnology. 2002;392:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barretina J, Caponigro G, Stransky N, et al. . The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;4837391:603–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Garnett MJ, Edelman EJ, Heidorn SJ, et al. . Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;4837391:570–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haverty PM, Lin E, Tan J, et al. . Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;5337603:333–37. [DOI] [PubMed] [Google Scholar]
- 21. Seashore-Ludlow B, Rees MG, Cheah JH, et al. . Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;511:1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;610:813–23. [DOI] [PubMed] [Google Scholar]
- 23. Iorio F, Knijnenburg TA, Vis DJ, et al. . A landscape of pharmacogenomic interactions in cancer. Cell. 2016;1663:740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jaeger S, Duran-Frigola M, Aloy P. Drug sensitivity in cancer cell lines is not tissue-specific. Mol Cancer. 2015;141:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van de Wetering M, Francies HE, Francis JM, et al. . Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;1614:933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res. 2014;749:2377–84. [DOI] [PubMed] [Google Scholar]
- 27. Witkiewicz AK, Balaji U, Eslinger C, et al. . Integrated patient-derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Rep. 2016;167:2017–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haibe-Kains B, El-Hachem N, Birkbak NJ, et al. . Inconsistency in large pharmacogenomic studies. Nature. 2013;5047480:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cancer Cell Line Encyclopedia Consortium, Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;5287580:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Safikhani Z, El-Hachem N, Quevedo R, et al. . Assessment of pharmacogenomic agreement. F1000Res. 2016;5:825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Safikhani Z, Smirnov P, Freeman M, et al. . Revisiting inconsistency in large pharmacogenomic studies. F1000Res. 2016;5:2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hatzis C, Bedard PL, Juul Birkbak N, et al. . Enhancing reproducibility in cancer drug screening: how do we move forward? Cancer Res. 2014;7415:4016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smirnov P, Safikhani Z, El-Hachem N, et al. . PharmacoGx: An R package for analysis of large pharmacogenomic datasets. Bioinformatics. 2016;328:1244–46. [DOI] [PubMed] [Google Scholar]
- 34. Yang W, Soares J, Greninger P, et al. . Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basu A, Bodycombe NE, Cheah JH, et al. . An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;1545:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bairoch A. ExPASy-Cellosaurus. Cellosaurus. http://web.expasy.org/cellosaurus/. 2015. Accessed January 26, 2016.
- 37. Anderson E, Veith GD, Weininger D. SMILES, a Line Notation and Computerized Interpreter for Chemical Structures. Duluth, MN: United States Environmental Protection Agency; 1987. [Google Scholar]
- 38. Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37(Web Server issue):W623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heller S, McNaught A, Stein S, Tchekhovskoi D, Pletnev I. InChI-the worldwide chemical structure identifier standard. J Cheminform. 2013;51:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Forbes SA, Beare D, Gunasekaran P, et al. . COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fallahi-Sichani M, Honarnejad S, Heiser LM, Gray JW, Sorger PK. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol. 2013;911:708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Geeleher P, Cox NJ, Huang RS. Cancer biomarker discovery is improved by accounting for variability in general levels of drug sensitivity in pre-clinical models. Genome Biol. 2016;171:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;10243:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Väremo L, Nielsen J, Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;418:4378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J Evol Biol. 2005;185:1368–73. [DOI] [PubMed] [Google Scholar]
- 46. Schröder MS, Culhane AC, Quackenbush J, Haibe-Kains B. Survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;2722:3206–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;571:289–300. [Google Scholar]
- 48. Harrell FE Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;154:361–87. [DOI] [PubMed] [Google Scholar]
- 49. Wishart DS, Knox C, Guo AC, et al. . DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krzywinski M, Schein J, Birol I, et al. . Circos: an information aesthetic for comparative genomics. Genome Res. 2009;199:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;4052:442–51. [DOI] [PubMed] [Google Scholar]
- 52. Sandve GK, Nekrutenko A, Taylor J, Hovig E. Ten simple rules for reproducible computational research. PLoS Comput Biol. 2013;910:e1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Belmann P, Dröge J, Bremges A, McHardy AC, Sczyrba A, Barton MD. Bioboxes: standardised containers for interchangeable bioinformatics software. Gigascience. 2015;4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klijn C, Durinck S, Stawiski EW, et al. . A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;333:306–12. [DOI] [PubMed] [Google Scholar]
- 55. Gupta S, Chaudhary K, Kumar R, et al. . Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Sci Rep. 2016;6:23857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li N, Ou W, Yang H, et al. . A randomized phase 2 trial of erlotinib versus pemetrexed as second-line therapy in the treatment of patients with advanced EGFR wild-type and EGFR FISH-positive lung adenocarcinoma. Cancer. 2014;1209:1379–86. [DOI] [PubMed] [Google Scholar]
- 57. Tanoue LT. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. Yearbook Pulmonary Dis. 2011;2011:93–95. [Google Scholar]
- 58. Paez JG. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;3045676:1497–500. [DOI] [PubMed] [Google Scholar]
- 59. Burris HA., 3rd Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 2004;9(Suppl 3):10–15. [DOI] [PubMed] [Google Scholar]
- 60. Din OS, Woll PJ. Treatment of gastrointestinal stromal tumor: focus on imatinib mesylate. Ther Clin Risk Manag. 2008;41:149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garnett MJ, McDermott U. The evolving role of cancer cell line–based screens to define the impact of cancer genomes on drug response. Curr Opin Genet Dev. 2014;24:114–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen B-J, Litvin O, Ungar L, Pe’er D. Context sensitive modeling of cancer drug sensitivity. PloS One. 2015;108:e0133850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goodspeed A, Heiser LM, Gray JW, Costello JC. Tumor-derived cell lines as molecular models of cancer pharmacogenomics. Mol Cancer Res. 2016;141:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Welsh M, Mangravite L, Medina MW, et al. . Pharmacogenomic discovery using cell-based models. Pharmacol Rev. 2009;614:413–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gillet J-P, Calcagno AM, Varma S, et al. . Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA. 2011;10846:18708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McDermott M, Eustace AJ, Busschots S, et al. . In vitro development of chemotherapy and targeted therapy drug-resistant cancer cell lines: a practical guide with case studies. Front Oncol. 2014;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kamb A. What’s wrong with our cancer models? Nat Rev Drug Discov. 2005;42:161–65. [DOI] [PubMed] [Google Scholar]
- 68. Horvath P, Aulner N, Bickle M, et al. . Screening out irrelevant cell-based models of disease. Nat Rev Drug Discov. 2016;1511:751–69. [DOI] [PubMed] [Google Scholar]
- 69. Almeida JL, Cole KD, Plant AL. Standards for cell line authentication and beyond. PloS Biol. 2016;146:e1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Freedman LP, Gibson MC, Ethier SP, Soule HR, Neve RM, Reid YA. Reproducibility: changing the policies and culture of cell line authentication. Nat Methods. 2015;126:493–97. [DOI] [PubMed] [Google Scholar]
- 71. Sarntivijai S, Lin Y, Xiang Z, et al. . CLO: The cell line ontology. J Biomed Semantics. 2014;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mumenthaler SM, Foo J, Choi NC, et al. . The impact of microenvironmental heterogeneity on the evolution of drug resistance in cancer cells. Cancer Inform. 2015;14(Suppl 4):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;9919:1441–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.